

Abstract
A middle-aged woman presented at our facility and was diagnosed after surgery with lung papillary adenocarcinoma. Seven years earlier, she had suffered from autoimmune hemolytic anemia (AIHA), which was refractory. Following lung surgery, the AIHA was cured.
Keywords: Autoimmune hemolytic anemia, lung papillary adenocarcinoma, paraneoplastic
Case report
A 49-year-old woman was admitted to our department in February 2004 with a history of fatigue and discoloured urine lasting more than 20 days. Upon physical examination, no other abnormality was found, except jaundice. Her blood count showed RBC 2.34 × 1012/L, Hb 79 g/L, Ret 16%, WBC 3.21 × 109/L, N 80%, L 19%, M 1%, PLT 123 × 109/L. Routine urine analysis showed urobilinogen (+). Liver function was total bilirubin (TBIL) 64 μmol/L, indirect bilirubin (IBIL) 48 μmol/L, and lactate dehydrogenase (LDH) 359 U/L. An immunologic test revealed anti-nuclear antibody 1:100, but no other autoantibody was detected. Congenital hemolysis tests were all negative. CD55 and CD59 on blood cells were normal. Coombs' test IgG1024, scored 106, and the titer of anti-C3 was 16, scored 28. The cold agglutinin test was negative and chest X-ray was normal. This patient was diagnosed with warm-type autoimmune hemolytic anemia (AIHA) and treated with HD-IVIG 0.4 g/kg/d for five days, combined with 1 mg/kg/d of prednisone and 100 mg/d of cyclosporine. After 30 days of therapy, hemoglobin levels increased to normal and IBIL returned to the upper limits of normal range, while reticulocyte stabilized between 3% and 4%. She was discharged and followed-up every five to 10 months. The disease relapsed twice, resulting from an upper respiratory tract infection and overwork, respectively. The first recurrence occurred in November 2004 (nine months after onset). The patient was admitted because of cough and pharyngalgia. Icteric sclera and mild splenomegaly (3 cm under the rib) were seen. The blood route test returned Hb118 g/L, Ret 4.78%, and other parameters were normal. Liver function was TBIL 34 μmol/L, IBIL 27.4 μmol/L, and LDH 300 U/L; Coombs IgG 1:2048. The patient was treated with 1 g of cyclophosphamide every 10 days (5 g in total) and 2 mg of vincristine every week (6 mg in total). The relapse AIHA was controlled and 5–7.5 mg/d of prednisone and 100–150 mg/d cyclosporine were administered as maintenance therapy. The second recurrence took place in June 2006 (two years after onset) after she had been overworking. She suffered from fatigue and black urine. The blood route test showed RBC 2.34 × 1012/L, Hb 79 g/L, Ret 11.21%, Ret 0.262 × 1012/L, WBC 3.21 × 1012/L. Liver function was ALT 62 u/L, GGT 64 u/L, TBIL 41.5 umol/L, DBIL 13.6 umol/L; Coombs IgG 1:1024. Cyclophosphamide was used again, 1 g every 10 days (6 g in total), 40 mg/d methylprednisolone, and 150 mg/d cyclosporine. Two months later the disease was controlled again. The patient took prednisone orally (5–7.5 mg/d) and cyclosporine (100–150 mg/d). The peripheral blood cell count was then normal and IBIL were also normal, while reticulocyte remained 3%-4% and LDH was 250 U/L. A bone marrow aspiration smear revealed hyperplastic anemia and erythroid cells accounted for 35%-65% nucleated cells. A B-cell subpopulation analysis returned CD19+ 4.5%, CD5+CD19+ 1.8%, CD5+CD19+/CD19+15.5%, and all were within normal range. A T-cell subpopulation analysis revealed that although CD3+CD4+ /CD3+CD8+ (1.7–1.8) was normal; CD3+ (77.9–80.4%), CD3+CD4+ (58–60.5%) and CD3+CD8+ (32.5–35.5%) were all above normal. The Coomb's test was negative. No antibody was detected via immunologic test and chest X-ray was also normal. Seven years after initial diagnosis, the patient returned with urinary infection. Upon physical examination, moist rales in the right lower lung could be heard and a chest computerized tomography (CT) scan revealed patchy enhancement in the right lower lung (Fig. 1). A lung cancer marker test revealed neuron-specific enolase (NSE) 33.01 μg/L (ULN 16.3 μg/L). After one week of antibacterial and antifungal therapy, the image on chest CT scan had not improved. In consultation with a chest surgeon, a diagnosis of right lower lung cancer was made. Therefore, cyclosporine was discontinued and pulmonary lobectomy was performed. The pathologic diagnosis was papillary adenocarcinoma (Fig. 2). One month after surgery, the bilirubin, LDH, blood cell count and reticulocyte count had all returned to normal. The patient was treated with chemotherapy once a month and the regimen was EP (VP-16 100 mg/m2 d1-3 and DDP 80∼100 mg/m2 d1) for four courses in total. Two months after the surgery the hemolytic parameters and T-cell subpopulation analysis were all normal, and prednisone was discontinued. At a 14-month follow-up after surgery, the image on chest CT scan and hemolytic parameters were normal.
Figure 1.

Lung computed tomography (CT).
Figure 2.
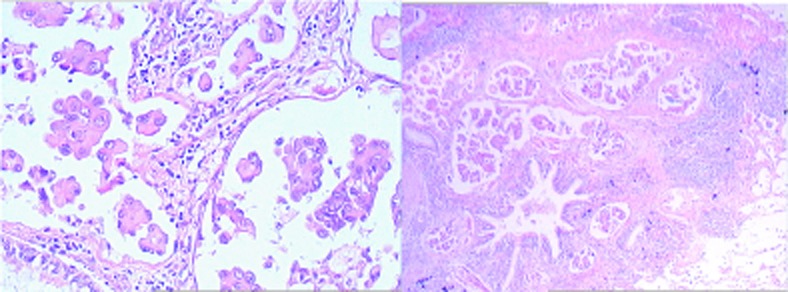
Lung pathology.
Discussion
The patient, a middle-aged woman, was diagnosed with warm-type AIHA. The disease relapsed twice and hemolysis was not controlled well: reticulocyte fluctuated between 3% and 4%, TBIL and IBIL levels were in the upper limits of normal and LDH>200 U/L. Seven years after initial diagnosis, right lower lung papillary adenocarcinoma was diagnosed and after pulmonary lobectomy, the AIHA was cured, thus, the patient was retrospectively diagnosed as paraneoplastic AIHA.
Paraneoplastic syndrome (PNS) refers to systemic manifestations, except for symptoms caused by oncothlipsis, invasion, and metastasis of tumor. The most common PNS of solid tumor, which involves the nervous system is cranial verve, spinal cord, and peripheral nerve dysfunction. Hematologic autoimmune diseases like AIHA and immune thrombocytopenic purpura (ITP) are the most common PNS of lymphoma.1,2
Paraneoplastic AIHA can be found in patients exhibiting all kinds of tumors. It occurs most commonly in tumors originating from the kidney, ovary, thymus gland and Kaposi sarcoma. The morbidity of paraneoplastic AIHA caused by lung cancer is relatively low, accounting for nine of all 52 cases reported by Puthenparambil;3 and all nine of these cases were non-small cell lung cancer. Paraneoplastic AIHA is divided into four types according to the order of AIHA and tumor: AIHA occurs before tumor, AIHA and tumor occur simultaneously, AIHA occurs when tumor relapses or AIHA occurs after complete remission of tumor. Wakata4 reported that AIHA might occur two to eight years prior to tumor. It is common that AIHA and tumor occur simultaneously, which means AIHA occurs before or after six months of the onset of tumor. The patient in our case was diagnosed with lung cancer in the seventh year of AIHA, with no respiratory symptoms. The lung abnormality was found by physical examination and chest CT scan. It was not until after surgery that the patient was finally diagnosed. Lung papillary adenocarcinoma (LPA) is an uncommon type of lung cancer, accounting for 0.84% of lung cancer. It relies on pathology to diagnose and lacks specific manifestations.5
Two thirds of paraneoplastic AIHA are warm-type, while one third are cold-type.6 Immunologic abnormalities existed constantly in this patient (who is warm-type AIHA) before she was diagnosed with lung cancer. T-cell subpopulation analysis showed that the ratio of CD3+ T-cell was above normal, CD4+ and CD8+ T-cell constantly above normal, and T-cell was always in an activated condition.
The therapeutic regimen of paraneoplastic AIHA is focused on treating the tumor, which is the protopathy, while the warm-type AIHA, which occurs prior to tumor, is treated mainly with glucocorticoid. When the tumor is controlled, the AIHA will subsequently be relieved. This patient's hemolytic parameters returned to normal gradually after pulmonary lobectomy. Two months after the surgery, prednisone application was ceased. After a 14-month follow-up, her AIHA condition remains stable.
Lung papillary adenocarcinoma, a subtype of lung adenocarcinoma, is rare and can produce endocrine hormone, antigens, and enzyme. It may be the cause of paraneoplastic AIHA.
Acknowledgments
This work was supported by grants from Tianjin Public Health Bureau (09KZ108) and National Natural Science Foundation of China (30971285).
Disclosure
No authors report any conflict of interest.
References
- Hauswirth AW, Skrabs C, Schützinger C, Gaiger A, Lechner K, Jäger U. Autoimmune hemolytic anemias, Evans syndromes, and pure red cell aplasia in non-Hodgkin lymphoma. Leuk Lymphoma. 2007;48:1139–1149. doi: 10.1080/10428190701385173. [DOI] [PubMed] [Google Scholar]
- Lechner K, Chen Y. Paraneoplastic autoimmune cytopenias in Hodgkin's lymphoma. Leuk Lymphoma. 2010;51(3):469–474. doi: 10.3109/10428190903556394. [DOI] [PubMed] [Google Scholar]
- Puthenparambil J, Lechner K, Kornek G. Autoimmune hemolytic anemia as a paraneoplastic phenomenon in solid tumors: a critical analysis of 52 cases reported in the literature. Wien Klin Wochenschr. 2010;122(7–8):229–236. doi: 10.1007/s00508-010-1319-z. [DOI] [PubMed] [Google Scholar]
- Wakata N, Kiyozuka T, Konno S, et al. Autoimmune thrombocytopenic purpura, autoimmune hemolytic anemia and gastric cancer appeared in a patient with myasthenia gravis. Intern Med. 2006;45:479–481. doi: 10.2169/internalmedicine.45.1496. [DOI] [PubMed] [Google Scholar]
- Jin Z, Li N, Zhang C, Chen C, Li K. Clinical Characteristics and Prognosis of 142 Cases with Papillary Adenocarcinoma of the Lung. Chin J Clin Oncol. 2011;38(19):1217–1220. [Google Scholar]
- Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136–151. doi: 10.1007/s00508-008-0945-1. [DOI] [PubMed] [Google Scholar]