

Abstract
β-Lactams are the most successful antibacterials, but their effectiveness is threatened by resistance, most importantly by production of serine- and metallo-β-lactamases (MBLs). MBLs are of increasing concern because they catalyze the hydrolysis of almost all β-lactam antibiotics, including recent-generation carbapenems. Clinically useful serine-β-lactamase inhibitors have been developed, but such inhibitors are not available for MBLs. l-Captopril, which is used to treat hypertension via angiotensin-converting enzyme inhibition, has been reported to inhibit MBLs by chelating the active site zinc ions via its thiol(ate). We report systematic studies on B1 MBL inhibition by all four captopril stereoisomers. High-resolution crystal structures of three MBLs (IMP-1, BcII, and VIM-2) in complex with either the l- or d-captopril stereoisomer reveal correlations between the binding mode and inhibition potency. The results will be useful in the design of MBL inhibitors with the breadth of selectivity required for clinical application against carbapenem-resistant Enterobacteriaceae and other organisms causing MBL-mediated resistant infections.
INTRODUCTION
The increasing problem of antibiotic resistance is a global health concern (1), with the World Health Organization (WHO) and the European Centre for Disease Prevention and Control (ECDPC) reporting that several million people are infected with antibiotic-resistant bacteria annually. It is estimated that >50,000 patients die each year due to infections caused by multidrug-resistant bacterial pathogens in the United States alone (2).
β-Lactam-containing compounds remain the most important antibiotics in clinical use, but their effectiveness is threatened by increasing resistance. β-Lactam resistance is most importantly mediated by serine- and zinc-dependent metallo-β-lactamases (SBLs and MBLs, respectively), which catalyze β-lactam hydrolysis (3). In combination with an appropriate penicillin antibiotic, class A SBL (penicillinase) inhibitors (i.e., clavulanic acid, tazobactam, and sulbactam) have been used widely in the clinic, and recently, a class C (cephalosporinase) SBL inhibitor (4), Avibactam, was approved for clinical use in combination with a cephalosporin (5). In contrast, there are no reports of clinically useful MBL inhibitors (6).
A challenge for the development of useful MBL inhibitors is achieving a breadth of inhibition against most MBL subtypes while avoiding inhibition of structurally related human MBL-fold enzymes (7). Crystal structures reveal that MBLs have a characteristic αβ/βα sandwich fold, that they possess conserved zinc ion binding sites, and that loops flanking the active site are involved in ligand binding (8). MBLs can be divided into three subclasses (B1, B2, and B3), based on the number of zinc ions in their metal binding sites and/or based on sequence and structural similarities (6). B1 MBLs (e.g., imipenemase [IMP], Verona integron-encoded MBL [VIM], and New Delhi MBL [NDM] types) are the most clinically relevant MBLs; these MBLs catalyze hydrolysis of almost all β-lactams, including the latest generations of cephalosporins and carbapenems (9). Several classes of known metalloenzyme inhibitors inhibit MBLs, including thiols, carboxylic acids, trifluoromethyl ketones, hydroxamic acids, and rhodanines (7, 10, 11) (see Fig. S1 in the supplemental material for structures).
(2S)-1-[(2S)-2-Methyl-3-sulfanylpropanoyl] pyrrolidine-2-carboxylic acid (commonly referred to as l-captopril) (Fig. 1) is a thiol-containing small molecule which was developed in the 1970s to target the zinc ion-utilizing human angiotensin-converting enzyme (ACE) (12, 13). l-Captopril was used successfully for several decades to control high blood pressure. The clinically used (2S,2S)-stereoisomer, i.e., l-captopril, inhibits several MBLs from all subclasses (14–18). However, the (2S,2R)-stereoisomer, (2S)-1-[(2R)-2-methyl-3-sulfanylpropanoyl] pyrrolidine-2-carboxylic acid (Fig. 1), commonly referred to as d-captopril, has been reported to be more active than l-captopril against some MBLs (e.g., NDM-1 [19], BcII [17], CcrA [20], and CphA [17]).
FIG 1.

Captopril has structural similarity to the hydrolyzed penicillin product of metallo-β-lactamase catalysis (penicillinoic acid). (A) Outline of the mode of action of metallo-β-lactamases (MBLs). (B) Structures of the four captopril stereoisomers (2S,2S, 2S,2R, 2R,2S, and 2R,2R) and of d- and l-MBP. (C) Binding mode of hydrolyzed ampicillin with NDM-1 (PDB code 4HL2). (D) Binding mode of d-captopril with IMP-1 (PDB code 4C1G [described in this study]).
Crystal structures have been reported for some MBLs in complex with l- or d-captopril, i.e., (i) in the case of the B1 subclass MBLs, for NDM-1 complexed with l-captopril (21) and for Chryseobacterium meningosepticum BlaB complexed with d-captopril (22); (ii) in the case of the B2 MBLs, for Aeromonas hydrophila CphA complexed with d-captopril (18); and (iii) in the case of the B3 MBLs, for the Fluoribacter gormanii MBL FEZ-1 complexed with d-captopril (23) and the Stenotrophomonas maltophilia MBL L1 complexed with d-captopril (15). Biophysical analyses employing extended X-ray absorption fine structure (EXAFS) and perturbed angular correlation of X-rays (PAC) spectroscopy have been reported for BcII and CphA complexed with d- and l-captopril (17). Molecular dynamic calculations on d- and l-captopril complexed with BcII and d-captopril complexed with NDM-1 have also been reported (20, 24). These analyses imply that both l- and d-captopril can bind with the thiol(ate) ligated to both active site Zn(II) ions (Fig. 2; see Fig. S2 to S4 in the supplemental material). Interestingly, despite BlaB and NDM-1 belonging to the same B1 MBL subclass, different binding modes were observed for the l- and d-captopril stereoisomers (19). In the case of the mono-Zn(II) ion-binding B2 subclass, a structure of the CphA–d-captopril complex (18) indicates that the d-captopril carboxylate, rather than the thiol(ate), binds to the single Zn(II) ion, a binding mode that possibly reflects the relatively weak inhibition of this enzyme by d-captopril (Ki = 72 μM). Finally, for the B3 MBL subclass, in a crystal structure of the FEZ-1–d-captopril complex (23), the binding of captopril was modeled such that neither the d-captopril thiol nor its carboxylate interacts with the active site Zn(II) ions, a binding mode that was also proposed to be consistent with the relatively weak inhibition observed in this case (Ki = 400 μM) (see Fig. S2 in the supplemental material). To date, there have been no reports on MBL inhibition by (2R)-1-[(2S)-2-methyl-3-sulfanylpropanoyl] pyrrolidine-2-carboxylic acid, subsequently referred to as epi-l-captopril (the 2R,2S-stereoisomer), or (2R)-1-[(2R)-2-methyl-3-sulfanylpropanoyl] pyrrolidine-2-carboxylic acid, subsequently referred to as epi-d-captopril (the 2R,2R-stereoisomer) (Fig. 1).
FIG 2.
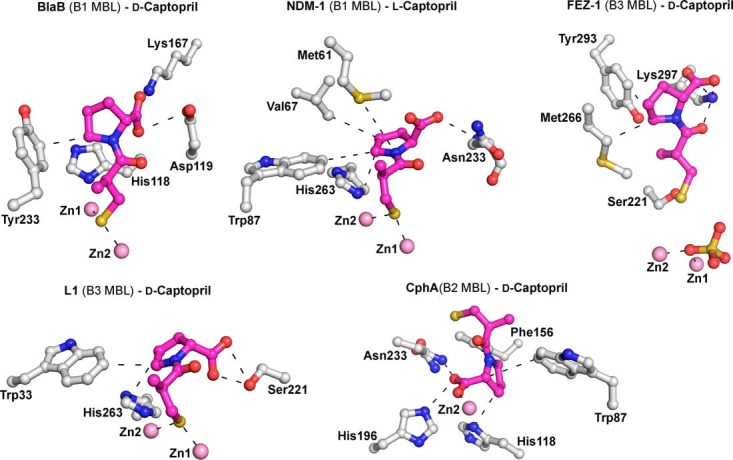
Crystallographic analysis reveals different binding modes for d- and l-captopril. Preliminary crystal structures are shown for BlaB, NDM-1, CphA, FEZ-1, and L1 complexed with l- and d-captopril (PDB entries 1M2X [1.50 Å], 4EXS [2.40 Å], 2QDS [1.66 Å], 1JT1 [1.65 Å], and 2FU8 [1.80 Å]). Zinc ions are represented by pink spheres, d- and l-captopril ligands are shown in magenta, and the amino acid residues interacting with captopril are depicted as gray stick models. Hydrogen bonds, zinc coordination bonds, and hydrophobic interactions are shown as thin black dashes.
We report systematic studies on the inhibition of four clinically relevant MBLs (IMP-1, VIM-2, SPM-1, and NDM-1) and the model MBL BcII by the four captopril stereoisomers and both enantiomers of a captopril derivative, 1-(2-mercaptobenzoyl)pyrrolidine-2-carboxylic acid (d- and l-MBP) (Fig. 1; see Table S1 in the supplemental material) (25, 26). The combined kinetic and structural studies clearly reveal different binding modes for different captopril stereoisomers and will help to enable the future development of broad-spectrum MBL inhibitors.
MATERIALS AND METHODS
Synthesis.
The different captopril isomers and captopril derivatives were prepared according to procedures in the literature (see Schemes S1 and S2 and the experimental section in the supplemental material).
Protein production and purification.
Recombinant forms of NDM-1, VIM-2, VIM-4, SPM-1, IMP-1, and BcII MBLs were produced in Escherichia coli as described previously (27, 28). Purified proteins were dialyzed into freshly prepared crystallization buffer (50 mM HEPES, pH 7.5, 150 mM NaCl containing 1 μg ZnCl2) and then concentrated (to 2 mM [BcII], 0.75 mM [IMP-1], and 0.36 mM [VIM-2]) before use in crystallization studies.
Crystallography.
Crystals were grown under the conditions stated in Table S2 in the supplemental material and were cryoprotected using well solution diluted with 25% glycerol before being flash cooled in liquid nitrogen. All data sets were collected at 100 K. All data were indexed, integrated, and scaled using HKL-3000 (29). The structures were solved by molecular replacement using Phaser (30). The structures were then refined using PHENIX (31) and COOT (32) until the Rwork and Rfree values no longer decreased. Data collection and refinement statistics are given in Tables S3 to S5 in the supplemental material.
Kinetic analyses.
Kinetic and inhibition assays with the bacterial MBLs, human angiotensin-converting enzyme 2 (hACE-2), and hydroxyacylglutathione hydrolase, human glyoxylase II (hHAGH), were performed as described previously (7, 27).
Nuclease assays.
Nuclease assays with DCLRE1A and DCLRE1B (DNA cross-link repair enzymes 1A and 1B) (33) were performed according to a previously described method (33), employing a 21-nucleotide DNA oligonucleotide with a fluorescein label at its 3′ end. In brief, exonuclease activity was measured using ΔN-DCLRE1A (3.5 ng; 8 nM) or ΔC-DCLRE1B (1.5 ng; 4 nM) mixed with 1 pmol (1 μM) of 3′-fluorescein-labeled DNA substrate in 10 μl of 20 mM HEPES, pH 7.9, 50 mM KCl, 10 mM MgCl2, 0.5 mM dithiothreitol (DTT), 0.05% Triton-X, 0.1 mg/ml bovine serum albumin (BSA), and 5% glycerol. Reaction mixtures were incubated at 37°C for 20 min with the indicated concentrations of d-captopril, l-captopril, d-MBP, or l-MBP (see Fig. S5 in the supplemental material), and reactions were then quenched by addition of 2 μl of 80% formamide-10 mM EDTA and heating at 95°C for 5 min. Following separation in a 20% polyacrylamide-7 M urea denaturing gel, substrate and product bands were visualized by use of a Typhoon Trio+ variable-model imager (excitation at 488 nm with a blue laser at 400 V).
MIC determinations.
The bacteria used were nonclonal international isolates whose mechanisms of carbapenem resistance have been defined genetically by sequencing. E. coli ATCC 25922 was used as a negative control. MBL genes were obtained by PCR using standard procedures and were inserted into a pK18 vector which was used to transform E. coli J53 (34). Transconjugants were obtained with MBL genes carried on native wild-type plasmids conjugated into J53 (35). Both transformants and transconjugants were confirmed by DNA sequencing and verified for MBL production by using MTS MBL strips (Liofilchem, Roseto, Italy). Additionally, 5 Escherichia coli (with NDM-1), 10 Klebsiella pneumoniae (5 with NDM-1, 3 with VIM-4, and 2 with IMP-4), 1 Serratia marcescens (with IMP-4), and 2 Pseudomonas aeruginosa (with VIM-2 and AIM-1) isolates and E. coli ATCC 25922 were used in the test panel. MICs were determined using the broth microdilution method according to CLSI guidelines. Strains were cultured and tested in cation-adjusted Mueller-Hinton agar and broth (Becton Dickinson).
Protein structure accession numbers.
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the following accession codes: for apo di-Zn(II)–BcII, PDB entry 4C09; for BcII–l-captopril, PDB entry 4C1H; for BcII–d-captopril, PDB entry 4C1C; for IMP-1–l-captopril, PDB entry 4C1F; for IMP-1–d-captopril, PDB entry 4C1G; for apo di-Zn(II)–VIM-2, PDB entry 4BZ3; for VIM-2–l-captopril, PDB entry 4C1D; and for VIM-2–d-captopril, PDB entry 4C1E. The URL for coordinate deposition is http://rcsb-deposit.rutgers.edu/.
RESULTS
MBL inhibition by captopril stereoisomers.
We first synthesized the four possible captopril stereoisomers (d-, l-, epi-d-, and epi-l-captopril) (see Schemes S1 and S2 and the experimental section in the supplemental material) and tested them as inhibitors against BcII and clinically relevant MBLs from the B1 subclass (IMP-1, VIM-2, SPM-1, and NDM-1) (Table 1 and Fig. 3) (9). Comparing the previously reported d-captopril and l-captopril inhibition values to our results for NDM-1, IMP-1, and BcII reveals relatively small differences (Table 1), likely due at least in part to different assay conditions (for d-captopril, NDM-1 IC50s of 20.1 μM versus 7.9 μM [36] and BcII IC50s of 10.7 μM versus 45 μM [Ki] [17]; and for l-captopril, NDM-1 IC50s of 157.4 μM versus 202 μM [36], IMP-1 IC50s of 7.2 μM versus 12.5 μM [Ki] [37], and BcII IC50s of 80.4 μM versus 65 μM [Ki] [17]). In all cases, d-captopril was the most potent of the four possible captopril stereoisomers (NDM-1 IC50, 20.1 ± 1.5 μM; IMP-1 IC50, 7.2 ± 1.2 μM; VIM-2 IC50, 0.072 ± 0.010 μM; SPM-1 IC50, 261.8 ± 1.3 μM; and BcII IC50, 10.7 ± 1.2 μM) (Table 1). d-Captopril was consistently more potent than l-captopril (∼7-fold for NDM-1, ∼3-fold for IMP-1, ∼60-fold for VIM-2, ∼2-fold for SPM-1, and ∼8-fold for BcII) (Table 1). Both the epi-l- and epi-d-captopril stereoisomers were poor inhibitors of BcII and SPM-1 (IC50s were all ≥500 μM), whereas for NDM-1 and IMP-1, unlike epi-l-captopril, epi-d-captopril showed some activity (NDM-1 IC50, 64 μM; and IMP-1 IC50, 173 μM). Relatively potent IC50s were observed for both epi-l- and epi-d-captopril against VIM-2 (IC50 = 5.5 μM). The captopril derivatives d- and l-MBP were less potent (IC50s of >500 μM) than d- and l-captopril against all MBLs (Table 1).
TABLE 1.
IC50s for the four captopril stereoisomers and derivatives of captopril (MBP) against different MBLs
| Compound | IC50 (μM)c |
||||
|---|---|---|---|---|---|
| BcII | IMP-1 | VIM-2 | SPM-1 | NDM-1 | |
| d-Captopril | 10.7 ± 1.2 (45a) | 7.2 ± 1.2 | 0.072 ± 0.01 | 261.8 ± 1.3 | 20.1 ± 1.5 (7.9b) |
| l-Captopril | 80.4 ± 1.1 (65a) | 23.3 ± 1.3(12.5a) | 4.4 ± 0.8 | >500 | 157.4 ± 1.3 (202b) |
| epi-d-Captopril | >500 | 173.2 ± 1.2 | 5.5 ± 0.7 | >500 | 64.6 ± 1.4 |
| epi-l-Captopril | 423.8 ± 1.5 | 436 ± 1.1 | 5.5 ± 1.5 | >500 | >500 |
| d-MBP | >500 | >500 | >500 | >500 | >500 |
| l-MBP | >500 | >500 | >500 | >500 | >500 |
Ki value from the literature.
IC50 from the literature.
All experiments were performed three or more times. Nonlinear regression analysis was used to calculate the IC50s and their corresponding 95% confidence intervals (GraphPad Prism). Results represent means ± standard deviations.
FIG 3.

IC50 curves for all captopril stereoisomers tested against BcII (A), IMP-1 (B), and VIM-2 (C) reveal different potencies. The l- and epi-l-captopril stereoisomers are represented by solid lines, and the d- and epi-d-captopril stereoisomers are depicted with dashed lines. R.A., remaining activity.
A key issue in work toward obtaining clinically relevant MBL inhibitors is the degree of selectivity toward the bacterial MBLs over human metalloenzymes, including MBL-fold enzymes. Although l-captopril is a well-studied ACE-2 inhibitor, there are no reports of its selectivity versus human MBL-fold enzymes. We tested l- and d-captopril as well as d- and l-MBP against the human MBL-fold enzymes DCLRE1A and DCLRE1B (33), hHAGH, and hACE-2 (zinc-dependent human metalloenzyme); no inhibition was observed at 100 μM under our standard assay conditions (see Fig. S5 in the supplemental material).
Pathogen susceptibility to d- and l-captopril.
Since the d- and l-captopril isomers were consistently the most potent stereoisomers against the tested MBLs (Table 2), we tested them against nonclonal multidrug-resistant bacteria expressing various MBLs. We used a variety of isolates with cloned MBLs, transconjugants, and wild-type clinical isolates. The panel of strains was tested for meropenem and ceftazidime MICs, with and without l- or d-captopril at 8 mg/liter (Table 2). In order to correlate the data with the results of cellular studies (see below), we determined the IC50s for d- and l-captopril against VIM-4 (VIM-4 IC50 of 1.7 ± 0.4 μM for d-captopril and 3.9 ± 0.5 μM for l-captopril). Pathogenic strains with different geographical origins (e.g., Greece [A-33; VIM-4] or India [IR60; NDM-1]) (38) and displaying resistance to β-lactams as well as fluoroquinolone and aminoglycoside antibiotics were selected for MIC tests (Table 2). While l-captopril showed potentiation with cloned and transconjugated MBLs, there was little synergy observed against wild-type strains, and it was generally less than that with d-captopril (Table 2). The addition of d-captopril potentiated the efficacy of meropenem against most of the VIM-2-, VIM-4-, IMP-4-, and NDM-1-producing strains tested, including E. coli, K. pneumoniae, S. marcescens, and P. aeruginosa (Table 2).
TABLE 2.
MICs of meropenem (MEM) and ceftazidime (CFZ), with and without d- and l-captopril (d- and l-CAP), versus various Gram-negative bacteria
| Strain (inhibitor) | Genotype | MIC |
|||||
|---|---|---|---|---|---|---|---|
| MEM | MEM + d-CAP (8 mg/liter) | MEM + l-CAP (8 mg/liter) | CFZ | CFZ + d-CAP (8 mg/liter) | CFZ + l-CAP (8 mg/liter) | ||
| E. coli 25922 | <0.125 | <0.125 | <0.125 | 0.125 | <0.125 | <0.125 | |
| E. coli J53 + NDM-1 clone | blaNDM-1 | 64 | 8 | 8 | 512 | 8 | 32 |
| E. coli J53 + NDM-1 transconjugant | blaNDM-1 | 128 | 16 | 16 | 512 | 16 | 32 |
| E. coli J53 + VIM-2 clone | blaVIM-2 | 8 | 2 | 4 | 32 | 8 | 16 |
| E. coli J53 + VIM-2 transconjugant | blaVIM-2 | 2 | 1 | 1 | 32 | 4 | 8 |
| E. coli J53 + IMP-1 clone | blaIMP-1 | 1 | 0.25 | 0.5 | 128 | 32 | 32 |
| K. pneumoniae IR16 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaSHV-1 blaNDM-1 | 128 | 8 | 128 | |||
| E. coli IR10 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaOXA-1 blaNDM-1 | 64 | 2 | 64 | |||
| K. pneumoniae IR8 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaSHV-1 blaNDM-1 | 16 | 2 | 16 | |||
| E. coli IR15 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaOXA-1 blaNDM-1 | 8 | 0.25 | 8 | |||
| K. pneumoniae IR19 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaSHV-1 blaNDM-1 | 8 | 1 | 8 | |||
| E. coli IR24 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaOXA-1 blaNDM-1 | 512 | 32 | 512 | |||
| E. coli IR60 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaOXA-1 blaNDM-1 | 128 | 32 | 128 | |||
| K. pneumoniae HR8 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaOXA-1 blaSHV-1 blaNDM-1 | 64 | 4 | 64 | |||
| K. pneumoniae N16 (NDM-1) | blaDHA-1 blaCTX-15 blaTEM-1 blaSHV-1 blaNDM-1 | 32 | 4 | 16 | |||
| K. pneumoniae A33 (VIM-4) | blaCTX-15 blaTEM-1 blaSHV-12a blaVIM-4 | 8 | 1 | 16 | |||
| K. pneumoniae A34 (VIM-4) | blaCTX-15 blaTEM-1 blaSHV-12a blaVIM-4 | 8 | 1 | 8 | |||
| K. pneumoniae A35 (VIM-4) | blaCTX-15 blaTEM-1 blaSHV-12a blaVIM-4 | 16 | 4 | 16 | |||
| K. pneumoniae B12 (IMP-4) | blaCTX-15 blaTEM-1 blaSHV-12a blaIMP-4 blaOXA-1 | 8 | 1 | 4 | |||
| S. marcescens B13 (IMP-4) | blaDHA-1 blaAMPC blaTEM-1 blaIMP-4 | 4 | 1 | 4 | |||
| K. pneumoniae B19 (IMP-4) | blaCTX-15 blaTEM-1 blaSHV-12a blaIMP-4 | 32 | 2 | 16 | |||
| P. aeruginosa 4470 (VIM-2) | blaAMPC blaVIM-2 | 512 | 512 | 512 | |||
| P. aeruginosa (AIM-1) | blaAMPC blaAIM-1 | 512 | 512 | 512 | |||
Structural analysis of captopril binding to IMP-1, VIM-2, and BcII.
We next investigated the mode of binding of the captopril stereoisomers to MBLs by using crystallography. We determined high-resolution crystal structures for d- and l-captopril in complex with IMP-1 (1.71- and 2.01-Å resolutions, respectively), VIM-2 (1.40- and 1.20-Å resolutions, respectively), and BcII (1.18- and 1.10-Å resolutions, respectively). For comparison, structures of di-Zn(II)–VIM-2 and di-Zn(II)–BcII without inhibitors were also determined, to 1.20- and 1.30-Å resolutions, respectively. For all structures, the crystal systems were similar to those reported previously (for VIM-2 [7], IMP-1 [39], and BcII [40]). (Note that we used the standard numbering scheme for class B β-lactamases, i.e., BBL numbering [41].)
As anticipated, in all cases the overall protein folds observed were characteristic αβ/βα MBL sandwich folds (2 β-sheets sandwiched with 2 helices buttressed against each external face of the sandwich) (8). The active sites, which are located at one end of the two β-sheets in a groove surrounded by several loops, were occupied in all cases by two zinc ions, as expected for B1 subclass MBLs. The L3 and L10 loops, which flank the active site, are located opposite each other and are involved in substrate binding (42). The L3 loop (residues 61 to 66 [BBL numbering]) is located between strands β3 and β4, and the L10 loop (residues 223 to 241) is located between strand β11 and helix α4, which includes Lys224/Arg228 and Asn233, whose side chains are directly involved in substrate and inhibitor binding (41, 43).
The electron density maps for the ligands in the various MBL-captopril complexes suggested various ligand occupancies and were carefully analyzed between rounds of refinement (see Fig. S6 in the supplemental material). For BcII, both the d- and l-captopril isomers were modeled and refined with 70% occupancy. For the VIM-2–d-captopril complex structure, the ligands were modeled and refined with 100% occupancy, and for the VIM-2–l-captopril complex structure, 80% occupancy was used. For the IMP-1–d-captopril and IMP-1–l-captopril structures, the ligands were modeled and refined with 100% occupancy in chain A, but the residual density present in chain B was interpreted as too weak (<50% occupancy) to include in the model.
Preliminary structural analysis indicated that partial oxidation of the metal-binding cysteine (Cys221) had occurred in both the crystallized BcII and VIM-2 proteins, in a manner similar to that observed in previous crystallographic studies of these B1 MBLs (40, 44). Due to the likelihood of active site cysteine oxidation (Cys221) interfering with the active site Zn(II) chemistry, and hence with our analysis of ligand binding, we worked to minimize Cys221 oxidation. In all cases, cysteine oxidation during crystallization could be prevented by the addition of tris(2-carboxyethyl)phosphine (TCEP) (40) (except for IMP-1, for which cysteine oxidation in the absence of TCEP was not observed).
Our MBL-captopril complex structures show an overall captopril binding mode similar to those previously observed for NDM-1–l-captopril (21), BlaB–d-captopril (22), and L1–d-captopril (15), but they differ significantly from the FEZ-1–d-captopril (23) and CphA–d-captopril complex structures (Fig. 3; see Fig. S2 in the supplemental material). Our MBL–l-captopril structures are most similar to the reported NDM-1–l-captopril complex structure (21), and our MBL–d-captopril structures are most similar to the L1–d-captopril complex structure (15) (see Fig. S2 in the supplemental material). Comparison of the MBL-captopril complex structures with the active site of apo-MBLs reveals that several water molecules are displaced upon binding of d- or l-captopril (see Fig. S7 in the supplemental material); these displacements likely contribute to the strength of inhibitor binding.
Captopril has distinct features that enable metalloprotein binding. The thiol acts as a metal binding ligand that displaces the proposed “hydrolytic” water molecule (or hydroxide) that bridges the two active site metal ions. An amide carbonyl group leads to the conformationally constrained prolyl ring, and a methyl group extends from the carbon bonded to the thiol. Both the l- and d-captopril diastereoisomers present two distinct binding faces (Fig. 4). One face is hydrophobic and is formed by the methyl group and the proline ring methylenes; the hydrophobic face interacts with residues from the L3 loop (Trp87BcII; Trp64IMP-1 and Val61IMP-1; and Trp87VIM-2, Phe61VIM-2, and Tyr67VIM-2) (Fig. 4). Such interactions are in agreement with the proposed role of the “mobile” L3 loop in interacting with the hydrophobic N-acyl substituents of cephalosporin and penicillin MBL substrates (45). The other face of captopril is more hydrophilic and is positioned to form hydrogen bonds to residues in the L10 loop. In all our MBL–d-captopril structures, the d-captopril carboxylate is positioned to form electrostatic and hydrogen-bonding interaction distances ranging from 2.3 Å for IMP-1 to 2.9 Å for BcII and VIM-2, with a conserved positively charged residue (Lys224IMP-1,BcII or Arg228VIM-2) which is predicted to be involved in binding the β-lactam substrate carboxylate (46, 47) (Fig. 4; Fig. S4 in the supplemental material). As observed for our MBL–d-captopril structures, the IMP-1–l-captopril structure has the inhibitor carboxylate positioned to form an electrostatic interaction with a conserved basic residue, i.e., Lys224. In contrast, in the BcII–l-captopril and VIM-2–l-captopril structures, the captopril carboxylate is oriented away from the conserved positively charged Lys/Arg residue (Lys224/Arg228). In all MBL–l-captopril structures (including those with IMP-1), the carboxylate is positioned to interact with the conserved asparagine (Asn233) from the L10 loop. IMP-1 is thus apparently a special case, i.e., both the l- and d-captopril isomers bind in similar modes with the inhibitor carboxylate positioned to interact with Lys224IMP-1. In the structures of BcII and VIM-2 complexed with d-captopril and VIM-2 complexed with l-captopril, the captopril amide carbonyl oxygen is positioned to interact with the conserved asparagine (Asn233) from the L10 loop (Fig. 4; see Fig. S4 in the supplemental material). In all cases, the L3 and L10 loops were observed to move slightly toward the inhibitors relative to their positions in the absence of inhibitor, consistent with an induced-fit mechanism during substrate binding. This is most clearly observed in the case of VIM-2 (Fig. 4).
FIG 4.

Crystallographic analyses reveal different binding modes for d- and l-captopril. The left column shows views of structures of BcII, IMP-1, and VIM-2 complexed with l- and d-captopril (PDB entries 4C1H [1.10 Å], 4C1C [1.18 Å], 4C1F [2.01 Å], 4C1G [1.71 Å], 4C1D [1.20 Å], and 4C1E [1.40 Å], respectively), highlighting residues involved in inhibitor-MBL complex formation. The right column shows an overlay of structures in the absence/presence of d- or l-captopril; these reveal L3 and L10 loop movements on inhibitor binding. With BcII, a comparison of the L3 loop was not possible, because some part of it was not modeled, but clear movement was identified for the L10 loop. In the case of IMP-1, we did not obtain a di-Zn(II) structure without an inhibitor; a comparison with published IMP-1 structures is difficult because of different crystallization conditions, but in the d- and l-captopril structures both the L3 and L10 loops display different conformations. Zinc atoms are represented by pink spheres, the d- and l-captopril ligands are shown in magenta, and the amino acid residues interacting with captopril are shown as gray stick models. The electron density maps (Fo-Fc) are contoured to 3.0 σ and shown in green. Hydrogen bonds, zinc coordination bonds, and hydrophobic interactions are shown with thin black dashes. The MBL backbone in the overlay plots is shown in gray, and the flexible active site loops are shown in different shades of gray (loops L3 and L10).
The observed binding modes for d-captopril carboxylate in our structures differ from the binding mode observed in the reported BlaB–d-captopril structure (22), despite a similar binding mode for the captopril thiol(ate) binding to the zinc ions. In the case of BlaB, the d-captopril carboxylate is rotated ∼180° relative to the thiolate mode of binding in our structures, such that it does not interact with the conserved Lys224BlaB but binds to Lys167BlaB from the L10 loop. The conserved Asn233BBL in the L10 loop is replaced by tyrosine in BlaB (22); this Asn-Tyr substitution likely contributes to the different d-captopril binding mode in BlaB compared to that in our structures. Thus, d-captopril is likely to bind to NDM-1, which contains an asparagine (Asn233BBL) rather than a tyrosine at this position, in a manner similar to that observed for BcII, IMP-1, and VIM-2 (rather than the BlaB binding mode) (21).
In all of our MBL-captopril structures, the distance from the Zn(II) atoms to the bridging thiolate sulfur atom is ∼2.3 Å. Similar distances have been reported for the BlaB (B1)– and L1 (B3)–d-captopril structures (PDB entries 1M2X and 2FU8), with a slightly shorter distance for the l-captopril–NDM-1 structure (PDB entry 4EXS) (21). Different values have been observed for the MBL intermetal distance in the absence of exogenous ligands, with reported values ranging from 2.5 to 4.5 Å (48); this may be dependent in part on the actual metals bound, which is not always possible to assign based on diffraction data alone (48). An interzinc distance of 3.5 Å was observed for both our di-Zn(II)–BcII and di-Zn(II)–VIM-2 apo structures, in which a bridging water was present as previously observed. The inter-zinc distances increased to 3.8 Å and 3.7 Å upon d- or l-captopril binding to BcII and VIM-2, respectively, with the binding thiolate. This observation is consistent with the different van der Waals atomic radii for sulfur (1.8 Å) and oxygen (1.5 Å). For IMP-1, the observed Zn-Zn distance (3.5 Å [PDB entry 1DDK] [39], as in our BcII and VIM-2 structures) increased to 3.7 Å on binding of either the l- or d-captopril stereoisomer.
DISCUSSION
Although once of little clinical relevance, MBLs now have increasing importance (6). The VIM-type B1 MBLs are a major problem in parts of Asia, being present in up to 99% of MBL-positive multidrug-resistant strains (49). Thus, there is a genuine need for a response to MBL-mediated resistance. The finding that d-captopril, a stereoisomer of the clinically used drug l-captopril, is consistently the most potent inhibitor among the captopril stereoisomers against MBLs and can potentiate the effects of meropenem against VIM-2- and other MBL-expressing pathogens is interesting.
Although several crystal structures of MBLs in combination with either d- or l-captopril have been reported, structures of the same MBL in complex with the d- and l-captopril isomers have not been reported previously. The captopril isomer binding modes that we observed are related in that they all involve thiol(ate) zinc chelation, as in most of the previously reported structures. An exception is the reported FEZ-1–d-captopril structure (23), which may not be representative of binding in solution due to its relatively poor quality.
Correlations can be made between the observed binding modes and the potency of inhibition. More potent inhibition was always observed when an interaction between the captopril carboxylate and the conserved basic Lys/Arg (Lys224BBL) involved in substrate binding was shown in the crystal structures. In all cases, VIM-2 manifested lower IC50s than those of the other tested MBLs—this correlates with the additional interactions observed between Arg228VIM-2 and the captopril carboxylate, as well as with additional interactions of the VIM-2 L3 loop with the hydrophobic face of captopril. Second, the observation of a decreased number of hydrogen bonding/electrostatic interactions for l- over d-captopril generally reflects weaker inhibition. l-Captopril was observed to be more potent against IMP-1 than against the other MBLs tested, and its IC50 was only 3-fold higher than that of d-captopril. This observation of relatively potent inhibition of IMP-1 by l-captopril correlates with the observation that for IMP-1, but none of the other MBLs, the l-captopril carboxylate favors binding to Lys224IMP-1.
Product inhibition is commonly observed for MBLs (16). Comparison of our MBL-captopril structural complexes with MBL–β-lactam product complexes (PDB entry 4HL2) (Fig. 1) shows that the most potent of the captopril isomers, d-captopril, has a mode of binding most similar to that of hydrolyzed β-lactams (50), especially penicillins, consistent with d-captopril being the most potent inhibitor (Table 1). However, d-captopril was significantly less potent (>20-fold) against SPM-1 than against all other MBLs tested; the other captopril stereoisomers did not inhibit SPM-1 (IC50s of >500 μM). This difference may reflect the unusual nature of SPM-1, as a proposed B1/B2 hybrid MBL (42). A challenge in MBL inhibition is to obtain breadth of selectivity toward the majority of prokaryotic MBLs, which often have relatively low sequence similarity (∼30% for the MBLs we used) (see Table S1 in the supplemental material), without inhibiting the related human MBL-fold enzymes. The combined structural and inhibition results reveal that captopril stereoisomers can potently inhibit B1 MBLs via related but sometimes different binding modes. These observations may be important in developing potent inhibitors with the required breadth of selectivity against different subtypes of MBLs, i.e., medicinal chemists may specifically aim to identify single compounds that bind differently to different MBL subtypes.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Medical Research Council (MRC)/Canadian grants G1100135 and MR/L007665/1 for support of J.B., S.Y.L., P.J.M., and C.J.S. Cancer Research UK (CRUK) is kindly acknowledged for the support of S.S.V.B. and C.J.S., and the Biotechnology and Biological Sciences Research Council (BBSRC; grant number BB/J014427/1) is acknowledged for the support of C.J.S. and D.Z.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01335-15.
REFERENCES
- 1.Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, Burgmann H, Sorum H, Norstrom M, Pons M-N, Kreuzinger N, Huovinen P, Stefani S, Schwartz T, Kisand V, Baquero F, Martinez JL. 2015. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13:310–317. doi: 10.1038/nrmicro3439. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. 2013. The antibiotic alarm. Nature 495:141. [DOI] [PubMed] [Google Scholar]
- 3.Bush K. 2013. Proliferation and significance of clinically relevant β-lactamases. Ann N Y Acad Sci 1277:84–90. doi: 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- 4.Bebrone C, Lassaux P, Vercheval L, Sohier JS, Jehaes A, Sauvage E, Galleni M. 2010. Current challenges in antimicrobial chemotherapy: focus on ss-lactamase inhibition. Drugs 70:651–679. doi: 10.2165/11318430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Ehmann DE, Jahić H, Ross PL, Gu RF, Hu J, Durand-Réville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornaglia G, Giamarellou H, Rossolini GM. 2011. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis 11:381–393. doi: 10.1016/S1473-3099(11)70056-1. [DOI] [PubMed] [Google Scholar]
- 7.Brem J, van Berkel SS, Aik W, Rydzik AM, Avison MB, Pettinati I, Umland KD, Kawamura A, Spencer J, Claridge TD, McDonough MA, Schofield CJ. 2014. Rhodanine hydrolysis leads to potent thioenolate mediated metallo-β-lactamase inhibition. Nat Chem 6:1084–1090. doi: 10.1038/nchem.2110. [DOI] [PubMed] [Google Scholar]
- 8.Carfi A, Pares S, Duee E, Galleni M, Duez C, Frère JM, Dideberg O. 1995. The 3-D structure of a zinc metallo-β-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J 14:4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karsisiotis AI, Damblon CF, Roberts GCK. 2014. A variety of roles for versatile zinc in metallo-β-lactamases. Metallomics 6:1181–1197. doi: 10.1039/c4mt00066h. [DOI] [PubMed] [Google Scholar]
- 10.Fast W, Sutton LD. 2013. Metallo-β-lactamase: inhibitors and reporter substrates. Biochim Biophys Acta 1834:1648–1659. doi: 10.1016/j.bbapap.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Buynak JD. 2013. β-Lactamase inhibitors: a review of the patent literature (2010–2013). Expert Opin Ther Pat 23:1469–1481. doi: 10.1517/13543776.2013.831071. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Xu Y, Xia Q, Bai C, Wang T, Wang L, He D, Xie N, Li L, Wang J, Zhou HG, Xu F, Yang C, Zhang Q, Yin Z, Guo Y, Chen Y. 2014. Simplified captopril analogues as NDM-1 inhibitors. Bioorg Med Chem Lett 24:386–389. doi: 10.1016/j.bmcl.2013.10.068. [DOI] [PubMed] [Google Scholar]
- 13.Day JA, Cohen SM. 2013. Investigating the selectivity of metalloenzyme inhibitors. J Med Chem 56:7997–8007. doi: 10.1021/jm401053m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma J, Cao Q, McLeod SM, Ferguson K, Gao N, Breeze AL, Hu J. 2015. Target-based whole-cell screening by 1H NMR spectroscopy. Angew Chem Int Ed Engl 54:4764–4767. doi: 10.1002/anie.201410701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nauton L, Kahn R, Garau G, Hernandez JF, Dideberg O. 2008. Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. J Mol Biol 375:257–269. doi: 10.1016/j.jmb.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 16.Badarau A, Page MI. 2006. The variation of catalytic efficiency of Bacillus cereus metallo-β-lactamase with different active site metal ions. Biochemistry 45:10654–10666. doi: 10.1021/bi060934l. [DOI] [PubMed] [Google Scholar]
- 17.Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. 2003. Coordination geometries of metal ions in d- or l-captopril-inhibited metallo-β-lactamases. J Biol Chem 278:20659–20666. doi: 10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]
- 18.Lienard BM, Garau G, Horsfall L, Karsisiotis AI, Damblon C, Lassaux P, Papamicael C, Roberts GC, Galleni M, Dideberg O, Frère JM, Schofield CJ. 2008. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org Biomol Chem 6:2282–2294. doi: 10.1039/b802311e. [DOI] [PubMed] [Google Scholar]
- 19.Rydzik AM, Brem J, van Berkel SS, Pfeffer I, Makena A, Claridge TD, Schofield CJ. 2014. Monitoring conformational changes in the NDM-1 metallo-β-lactamase by 19F NMR spectroscopy. Angew Chem Int Ed Engl 53:3129–3133. doi: 10.1002/anie.201310866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antony J, Gresh N, Olsen L, Hemmingsen L, Schofield CJ, Bauer R. 2002. Binding of d- and l-captopril inhibitors to metallo-β-lactamase studied by polarizable molecular mechanics and quantum mechanics. J Comput Chem 23:1281–1296. doi: 10.1002/jcc.10111. [DOI] [PubMed] [Google Scholar]
- 21.King DT, Worrall LJ, Gruninger R, Strynadka NC. 2012. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J Am Chem Soc 134:11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Saez I, Hopkins J, Papamicael C, Franceschini N, Amicosante G, Rossolini GM, Galleni M, Frère JM, Dideberg O. 2003. The 1.5-A structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, d-captopril. J Biol Chem 278:23868–23873. doi: 10.1074/jbc.M301062200. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Saez I, Mercuri PS, Papamicael C, Kahn R, Frère JM, Galleni M, Rossolini GM, Dideberg O. 2003. Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-β-lactamase from Fluoribacter gormanii, in native form and in complex with d-captopril. J Mol Biol 325:651–660. doi: 10.1016/S0022-2836(02)01271-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang YT, Lu CY, Hour TC, Cheng TL. 2014. Inhibitor and substrate binding by New Delhi metallo-β-lactamase-1: a molecular dynamics studies. Curr Comput Aided Drug Des 10:197–204. doi: 10.2174/1574886309666141126145225#sthash.fxKtMHtA.dpuf. [DOI] [PubMed] [Google Scholar]
- 25.Menard PR, Suh JT, Jones H, Loev B, Neiss ES, Wilde J, Schwab A, Mann WS. 1985. Angiotensin converting enzyme inhibitors. (Mercaptoaroyl)amino acids. J Med Chem 28:328–332. doi: 10.1021/jm00381a012. [DOI] [PubMed] [Google Scholar]
- 26.Skiles JW, Suh JT, Williams BE, Menard PR, Barton JN, Loev B, Jones H, Neiss ES, Schwab A. 1986. Angiotensin-converting enzyme inhibitors: new orally active 1,4-thiazepine-2,5-diones, 1,4-thiazine-2,5-diones, and 1,4-benzothiazepine-2,5-diones possessing antihypertensive activity. J Med Chem 29:784–796. doi: 10.1021/jm00155a032. [DOI] [PubMed] [Google Scholar]
- 27.van Berkel SS, Brem J, Rydzik AM, Salimraj R, Cain R, Verma A, Owens RJ, Fishwick CWG, Spencer J, Schofield CJ. 2013. Assay platform for clinically relevant metallo-β-lactamases. J Med Chem 56:6945–6953. doi: 10.1021/jm400769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lassaux P, Traore DA, Loisel E, Favier A, Docquier JD, Sohier JS, Laurent C, Bebrone C, Frere JM, Ferrer JL, Galleni M. 2011. Biochemical and structural characterization of the subclass B1 metallo-β-lactamase VIM-4. Antimicrob Agents Chemother 55:1248–1255. doi: 10.1128/AAC.01486-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengerová B, Allerston CK, Abu M, Lee SY, Hartley J, Kiakos K, Schofield CJ, Hartley JA, Gileadi O, McHugh PJ. 2012. Characterization of the human SNM1A and SNM1B/Apollo DNA repair exonucleases. J Biol Chem 287:26254–26267. doi: 10.1074/jbc.M112.367243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR. 2009. Characterization of a new metallo-β-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 53:5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Salabi A, Borra PS, Toleman MA, Samuelsen O, Walsh TR. 2012. Genetic and biochemical characterization of a novel metallo-β-lactamase, TMB-1, from an Achromobacter xylosoxidans strain isolated in Tripoli, Libya. Antimicrob Agents Chemother 56:2241–2245. doi: 10.1128/AAC.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, Rao Z. 2011. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2:384–394. doi: 10.1007/s13238-011-1055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vella P, Hussein WM, Leung EWW, Clayton D, Ollis DL, Mitić N, Schenk G, McGeary RP. 2011. The identification of new metallo-β-lactamase inhibitor leads from fragment-based screening. Bioorg Med Chem Lett 21:3282–3285. doi: 10.1016/j.bmcl.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 38.Davey MS, Tyrrell JM, Howe RA, Walsh TR, Moser B, Toleman MA, Eberl M. 2011. A promising target for treatment of multidrug-resistant bacterial infections. Antimicrob Agents Chemother 55:3635–3636. doi: 10.1128/AAC.00382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frère JM, Payne DJ, Bateson JH, Abdel-Meguid SS. 2000. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 40.Davies AM, Rasia RM, Vila AJ, Sutton BJ, Fabiane SM. 2005. Effect of pH on the active site of an Arg121Cys mutant of the metallo-β-lactamase from Bacillus cereus: implications for the enzyme mechanism. Biochemistry 44:4841–4849. doi: 10.1021/bi047709t. [DOI] [PubMed] [Google Scholar]
- 41.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM. 2001. Standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brem J, Struwe WB, Rydzik AM, Tarhonskaya H, Pfeffer I, Flashman E, van Berkel SS, Spencer J, Claridge TD, McDonough MA, Benesch JL, Schofield CJ. 2015. Studying the active-site loop movement of the Sao Paolo metallo-β-lactamase-1. Chem Sci 6:956–963. doi: 10.1039/C4SC01752H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garau G, Garcia-Saez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frere JM, Dideberg O. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. 2008. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J Mol Biol 375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, Hao Q. 2011. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J 25:2574–2582. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 46.Merino M, Perez-Llarena FJ, Kerff F, Poza M, Mallo S, Rumbo-Feal S, Beceiro A, Juan C, Oliver A, Bou G. 2010. Role of changes in the L3 loop of the active site in the evolution of enzymatic activity of VIM-type metallo-β-lactamases. J Antimicrob Chemother 65:1950–1954. doi: 10.1093/jac/dkq259. [DOI] [PubMed] [Google Scholar]
- 47.Mojica MF, Mahler SG, Bethel CR, Taracila MA, Kosmopoulou M, Papp-Wallace KM, Llarrull LI, Wilson BM, Marshall SH, Wallace CJ, Villegas MV, Harris ME, Vila AJ, Spencer J, Bonomo RA. 2015. Exploring the role of residue 228 in substrate and inhibitor recognition by VIM metallo-β-lactamases. Biochemistry 54:3183–3196. doi: 10.1021/acs.biochem.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cadag E, Vitalis E, Lennox KP, Zhou CL, Zemla AT. 2012. Computational analysis of pathogen-borne metallo β-lactamases reveals discriminating structural features between B1 types. BMC Res Notes 5:96. doi: 10.1186/1756-0500-5-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edelstein MV, Skleenova EN, Shevchenko OV, D'Souza JW, Tapalski DV, Azizov IS, Sukhorukova MV, Pavlukov RA, Kozlov RS, Toleman MA, Walsh TR. 2013. Spread of extensively resistant VIM-2-positive ST235 Pseudomonas aeruginosa in Belarus, Kazakhstan, and Russia: a longitudinal epidemiological and clinical study. Lancet Infect Dis 13:867–876. doi: 10.1016/S1473-3099(13)70168-3. [DOI] [PubMed] [Google Scholar]
- 50.Meini MR, Llarrull LI, Vila AJ. 20 August 2015. Overcoming differences: the catalytic mechanism of metallo-β-lactamases. FEBS Lett doi: 10.1016/j.febslet.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.