

Abstract
Background
This study assesses whether MET expression in tumor tissue is associated with an increased sensitivity to epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) in non-small-cell lung cancer (NSCLC) patients.
Methods
This retrospective study included 69 NSCLC participants with available tumor tissue and data on treatment response and survival. MET and hepatocyte growth factor expression in tumor tissue were evaluated by immunohistochemistry.
Results
Positive tMET expression correlated with a shorter progression-free survival (PFS; P = 0.003) and overall survival (OS; P = 0.05). Positive pY1234/1235 expression was significantly associated with a longer PFS (P = 0.031) and OS (P = 0.012). In multivariable analyses, tMET and pY1234/1235 expression were independent factors for PFS and OS, respectively. (tMET, PFS; P = 0.02, OS; P = 0.0007 and pY1234/1234, PFS; P = 0.01, OS; P = 0.004).
Conclusions
This study suggests that total and phosphorylated MET expression in tumor tissue is potentially useful for the selection of NSCLC patients who are likely to benefit from EGFR-TKIs, irrespective of their EGFR status.
Keywords: Epidermal growth factor receptor, MET, non-small cell lung cancer, tyrosine kinase inhibitor
Introduction
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) provide substantial clinical benefit to non-small cell lung carcinoma (NSCLC) patients,1,2 especially NSCLC patients with an activated EGFR mutation.3 Recent phase III trials targeting NSCLC adenocarcinoma patients with EGFR mutations showed that the EGFR-TKI cohort experienced significantly longer progression-free survival (PFS) than the platinum-doublet therapy group.4,5 However, not all patients with activating EGFR mutations respond to EGFR-TKI treatment. Approximately 30% of patients with activating EGFR mutations show primary resistance to EGFR-TKIs.4,5 Several previous studies focused on identifying biomarkers associated with primary resistance to EGFR-TKIs, but none of these studies successfully identified a major resistance biomarker.6–9
EGFR-TKIs prescribed as a second- or third-line therapy benefit approximately 50% of NSCLC patients by stabilizing disease, even though EGFR activating mutations are only found in 10 to 15% of all NSCLC patients.10,11 Therefore, NSCLC patients with wild-type EGFR can benefit from EGFR-TKI therapy. Biomarkers that predict the clinical benefit of EGFR-TKIs in patients expressing wild-type EGFR have not been fully characterized.12,13
The MET receptor tyrosine kinase is a critical regulator of cancer migration, invasion, and proliferation; therefore, altered MET function is a major contributor to metastasis. Dysregulation of MET signaling through gene amplification, mutation, or autocrine or paracrine activation occurs in several human cancers, including NSCLC.14,15 MET activation induces phosphorylation of several tyrosine residues (Y1003-Y1313-Y1230/1234/1235-Y1349-Y1356) on EGFR, which, consequently, activates multiple downstream signaling pathways, including the RAS/extracellular-signal-regulated-kinase (ERK) and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathways.16 Activation of MET signaling is associated with advanced cancer stage and shorter survival time in NSCLC patients.17 In addition, MET amplification correlates with acquired resistance to EGFR-TKIs. Engelman et al.18 showed that MET amplification induces gefitinib resistance via promotion of ERBB3-dependent activation of PI3K. Moreover, MET inhibition with a MET-TKI restored gefitinib sensitivity. Bean et al.19 demonstrated that there was MET amplification in 21% of patients with acquired resistance to EGFR-TKIs and only amplification in 3% of TKI-naive patients. These results suggest that cMET amplification could be an acquired resistance mechanism to EGFR-TKIs, rather than a primary resistance mechanism. However, other MET activation mechanisms in addition to amplification have not been extensively studied in the context of primary resistance to EGFR-TKIs in NSCLC patients.
In this study, we investigated whether the activation of MET is associated with primary resistance to EGFR-TKIs in NSCLC patients, irrespective of their EGFR mutation status.
Materials and methods
Participants
Sixty-nine consecutive patients who received gefitinib (250 mg/d) or erlotinib (150 mg/d) monotherapy for metastatic or recurrent disease at Severance Hospital (Seoul, Korea) between December 2005 and June 2011 were selected for this study, regardless of previous treatment. Inclusion criteria included available response assessment data, survival data, and paraffin-embedded tissue samples for molecular analysis.
Tumor histology was classified according to World Health Organization criteria.20 Data on clinicopathological characteristics, including age, gender, Eastern Cooperative Oncology Group (ECOG) performance status, tumor node metastasis stage, smoking status, previous anti-cancer treatment, EGFR mutation status, and KRAS mutation status, were extracted from patient medical records. Non-smokers were defined as individuals who had smoked less than 100 cigarettes in their lifetime. Tumor assessments were classified according to the Response Evaluation Criteria in Solid Tumors (RECIST version 1.0). The disease control rate (DCR) was defined as the rate of complete response (CR), partial response (PR) and stable disease (SD). The Severance Hospital Institutional Review Board approved this study (No. 4-2011-0088). All participants provided written informed consent for molecular analysis of their tumor tissue.
Immunohistochemical analysis
Four-micron tissue sections from formalin-fixed, paraffin-embedded samples were stained with antibodies against total MET (sc-8057; Santa Cruz Biotechnology, Inc., dilution 1:100), phosphorylated MET at Y1003 (ab61024; Abcam, dilution 1:50) and Y1234/1235 (#3077, Cell Signaling, dilution 1:50), and hepatocyte growth factor (HGF) (sc-7949, Santa Cruz Biotechnology, Inc., dilution 1:100).
Currently, there are no validated scoring systems for interpreting immunohistochemical (IHC) staining for total MET (tMET), phosphorylated MET at Y1003 (pY1003) and Y1234/1235 (pY1234/1235), and HGF. IHC scoring was performed using the modified Histo-score (H-score), which involves semiquantitative assessment of both the staining intensity (graded as 1–3) and the percentage of positive cells.21 The possible scores range from 0 to 300. The median H-score of tMET, pY1003, pY1234/1235, and HGF was used as the cut-off criterion. Tumor samples with an H-score less than the cut-off were classified as negative, whereas scores greater than or equal to the cut-off were designated as positive. IHC staining of tMET and HGF was mainly localized to the membranes and cytoplasm of cancer cells; pY1003 and pY1234/1234 staining was specifically localized to tumor cell membranes.
cMET amplification
cMET amplification analysis was performed using the StepOnePlus real-time polymerase chain reaction (RT-PCR) system (Applied Biosystems, Foster City, CA) using the following cycling parameters: 95°C for 10 minutes (one cycle), followed by 95°C for 15 seconds and 60°C for one minute (40 cycles). The quantative PCR reaction mix contained 5 μL 2× TaqMan Genotyping Master Mix, 0.5 μL TaqMan Copy Number Target Assay, 0.5 μL TaqMan Copy Number Reference Assay (RNase P, which exists only in two copies in a diploid genome), 2.0 μL nuclease-free water, and 2 μL DNA (diluted to a concentration of 5 ng/μL). Each sample was run in triplicate. Following amplification, the cycle threshold values for the copy number and reference assay were imported into the CopyCaller Software (Applied Biosystems, Foster City, CA) for post-PCR data analysis. Copy numbers greater than five were considered high-level amplification.22
Analysis of epidermal growth factor receptor (EGFR) and KRAS mutations
EGFR and KRAS mutation analysis was performed on 69 (100%) and 55 (79.7%) patient specimens, respectively. DNA was extracted from areas of paraffin-embedded tissue samples containing greater than 70% tumor. Mutational analyses of EGFR exons 18–21 and KRAS exons 12–13 were performed using a PCR-based assay.
Statistical analysis
Patient groups were compared using the Fisher's exact or χ2 test where appropriate. Correlations were considered significant at the <0.05 level (two-tailed). PFS was measured from the start of EGFR-TKI treatment until progression or death. Overall survival (OS) was measured from the start of treatment with TKIs until death from any cause. Survival curves were constructed using the Kaplan-Meier method, and the differences between groups were analyzed using the log-rank test. Multivariate analysis was carried out using the Cox proportional hazards model with a significance level of 0.05. Only variables at the P < 0.1 level in univariate analyses were entered into the Cox regression analysis. All statistical analyses were carried out using SPSS software (version 14.0; SPSS Inc., Chicago, IL).
Results
Patients' characteristics
Characteristics for the 69 participants are provided in Table 1. The study population was predominantly male (62.3%) and had a median age of 63 years. Adenocarcinoma was the most frequent histologic type (76.8%). ECOG performance status scores were 0 or 1 in 52% of cases, and 27 participants were non-smokers (39.1%). There were 44 participants (63.8%) who received EGFR-TKIs as a first- or second-line therapy. There were 24 participants (34.8%) who harbored an active EGFR mutation (exon 19 deletion in 16 and exon 21 point mutation in eight cases). There were three patients who had a KRAS mutation (G12D in two and G13D in one case).
Table 1.
Patient characteristics and clinical outcomes
| Patient characteristics | No. | % | OR (No.) | % | P | Median PFS (mo) | P | Median OS (mo) | P |
|---|---|---|---|---|---|---|---|---|---|
| Total | 69 | 100 | 18 | 26.1 | – | 2.9 | – | 11.5 | – |
| Gender | |||||||||
| Male | 43 | 62.3 | 10 | 23.2 | 0.49 | 2.3 | 0.25 | 7.5 | 0.15 |
| Female | 26 | 37.7 | 8 | 30.7 | 4.0 | 21.0 | |||
| Age | |||||||||
| ≤60 years | 30 | 43.5 | 8 | 26.6 | 1.0 | 2.4 | 0.15 | 17.7 | 0.050** |
| >60 years | 39 | 56.5 | 10 | 25.6 | 2.9 | 7.6 | |||
| Stage | |||||||||
| IIIb | 5 | 7.2 | 1 | 20.0 | 0.74 | 1.6 | 0.06** | 21.8 | 0.7 |
| IV | 64 | 92.8 | 17 | 26.5 | 3.3 | 11.5 | |||
| Pathology | |||||||||
| Adenocarcinoma | 53 | 76.8 | 15 | 28.3 | 0.44 | 3.3 | 0.86 | 12.7 | 0.5 |
| Other | 16 | 23.2 | 3 | 18.8 | 2.0 | 7.5 | |||
| Performance | |||||||||
| 0, 1 | 52 | 75.4 | 13 | 25.0 | 0.75 | 2.9 | 0.31 | 12.7 | 0.01* |
| ≥2 | 17 | 24.6 | 5 | 29.4 | 2.5 | 5.1 | |||
| Smoking | |||||||||
| Never | 27 | 39.1 | 9 | 33.3 | 0.27 | 5.6 | 0.03* | 21.0 | 0.005* |
| Ever | 42 | 60.9 | 9 | 21.4 | 2.3 | 7.2 | |||
| Previous treatment | |||||||||
| ≤1 | 44 | 63.8 | 12 | 27.2 | 0.65 | 2.7 | 0.77 | 8.4 | 0.21 |
| ≥2 | 25 | 36.2 | 6 | 24.0 | 3.4 | 14.4 | |||
| EGFR | |||||||||
| Mutated | 24 | 34.8 | 14 | 58.3 | <0.001* | 7.2 | <0.001* | 17.7 | 0.06** |
| Wild type | 45 | 65.2 | 4 | 8.9 | 1.9 | 8.6 | |||
| KRAS | |||||||||
| Mutated | 3 | 5.5 | 1 | 33.3 | 0.86 | 2.5 | 0.66 | 3.4 | 0.08** |
| Wild type | 52 | 94.5 | 15 | 28.8 | 3.4 | 11.5 |
P < 0.05
P < 0.1. EGFR, epidermal growth factor receptor; OR, objective response; OS, overall survival; PFS, progression-free survival.
Assessment of MET and hepatocyte growth factor (HGF) expression
IHC results for tMET, pY1003, pY1234/235, and HGF could be obtained for 63 (91%), 61 (88%), 65 (94%), and 64 (92%) participants, respectively. The median H-scores for tMET, pY1003, pY1234/1235, and HGF were 200 (range: 10–290), 90 (range: 0–280), 0 (range: 0–290) and 200 (range: 40–300), respectively (Fig. 1). tMET, pY1003, and pY1234/1235 positivity were observed in 15 (23.8%), 16 (23.2%) and 13 (20.0%) cases, respectively. HGF positivity was detected in 29 (45.3%) cases. The tMET-positive participants demonstrated a significantly lower EGFR-mutation rate than the tMET-negative patients (9.0% vs. 31.7%, P = 0.045). HGF-positive participants exhibited a higher number of previous chemotherapy regimens (% of ≥2 regimes, 71.4% vs. 32.5%, P = 0.003). The other evaluated clinicopathological factors were not associated with the tMET, pY1003, pY1234/235, and HGF expression levels.
Figure 1.

Representative immunohistochemistry for (a) tMET, (b) pY1003, (c) pY1234/1235, and (d) hepatocyte growth factor (HGF). Digital pictures were taken at ×500 magnification.
Quantitative RT-PCR for MET amplification was successful for 59 (85.5%) of the participants. The median gene copy number for MET was 2.22 (range; 0.95–4.10). None of the 59 cases exhibited MET amplification.
Tumor response and survival outcomes
Of the 69 participants evaluated for response, 18 (26.1%) had PR, 25 (35.9%) displayed SD, and 26 (37.7%) exhibited progressive disease as their best tumor response. Therefore, the objective response rate was 26.1%, and the DCR was 62.0%. EGFR mutation status was the only factor identified as predicting a response to EGFR-TKI treatment (Tables 1 and 2). Participants with activating EGFR mutations showed a significantly higher response rate than participants with wild-type EGFR (58.3% vs. 8.9%, P < 0.001). tMET, pY1003, pY1234/1235, and HGF expression were not associated with response rate.
Table 2.
MET and HGF expression and clinical outcomes
| Patient characteristics | No. | % | OR (No.) | % | P | Median PFS (mo) | P | Median OS (mo) | P |
|---|---|---|---|---|---|---|---|---|---|
| tMET | |||||||||
| Negative | 48 | 76.1 | 15 | 31.3 | 0.31 | 3.5 | 0.003* | 12.7 | 0.05** |
| Positive | 15 | 23.8 | 2 | 13.3 | 1.6 | 5.5 | |||
| pY1003 | |||||||||
| Negative | 53 | 76.8 | 7 | 21.2 | 0.25 | 2.4 | 0.027* | 7.5 | 0.3 |
| Positive | 16 | 23.2 | 10 | 35.7 | 4.5 | 11.7 | |||
| pY1234/1235 | |||||||||
| Negative | 52 | 80.0 | 12 | 23.1 | 0.29 | 2.3 | 0.029* | 7.6 | 0.009* |
| Positive | 13 | 20.0 | 5 | 38.5 | 8.4 | 25.0 | |||
| HGF | |||||||||
| Negative | 35 | 54.6 | 8 | 22.9 | 0.57 | 2.7 | 0.29 | 11.5 | 0.83 |
| Positive | 29 | 45.3 | 9 | 31.0 | 4.0 | 11.7 |
P < 0.05
P < 0.1. EGFR, epidermal growth factor receptor; OR, objective response; OS, overall survival; PFS, progression-free survival.
For the entire study population, the median PFS and OS were 2.9 months (95% confidence interval [CI]: 1.8–4.0) and 11.5 months (95% CI: 7.1–15.8), respectively. Analysis of PFS indicated that smoking status, EGFR mutation status, tMET, pY1003, and pY1234/1235 expression were significant predictors of PFS following EGFR-TKI treatment (Tables 1 and 2). The same trend was observed when only EGFR wild-type patients were analyzed (tMET+ vs. tMET−, 1.0 months vs. 1.9 months, P = 0.10; pY1234/1235+ vs. pY1234/1235−, 3.4 months vs. 1.7 months, P = 0.066). Multivariate analysis demonstrated that EGFR mutation, tMET, and pY1234/1235 were independent factors of PFS. EGFR mutation was an independent factor for improved PFS (hazard ratio [HR] 0.50; 95% CI: 0.26–0.93, P = 0.029; Table 3). The tMET-positive participants were at a higher risk of progression than the tMET-negative participants (HR 2.19; 95% CI: 1.10–4.32, P = 0.02; Fig. 2). In contrast, pY1234/1235 positive participants were at a lower risk of progression than the pY1234/1235-negative participants (HR 0.43; 95% CI: 0.21–0.85, P = 0.01; Fig. 2).
Table 3.
Multivariable analysis on progression-free and overall survival
| Variable | PFS | OS | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Smoking | ||||||
| Never | 1 | 0.91–2.90 | 0.10 | 1 | 1.34–6.28 | 0.007* |
| Ever | 1.62 | 2.90 | ||||
| EGFR mutation | ||||||
| Mutated | 1 | 1.07–3.71 | 0.02* | 1 | 0.73–3.35 | 0.24 |
| Wild | 1.99 | 1.56 | ||||
| tMET | ||||||
| Negative | 1 | 1.10–4.32 | 0.02* | 1 | 1.12–5.81 | 0.007* |
| Positive | 2.19 | 2.55 | ||||
| pY1234/1235 | ||||||
| Negative | 1 | 0.21–0.85 | 0.01* | 1 | 0.10–0.65 | 0.004* |
| Positive | 0.43 | 0.26 | ||||
P < 0.05. CI, confidence interval; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Figure 2.
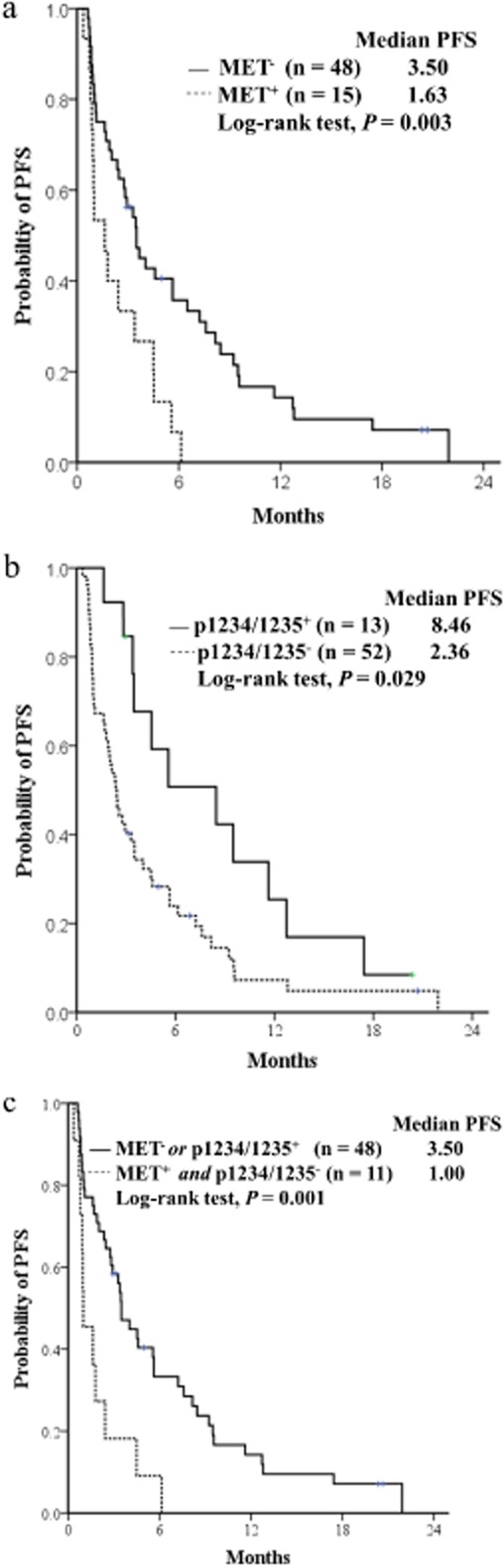
Progression-free survival (PFS) curves for the two groups according to (a) tMET status, (b) pY1234/1235, and (c) the combined criteria of tMET and pY1234/1234 expression in non-small cell lung cancer (NSCLC) patients receiving epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs).
Analysis of OS demonstrated that ECOG performance status, smoking history, and pY1234/1235 were significant predictors of OS (Tables 1 and 2). Age, EGFR mutation, and tMET positivity showed borderline statistical significance (Tables 1 and 2). Similar results were observed when the analysis was restricted to the EGFR wild-type patients (tMET+ vs. tMET−, 3.6 months vs. 12.7 months, P = 0.054; pY1234/1235+ vs. pY1234/1235−, 28.1 months vs. 5.5 months, P = 0.038). In multivariate analysis of OS, smoking history, tMET, and pY1234/1235 remained independent factors of OS (Table 3). Smoking history was an independent factor for poor OS (HR 2.90; 95% CI: 1.34–6.28, P = 0.007). The tMET-positive patients were at a higher risk of death than the tMET-negative patients (HR 2.55; 95% CI: 1.12–5.81, P = 0.007). In contrast, pY1234/1235-positive patients were at a lower risk of death than the pY1234/1235-negative patients (HR 0.26; 95% CI: 0.10–0.65, P = 0.004).
Combined total and phosphorylated MET expression
Because tMET and pY1234/1235 expression correlated not only with PFS, but also significantly with OS, the treatment outcomes in the subgroups were classified according to the combined criteria of tMET and pY1234/1235 expression (Table 4 and Fig. 2). The group of tMET-positive and pY1234/1235-negative patients exhibited poorer clinical outcomes than the group with tMET-negative or pY1234/1235-positive expression. There was a shorter median PFS (1.0 months [95% CI: 0.24–1.75] vs. 3.5 months [95% CI: 2.56–4.43, respectively; P = 0.009]), and shorter median OS (3.66 months [95% CI: 1.45–5.87] vs. 12.8 months [95% 3.52–22.21], respectively; P = 0.011).
Table 4.
Treatment outcome by combined total and phosphorylated cMET expression
| tMET+ and pY1234/1235− | tMET+ and pY1234/1235+ | tMET− and pY1234/1235− | tMET− and pY1234/1235+ | tMET− or pY1234/1235+ | P† | |
|---|---|---|---|---|---|---|
| No. responder/total | 2/11 | 0/3 | 9/35 | 5/10 | 14/48 | |
| ORR, % | 18.1 | 0 | 25.7 | 50 | 29.1 | 0.71‡ |
| Median PFS, mo (95%CI) | 1.00 (0.24–1.75) | 4.53 (2.72–6.34) | 2.93 (1.87–3.99) | 9.50 (4.98–14.0) | 3.50 (2.56–4.43) | – |
| HR of progression (95% CI) | 1.00 | 0.37 (0.10–1.14) | 0.43 (0.20–0.93) | 0.20 (0.07–0.54) | 0.37 (0.17–0.78) | 0.009§ |
| Median OS, mo (95% CI) | 3.66 (1.45–5.87) | 12.8 (not calculated) | 11.7 (3.15–20.3) | 28.0(12.0–44.1) | 12.8 (3.52–22.2) | – |
| HR of death (95% CI) | 1.00 | 0.29 (0.05–1.44) | 0.40 (0.16–0.99) | 0.10 (0.02–0.38) | 0.31 (0.12–0.77) | 0.011 |
Statistical difference between two groups ([tMET+ and pY1234/1235−] and [tMET− or pY1234/1235+]).
Tested with Fisher's exact test.
Tested with the Cox proportional hazards model adjusted with for smoking history and epidermal growth factor receptor (EGFR) mutation. CI, confidence interval; HR, hazard ration; ORR, objective response rate; PFS, progression-free survival.
Discussion
We investigated several forms of MET as potential biomarkers for clinical outcomes of EGFR-TKIs in NSCLC patients. We showed that MET over-expression and activation, which was indicated by tMET and pY1234/1235 over-expression, respectively, was significantly associated with PFS and OS in participants treated with EGFR-TKIs. MET over-expression correlated with a worse clinical outcome, while activated MET correlated with improved survival. The robust association of these parameters with outcome, regardless of EGFR mutation status, supports the theory that MET expression and activation could be useful for predicting the individual clinical outcome of EGFR-TKI treatment.
MET over-expression has been identified in the majority of human cancers and represents the most common type of MET deregulation.23 Several clinical studies of the HGF/MET pathway in NSCLC have shown that MET expression correlates with poor survival in NSCLC patients.17,24,25 In those study participants with high tMET expression, there was an increased risk of progression and death in the EGFR-TKI treatment group, which was significant after appropriate adjustments were made for EGFR mutation status and smoking history. This observation is consistent with the results of the MetMab plus erlotinib clinical trial, which demonstrated that placebo-treated patients with high tMET expression showed decreased PFS and OS, compared with those with low tMET expression.26 Therefore, our study supports that NSCLC patients with high tMET expression could benefit from a combination approach with tMET to maximize the efficacy of EGFR-TKIs. In fact, MetMab plus erlotinib improved PFS (HR 0.47) and OS (HR 0.37) in tMET high-expressing NSCLC patients.26
Interestingly, our analyses demonstrated a relationship between phosphorylated MET at Y1234/1235 and improved survival, regardless of the EGFR mutation status. This is consistent with the Matsubara et al.27 in vitro study, which demonstrated that lung cancer cell lines with activation of EGFR and MET were more sensitive to gefitinib than other cell lines with little to no phosphorylation of MET and EGFR.
Although we do not have any definitive biological data to address the mechanism of how high expression of phosphorylated MET is associated with a good clinical outcome after EGFR-TKI treatment, the results from several previous studies provide some insights. EGFR ligands induce a delayed accumulation of tyrosine-phosphorylated MET receptors,28 and EGFR-TKIs decrease the expression of MET.29 In addition, extensive overlap in the phosphoproteome profiles between EGFR mutant lung adenocarcinoma cell lines and a MET-amplified gastric cell line was observed.29 Moreover, MET is a central downstream mediator of EGFR-induced invasiveness in NSCLC cells with an activating EGFR mutation, as well as those expressing wild-type EGFR.28,30 These studies indicate that EGFR in specific lung cancer cells sits at the top of a hierarchical oncogenic signaling network that drives the phosphorylation and activity of MET.
Therefore, EGFR-induced invasion and motility is MET dependent; lung cancer cells with high levels of phosphorylated MET expression might be more invasive and motile, but their EGF-induced invasion and motility might be reduced by EGFR-TKIs more than those with low phosphorylated MET expression. This, subsequently, improves PFS and OS. In addition, this study showed that MET amplification is a rare event and genomic gain for MET may not contribute to the primary resistance of EGFR-TKIs in advanced NSCLC patients.
One study limitation is that our conclusions are derived from a single retrospective study with a relatively small number of participants. Our study needs to be performed on a larger cohort of participants, in a prospective manner.
Conclusion
In conclusion, total and phosphorylated MET expression in tumor tissue could be a useful biomarker to assist with selecting NSCLC patients who are most likely to benefit from EGFR-TKIs. A prospective study is planned to validate the ability of assessment of MET expression in tumor tissue to predict the prognosis of NSCLC patients treated with EGFR-TKIs.
Acknowledgments
This study was supported by the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea [A110080]; and the National Research Foundation of Korea Grant funded by the Korean Government [313-2007-2-E00373].
Disclosure
No authors report any conflict of interest.
References
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9:2316–2326. [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–4346. [PubMed] [Google Scholar]
- Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–950. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372:1809–1818. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Arao T, Sakai K, et al. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin Cancer Res. 2010;16:4616–4624. doi: 10.1158/1078-0432.CCR-10-0383. [DOI] [PubMed] [Google Scholar]
- Lee Y, Shim HS, Park MS, et al. High EGFR gene copy number and skin rash as predictive markers for EGFR tyrosine kinase inhibitors in patients with advanced squamous cell lung carcinoma. Clin Cancer Res. 2012;18:1760–1768. doi: 10.1158/1078-0432.CCR-11-2582. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. FASEB J. 2006;20:1611–1621. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- Masuya D, Huang C, Liu D, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer. 2004;90:1555–1562. doi: 10.1038/sj.bjc.6601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Semin Roentgenol. 2005;40:90–97. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Arriola E, Cañadas I, Arumí-Uría M, et al. MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines. Br J Cancer. 2011;105:814–823. doi: 10.1038/bjc.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Yamamoto H, Lockwood WW, et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778–1784. doi: 10.1002/ijc.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou HY, Li Q, Lee JH, et al. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol Cancer Ther. 2012;11:1036–1047. doi: 10.1158/1535-7163.MCT-11-0839. [DOI] [PubMed] [Google Scholar]
- Ichimura E, Maeshima A, Nakajima T, Nakamura T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. 1996;87:1063–1069. doi: 10.1111/j.1349-7006.1996.tb03111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanami I, Tanana F, Hashizume T, et al. Hepatocyte growth factor and c-Met/hepatocyte growth factor receptor in pulmonary adenocarcinomas: an evaluation of their expression as prognostic markers. Oncology. 1996;53:392–397. doi: 10.1159/000227594. [DOI] [PubMed] [Google Scholar]
- Spigel DR, Ervin TJ, Ramlau R, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol. 2011;29 2011 ASCO Annual Meeting Proceedings: abstract 7505. [Google Scholar]
- Matsubara D, Ishikawa S, Sachiko O, Aburatani H, Fukayama M, Niki T. Co-activation of epidermal growth factor receptor and c-MET defines a distinct subset of lung adenocarcinomas. Am J Pathol. 2010;177:2191–2204. doi: 10.2353/ajpath.2010.100217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulak AM, Gubish CT, Stabile LP, Henry C, Siegfried JM. HGF-independent potentiation of EGFR action by c-Met. Oncogene. 2011;30:3625–3635. doi: 10.1038/onc.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Villén J, Kornhauser J, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–697. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Nilsson MB, Saintigny P, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene. 2010;29:2616–2627. doi: 10.1038/onc.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]