

Abstract
Tenofovir alafenamide fumarate (TAF) is an oral phosphonoamidate prodrug of the HIV reverse transcriptase nucleotide inhibitor tenofovir (TFV). Previous studies suggested a principal role for the lysosomal serine protease cathepsin A (CatA) in the intracellular activation of TAF. Here we further investigated the role of CatA and other human hydrolases in the metabolism of TAF. Overexpression of CatA or liver carboxylesterase 1 (Ces1) in HEK293T cells increased intracellular TAF hydrolysis 2- and 5-fold, respectively. Knockdown of CatA expression with RNA interference (RNAi) in HeLa cells reduced intracellular TAF metabolism 5-fold. Additionally, the anti-HIV activity and the rate of CatA hydrolysis showed good correlation within a large set of TFV phosphonoamidate prodrugs. The covalent hepatitis C virus (HCV) protease inhibitors (PIs) telaprevir and boceprevir potently inhibited CatA-mediated TAF activation (50% inhibitory concentration [IC50] = 0.27 and 0.16 μM, respectively) in vitro and also reduced its anti-HIV activity in primary human CD4+ T lymphocytes (21- and 3-fold, respectively) at pharmacologically relevant concentrations. In contrast, there was no inhibition of CatA or any significant effect on anti-HIV activity of TAF observed with cobicistat, noncovalent HIV and HCV PIs, or various prescribed inhibitors of host serine proteases. Collectively, these studies confirm that CatA plays a pivotal role in the intracellular metabolism of TAF, whereas the liver esterase Ces1 likely contributes to the hepatic activation of TAF. Moreover, this work demonstrates that a wide range of viral and host PIs, with the exception of telaprevir and boceprevir, do not interfere with the antiretroviral activity of TAF.
INTRODUCTION
Tenofovir (TFV), an acyclic nucleotide analog of dAMP, is an antiretroviral agent with activity against HIV-1, HIV-2, and hepatitis B virus (HBV) (1, 2). It contains a stable phosphonic acid moiety and is sequentially phosphorylated by intracellular AMP kinase and nucleoside diphosphate kinase to form the active species, tenofovir diphosphate (TFV-DP) (3). TFV-DP acts as a potent HIV-1 reverse transcriptase (RT) inhibitor through an obligatory chain termination of viral DNA synthesis (4).
The presence of two negative charges on the TFV molecule limits its cellular permeativity and precludes oral administration due to low intestinal absorption. To overcome these limitations, various TFV prodrugs containing lipophilic groups masking the charged phosphonate moiety have been designed. Tenofovir disoproxil fumarate (TDF) (Viread) is an ester prodrug of TFV with increased cellular permeativity and oral bioavailability compared to the parent TFV. Because of its favorable resistance profile and long-term tolerability, TDF therapy is broadly used in both treatment-naive and -experienced HIV-infected patients (5).
Tenofovir alafenamide fumarate (TAF) (formerly GS-7340) is an amidate prodrug of TFV with good oral bioavailability and increased plasma stability compared to TDF (6, 7). TAF exhibits >600-fold-enhanced in vitro antiviral activity against HIV-1 compared to the parent TFV (6, 8). Phase 1b 10-day monotherapy studies in HIV-infected patients demonstrated a higher magnitude of viral suppression at substantially lower doses of TAF compared to TDF. Median HIV-1 RNA levels (copies per milliliter) were reduced by 1.59 and 0.97 log10 for the 25-mg TAF and 300-mg TDF doses, respectively (9). The increased clinical efficacy of TAF correlated with higher concentrations of TFV-DP in peripheral blood mononuclear cells (PBMCs) from treated subjects. At the same time, the reduced dose of TAF relative to TDF resulted in proportionally reduced systemic levels of parent TFV. Subsequently, a phase 2 study assessing TAF in combination with emtricitabine (FTC), elvitegravir (EVG), and the pharmacokinetic enhancer cobicistat coformulated as a single tablet regimen (E-C-F-TAF) demonstrated clinical efficacy similar to that for Stribild (E-C-F-TDF) following up to 48 weeks of therapy in treatment-naive patients (10). Recently published phase III data demonstrated that E-C-F-tenofovir alafenamide provided noninferior virological suppression compared to E-C-F-tenofovir disoproxil fumarate. Furthermore, compared with TDF, TAF showed significantly more favorable effects on renal and bone parameters. These effects were likely related to the markedly lower plasma concentrations of tenofovir reported with tenofovir alafenamide compared to tenofovir disoproxil fumarate (11).
The pharmacological advantages provided by TAF are attributed mostly to its unique activation mechanism, which is distinct from that of TDF. Previous studies have implicated the serine protease cathepsin A (CatA) as a major hydrolase involved in the intracellular activation of TAF in human PBMCs (12, 13). CatA is a lysosomal enzyme with deamidase, esterase, and carboxypeptidase activities (14–17). After TAF penetrates into cells, CatA cleaves the carboxyester bond in the prodrug moiety to release a metastable metabolite, from which the phenol group is eliminated via intramolecular cyclization and hydrolysis to form TFV-Ala conjugate (18, 19). Conversion of the TFV-Ala intermediate to the parent TFV occurs spontaneously due to the acidic pH within the lysosomes (20) (Fig. 1).
FIG 1.

Mechanism of the intracellular activation of TAF.
In this study, we further investigated the role of CatA and other human hydrolases in the intracellular activation of TAF. We knocked down and overexpressed CatA and other human hydrolases to assess their effect on the intracellular activation of TAF. We also examined the effects of various therapeutic viral and host protease inhibitors (PIs) on in vitro CatA-mediated activation of TAF using purified CatA enzyme and determined the effect of selected PIs on the antiretroviral activity of TAF in HIV-infected primary human CD4+ T lymphocytes. Since some patients undergoing antiretroviral therapy might require concomitant treatment with inhibitors of HIV/HCV viral proteases or agents targeting various host serine proteases, such as those prescribed for type 2 diabetes or as anticoagulants, our studies provide an initial assessment of the potential for pharmacological interactions between TAF and therapeutic PIs via their interference with CatA-mediated enzyme activity.
MATERIALS AND METHODS
Materials.
Antibody against human cathepsin A (CatA) and cathepsin X/Z (CatX/Z) were obtained from R&D Systems (Minneapolis, MN). Rabbit antibody against human carboxylesterase 1 (Ces1) and carboxylesterase 2 (Ces2) were custom made by Life Sciences (Grand Island, NY).
Antibody against serine protease 1 (SCPEP1) (catalog no. sc-55754) and vitellogenin-like carboxypeptidase (CPVL) (catalog no. sc-100686) were from Santa Cruz Biotechnology (Dallas, TX). Antibody against paraoxonase 1 (Pon1) (catalog no. ab24261), paraoxonase 2 (Pon2) (catalog no. ab40969), paraoxonase 3 (Pon3) (catalog no. ab42322), and carboxylesterase 3 (Ces3) (catalog no. ab69140) were from Abcam (Cambridge, MA). Expression constructs for CatA, CatX/Z, SCPEP1, CPVL, Pon1, Pon2, Pon3, Ces3, and Ces7 were acquired from Origene (Rockville, MD) (catalog no. SC112902, SC107803, SC124414, SC100686, SC107037, SC119866, SC119958, SC100049, and SC305193).
Expression plasmids for Ces1 and Ces2 were kindly provided by Philip Potter (St. Jude Children's Research Hospital, Memphis, TN). Recombinant Ces1 and Ces2 were supplied by R&D Systems (Minneapolis, MN). CatA was purified from human peripheral blood mononuclear cells using previously published protocol (12).
Compounds.
TAF (GS-7340), TDF (Viread), TFV, cobicistat (GS-9350), GS-9256, GS-9451, MK-5172, faldaprevir (BI-201335), and simeprevir (TMC-435) were synthesized by the Department of Medicinal Chemistry at Gilead Sciences (Foster City, CA). Telaprevir and boceprevir were purchased from Acme Bioscience (Palo Alto, CA). Apixaban and zidovudine (AZT) were purchased from Fisher Scientific (Pittsburgh, PA) and Sigma Chemicals (St. Louis, MO), respectively. Darunavir, atazanavir, lopinavir, ritonavir, argatroban, dabigatran, sitagliptin, and rivaroxaban were each purchased from Toronto Research Chemicals (Toronto, Ontario, Canada).
Overexpression of hydrolases in HEK293T cells.
Human embryonic kidney HEK293T cells (ATCC, Manassas, VA) were seeded into 24-well plates coated with poly-d-lysine (BD Biosciences, San Jose, CA) at a density of 1.5 × 105 cells/well in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transfections were performed the next day with 0.5 μg expression plasmids and 1 μl/well TransIT-293 transfection reagent (Mirus, Madison, WI). Crude cell lysates were prepared 2 days later, subjected to Western blotting, and analyzed on a phosphorimager (GE Healthcare Bio-Sciences, Pittsburgh, PA) to determine the fold increase in expression of each hydrolase relative to the mock-transfected control. Enzymatic activities of the overexpressed hydrolases in crude cellular extracts were monitored as described below.
Quantification of TAF metabolites.
Metabolism of TAF in mock- or hydrolase-transfected HEK292T cells was performed 48 h after transfection. The cells were incubated with 10 μM [3H]TAF (0.5 μCi/ml) for 45 and 90 min in DMEM with 10% FBS. Following incubation, the proteins in medium were precipitated with 4 volumes of ice-cold 100% methanol. The cells were washed with phosphate-buffered saline (PBS) and detached by incubation with trypsin-EDTA in PBS. Trypsin was neutralized with cell culture medium, and the cells were harvested by centrifugation for 5 min at 1,500 rpm (Beckman GPR, 4°C). The cell pellets were washed with ice-cold PBS, and the TAF metabolites were extracted from cells by adding 400 μl of 80% methanol. The cell extracts and precipitated medium were centrifuged at 13,000 × g for 30 min at 4°C, and the supernatants were evaporated under a vacuum and resuspended in 100 μl of buffer A (25 mM phosphate buffer [pH 6] and 5 mM tetrabutylammonium bromide). Aliquots of the cell extracts were analyzed for their 3H radioactivity content following the addition of 5.0 ml Ready Safe scintillation fluid (Beckman Instruments, Fullerton, CA).
The column chromatography was performed using a Prodigy column (5 μm, octadecyl silica 3 [ODS-3], 150 by 4 mm; Phenomenex) on a Waters high-performance liquid chromatography (HPLC) system connected to a Radiomatic Flo-One/beta liquid scintillation detector (Packard series A-500). To separate the TAF metabolites, a gradient elution profile from buffer A (flow rate, 1.0 ml/min at 40°C) to 45% acetonitrile in buffer A for 30 min, followed by a 5-min column wash with 65% acetonitrile in buffer A, was used. The amount of metabolites was calculated from the calibration curve for TAF.
CatA knockdown in HeLa cells.
HeLa cells (5 × 105 per well) were transfected with 75 ng Flexitube gene solution small interfering RNA (siRNA) mixture specific for CatA (Hs_PPGB_2, Hs_PPGB_3, Hs_PPGB_5, and Hs_PPGB_6; Qiagen, Valencia, CA) or with All Stars negative-control siRNA (Qiagen) according to the manufacturer's protocol or were left untreated. At day 2 posttransfection, the crude cell lysates were normalized on the total protein and separated by PAGE. The level of CatA knockdown was analyzed by quantitative immunoblot analysis using a phosphorimager and a serial dilution of recombinant CatA protein of known concentration as a reference standard. At day 3 posttransfection, the cells were incubated with 10 μM [14C]TAF (0.5 μCi/ml) for various periods of time (0, 0.5, 1, 2, and 4 h). The cells were washed 3 times with ice-cold PBS before being harvested. The total level of TAF metabolites in cells was determined using liquid scintillation as described above.
Enzymatic assays.
The rate of hydrolysis of amidate prodrugs of tenofovir with CatA was measured in a reaction buffer containing 25 mM morpholineethanesulfonic acid (MES) (pH 6.5), 100 mM NaCl, 1 mM dithiothreitol (DTT), 0.1% NP-40, and 1 μg/ml enzyme at 37°C. Inhibition of CatA-mediated TAF hydrolysis by protease inhibitors was performed by preincubation of the enzyme with serially diluted inhibitors for 10 min at 37°C. Ces1 and Ces2 (5 μg/ml) were assayed in 50 mM HEPES buffer (pH 7.2). The reactions were initiated by adding substrates to the reaction mixtures to the final concentration of 30 μM. At various time points, 100-μl aliquots were mixed with 200 μl of ice-cold methanol to stop the reactions. The samples were incubated at −20°C for 30 min and spun at 13,000 × g for 30 min at 4°C to remove denatured proteins. The supernatants were evaporated under vacuum, resuspended in 100 μl buffer A (25 mM potassium phosphate [pH 6.0], 5 mM tetrabutylammonium bromide) and injected onto a C18 reverse-phase column (5 μm, 2.1 by 100 mm, octadecyl silica 2 [ODS-2]; Phenomenex, Torrance, CA) equilibrated with buffer A. The substrates and reaction products were eluted using a linear gradient of acetonitrile (0% to 65%, 10 min, 0.25 ml/min) in buffer A.
Primary cell isolation and culture.
Peripheral blood mononuclear cells (PBMCs) were purchased from All Cells Inc. (Alameda, CA) and were obtained from volunteers participating in an Institute Review Board (IRB)-approved donor program under informed consent. Donors were negative for HIV-1, hepatitis B virus, and hepatitis C virus infections. Primary CD4+ T lymphocytes (CD4s) were prepared from donor PBMCs using an Automacs Pro Separator (Miltenyi Biotech, Auburn, CA). CD4s were isolated by negative selection using a magnetically labeled antibody cocktail (CD4+ T cell isolation kit II; Miltenyi Biotech, Auburn, CA) and activated with 1 μg/ml phytohemagglutinin (PHA) (Sigma, St. Louis, MO) and 5 ng/ml interleukin-2 (IL-2) (Roche Diagnostics, Indianapolis, IN) for 48 h in a humidified 37°C incubator containing 5% CO2. The enrichment of CD4s was confirmed by fluorescence-activated cell sorter (FACS) staining with anti-CD4 antibody (BD Biosciences, San Jose, CA).
Antiviral assays.
The antiviral activities of TAF and other phosphonoamidate prodrugs of TFV were determined using a 5-day cytoprotection antiviral assay in MT-4 cells infected with HIV-1 IIIB, as previously described (21). For primary cell studies, activated CD4s were infected for 3 h with vesicular stomatitis virus (VSV) G-pseudotyped HIV-Luc reporter virus (22), washed with complete RPMI medium, and then seeded (2 × 105 cells per well) onto white, solid-bottom 96-well plates (Costar, Corning, NY). Infected CD4s were pretreated with dimethyl sulfoxide (DMSO) or test compounds at their respective clinical maximum concentrations (Cmax) for 1 h before addition of RPMI medium containing serially diluted TAF or TDF. At 3 days postinfection, 100 μl One-Glo reagent (Promega, Madison, WI) was added per well, and the luciferase signal measured using an Envision plate reader (Perkin-Elmer, Waltham, MA). Data analysis was done using XL-fit to calculate the 50% effective concentration (EC50) from an 8-point dose-response curve and 3-fold compound serial dilutions. The fold change in TAF and TDF EC50 was determined in CD4 cells from two independent donors as a ratio of their activity in the presence and absence of tested compounds.
RESULTS
Effect of overexpression of hydrolases on intracellular activation of TAF.
Metabolism of TAF is initiated by hydrolysis of the isopropyl ester in the 5′-phosphonoamidate prodrug moiety, and CatA was indicated to be responsible for this activation step in PBMCs (12). However, other proteases and esterases were also shown to cleave this carboxylester bond in vitro (13). We therefore evaluated the role of liver esterases Ces1, Ces2, Ces3, Ces7, Pon1, Pon2, and Pon3 and ubiquitous lysosomal carboxylesterases CatA, CatZ/X, SCPEP1, and CPVL in the intracellular activation of TAF by transiently overexpressing these enzymes in HEK293T cells. All enzymes were overexpressed by at least 10-fold compared to in the mock-transfected control cells based on quantitative Western blot analysis (data not shown). Total intracellular metabolites were monitored following the incubation of the cells with TAF for 45 or 90 min. In CatA- or Ces1-overexpressing cells, approximately 2- and 5.5-fold more intracellular metabolites were formed than in the mock-transfected control cells, respectively (Fig. 2). Increased levels of the metabolites were found in the medium of the cells overexpressing CatA and Ces1, likely due to efflux of the metabolites (data not shown). The overexpression of the other hydrolytic enzymes did not significantly affect the intracellular levels of the TAF metabolites (Fig. 2). In vitro enzymatic assays confirmed the ability of Ces1, but not of Ces2, to hydrolyze TAF in vitro (data not shown). These results indicate that in addition to CatA, Ces1 contributes to the intracellular metabolism of TAF in liver, where it is abundantly expressed.
FIG 2.
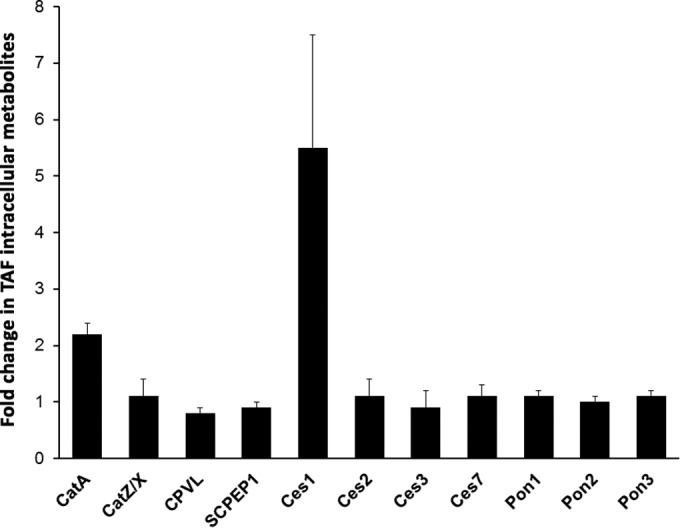
Effect of transient overexpression of hydrolases on TAF metabolism. HEK293T cells were transiently transfected with empty vector or with plasmids containing full-length cDNAs encoding the indicated hydrolases. At 2 days posttransfection, cells were incubated with 10 μM [3H]TAF, and the intracellular concentration of total TAF metabolites was determined by liquid scintillation. The magnitude of each bar represents the mean (± standard deviation) of TAF metabolites observed in cells overexpressing a hydrolase relative to cells transfected with the empty vector as determined from four independent experiments.
Effect of CatA knockdown of on intracellular activation of TAF.
TAF is metabolized approximately 7.5-fold less efficiently in CatA-deficient fibroblasts derived from patients with the genetic disorder galactosialidosis than in normal control fibroblasts (12). We wished to further confirm the importance of CatA in the intracellular hydrolysis of TAF by using RNA interference (RNAi). On day 3 posttransfection, the knockdown of CatA in HeLa cells resulted in a 4-fold reduction of the intracellular levels of TAF metabolites compared to that in cells transfected with control siRNA (Fig. 3). The quantification of the CatA protein level in cells by immunoblot analysis showed that the siRNA knockdown reduced CatA expression approximately 7-fold compared to that in control cells (Fig. 3). Taken together, these data further support the central role of CatA in the intracellular activation of TAF.
FIG 3.
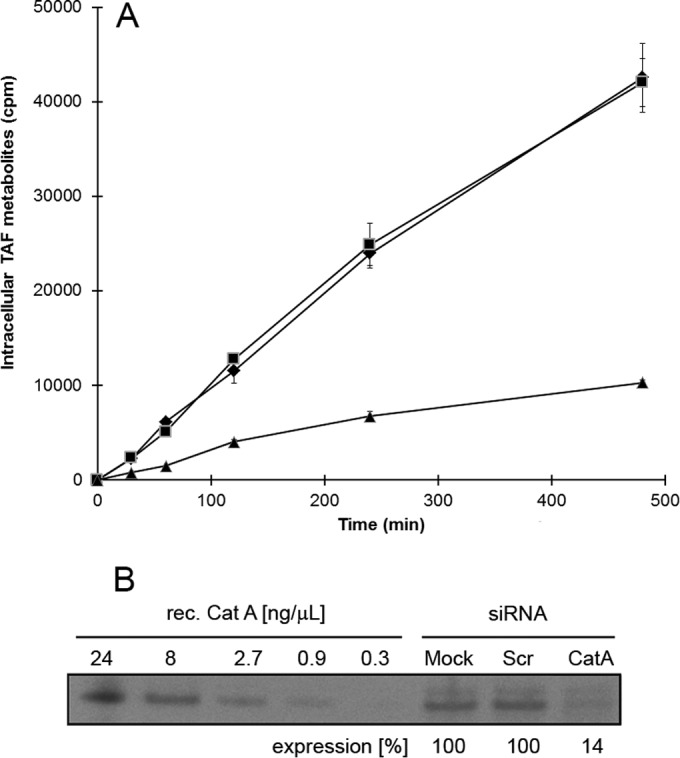
CatA knockdown reduces the intracellular accumulation of TAF metabolites. (A) HeLa cells were mock transfected (◆) or transfected with CatA-specific (▲) or scrambled (■) siRNAs and incubated with 10 μM [3H]TAF at 3 days posttransfection. The intracellular concentration of TAF metabolites in cell lysates was determined by scintillation counting. (B) CatA knockdown efficiency in HeLa cells. At 2 days posttransfection, crude cell lysates were prepared, normalized for total protein content, and analyzed by anti-CatA immunoblotting and phosphorimager analyses. A representative immunoblot from three independent experiments is shown.
Correlation between antiretroviral activity and CatA-mediated hydrolysis of phosphonoamidate prodrugs of TFV.
Phosphonoamidate prodrugs of TFV exist in two different stereochemical configurations due to the presence of a stereocenter at the phosphorus atom. To extend the assessment of the role of CatA in enabling the antiretroviral activity of a wide range of TFV phosphonoamidate prodrugs, we measured the rate of hydrolysis of (S)-stereoisomers of 48 prodrugs of TFV containing structurally diverse amino acids and alcohols in the prodrug moieties and correlated these rates with antiretroviral activity of the prodrugs in HIV-1-infected MT-4 cells. We found a strong correlation (r2 = 0.71, P < 0.001) between the two measured parameters (Fig. 4). Importantly, this correlation was substantially weaker (r2 = 0.34) when corresponding (R)-stereoisomers were included in the analysis (data not shown). This was due to the fact that the (R)-stereoisomers are not CatA substrates but still exhibit antiviral activity about 10-fold lower than that of their corresponding (S) counterparts, indicating divergent metabolic pathways being involved in the intracellular activation of the (S)- and (R)-stereoisomers of TFV amidate prodrugs.
FIG 4.
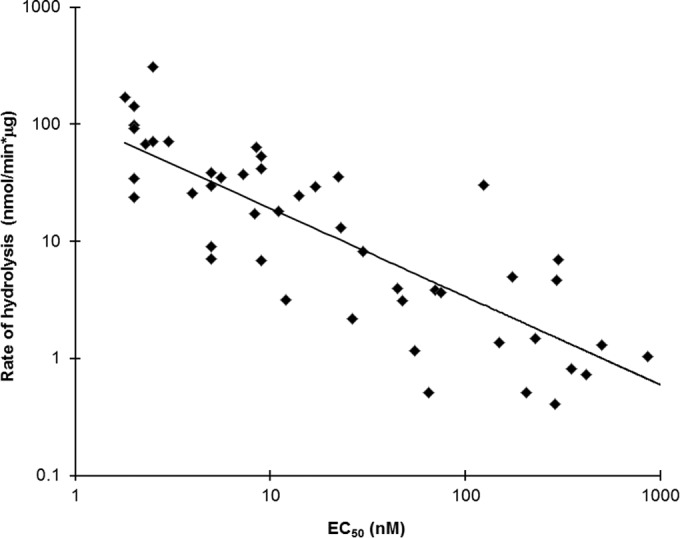
Correlation between CatA-mediated hydrolysis and antiretroviral activity of phosphonoamidate prodrugs of TFV. TAF and a panel of 48 different phosphonoamidate prodrugs of TFV were incubated with CatA to determine the rate of hydrolysis of the prodrugs. The corresponding antiviral activity for each compound was determined from quadruplicate measurements using a 5-day cytopathic assay in HIV-1-infected MT-4 cells. EC50s, determined from at least two independent experiments, represent the mean drug concentration providing 50% protection against the cytopathic effects associated with HIV-1 (IIIb) replication.
Effect of viral and host protease inhibitors on CatA-mediated TAF activation.
Because the proteolytic function of CatA is required for TAF activation, there may be a potential for antagonistic drug-drug interactions between TAF and inhibitors of viral or host proteases of clinical relevance that may be coadministered to patients. We therefore conducted experiments to determine the inhibitory effect of various HIV and HCV protease inhibitors (PIs) and host serine PIs, as well as the pharmacological boosting agent cobicistat, on CatA-mediated hydrolysis of TAF. The HIV PIs darunavir, atazanavir, lopinavir, and ritonavir and the boosting agent cobicistat had no effect on CatA-mediated TAF hydrolysis up to the highest tested concentration of 50 μM (Table 1). This concentration is well above the free and total drug levels at the clinical peak plasma exposure (Cmax) following the administration of the approved doses of each tested drug. Similarly, the HCV PIs simeprevir, faldaprevir, MK-5172, GS-9256, and GS-9451 also showed no effect on the enzymatic activity of CatA up to the highest concentrations tested. On the other hand, HCV PIs telaprevir and boceprevir were found to be potent inhibitors of CatA-mediated hydrolysis of TAF, with 50% inhibitory concentrations (IC50s) of 0.27 and 0.16 μM, respectively (Table 1). These observations are consistent with previously demonstrated inhibition of CatA-mediated hydrolysis of sofosbuvir (formerly PSI-7977) by telaprevir (23). Importantly, the determined IC50s for these two compounds are 6- to 8-fold below their free drug levels at the clinical Cmax, indicating a clinically relevant potential to affect the intracellular metabolism of TAF. Finally, host serine PI classes of compounds commonly used in the clinic, such as inhibitors of factor Xa (apixaban and rivaroxaban), thrombin (argatroban and dabigatran), or DPP4 (sitagliptin), did not interfere with the in vitro CatA-mediated hydrolysis of TAF.
TABLE 1.
Effect of HIV and HCV PIs, cobicistat, and inhibitors of host serine proteases on CatA-mediated activation of TAF in vitroa
| Inhibitor class | Compound | IC50 (μM)b | Cmax (μM)c |
|---|---|---|---|
| HIV PI | Darunavir | >50 | 8.9 |
| Atazanavir | >50 | 6.3 | |
| Lopinavir | >50 | 15.2 | |
| Ritonavir | >50 | 1.3 | |
| HCV PI | Telaprevir | 0.27 ± 0.17 | 5.2 |
| Boceprevir | 0.16 ± 0.02 | 3.3 | |
| Simeprevir | >50 | 13.3 | |
| Faldaprevir | 25 ± 7 | 20.0 | |
| MK-5172 | >50 | 2.5 | |
| GS-9256 | >50 | 10.6 | |
| GS-9451 | >50 | 1.7 | |
| CYP3A | Cobicistat | >50 | 2.2 |
| Factor Xa | Apixaban | >50 | 0.2 |
| >50 | 0.3 | ||
| Thrombin | Argatroban | >50 | 0.4 |
| Dabigatran | >50 | 0.4 | |
| DPP4 | Sitagliptin | >50 | 1.0 |
TAF (30 μM) was incubated with purified CatA in the presence of various protease inhibitors, and the rate of TFV-Ala conjugate formation was monitored by HPLC.
Data represent mean values ± standard deviations from at least 2 independent experiments.
Effect of viral and host protease inhibitors on TAF antiviral activity.
To further confirm the above observations from the CatA enzymatic inhibition assays, we tested the effects of a selected subset of the most relevant PIs on the antiviral activity of TAF in HIV-1-infected primary human CD4+ T lymphocytes. Following the infection with a single-cycle HIV-1 reporter virus, the cells were pretreated with tested PIs at concentrations corresponding to their clinical peak plasma levels (Cmax) before adding serial dilutions of either TAF or TDF. TDF served as a control for CatA-independent prodrug activation. Baseline antiviral activities (mean EC50 ± standard deviation) obtained in the absence of a PI were determined from 6 independent donors for TAF (EC50 = 5.3 ± 1.2 nM) and TDF (EC50 = 2.8 ± 1.1 nM) and were used to calculate fold change EC50s from four independent donors under conditions of PI pretreatment. Consistent with the results obtained in vitro using recombinant CatA enzyme, TAF antiviral activity in CD4+ T lymphocytes was reduced 21-fold and 2.7-fold in the presence of the covalent anti-HCV PIs telaprevir and boceprevir, respectively. In contrast, neither the noncovalent HIV and HCV PIs nor cobicistat or the tested host serine PIs showed any significant effect (≤2-fold change) on the antiretroviral activity of TAF in primary CD4+ T cells. These results further underscore the low potential of pharmacological drug-drug interactions between the vast variety of therapeutic PIs and TAF due to the interference with CatA enzymatic activity.
DISCUSSION
Since first described in early 1990s (24–28), amidate prodrugs of nucleotides have been the most successfully explored class of prodrugs to bypass the first phosphorylation step of nucleosides or to deliver nucleoside phosphonates into cells (29). TAF is the first nucleotide phosphonoamidate prodrug to successfully complete a proof-of-concept clinical efficacy study (30). The clinical candidates GS-9131 and GS-9191, explored for the treatment of HIV and human papillomavirus (HPV), respectively, are other examples of amidate prodrug technology applied to nucleoside phosphonates (31, 32). Besides HIV and HPV, amidates have also been extensively explored as liver-targeted antiviral prodrugs of nucleoside monophosphates for the treatment of HCV, with sofosbuvir being the first FDA-approved amidate prodrug (33).
Because amidate prodrugs are lipophilic cell-permeative compounds, the hydrolysis of the carboxylester bond in the prodrug moieties is a critical event in their intracellular activation. Purified CatA enzyme has been shown to cleave the carboxylester bond of TAF and other nucleotide amidates (12, 13, 23), and CatA-deficient fibroblasts exhibited impaired metabolism of TAF, GS-9131, and GS-9191 (12, 20). Here we provide additional supporting evidence implicating CatA as the major hydrolase responsible for the intracellular activation of TAF. First, we showed that CatA knockdown in HeLa cells results in impaired TAF metabolism. Second, cotreatment of cells with telaprevir, a potent CatA inhibitor, increases EC50s for TAF in HIV-infected CD4+ cells by approximately 20-fold (Tables 1 and 2). Finally, we observed a strong correlation between antiretroviral activity and CatA-mediated hydrolysis of an extensive structurally diverse set of phosphonoamidate prodrugs of TFV.
TABLE 2.
Effect of HIV and HCV PIs, cobicistat, and inhibitors of host serine proteases on TAF antiviral activity in primary human CD4+ T cells
| Inhibitor class | Compound (concn tested, μM)a | EC50, nM (fold change)b |
|
|---|---|---|---|
| TAF | TDF | ||
| HIV PI | Darunavir (8.9) | 4.2 ± 0.8 (0.8) | 2.7 ± 1.0 (1.0) |
| Atazanavir (6.3) | 5.4 ± 2.1 (1.0) | 3.0 ± 1.0 (1.1) | |
| HCV PI | Telaprevir (5.2) | 110.5 ± 51.2 (20.9) | 3.3 ± 1.0 (1.2) |
| Boceprevir (3.3) | 14.2 ± 3.9 (2.7) | 2.6 ± 0.4 (0.9) | |
| Simeprevir (13.3) | 5.2 ± 1.5 (1.0) | 3.2 ± 1.6 (1.1) | |
| CYP3A | Cobicistat (2.2) | 4.4 ± 0.5 (0.8) | 3.4 ± 1.5 (1.2) |
| Factor Xa | Apixaban (0.2) | 10.4 ± 1.2 (2.0) | 4.0 ± 0.6 (1.4) |
| Rivaroxaban (0.3) | 7.2 ± 3.0 (1.4) | 4.2 ± 0.4 (1.5) | |
| Thrombin | Argatroban (1.0) | 8.9 ± 4.7 (1.7) | 4.6 ± 0.4 (1.6) |
| Dabigatran (0.4) | 9.1 ± 4.0 (1.7) | 4.2 ± 1.5 (1.5) | |
| DPP4 | Sitagliptin (1.0) | 8.3 ± 6.7 (1.6) | 3.6 ± 0.1 (1.3) |
The tested inhibitors were used at concentrations corresponding to their clinical peak plasma levels (Cmax).
Baseline TAF and TDF EC50 values (mean ± standard deviation), determined from triplicate infectivity measurements within CD4+ T cell populations from six independent donors, were 5.3 ± 1.2 nM and 2.8 ± 1.1 nM, respectively. The average fold changes in TAF and TDF EC50s were obtained from triplicate measurements in four independent donors after a 1-h pretreatment of infected cells at their clinical Cmax prior to addition of serially diluted TAF or TDF.
It is likely that in addition to CatA, other cellular hydrolases may play a significant role in the metabolic activation of TAF in various tissues and body compartments. In CatA-deficient fibroblasts, some level of TAF metabolism is still observed, and porcine liver carboxylesterase as well as a variety of serine proteases with a preference for alanine in the P1 position of their peptide substrates could hydrolyze TAF in vitro, albeit with dramatically lower efficiency than CatA (12, 13). In vitro enzymatic assays may not necessarily reflect the true intracellular activity of hydrolases due to many factors, including enzyme quality, stability, and/or reaction buffer conditions. Consequently, these biochemical assessments may under- or overestimate the potential role of a given enzyme in the intact cells or tissues. To address these experimental limitations, we tested the effect of overexpressing selected serine carboxylpeptidases and esterases in intact cells on the intracellular metabolism of TAF. Among 11 tested enzymes, only CatA and Ces1 overexpression increased the intracellular metabolism of TAF. Interestingly, the overexpression of CatA in HEK293T cells resulted in a much smaller effect on TAF conversion than the overexpression of Ces1 (Fig. 2). This might be due to the fact that CatA forms a complex with lysosomal neuraminidase and β-galactosidase, which were not coexpressed together with CatA in our experiments (14–17). Ces1 has a restricted tissue expression, with the highest levels being present in the liver (34). While CatA is the main TAF-activating enzyme in most tissues, Ces1 may play an important role in metabolism of TAF in the liver. Indeed, Ces1 has recently been shown to be the main hydrolase responsible for TAF activation in the liver, with a minor contribution coming from CatA (35). Those authors showed an impaired metabolism of TAF in primary human hepatocytes in the presence of bis(4-nitrophenyl)phosphate (BNPP), a selective inhibitor of human carboxylesterase 1. Telaprevir on its own did not change TAF metabolism in these cells; however, a combination of both BNPP and telaprevir further decreased TAF metabolism. Therefore, Ces1 is an important contributor to the enhanced anti-HBV efficacy of TAF compared to the standard 300-mg TDF dose, as seen in a recent 28-day dose-ranging study (36).
Given that TAF has shown clinical efficacy in HIV-infected patients, it might be coadministered with other therapeutically relevant inhibitors of viral and host proteases. To understand the potential for drug-drug interactions due to the interference with CatA enzymatic activity, the in vitro effect of clinically relevant inhibitors of HIV, HCV, and host cell proteases, as well as the boosting agent cobicistat, toward CatA was determined. None of the HIV PIs tested here, or the boosting agent cobicistat, showed any effect on CatA-mediated activation of TAF when tested in an in vitro enzyme inhibition assay. Similarly, the majority of tested HCV PIs, including simeprevir, faldaprevir, MK-5172, GS-9256, and GS-9451, showed little to no inhibition of CatA at levels significantly exceeding their clinical peak plasma levels. In contrast, the covalent HCV PIs telaprevir and boceprevir emerged as potent inhibitors of CatA-mediated TAF activation in vitro. In agreement with these data, the antiviral potency of TAF in CD4+ T lymphocytes was significantly compromised in the presence of telaprevir and boceprevir but remained unchanged in the presence of cobicistat and all the other tested inhibitors of HIV, HCV, and host cell proteases. The inhibitory effect of both telaprevir and boceprevir is likely a consequence of their covalent mechanism of inhibition due to the presence of a ketoamide group in the C-terminal part of these molecules (37, 38). Because CatA is a carboxypeptidase (14–17), telaprevir and boceprevir likely form a covalent bond with the active-site serine residue of the enzyme and function as covalent inhibitors of the enzyme. Hence, coadministration of telaprevir or boceprevir together with TAF in HIV/HCV-coinfected individuals has a potential to adversely affect the intracellular activation and clinical antiviral efficacy of TAF.
In summary, this study expanded the evidence for the central role of CatA in the intracellular activation of TAF. In addition, several experimental approaches were taken to consistently demonstrate a low potential of multiple therapeutically relevant inhibitors of both viral and host proteases to adversely affect the antiretroviral activity of TAF due to the interference with its intracellular activation mediated by CatA.
REFERENCES
- 1.Barditch-Crovo P, Deeks SG, Collier A, Safrin S, Coakley DF, Miller M, Kearney BP, Coleman RL, Lamy PD, Kahn JO, McGowan I, Lietman PS. 2001. Phase I/II trial of the pharmacokinetics, safety, and antiretroviral activity of tenofovir disoproxil fumarate in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother 45:2733–2739. doi: 10.1128/AAC.45.10.2733-2739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan J, Degertekin B, Wong SN, Husain M, Oberhelman K, Anna SF. 2008. Tenofovir monotherapy is effective in hepatitis B patients with antiviral treatment failure to adefovir in the absence of adefovir-resistant mutations. J Hepatol 48:391–398. doi: 10.1016/j.jhep.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Robbins BL, Wilcox CK, Fridland A, Rodman JH. 2003. Metabolism of tenofovir and didanosine in quiescent or stimulated human peripheral blood mononuclear cells. Pharmacotherapy 23:695–701. doi: 10.1592/phco.23.6.695.32189. [DOI] [PubMed] [Google Scholar]
- 4.Suo Z, Johnson KA. 1998. Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-phosphonylmethoxypropyl)adenine. J Biol Chem 273:27250–27258. doi: 10.1074/jbc.273.42.27250. [DOI] [PubMed] [Google Scholar]
- 5.Grim SA, Romanelli F. 2003. Tenofovir disoproxil fumarate. Ann Pharmacother 37:849–859. doi: 10.1345/aph.1C388. [DOI] [PubMed] [Google Scholar]
- 6.Lee WA, He GX, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. 2005. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother 49:1898–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenberg EJ, He GX, Lee WA. 2001. Metabolism of GS-7340, a novel phenyl monophosphoramidate intracellular prodrug of PMPA, in blood. Nucleosides Nucleotides Nucleic Acids 20:1091–1098. doi: 10.1081/NCN-100002496. [DOI] [PubMed] [Google Scholar]
- 8.Lee WA, He G, Mulato A, Delaney W, Eisenberg E, Cihlar T, Xiong S, Miller M, Gill S, Shibata R, Gibbs C. 2002. Abstr 9th Conf Retroviruses Opportun Infect, abstr 384-T. [Google Scholar]
- 9.Ruane PJ, DeJesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, Zhong L, Ramanathan S, Rhee MS, Fordyce MW, Yale K. 2013. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide (TAF) as 10-day monotherapy in HIV-1-positive adults. J Acquir Immune Defic Syndr 63:449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
- 10.Sax PE, Zolopa A, Brar I, Elion R, Ortiz R, Post F, Wang H, Callebaut C, Martin H, Fordyce MW, McCallister S. 2014. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr 67:52–58. doi: 10.1097/QAI.0000000000000225. [DOI] [PubMed] [Google Scholar]
- 11.Sax PE, Wohl D, Yin MT, Post F, DeJesus E, Saag M, Pozniak A, Thompson M, Podzamczer D, Molina JM, Oka S, Koenig E, Trottier B, Andrade-Villanueva J, Crofoot G, Custodio JM, Plummer A, Zhong L, Cao H, Martin H, Callebaut C, Cheng AK, Fordyce MW, McCallister S, GS-US-292-0104/0111 Study Team . 2015. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 385:2606–2615. doi: 10.1016/S0140-6736(15)60616-X. [DOI] [PubMed] [Google Scholar]
- 12.Birkus G, Wang R, Liu X, Kutty N, MacArthur H, Cihlar T, Gibbs C, Swaminathan S, Lee W, McDermott M. 2007. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother 51:543–550. doi: 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T. 2008. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol Pharmacol 74:92–100. doi: 10.1124/mol.108.045526. [DOI] [PubMed] [Google Scholar]
- 14.D'Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H. 1982. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci U S A 79:4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pshezhetsky AV, Ashmarina M. 2001. Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog Nucleic Acid Res Mol Biol 69:81–114. doi: 10.1016/S0079-6603(01)69045-7. [DOI] [PubMed] [Google Scholar]
- 16.Jackman HL, Tan FL, Tamei H, Beurling-Harbury C, Li XY, Skidgel RA, Erdos EG. 1990. A peptidase in human platelets that deamidates tachykinins: probable identity with the lysosomal “protective protein.” J Biol Chem 265:11265–11272. [PubMed] [Google Scholar]
- 17.Tranchemontagne J, Michaud L, Potier M. 1990. Deficient lysosomal carboxypeptidase activity in galactosialidosis. Biochem Biophys Res Commun 168:22–29. doi: 10.1016/0006-291X(90)91669-J. [DOI] [PubMed] [Google Scholar]
- 18.Balzarini J, Karlsson A, Aquaro S, Perno CF, Cahard D, Naesens L, De Clercq E, McGuigan C. 1996. Mechanism of anti-HIV action of masked alaninyl d4T-MP derivatives. Proc Natl Acad Sci U S A 93:7295–7299. doi: 10.1073/pnas.93.14.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valette G, Pompon A, Girardet JL, Cappellacci L, Franchetti P, Grifantini M, La Colla P, Loi AG, Perigaud C, Gosselin G, Imbach JL. 1996. Decomposition pathways and in vitro HIV inhibitory effects of isoddA pronucleotides: toward a rational approach for intracellular delivery of nucleoside 5′-monophosphates. J Med Chem 39:1981–1990. doi: 10.1021/jm9507338. [DOI] [PubMed] [Google Scholar]
- 20.Birkus G, Kutty N, Frey CR, Shribata R, Chou T, Wagner C, McDermott M, Cihlar T. 2011. Role of cathepsin A and lysosomes in the intracellular activation of novel antipapillomavirus agent GS-9191. Antimicrob Agents Chemother 55:2166–2173. doi: 10.1128/AAC.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones GS, Yu F, Zeynalzadegan A, Hesselgesser J, Chen X, Chen J, Jin H, Kim CU, Wright M, Geleziunas R, Tsiang M. 2009. Preclinical evaluation of GS-9160, a novel inhibitor of HIV-1 integrase. Antimicrob Agents Chemother 53:1194–1203. doi: 10.1128/AAC.00984-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bam RA, Birkus G, Babusis D, Cihlar T, Yant SR. 2014. Metabolism and antiretroviral activity of tenofovir alafenamide in CD4+ T-cells and macrophages from demographically diverse donors. Antiviral Ther 19:669–677. doi: 10.3851/IMP2767. [DOI] [PubMed] [Google Scholar]
- 23.Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA. 2010. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem 285:34337–34347. doi: 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devine KG, McGuigan C, O'Connor TJ, Nicholls SR, Kinchington D. 1990. Novel phosphate derivatives of zidovudine as anti-HIV compounds. AIDS 4:371–373. doi: 10.1097/00002030-199004000-00019. [DOI] [PubMed] [Google Scholar]
- 25.Molema G, Jansen RW, Pauwels R, de Clercq E, Meijer DK. 1990. Targeting of antiviral drugs to T4-lymphocytes. Anti-HIV activity of neoglycoprotein-AZTMP conjugates in vitro. Biochem Pharmacol 40:2603–2610. [DOI] [PubMed] [Google Scholar]
- 26.Jones BCNM, McGuigan C, O'Conner TJ, Jeffries DJ, Kinchington D. 1991. Synthesis and anti-HIV activity of some novel phosphorodiamidate derivatives of 3′-azido-3′-deoxythymidine (AZT). Antiviral Chem Chemother 2:35–39. doi: 10.1177/095632029100200106. [DOI] [Google Scholar]
- 27.McGuigan C, Devine KG, O'Connor TJ, Kinchington D. 1991. Synthesis and anti-HIV activity of some haloalkyl phosphoramidate derivatives of 3′-azido-3′-deoxythymidine (AZT): potent activity of the trichloroethyl methoxyalaninyl compound. Antiviral Res 15:255–263. doi: 10.1016/0166-3542(91)90071-X. [DOI] [PubMed] [Google Scholar]
- 28.McGuigan C, Jones BCNM, Devine KG, Nicholls SR, O'Conner TJ, Kinchington D. 1991. Attempts to introduce chemotherapeutic nucleotides into cells: studies on the anti-HIV agent FDT. Biorganic Med Chem Lett 1:729–732. doi: 10.1016/S0960-894X(01)81057-X. [DOI] [Google Scholar]
- 29.Ray AS, Hostetler KY. 2011. Application of kinase bypass strategies to nucleoside antivirals. Antiviral Res 92:277–291. doi: 10.1016/j.antiviral.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 30.Markowitz M, Zolopa A, Ruane P, Squires K, Zhong L, Kearney BP, Lee W. 2011. Abstr 18th Conf Retroviruses Opportun Infect, abstr 152LB. [Google Scholar]
- 31.Cihlar T, Ray AS, Boojamra CG, Zhang L, Hui H, Laflamme G, Vela JE, Grant D, Chen J, Myrick F, White KL, Gao Y, Lin KY, Douglas JL, Parkin NT, Carey A, Pakdaman R, Mackman RL. 2008. Design and profiling of GS-9148, a novel nucleotide analog active against nucleoside-resistant variants of human immunodeficiency virus type 1, and its orally bioavailable phosphonoamidate prodrug, GS-9131. Antimicrob Agents Chemother 52:655–665. doi: 10.1128/AAC.01215-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfgang GH, Shibata R, Wang J, Ray AS, Wu S, Doerrfler E, Reiser H, Lee WA, Birkus G, Christensen ND, Andrei G, Snoeck R. 2009. GS-9191 is a novel topical prodrug of the nucleotide analog 9-(2-phosphonylmethoxyethyl)guanine with antiproliferative activity and possible utility in the treatment of human papillomavirus lesions. Antimicrob Agents Chemother 53:2777–2784. doi: 10.1128/AAC.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sofia MJ. 2013. Nucleotide prodrugs for the treatment of HCV infection. Adv Pharmacol 67:39–73. doi: 10.1016/B978-0-12-405880-4.00002-0. [DOI] [PubMed] [Google Scholar]
- 34.Imai T. 2006. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab Pharmacokinet 21:173–185. doi: 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- 35.Murakami E, Wang T, Park Y, Hao J, Lepist EI, Babusis D, Ray AS. 2015. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob Agents Chemother 59:3563–3569. doi: 10.1128/AAC.00128-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal K, Fung SK, Nguyen TT, Cheng W, Sicard E, Ryder SD, Flaherty JF, Lawson E, Zhao S, Subramanian GM, McHutchinson JG, Gane EJ, Foster GR. 2015. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol 62:533–540. doi: 10.1016/j.jhep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 37.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother 50:899–909. doi: 10.1128/AAC.50.3.899-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Markowitz M, Zolopa Ruane AP, Squires K, Zhong L, Kearney BP, Lee W. 2006. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother 50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bristol-Myers Squibb Company. 2011. Reyataz® (atazanavir sulfate) capsules. US prescribing information; Bristol-Myers Squibb Company, Princeton, NJ, USA. [Google Scholar]
- 40.Schering Corporation. 2011. Victrelis® (boceprevir) capsules. US prescribing information. Schering Corporation, Whitehouse Station, NJ, USA. [Google Scholar]
- 41.Vertex Pharmaceuticals Incorporated. 2011. IncivekTM (telaprevir) film coated tablets. US prescribing information. Vertex Pharmaceuticals Incorporated, Cambridge, MA, USA. [Google Scholar]
- 42.Bristol-Myers Squibb Company. 2011. Eliquis® (apixaban) tablets. Australian therapeutic goods administration; Bristol-Myers Squibb Company, Princeton, NJ, USA. [Google Scholar]
- 43.Electronic Medicines Compendium. 2011. Xareto® (rivaroxaban) tablets. Electronic medicines compendium. Bayer House, Strawberry Hill, Newbury, Berkshire, United Kingdom. [Google Scholar]
- 44.Stangier J, Eriksson BI, Dahl OE, Ahnfelt L, Nehmiz G, Stähle H, Rathgen K, Svärd R. 2005. Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement. J Clin Pharmacol 45:555–563. doi: 10.1177/0091270005274550. [DOI] [PubMed] [Google Scholar]
- 45.Merck. 2011. Januvia® (sitagliptin) tablets. US prescribing information. Merck, Whitehouse Station, NJ, USA. [Google Scholar]
- 46.Swan SK, Hursting MJ. 2000. The pharmacokinetics and pharmacodynamics of argatroban: effects of age, gender, and hepatic or renal dysfunction. Pharmacotherapy 20:318–329. doi: 10.1592/phco.20.4.318.34881. [DOI] [PubMed] [Google Scholar]