

Abstract
The activity of daptomycin (DAP) against methicillin-resistant Staphylococcus aureus (MRSA) is enhanced in the presence of β-lactam antibiotics. This effect is more pronounced with β-lactam antibiotics that exhibit avid binding to penicillin binding protein 1 (PBP1). Here, we present evidence that PBP1 has a significant role in responding to DAP-induced stress on the cell. Expression of the pbpA transcript, encoding PBP1, was specifically induced by DAP exposure whereas expression of pbpB, pbpC, and pbpD, encoding PBP2, PBP3, and PBP4, respectively, remained unchanged. Using a MRSA COL strain with pbpA under an inducible promoter, increased pbpA transcription was accompanied by reduced susceptibility to, and killing by, DAP in vitro. Exposure to β-lactams that preferentially inactivate PBP1 was not associated with increased DAP binding, suggesting that synergy in the setting of anti-PBP1 pharmacotherapy results from increased DAP potency on a per-molecule basis. Combination exposure in an in vitro pharmacokinetic/pharmacodynamic model system with β-lactams that preferentially inactivate PBP1 (DAP-meropenem [MEM] or DAP-imipenem [IPM]) resulted in more-rapid killing than did combination exposure with DAP-nafcillin (NAF) (nonselective), DAP-ceftriaxone (CRO) or DAP-cefotaxime (CTX) (PBP2 selective), DAP-cefaclor (CEC) (PBP3 selective), or DAP-cefoxitin (FOX) (PBP4 selective). Compared to β-lactams with poor PBP1 binding specificity, exposure of S. aureus to DAP plus PBP1-selective β-lactams resulted in an increased frequency of septation and cell wall abnormalities. These data suggest that PBP1 activity may contribute to survival during DAP-induced metabolic stress. Therefore, targeted inactivation of PBP1 may enhance the antimicrobial efficiency of DAP, supporting the use of DAP–β-lactam combination therapy for serious MRSA infections, particularly when the β-lactam undermines the PBP1-mediated compensatory response.
INTRODUCTION
The presence of subinhibitory concentrations of β-lactam antibiotics increases daptomycin (DAP) activity against both DAP-susceptible and -nonsusceptible (DNS) methicillin-resistant Staphylococcus aureus (MRSA) (1, 2). The mechanisms for this are not fully understood, but some β-lactams have been observed to increase binding of DAP to bacterial cell membranes (3, 4) or to target binding to membrane regions where DAP is most effective (5). This is consistent with reports of a DAP–β-lactam “seesaw effect,” whereby S. aureus frequently gains susceptibility to β-lactams upon acquisition of the DAP-nonsusceptible (DNS) phenotype (6). Collectively, these in vitro observations have been translated to antimicrobial therapy combinations to successfully manage difficult-to-treat MRSA infections (1, 3, 6, 7).
The targets of β-lactam antibiotics are the penicillin binding proteins (PBPs) that assemble and cross-link the bacterial cell wall through transglycosylation and transpeptidation. S. aureus produces four PBPs, all of which retain transpeptidase activity but only one of which (PBP2) also demonstrates transglycosylase activity (8). The mecA element found in methicillin-resistant S. aureus (MRSA) encodes a fifth PBP (PBP2a), a transpeptidase that is resistant to β-lactam inactivation (9, 10). The relative affinity of different β-lactams for PBPs varies: some β-lactams bind to and inactivate one PBP preferentially, while other β-lactams demonstrate relatively nonselective binding to multiple PBPs (11, 12). Previous work has suggested that the degree to which different β-lactams potentiate the anti-MRSA activity of DAP may be associated with the relative affinity for PBP1 (13). This specific differential effect echoes other recent studies that observed increased toxin expression (12) and induction of DNA repair systems (14) upon inhibition of PBP1 but not upon inhibition of other PBPs (12, 15–17).
We hypothesize that the components of the cellular divisome, including PBP1, may form a critical adaptive response to DAP-mediated surface injury and that β-lactams which compromise PBP1 activity may enhance the efficiency of DAP killing without necessarily increasing DAP binding.
Importantly, PBP1 of S. aureus is homologous to PBP2x of Streptococcus pneumoniae and PBP3 of Escherichia coli, each comprised of a C-terminal transpeptidase domain and an N-terminal structural domain recently identified as a critical component in the bacterial divisome complex responsible for mediating cell division (18, 19). In fact, these homologous PBPs, including PBP1 in S. aureus, are located in division and cell wall synthesis clusters on their respective bacterial chromosomes. Furthermore, mediation of cell division appears to supersede peptidoglycan synthesis in the hierarchy of PBP1 function, given that depletion of PBP1 has been shown to induce abnormal cell morphology and incomplete septation but does not appear to alter peptidoglycan cross-linking (18), whereas depletion of PBP2 or PBP4 results in significantly altered peptidoglycan (10, 20).
This study provides evidence that PBP1 contributes to survival in the presence of DAP and that modulation of PBP1 activity can alter the in vitro efficacy of DAP killing.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. aureus strains D592 and D712 are paired isogenic MRSA isolates from a clinical series using DAP plus antistaphylococcal β-lactam in refractory bacteremia (3). Strain D592 is the DAP-susceptible index isolate, whereas D712 is a subsequent isolate that had become DNS as well as vancomycin intermediate following multiple unsuccessful antimicrobial regimens, as previously characterized (13). The genetic relatedness of the two patient isolates was confirmed by whole-genome sequencing (21). S. aureus strain COL is a prototypical MRSA strain that is DAP susceptible (18, 22), and constructs placing pbpA under an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter have been previously described (18). All antibacterials were purchased as commercial agents. MICs were determined by broth microdilution per the Clinical and Laboratory Standards Institute guidelines and with S. aureus ATCC 29213 for quality control (23). S. aureus was propagated in calcium-supplemented (50-mg/liter) Mueller-Hinton broth. For each antibiotic, the fCavg is here defined as the average free concentration of drug achieved in serum during a typical dosing interval, selected with the formula fCavg = (fCmax + fCmin)/2, where fCmax and fCmin are maximum and minimum free concentrations of drug in serum, respectively.
Cathelicidin LL37 killing assays.
Cationic peptides have an antibiotic mechanism similar to that of DAP, and yet distinct differences in potency and membrane activity have been identified (24). Therefore, the primary antimicrobial peptide LL37 was evaluated in combination with β-lactams. S. aureus D712 was grown in antibiotic-free LB broth or broth containing 5 mg/liter of the test β-lactam antibiotic overnight (15 to 18 h), resuspended in phosphate-buffered saline (PBS), and subjected to killing assays in 128 μM LL37, as previously described (25).
DAP binding assays.
Enhanced DAP binding in the presence of β-lactams is one proposed mechanism for synergy, but only select β-lactams have been previously evaluated. This study provides a more comprehensive evaluation of β-lactams with distinct PBP inhibition profiles. S. aureus D712 was grown in antibiotic-free LB broth or broth containing 5 mg/liter of the test β-lactam antibiotic to an optical density at 600 nm (OD600) of 0.4 to 0.6, stained for 30 min with dipyrromethene boron difluoride (BODIPY)-DAP (16 mg/liter) and Ca2+ (50 mg/liter), and visualized microscopically as previously described (25). DAP binding was evaluated visually and measured by binding intensity and spots per cell to determine the quality and focus of binding. A β-lactam concentration of 5 mg/liter was used for the binding and LL37 killing assays in order to study this effect at physiologically relevant concentrations and provide relative comparisons among the β-lactams.
Autolysis assays.
Resistance to cationic peptides has been linked to reduced autolysis (26), so it is of interest to evaluate autolysis during β-lactam exposure to understand how β-lactams might alter DAP activity. S. aureus D712 was grown in antibiotic-free LB broth or broth containing 5 mg/liter of the test β-lactam antibiotic overnight (15 to 18 h), resuspended in 0.1% Triton X-100 in PBS to an OD600 of 0.8, and measured spectrophotometrically over time. Results were expressed graphically as percent OD remaining versus time zero.
FITC-labeled PLL binding.
DAP activity has been previously associated with alterations in cell surface charge (26), although such changes are not observed in all DAP-nonsusceptible S. aureus isolates (24). Fluorescein isothiocyanate (FITC)-labeled poly-l-lysine (PLL) binding studies were performed to evaluate surface charge in the presence of β-lactams. Assays were performed using a flow cytometric method as previously described (27). PLL is a polycationic molecule used to study the interactions of cationic peptides with charged bacterial envelopes. In this analysis, the extent of bacterium-bound FITC-labeled PLL inversely reflects the relative surface positive charge. A total of 10,000 events were counted and analyzed using a BD FACSCalibur system (Becton Dickinson Labware, San Jose, CA). At least two independent experiments of triplicate samples were performed.
qPCR.
Reverse transcription-PCR (RT-PCR) was performed to identify the effect of DAP exposure on PBP regulation in S. aureus. Samples were prepared and analyzed as previously described (12). Briefly, overnight cultures were diluted 1:100 in fresh medium and propagated at 37°C (250 rpm) for 6 h in the presence or absence of 0.125 mg/liter DAP. Total RNA from pelleted cultures was isolated using phenol-chloroform and treated with DNase (Life Technologies, Madison, WI, USA) per manufacturer recommendations. Purified RNA (500 ng) was reverse transcribed (iScript; Bio-Rad, Hercules, CA, USA), and the resulting cDNA was used as the template for real-time PCR (StepOne Plus; Applied Biosystems, Foster City, CA, USA) with Kapa Sybr green universal quantitative CPR (qPCR) mix (Kapa Biosystems, Wilmington, MA, USA) using primers as described by Dumitrescu et al. (12). Relative expression levels were normalized to gyrB (StepOne Plus software; Applied Biosystems).
Effects of PBP1 expression modulation on DAP activity.
A strain of MRSA COL with pbpA under the control of a spac1 promoter with the corresponding relative PBP1 expression previously described (18) was grown overnight (14 to 18 h) in LB containing erythromycin at 10 mg/liter and IPTG at 35, 100, or 1,000 μM and then diluted 20× into fresh LB containing the same IPTG concentration plus 50 mg/liter Ca2+ and either 16 mg/liter DAP or no antibiotic. CFU were enumerated at time zero and 90 min by plating 10-μl aliquots in triplicate. The experiment was performed 3 times, and all results were pooled prior to analysis.
In vitro PK/PD model.
A previously described in vitro pharmacokinetic/pharmacodynamic (PK/PD) model was used for simulating one-compartment antibiotic exposures of DAP and/or study β-lactams (28, 29). All model experiments were performed in duplicate. Overnight cultures of DNS MRSA strain D712 were adjusted to obtain a starting model inoculum of ∼106 CFU/ml. The following antibiotic regimens were evaluated: (i) DAP at 6 mg/kg of body weight every 24 h (targeted maximum free drug concentration [fCmax], 7.2 mg/liter; half-life, 8 h), (ii) imipenem (IPM) at 1,000 mg every 8 h (fCmax, 50 mg/liter; half-life, 1 h), (iii) nafcillin (NAF) at 2,000 mg every 4 h (fCmax, 5 mg/liter; half-life, 1 h), (iv) cefotaxime (CTX) at 2,000 mg every 6 h (fCmax, 128 mg/liter; half-life, 1.2 h), (v) cefaclor (CEC) at 500 mg every 8 h (fCmax, 13 mg/liter; half-life, 1 h), (vi) cefoxitin (FOX) at 2,000 mg every 6 h (fCmax, 43 mg/liter; half-life, 1 h), (vii) meropenem (MEM) at 1,000 mg every 8 h (fCmax, 110 mg/liter; half-life, 1 h), and (viii) ceftriaxone (CRO) at 1,000 mg every 24 h (fCmax, 20 mg/liter; half-life, 8 h). Modeling of antibiotics in combination with two different elimination rates was performed according to the methods described by Blaser (30). Areas under the growth curve were calculated using the trapezoidal method.
Electron microscopy.
Transmission electron microscopy (TEM) images were obtained as described previously (31). Samples were collected following overnight growth in β-lactam antibiotic at the average unbound serum concentration obtained from the regimens modeled in the in vitro pharmacokinetic/pharmacodynamic experiment. Cell wall thickness measurements were determined using ImageJ 1.39t software on a minimum of 25 cells per treatment using four separate quadrants of each cell. Septation frequency was determined by examination of a minimum of 25 cells per treatment.
Statistical analysis.
Areas under the growth curve for in vitro model cultures were assessed via one-way analysis of variance (ANOVA) at predefined time points (4 h, 12 h, 24 h, and 48 h), and combination exposures were compared using Tukey's honestly significant difference (HSD) post hoc test. Groupings of combination exposures comparing PBP1-selective to non-PBP1-selective β-lactams were assessed using Student's t test. All statistical analyses were performed using GraphPad Prism version 6.05 (GraphPad Software, Inc., La Jolla CA).
RESULTS
Antibiotic susceptibilities.
Results of susceptibility testing with study antibiotics are reported in Table 1. All study strains were resistant to β-lactam antibiotics; however, the presence of β-lactam antibiotics reduced the amount of DAP required to inhibit organism growth regardless of the PBP binding profile of the β-lactam or the strain background.
TABLE 1.
MICs of single agents and of DAP in the presence of β-lactams
| Strain | MIC (mg/liter) ofa: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Single agent: |
DAP in presence of β-lactam (fCavg): |
||||||||||||||
| DAP | IPM | MEM | ERT | NAF | CTX | CEC | FOX | IPM (11) | MEM (55) | ERT (4) | NAF (2.7) | CTX (65) | CEC (6.5) | FOX (16) | |
| D592 | 0.5 | 64 | 128 | 64 | 256 | 256 | 128 | 256 | 0.13 | 0.03 | 0.13 | 0.13 | 0.13 | 0.13 | 0.13 |
| D712 | 2 | 32 | 32 | 32 | 256 | 256 | 64 | 256 | 0.5 | NG | 0.5 | 0.5 | 0.5 | 1 | 1 |
| COL | 0.5 | 16 | 16 | 64 | 64 | 256 | 128 | 256 | |||||||
Abbreviations: ERT, ertapenem; NG, no growth.
Effects of DAP on PBP expression in S. aureus D712.
To identify potential effects of DAP exposure on PBP transcription, qPCR was performed on S. aureus D712 exposed to subinhibitory DAP. Results are presented in Fig. 1. We observed that DAP exposure differentially affected expression of PBPs. Expression of pbpA (encoding PBP1) in cultures exposed to DAP increased 5.3-fold compared to a no-antibiotic control (P < 0.01). In contrast, exposure to DAP did not result in significant changes to expression of transcripts encoding other PBPs (pbpB [PBP2], 1.1-fold; pbpC [PBP3], 0.8-fold; pbpD [PBP4], 1.9-fold; P > 0.05 for all comparisons).
FIG 1.
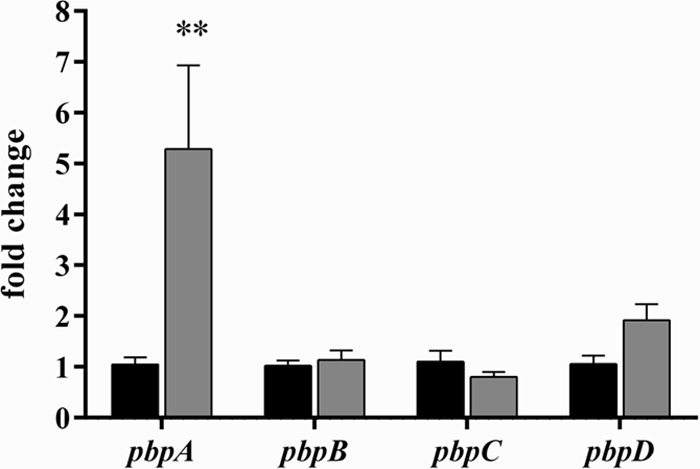
PBP expression profile following exposure to subinhibitory daptomycin. Black bars, no-antibiotic control; gray bars, 1/4× daptomycin MIC. Values marked with an asterisk denote statistically significant differences between daptomycin exposure and the no-antibiotic control (**, P < 0.01).
Effects of inducible PBP1 production on DAP susceptibility.
To assess if PBP1 expression contributes to DAP nonsusceptibility, the MIC of DAP was determined in S. aureus strain COLspacP1 in the presence of increasing IPTG concentrations. COLspacP1 is a derivative of COL in which expression of pbpA (encoding PBP1) is controlled by the IPTG-inducible Pspac promoter (18). Results are presented in Table 2. DAP MICs increased in an IPTG dose-dependent manner, resulting in DAP nonsusceptibility with IPTG concentrations in excess of 100 μM. DAP bacterial killing is presented in Fig. 2. Recoverable CFU after 90 min of DAP exposure increased significantly in an IPTG dose-dependent manner, suggesting that DAP is less effective under conditions of high PBP1 production.
TABLE 2.
Increase in daptomycin MICs in S. aureus COL upon pbp1 induction
| IPTG concn added (μM) | DAP MIC (mg/liter) in strain: |
|
|---|---|---|
| S. aureus COL | S. aureus COLPspacPBP1 | |
| 1 | 0.5 | 0.5 |
| 10 | 0.5 | 1 |
| 100 | 0.5 | 4 |
| 1,000 | 0.5 | 4 |
FIG 2.
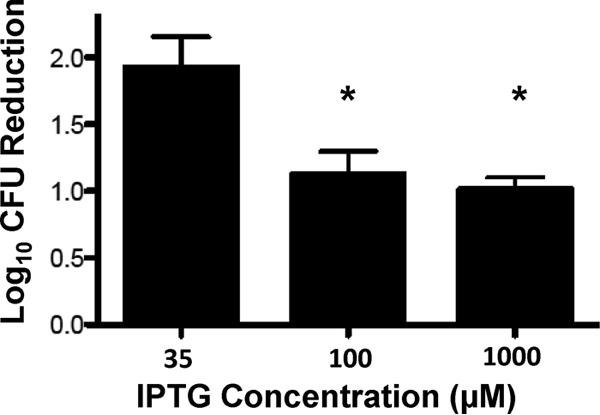
Bacterial recovery after daptomycin exposure with varied amounts of IPTG inducer. Data represent the differences in the number of recoverable CFU evaluated at 90 min relative to the number of recoverable CFU at baseline. Values marked with an asterisk denote statistically significant differences between the respective treatment arm and the 35 μM IPTG control required for basal-level expression of pbpA (*, P = 0.03).
Effect of adjunctive β-lactams on DAP activity in an in vitro PK/PD model.
β-Lactams with different specificities for S. aureus PBPs were modeled with DAP in an in vitro PK/PD simulation to investigate if inactivation of PBP1 chemically would result in enhanced antimicrobial activity. Results are presented in Fig. 3 and Table 3. Addition of any β-lactam enhanced the efficacy of DAP, and this activity was particularly relevant during the initial synergy phase of exposure in terms of both area under the concentration-time curve from 0 to 4 h (AUC0–4) and time to 3-log reduction in recoverable CFU per milliliter. This was anticipated, as the initial β-lactam concentration following a dose (the fCmax) was predicted to exceed the 50% inhibitory concentration (IC50) for all four PBPs. However, the duration of PBP blockade during the dosing interval will depend on the specificity of the individual β-lactam for different PBPs. Comparing DAP simulations containing PBP1-specific β-lactams (DAP-MEM and DAP-IPM) and simulations containing non-PBP1-specific β-lactams (DAP-NAF, DAP-CRO, DAP-CTX, DAP-CEC, and DAP-FOX), simulations containing PBP1-specific β-lactams were consistently superior in terms of lower AUCs throughout the simulation and times to 3-log reductions in recoverable CFU per milliliter (P < 0.05 at all time points assessed).
FIG 3.

Activity of daptomycin and/or β-lactam in a 48-hour in vitro pharmacokinetic/pharmacodynamic model. Dashed line, growth control; small black circles, daptomycin; white symbols, β-lactam monotherapy; gray symbols, daptomycin–β-lactam.
TABLE 3.
Effectiveness of DAP with or without β-lactam in in vitro pharmacokinetic/pharmacodynamic modela
| Antibacterial(s) | AUC (log10 CFU · h/ml) |
Time to achieve 3-log10 reduction (h) | |||
|---|---|---|---|---|---|
| 0–4 h | 0–12 h | 0–24 h | 0–48 h | ||
| None (growth) | 24.0 ± 0.7 | 78.2 ± 5.7 | 174.3 ± 14.0 | 384.8 ± 19.8 | |
| DAP | 19.2 ± 0.6 | 42.2 ± 1.4 | 84.4 ± 5.4 | 191.4 ± 31.9 | 6.93 ± 0.30 |
| DAP + IPM | 10.1 ± 0.4 | 25.2 ± 4.3 | 58.5 ± 10.3 | 147.9 ± 33.4 | 1.10 ± 0.02 |
| DAP + MEM | 10.7 ± 0.1 | 28.3 ± 0.0 | 63.6 ± 11.3 | 140.1 ± 19.8 | 1.08 ± 0.18 |
| DAP + NAF | 13.7 ± 0.1 | 35.8 ± 3.9 | 84.7 ± 6.6 | 212.8 ± 21.4 | 1.76 ± 0.08 |
| DAP + CTX | 13.6 ± 0.2 | 39.0 ± 5.4 | 85.6 ± 18.9 | 192.6 ± 46.9 | 1.71 ± 0.04 |
| DAP + CRO | 15.5 ± 0.2 | 39.4 ± 1.6 | 93.0 ± 8.6 | 208.8 ± 22.9 | 1.74 ± 0.03 |
| DAP + CEC | 14.1 ± 1.3 | 29.5 ± 4.7 | 68.2 ± 14.0 | 171.6 ± 28.9 | 2.68 ± 0.56 |
| DAP + FOX | 12.9 ± 0.1 | 35.2 ± 2.0 | 95.1 ± 1.9 | 248.9 ± 16.4 | 1.83 ± 0.12 |
Values represent the average ± standard deviation from two independent experiments.
Effect of adjunctive β-lactams on DAP binding, autolysis, and killing by cathelicidin LL37.
S. aureus D712 was grown in medium containing 5 mg/liter of antibiotic selected from a diverse panel of β-lactams with differential specificities in PBP binding, and the effects of β-lactam exposure on DAP binding, autolysis, PLL binding, and susceptibility to LL37 killing were measured. As shown in Fig. 4, exposure to most β-lactams increased both the number of BODIPY-DAP spots per cell and the intensity of binding at those spots. The exception to this trend was IPM, a β-lactam with high specificity for PBP1. Treatment with IPM resulted in no significant increase in the number of binding foci on the surface of the bacteria, even though it was one of the most effective synergistic agents in combination with DAP (Fig. 4B). This finding paralleled the results of the other experiments whereby IPM was unique among the β-lactams tested in that exposure did not significantly alter surface charge (see Fig. S1 in the supplemental material), autolysis in Triton X-100 (Fig. 5A), or sensitization to killing by LL37 (Fig. 5B).
FIG 4.

Binding of BODIPY-daptomycin to S. aureus D712 following overnight β-lactam exposure. (A) Fluorescence microscopy of S. aureus pretreated for 24 h in the presence of 5 mg/liter of the β-lactam indicated and subsequently exposed to 16 mg/liter BODIPY-daptomycin for 20 min. (B) Number of BODIPY-daptomycin spots per cell. (C) Signal intensity of BODIPY-daptomycin bound to S. aureus membranes.
FIG 5.

Effects of pretreatment with subinhibitory β-lactam on S. aureus autolysis. (A) Triton X-100 autolysis. Bacteria were pretreated overnight with β-lactam. Antibiotic was removed at time zero, and bacteria were resuspended in 0.1% Triton X-100. Data represent the percentages of the initial OD600 reading evaluated at preset time points following Triton X-100 exposure. (B) Cathelicidin (LL37)-mediated lysis. Bacteria were pretreated overnight with β-lactam. Antibiotic was removed at time zero, and bacteria were resuspended in PBS containing 128 μM LL37. Data represent the percentages of recoverable CFU remaining after 2 h of incubation with LL37.
Effect of adjunctive β-lactams on cell morphology.
Electron microscopy images of S. aureus D712 exposed to β-lactam were examined to investigate if specific chemical inactivation of different PBPs would result in distinct alterations in cell morphology. Results are presented in Fig. 6 and Table 4. Addition of β-lactam with affinity for PBP1 significantly increased the frequency of cells undergoing septation events relative to exposure to DAP alone (P < 0.001). Addition of β-lactam with nonspecific PBP binding also significantly increased the frequency of septation (P < 0.05). In contrast, addition of β-lactam with affinity for PBP2, PBP3, or PBP4 resulted in no difference in the septation frequency (P > 0.05). S. aureus cell walls demonstrated significant thickening upon exposure to β-lactam regardless of PBP specificity, with the exception of the PBP3-specific drug CEC, which resulted in significantly thinner cell walls (P < 0.01 for all comparisons). Exposure to β-lactams with affinity for either PBP1 or PBP2 resulted in larger cell diameters (P < 0.01), whereas exposure to PBP3- or PBP4-specific agents had no effect on cell size (P > 0.05).
FIG 6.
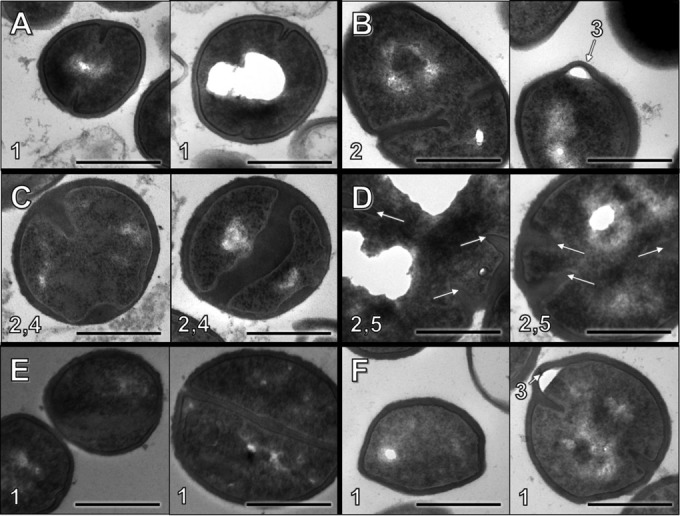
Cell morphology observations following overnight β-lactam exposure at fCavg concentration. Each panel consists of two representative images of S. aureus D712 exposed to the following conditions: antibiotic-free control (A), NAF (B), MEM (C), CTX (D), CEC (E), and FOX (F). Notable features: 1, well-defined borders and/or septa; 2, abnormal septation/separation; 3, membrane invagination; 4, inconsistent cell wall thickness; 5, atypical cell shape/size. Bar, 500 nm.
TABLE 4.
Differential alteration of cell morphology characteristics by β-lactam inhibition of discrete PBPsa
| Exposure | S. aureus PBP specificity (12, 13) | % of cells containing septa | Cell wall thickness (nm) | Cell diam (nm) |
|---|---|---|---|---|
| Medium-only control | 12.8 | 26.7 ± 5.36 | 662 ± 61 | |
| NAF | Nonspecific | 38.9* | 35.4 ± 5.65** | 914 ± 155** |
| MEM | PBP1 | 73.9** | 33.7 ± 6.07** | 1,002 ± 135** |
| CTX | PBP2 | 37.5** | 47.1 ± 11.86** | 1,307 ± 215** |
| CEC | PBP3 | 24.0 | 21.7 ± 4.65** | 729 ± 99 |
| FOX | PBP4 | 25.4 | 34.1 ± 4.70** | 748 ± 92* |
Values marked with asterisks denote statistically significant differences between β-lactam exposure and medium-only control (*, P < 0.05; **, P < 0.01).
DISCUSSION
Several published studies have noted synergy between DAP and β-lactam antibiotics, both in vitro (2, 6, 32) and in vivo (1, 3, 7). This improved activity has largely been attributed to enhanced DAP binding to S. aureus in the presence of β-lactam antibiotics (3). However, alterations to the cell membrane surface charge thought to result in enhanced binding are not observed in all strains that show synergy (33). The demonstrated effectiveness of this combination therapy despite heterogeneity in the specific β-lactam employed reflects the possibility that DAP–β-lactam synergy may be a β-lactam class effect. However, a recent study observed significant diversity among different β-lactams in their relative efficacies in combination with DAP and reported that synergy is dependent on the relative affinity of a β-lactam for PBP1 (13).
Here, we have established several additional lines of evidence that increased production of PBP1 is a crucial compensatory response to DAP injury and that undermining this response is a viable strategy in enhancing DAP activity pharmacodynamically using currently available β-lactams in combination therapy: (i) exposure to subinhibitory DAP resulted in increased transcription of pbpA, the gene encoding PBP1, but not of genes encoding other S. aureus PBPs; (ii) using a previously characterized bacterial construct placing pbpA under control inducible by IPTG in MRSA COL, increased pbpA transcription was accompanied by reduced susceptibility to, and killing by, DAP in vitro; (iii) exposure to β-lactams exhibiting high selectivity for PBP1 resulted in more-rapid bactericidal activity and lower areas under the inhibitory curve (AUICs) relative to β-lactams with low selectivity for PBP1 in pharmacokinetic/pharmacodynamic modeling of DAP; (iv) the PBP1-selective β-lactams enhanced DAP killing without enhancing DAP binding, suggesting a mechanism of enhancing the efficiency of DAP killing per molecule of DAP membrane insertion, rather than increasing the number of DAP molecules binding to the membrane; (v) supporting the latter, PBP1-selective β-lactams, unlike β-lactams binding selectively to other PBPs, did not enhance PLL binding (i.e., reduce net surface charge) or increase autolysis. These findings are supportive of prior work demonstrating that mediation of cell division appears to be a primary role for PBP1 in S. aureus that supersedes its C-terminal transpeptidase function (18). One response of bacteria to DAP exposure appears to be an increased rate of septation and cell division (5). Interference with this aspect of PBP1 function, therefore, appears to dominate the mechanism of synergy with DAP in MRSA, given that DAP binding is not enhanced by PBP1-selective agents such as the carbapenems. To this point, there was an overall lack of cell surface charge alteration with β-lactams, which is in contrast to previous studies with nafcillin and cephalosporins (3, 34). This effect may play a role in moderately enhancing DAP activity, but the PBP1 target is essential for synergistic effect.
In addition, these data demonstrate that PBP1-specific β-lactams appear to discriminate between enhancement of DAP and of cationic antimicrobial peptides such as cathelicidin (LL37). We have previously shown that penicillins and cephalosporins sensitize MRSA to killing by LL37 and other host defense peptides, but we did not include carbapenems in that analysis (25). Interestingly, in this study we found that growth of MRSA in the PBP1-specific carbapenems did not enhance LL37 killing, despite potentiation of DAP activity. This paralleled their inability to induce autolysis or alter surface charge. Enhanced autolysis has been consistently linked to increased susceptibility to host defense peptide killing, with bacterial strains that show a relative resistance to host defense peptide killing having a reduced autolysis phenotype (26).
It appears, therefore, that while killing by the DAP-calcium complex parallels that of host defense peptides, there are critical differences that are distinguished by carbapenems, presumably due to the effects on PBP1. The activity of DAP is dependent not only on the quantity of binding but also on the quality of binding, as some β-lactams induce marked increases in DAP surface binding but do not increase DAP potency. This feature has been recently highlighted in the mechanism of DAP resistance in Enterococcus faecalis, whereby DAP is bound and diverted to membrane sites away from the septum and, therefore, bound drug is sequestered to a site where it is functionally inactive in killing the bacterium (35).
NAF, which is considered to be the optimal agent against serious methicillin-susceptible S. aureus infections, appears to exhibit a “best of both worlds” phenotype of potentiating the activity of both DAP and LL37. Carbapenems are effective in potentiating DAP activity but offer no enhancement of killing by host defense peptides of the innate immune system, whereas cephalosporins do potentiate innate host defense peptides. While DAP binding is enhanced by NAF, it may be that its effects on PBP1 through its broader PBP binding profile may actually be responsible for its potentiation of DAP activity, rather than the increased DAP binding. These findings highlight that the potency of antimicrobial therapy should be gauged not simply through in vitro killing but rather through a concerted effect of direct killing and the enhancement of defense peptides that are already being produced in the host.
Differences in morphology identified following exposure to β-lactams with different PBP specificities may illuminate why β-lactams that inactivate PBP1 appear to have the strongest synergy with DAP. Recent work in Bacillus subtilis suggests that DAP preferentially binds to regions of extreme membrane strain, such as those found around nascent septa (5). Upon DAP damage of the cell membrane in B. subtilis, relocalization of proteins involved in cell division and cell wall synthesis occurs and results in defects in cell wall and cell membrane formation. We hypothesize that DAP exposure in S. aureus results in a similar disruption in essential proteins involved in cell division, and selective or nonselective β-lactam inhibition of PBP1, which is the PBP in S. aureus responsible for cell division, leads to enhanced lethality by disrupting this important response mechanism. In our study, strains exposed to β-lactams that inactivate PBP1 show a significantly increased prevalence of cells in the process of nascent septation. Additionally, S. aureus exposed to β-lactams that inactivate PBP1 may experience further membrane strain due to the significant inconsistencies in cell wall thickness within a single cell. While exposure to most β-lactams tested resulted in thickened cell walls, the significance of this finding is unclear, as previous studies have demonstrated that cell wall thickness has no significant direct effect on DAP activity (36). However, for isolates exposed to β-lactams specific for PBP1, the cell wall thickening was not uniform within a single cell, resulting in a wavy cell wall-membrane interface. These extremes of cell wall thickening and thinning were not observed following exposure to β-lactams with alternative binding profiles (both specific and nonspecific) and may result in strained membranes providing targeted sites for more-effective DAP binding rather than less-effective diffuse binding.
In summary, S. aureus PBPs differ in their role with respect to cell wall maintenance and response to cell injury. Their common property of binding β-lactam antibiotics has led some to presume that they are variations of the same theme and represent bacterial redundancy. On the contrary, we have found that PBP1 of S. aureus stands out as a protein whose production is enhanced by DAP-induced cell injury and that increasing PBP1 renders MRSA less susceptible to DAP killing and growth inhibition. Carbapenems that preferentially bind PBP1 allow for enhanced DAP activity by not necessarily increasing DAP binding, presumably because of efficiency in killing. In contrast, cephalosporins appear to increase DAP binding but not necessarily activity. NAF stands out as a β-lactam that enhances DAP binding and host defense peptide killing, perhaps laying the foundation of its excellent clinical performance among the other β-lactams in serious S. aureus infections. In order to further explore this PBP1 target and mechanism, we are pursuing additional studies on how individual regulation and selective inactivation of the PBPs in S. aureus impact DAP activity. The effect of DAP exposure on the localization of cell wall and membrane proteins, including PBP1, is also being investigated. Further studies are necessary to reveal the maximal benefits of β-lactam antibiotics as adjunctive therapies against MRSA.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alexander Tomasz (The Rockefeller Institute) for the generous donation of strains used in this study.
Funding Statement
A.D.B. is supported by a Pharmacy Practice research fellowship award from UW–Madison. E.T. was supported by a Medical Sciences Training Program at UW–Madison (T32 GM008692). This work was supported in part by a grant by the National Institutes of Health, Division of National Institute of Child Health and Human Development (NICHD), grant U54 HD071600-01 (09/26/2011-06/30/2016 Developmental and Translational Pharmacology of Pediatric Antimicrobial Therapy [G.S. and V.N.]).The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02071-15.
REFERENCES
- 1.Rose WE, Schulz LT, Andes D, Striker R, Berti AD, Hutson PR, Shukla SK. 2012. Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob Agents Chemother 56:5296–5302. doi: 10.1128/AAC.00797-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rand KH, Houck HJ. 2004. Synergy of daptomycin with oxacillin and other beta-lactams against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 48:2871–2875. doi: 10.1128/AAC.48.8.2871-2875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dhand A, Bayer AS, Pogliano J, Yang SJ, Bolaris M, Nizet V, Wang G, Sakoulas G. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis 53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Werth BJ, Sakoulas G, Rose WE, Pogliano J, Tewhey R, Rybak MJ. 2013. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 57:66–73. doi: 10.1128/AAC.01586-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol 194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang SJ, Xiong YQ, Boyle-Vavra S, Daum R, Jones T, Bayer AS. 2010. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”). Antimicrob Agents Chemother 54:3161–3169. doi: 10.1128/AAC.00487-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moise PA, Amodio-Groton M, Rashid M, Lamp KC, Hoffman-Roberts HL, Sakoulas G, Yoon MJ, Schweitzer S, Rastogi A. 2013. Multicenter evaluation of the clinical outcomes of daptomycin with and without concomitant β-lactams in patients with Staphylococcus aureus bacteremia and mild to moderate renal impairment. Antimicrob Agents Chemother 57:1192–1200. doi: 10.1128/AAC.02192-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgopapadakou NH, Liu FY. 1980. Penicillin-binding proteins in bacteria. Antimicrob Agents Chemother 18:148–157. doi: 10.1128/AAC.18.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartman BJ, Tomasz A. 1984. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J Bacteriol 158:513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinho MG, Filipe SR, de Lencastre H, Tomasz A. 2001. Complementation of the essential peptidoglycan transpeptidase function of penicillin-binding protein 2 (PBP2) by the drug resistance protein PBP2A in Staphylococcus aureus. J Bacteriol 183:6525–6531. doi: 10.1128/JB.183.22.6525-6531.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar KM, Anbarasu A, Ramaiah S. 2014. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol Biosyst 10:891–900. doi: 10.1039/c3mb70537d. [DOI] [PubMed] [Google Scholar]
- 12.Dumitrescu O, Choudhury P, Boisset S, Badiou C, Bes M, Benito Y, Wolz C, Vandenesch F, Etienne J, Cheung AL, Bowden MG, Lina G. 2011. Beta-lactams interfering with PBP1 induce Panton-Valentine leukocidin expression by triggering sarA and rot global regulators of Staphylococcus aureus. Antimicrob Agents Chemother 55:3261–3271. doi: 10.1128/AAC.01401-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berti AD, Sakoulas G, Nizet V, Tewhey R, Rose WE. 2013. β-Lactam antibiotics targeting PBP1 selectively enhance daptomycin activity against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:5005–5012. doi: 10.1128/AAC.00594-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plata KB, Riosa S, Singh CR, Rosato RR, Rosato AE. 2013. Targeting of PBP1 by β-lactams determines recA/SOS response activation in heterogeneous MRSA clinical strains. PLoS One 8:e61083. doi: 10.1371/journal.pone.0061083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muthaiyan A, Silverman JA, Jayaswal RK, Wilkinson BJ. 2008. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob Agents Chemother 52:980–990. doi: 10.1128/AAC.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuroda H, Kuroda M, Cui L, Hiramatsu K. 2007. Subinhibitory concentrations of beta-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol Lett 268:98–105. doi: 10.1111/j.1574-6968.2006.00568.x. [DOI] [PubMed] [Google Scholar]
- 17.Utaida S, Dunman PM, Macapagal D, Murphy E, Projan SJ, Singh VK, Jayaswal RK, Wilkinson BJ. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719–2732. doi: 10.1099/mic.0.26426-0. [DOI] [PubMed] [Google Scholar]
- 18.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2007. Role of PBP1 in cell division of Staphylococcus aureus. J Bacteriol 189:3525–3531. doi: 10.1128/JB.00044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2009. Evidence for a dual role of PBP1 in the cell division and cell separation of Staphylococcus aureus. Mol Microbiol 72:895–904. doi: 10.1111/j.1365-2958.2009.06687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Łeski TA, Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol 187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berti AD, Baines SL, Howden BP, Sakoulas G, Nizet V, Proctor RA, Rose WE. 2015. Heterogeneity of genetic pathways toward daptomycin nonsusceptibility in Staphylococcus aureus determined by adjunctive antibiotics. Antimicrob Agents Chemother 59:2799–2806. doi: 10.1128/AAC.04990-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cassat JE, Dunman PM, McAleese F, Murphy E, Projan SJ, Smeltzer MS. 2005. Comparative genomics of Staphylococcus aureus musculoskeletal isolates. J Bacteriol 187:576–592. doi: 10.1128/JB.187.2.576-592.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clinical and Laboratory Standards Institute. 2010. Performance standards for antimicrobial susceptibility testing; approved standard, 20th informational supplement M100-S20 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 24.Mishra NN, McKinnell J, Yeaman MR, Rubio A, Nast CC, Chen L, Kreiswirth BN, Bayer AS. 2011. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 55:4012–4018. doi: 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V. 2014. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 92:139–149. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones T, Yeaman MR, Sakoulas G, Yang SJ, Proctor RA, Sahl HG, Schrenzel J, Xiong YQ, Bayer AS. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother 52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakoulas G, Bayer AS, Pogliano J, Tsuji BT, Yang SJ, Mishra NN, Nizet V, Yeaman MR, Moise PA. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:838–844. doi: 10.1128/AAC.05551-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose WE, Rybak MJ, Kaatz GW. 2007. Evaluation of daptomycin treatment of Staphylococcus aureus bacterial endocarditis: an in vitro and in vivo simulation using historical and current dosing strategies. J Antimicrob Chemother 60:334–340. doi: 10.1093/jac/dkm170. [DOI] [PubMed] [Google Scholar]
- 29.Akins RL, Rybak MJ. 2000. In vitro activities of daptomycin, arbekacin, vancomycin, and gentamicin alone and/or in combination against glycopeptide intermediate-resistant Staphylococcus aureus in an infection model. Antimicrob Agents Chemother 44:1925–1929. doi: 10.1128/AAC.44.7.1925-1929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blaser J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J Antimicrob Chemother 15(Suppl A):125–130. doi: 10.1093/jac/15.suppl_A.125. [DOI] [PubMed] [Google Scholar]
- 31.Rose WE, Knier RM, Hutson PR. 2010. Pharmacodynamic effect of clinical vancomycin exposures on cell wall thickness in heterogeneous vancomycin-intermediate Staphylococcus aureus. J Antimicrob Chemother 65:2149–2154. doi: 10.1093/jac/dkq292. [DOI] [PubMed] [Google Scholar]
- 32.Barber KE, Werth BJ, Ireland CE, Stone NE, Nonejuie P, Sakoulas G, Pogliano J, Rybak MJ. 2014. Potent synergy of ceftobiprole plus daptomycin against multiple strains of Staphylococcus aureus with various resistance phenotypes. J Antimicrob Chemother 69:3006–3010. doi: 10.1093/jac/dku236. [DOI] [PubMed] [Google Scholar]
- 33.Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, Yang SJ. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One 9:e107426. doi: 10.1371/journal.pone.0107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehta S, Singh C, Plata KB, Chanda PK, Paul A, Riosa S, Rosato RR, Rosato AE. 2012. β-Lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob Agents Chemother 56:6192–6200. doi: 10.1128/AAC.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. 2013. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. mBio 4:e00281-13. doi: 10.1128/mBio.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang SJ, Nast CC, Mishra NN, Yeaman MR, Fey PD, Bayer AS. 2010. Cell wall thickening is not a universal accompaniment of the daptomycin nonsusceptibility phenotype in Staphylococcus aureus: evidence for multiple resistance mechanisms. Antimicrob Agents Chemother 54:3079–3085. doi: 10.1128/AAC.00122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.