

Key points
In peripheral myelinated axons of mammalian spinal motor neurons, Ca2+ influx was thought to occur only in pathological conditions such as ischaemia.
Using Ca2+ imaging in mouse large motor axons, we find that physiological stimulation with trains of action potentials transiently elevates axoplasmic [Ca2+] around nodes of Ranvier.
These stimulation‐induced [Ca2+] elevations require Ca2+ influx, and are partially reduced by blocking T‐type Ca2+ channels (e.g. mibefradil) and by blocking the Na+/Ca2+ exchanger (NCX), suggesting an important contribution of Ca2+ influx via reverse‐mode NCX activity.
Acute disruption of paranodal myelin dramatically increases stimulation‐induced [Ca2+] elevations around nodes by allowing activation of sub‐myelin L‐type (nimodipine‐sensitive) Ca2+ channels.
The Ca2+ that enters myelinated motor axons during normal activity is likely to contribute to several signalling pathways; the larger Ca2+ influx that occurs following demyelination may contribute to the axonal degeneration that occurs in peripheral demyelinating diseases.
Abstract
Activity‐dependent Ca2+ signalling is well established for somata and terminals of mammalian spinal motor neurons, but not for their axons. Imaging of an intra‐axonally injected fluorescent [Ca2+] indicator revealed that during repetitive action potential stimulation, [Ca2+] elevations localized to nodal regions occurred in mouse motor axons from ventral roots, phrenic nerve and intramuscular branches. These [Ca2+] elevations (∼0.1 μm with stimulation at 50 Hz, 10 s) were blocked by removal of Ca2+ from the extracellular solution. Effects of pharmacological blockers indicated contributions from both T‐type Ca2+ channels and reverse mode Na+/Ca2+ exchange (NCX). Acute disruption of paranodal myelin (by stretch or lysophosphatidylcholine) increased the stimulation‐induced [Ca2+] elevations, which now included a prominent contribution from L‐type Ca2+ channels. These results suggest that the peri‐nodal axolemma of motor axons includes multiple pathways for stimulation‐induced Ca2+ influx, some active in normally‐myelinated axons (T‐type channels, NCX), others active only when exposed by myelin disruption (L‐type channels). The modest axoplasmic peri‐nodal [Ca2+] elevations measured in intact motor axons might mediate local responses to axonal activation. The larger [Ca2+] elevations measured after myelin disruption might, over time, contribute to the axonal degeneration observed in peripheral demyelinating neuropathies.
Key points
In peripheral myelinated axons of mammalian spinal motor neurons, Ca2+ influx was thought to occur only in pathological conditions such as ischaemia.
Using Ca2+ imaging in mouse large motor axons, we find that physiological stimulation with trains of action potentials transiently elevates axoplasmic [Ca2+] around nodes of Ranvier.
These stimulation‐induced [Ca2+] elevations require Ca2+ influx, and are partially reduced by blocking T‐type Ca2+ channels (e.g. mibefradil) and by blocking the Na+/Ca2+ exchanger (NCX), suggesting an important contribution of Ca2+ influx via reverse‐mode NCX activity.
Acute disruption of paranodal myelin dramatically increases stimulation‐induced [Ca2+] elevations around nodes by allowing activation of sub‐myelin L‐type (nimodipine‐sensitive) Ca2+ channels.
The Ca2+ that enters myelinated motor axons during normal activity is likely to contribute to several signalling pathways; the larger Ca2+ influx that occurs following demyelination may contribute to the axonal degeneration that occurs in peripheral demyelinating diseases.
Abbreviations
- CPA
cyclopiazonic acid
- ER
endoplasmic reticulum
- FM4‐64
N‐(3‐triethylammoniumpropyl)‐4‐(p‐diethylaminophenylhexatrienyl) pyridinium dibromide
- HVA
high voltage‐activated Ca2+ channel
- LPC
lysophosphatidylcholine
- LVA
low voltage‐activated Ca2+ channel
- NCX
plasmalemmal Na+/Ca2+ exchanger
- OG‐1
Oregon green 488 BAPTA1
- PMCA
plasma membrane Ca2+‐ATPase
Introduction
In mammalian spinal motor neurons, action potentials generated in the soma/initial segment travel via peripheral myelinated axons to motor nerve terminals. In both soma and terminals, action potentials activate Ca2+ influx, which modulates excitability and/or transmitter release (Catterall et al. 2005). Do action potentials also activate Ca2+ influx in myelinated axons? Work from the laboratory of Dr. S. Y. Chiu found that stimulation does increase axoplasmic [Ca2+], but the pattern of [Ca2+] elevation differed in the investigated central and peripheral axons. In mouse optic nerve stimulation elevated [Ca2+] in a spatially uniform manner (Zhang et al. 2006), whereas in large frog sciatic nerve axons stimulation produced non‐uniform [Ca2+] elevations localized around nodes of Ranvier (Zhang et al. 2010). Work presented here addressed this question for mammalian (mouse) motor axons located in ventral roots, phrenic nerve and muscle. Measurements using an iontophoretically‐injected fluorescent [Ca2+] indicator demonstrated that trains of action potentials (50 Hz) induce [Ca2+] elevations that are localized around nodes.
What mechanisms underlie these nodal [Ca2+] elevations? In mammalian peripheral axons Ca2+ influx has previously been documented only during severe energy depletion, induced by anoxia or by chemical blockade of ATP generation, also thought to be mediated by reverse mode transport via the plasmalemmal Na+/Ca2+ exchanger (NCX) (Lehning et al. 1996; Petrescu et al. 2007). These conclusions follow from the properties of NCX‐mediated transport: NCX transport is electrogenic (ratio 3 Na+:1 Ca2+), so the net direction of transport is determined by both membrane potential and transmembrane [Na+] and [Ca2+] gradients. Reverse mode operation (resulting in Ca2+ influx and Na+ efflux) would be favoured by axonal depolarization and elevated intracellular [Na+], both resulting from energy depletion. These conditions can also be produced during trains of action potentials, and we present evidence that reverse mode operation of NCX accounts for a portion of the stimulation‐induced nodal [Ca2+] elevations in mouse motor axons. We also find evidence for a substantial contribution from Ca2+ influx via T‐type (low voltage‐activated, Cav3.x) Ca2+ channels.
In myelinated axons some depolarization‐activated ion channels (e.g. K+ channels) are located in the sub‐myelin juxtaparanodal axolemma (reviewed in Stathopoulos et al. 2015). Ion channels in this location would experience only a small fraction of the total action potential depolarization. Disruption of paranodal myelin would increase the depolarization experienced by such channels, and might thereby disclose the existence of any sub‐myelin depolarization‐activated Ca2+ entry pathways. We report that acute disruption of paranodal myelin (via mechanical stretch or detergent application) greatly increases the stimulation‐induced nodal Ca2+ elevations, and that this increase has pharmacological properties characteristic of L‐type (high voltage‐activated) channels.
Thus mouse motor axons exhibit nodal Ca2+ elevations during trains of action potentials, mediated by at least three Ca2+ entry pathways. When myelin is intact, Ca2+ enters via T‐type Ca2+ channels and reverse mode NCX activity. Disruption of myelin permits additional Ca2+ influx via L‐type Ca2+ channels. Implications of this Ca2+ entry for axonal physiology and pathophysiology are discussed.
Methods
Ethical approval
Experiments used tissues from adult C57BL/6 mice, housed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Experimental protocols were approved by the University of Miami Animal Care and Use Committee, according to National Institutes of Health guidelines (assurance number A‐3224‐01). Mice were killed with 100% CO2 followed by cervical dislocation, according to American Veterinary Medical Association guidelines.
Nerve isolation and stimulation
Experiments used phrenic nerves, ventral roots and motor nerves innervating the levator auris muscle, all dissected rapidly following death. The phrenic nerve comprised a 10–15 mm nerve segment between the thoracic inlet and the attachment to the diaphragm. Perineural and epineural sheaths were removed with fine forceps to make axons accessible to bath‐applied drugs and to the microelectrode used for iontophoretic injection of the Ca2+ indicator (see below). Ventral roots were isolated by cleaning overlying muscles off the lumbar spinal cord, followed by dorsal laminectomy and exposure of the cord from the mid‐thoracic to the sacral area. The lumbar cord was isolated together with ventral and dorsal roots, and 5 mm long segments of L4 and L5 ventral root were isolated in a dissection dish. Desheathing was not necessary for ventral roots, which lack perineural sheaths. Ventral roots are myelinated by Schwann cells (Coulpier et al. 2010). The thin levator auris neuromuscular preparation was dissected as described by Angaut‐Petit et al. (1987).
Dissected nerves were placed in a 10 × 20 mm chamber, constructed with a no. 1 glass coverslip bottom and 3–4 mm tall silicon walls. Nerves were stabilized using overlying flexible glass‐wool fibres (10–20 μm diameter, 10–20 mm long) whose ends were secured to the base of the chamber walls. During the experiment, the chamber volume (0.5 ml) was perfused at a rate of 0.5–1 ml min−1 with a physiological saline solution containing (in mM): NaCl 137, NaHCO3 15, KCl 4, CaCl2 1.8, MgCl2 1.1, glucose 11.2 and NaH2PO4 0.33. The solution was aerated with 95% O2/5% CO2 and maintained at 25–27°C and pH 7.4. Under these conditions nerve preparations yielded stable responses for up to 9 h. For the zero‐Ca2+ solutions used in Fig. 5, Ca2+ was replaced with an equivalent concentration of Mg2+.
Figure 5. Stimulation‐induced elevations in nodal [Ca2+] are reversibly abolished by removal of bath [Ca2+], and are inhibited by blockers of T‐type Ca2+ channels and NCX .
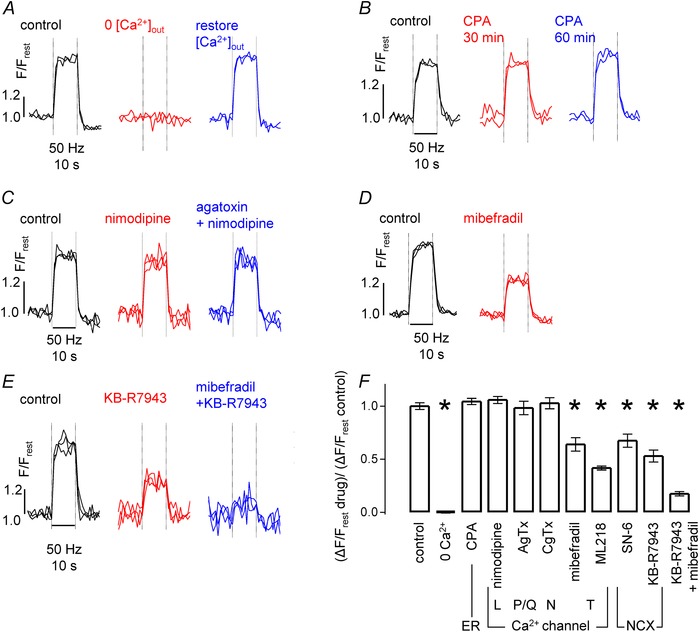
A–E, each panel illustrates a control response to stimulation (50 Hz, 10 s), and a response to the indicated treatment/drug or drug combination. All recordings from phrenic nerve, with multiple traces superimposed in each record. Drug targets are indicated in F. A, reversible inhibition of [Ca2+] response after removal of extracellular Ca2+ (replaced by equimolar Mg2+). B, lack of effect of 30 and 60 min exposure to CPA. C, lack of effect of nimodipine and of nimodipine + agatoxin. D, partial inhibition with mibefradil. E, partial inhibition with KB‐R7943, and nearly complete inhibition with KB‐R7943 + mibefradil combination. F, summary of the mean (± SEM) results of similar experiments. Each drug/treatment response (ΔF/F rest) was normalized to control ΔF/F rest; 3–13 axons per group. *Significant difference from control at P < 0.001, after correction for multiple comparisons to control. Concentrations: CPA (10 μm), nimodipine (4 μm), ω‐agatoxin IVA (AgTx, 1 μm), ω‐conotoxin GVIA (CgTx, 3 μm), mibefradil (2 μm), ML218 (6 μm), SN‐6 (10 μm), KB‐R7943 (10 μm).
Action potentials were evoked with brief, supra‐threshold depolarizing current pulses (0.3 ms, 0.5–1 mA) delivered via a suction electrode at the proximal end of the nerve trunk. Most stimulation protocols consisted of 500 stimuli delivered at 50 Hz, controlled by a digital stimulator (Master‐8, AMPI, Jerusalem, Israel).
Injection of Ca2+ indicator, imaging techniques and analysis
In all preparations the internodal region of a visualized myelinated axon was injected iontophoretically with the membrane‐impermeant hexapotassium salt of a fluorescent Ca2+ indicator, Oregon green 488 BAPTA1 (OG‐1) (David et al. 1997 b; David & Barrett, 2000). Microelectrodes were back‐filled with a solution of 4.5 mm OG‐1 and had a resistance of 100–150 MΩ. After microelectrode insertion into an internode, OG‐1 was iontophoresed with negative current pulses (0.2–0.5 nA, 0.5 s) delivered every second for 5–15 min; the microelectrode was then withdrawn and OG‐1 diffused throughout the axon. This procedure restricts the indicator to the axoplasm, eliminating artifact due to possible [Ca2+] changes in Schwann cells (Lev Ram & Ellisman, 1995). Axons studied here were not subjected to mechanical or chemical procedures traditionally used for isolating single axons for voltage clamp or immunohistochemical studies. Rather, OG1‐injection of only individual axons in their native nerve trunks allowed us to trace axons along 3–4 mm, allowing comparison of [Ca2+] responses at different nodal and internodal regions along the same axon (e.g. Fig. 1 B and C). Analysed data came from axonal regions at least two nodes distant from the injection site, i.e. from nodes adjoining non‐impaled Schwann cells. The OG‐1 concentration in axoplasm, estimated from the fluorescence of known dye concentrations in glass capillaries, was 1.4 ± 0.2 μm (SEM, n = 29 axons).
Figure 1. Stimulation‐induced elevations of [Ca2+] originate at nodes of Ranvier .
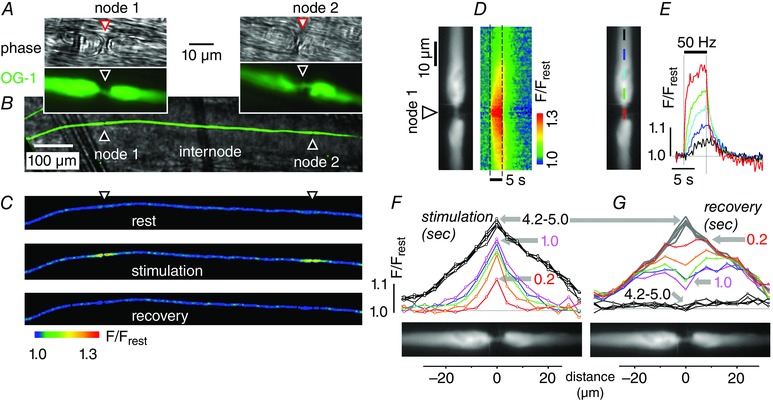
All records are from the phrenic nerve axon illustrated in A–C, which was iontophoretically injected with the Ca2+ indicator OG‐1 and stimulated at 50 Hz for 5 s. The low magnification field in Fig. 1 B shows a fluorescence image of the injected axon (green) superimposed on a phase image (grey) of the phrenic nerve trunk. C shows 3 fluorescence micrographs (coded as F/F rest) of this axon before, during and after stimulation. In the pseudocoloured images of C and D, blue indicates low resting [Ca2+] and warmer colours indicate elevated [Ca2+]. The stimulated image in C shows two areas of elevated [Ca2+], both of which were identified as nodes in the phase (greyscale) and fluorescence (green) images shown in A and B. Distance calibration bar in B also applies to C. Records in D–G came from node no. 1; each plot is shown adjacent to a greyscale fluorescence image defining nodal and paranodal regions. D, pseudocoloured image shows the time course of [Ca2+] changes (measured as F/F rest) before, during and after the stimulus train (duration indicated by dashed lines) along the length of the axon (vertical axis indicates location, horizontal axis indicates time). E, superimposed time courses of stimulation‐induced F/F rest changes at different distances from the node. Coloured bars in the fluorescence image indicate the location at which each F/F rest trace was measured. F and G, spatial plots of F/F rest for the indicated times (0.2–5 s) after the onset (F) and termination (G) of stimulation. Recovery curves in G began at the peak of the stimulation response shown in F.
All imaged axons had diameters > 5 μm. Identification of large axons as motor was assured for ventral roots and for intramuscular axons (where injected indicator reached motor nerve terminals, Fig. 4 B). OG‐1‐labelled axons in the thoracic trunk of phrenic nerves had diameters of 5.0–8.2 μm (mean 6.2 ± 0.1 μm (SEM, n = 25)). Most (82%) of phrenic nerve axons of this size are motor (Gottschall, 1981; Friede et al. 1984; Schmalbruch et al. 1991; Fazan et al. 2009), so it is likely that the stimulation‐induced Ca2+ elevations demonstrated in Fig. 1 originated from motor axons.
Figure 4. Stimulation‐induced elevations of nodal [Ca2+] in a ventral root axon (A) and in a motor axon coursing through the levator auris muscle (B) .
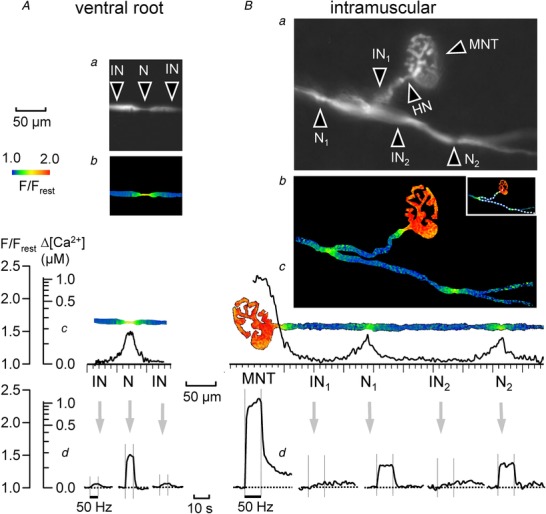
Both A and B show fluorescence images of an OG‐1‐filled axon (a), an elevation of nodal [Ca2+] during stimulation at 50 Hz, coded as F/F rest (b), the spatial extent of the Δ[Ca2+] response (c), and the time course of the response at nodal and flanking internodal regions (d). The intramuscular axon had 2 nodes, each at a branch point, and a heminode (HN) at the junction between the myelinated axon and the motor nerve terminal (MNT). Bb–d, also shows the much larger, prolonged [Ca2+] elevation averaged over the motor nerve terminal. For analysis the intramuscular axon (dotted lines in Bb inset) was digitally straightened to yield the image shown in Bc, as described in Methods. The spatial distributions in Ab and c and Bb and c were taken at the 5th and 10th second after beginning of stimulation at 50 Hz, respectively.
Preparations were mounted on a microscope stage (TE‐2000) and imaged using 60× (Nikon water immersion, NA 1.2) or 10× (Nikon, NA 0.5) objectives. OG‐1 was excited at 488 nm (Delta‐Ram Monochromator, Photon Technology International, Birmingham, NJ, USA), and emission at 520–540 nm was collected with a CCD camera (Retiga EXI, QImaging, Surrey, BC, Canada) with a 1360 × 1036 pixel sensor. Image acquisition software and custom scripts (IPLAB) were used to collect time series stacks of images (1–5 Hz), and to trigger nerve stimulation trains (after acquisition of control, pre‐stimulation images). Analysis used ImageJ software, macros and plugins (Schneider et al. 2012), and consisted of the following steps: (i) alignment in the XY plane using the Turboreg plugin (Thévenaz, 1998); (ii) background subtraction; (iii) construction of F/F rest images, by dividing the fluorescence values in each image (F) by the average of 20 pre‐stimulation images (F rest); (iv) measurement, in each image, of F/F rest along a multi‐segmented line drawn along the midline of the axon (spatial profile), and (v) side‐by‐side combination of line measurements from the whole stack to form a spatio‐temporal map of F/F rest (e.g. Fig. 1 D, right). The same multi‐segmented line was used to straighten the fluorescence image of the axon (straighten macro in ImageJ), so that the location of the F/F rest measurements matched the location of the morphological features in the adjacent fluorescence image (e.g. Fig. 1 D, left). In Fig. 4 the dotted line in Fig. 4 Bb inset was used to straighten the structures in Fig. 4 Bb, transforming it into the picture in Fig. 4 Bc. F/F rest was plotted vs. time at different locations, or vs. space at different times, using Prism software (GraphPad Software, La Jolla, CA, USA).
OG‐1 is a high‐affinity Ca2+ indicator with a dissociation constant (K d) of 0.4 μm in cytoplasm (Thomas et al. 2000). For data in Figs 2 and 4 resting [Ca2+] and the stimulation‐induced change in [Ca2+] (∆[Ca2+] = [Ca2+] − [Ca2+]rest) were estimated using the expressions below (Maravall et al. 2000; F/F rest is denoted by δ for simplicity):
| (1) |
| (2) |
R f is the dynamic range of OG‐1 (fluorescence at saturating [Ca2+]/fluorescence at 0 Ca2+); δ is F/F rest; and δmax is the maximal δ obtainable by increasing [Ca2+]. We estimated δmax (2.5–3.5) in OG1‐labelled axons by inducing large (unphysiological) [Ca2+] elevations with extracellularly‐applied electroporating current pulses delivered to the nodal axolemma. Determination of K d and R f in axoplasm would require extensive permeabilization of the axolemma (to Ca2+), but the myelin sheath limits the access of ionophores required for this procedure. Thus the values used for K d (0.43 μm) and R f (6.0) are based on reported in vitro and in situ measurements with OG‐1 (Thomas et al. 2000).
Figure 2. Responses to stimulation averaged for 11 phrenic nerve axons .
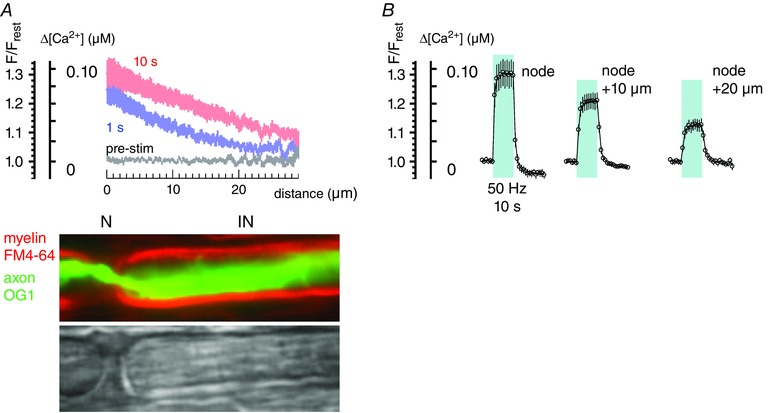
A, average change in F/F rest and Δ[Ca2+] as a function of distance, calculated for the 1st and 10th second of stimulation trains at 50 Hz. Phase (greyscale) and coloured fluorescence images indicate the nodal (N) and internodal (IN) locations corresponding to the plotted distances. B, averaged time course of changes in F/F rest and Δ[Ca2+] for nodes and locations 10 and 20 μm distant from nodes. Shading indicates duration of stimulation. Bars indicate ± SEM. Equations used to convert F/F rest measurements into estimates of Δ[Ca2+] are given in Methods.
Myelin staining and acute paranodal demyelination
To visualize myelin in living axons injected with OG‐1 (Fig. 2 A), nerves were incubated for 60 min in physiological saline containing 10 μm N‐(3‐triethylammoniumpropyl)‐4‐(p‐diethylaminophenylhexatrienyl) pyridinium dibromide (FM4‐64), a fluorescent lipophilic styryl dye that partitions into, but does not cross, membranes (Mattei et al. 1998). For dual imaging of OG‐1 (axon) and FM4‐64 (myelin) we excited at 488 nm, and collected emissions at 520–540 nm for OG‐1 and above 610 nm for FM4‐64.
Acute paranodal demyelination was produced by detergent application or mechanical stretch. The detergent lysophosphatidylcholine (LPC) causes retraction of paranodal Schwann cell loops, thereby exposing the underlying axolemma (Chiu & Ritchie, 1980, 1981). LPC (1 mg mL–1) was dissolved in normal saline by heating to 40°C and vigorous shaking for 15 min. Preparations were perfused with freshly prepared LPC solution for 5–10 min, followed by washout of LPC. The duration of exposure was critical, since axons exposed to LPC for longer than ∼15 min often lost their membrane integrity, likely to be due to lysis of axonal membranes.
Mechanical stretch of mammalian myelinated axons also produces acute paranodal demyelination (Chiu & Ritchie, 1980; 1981). After obtaining control records of stimulation‐induced [Ca2+] responses, the phrenic nerve containing the imaged axon was immobilized at one end and the other end was pulled longitudinally with a micromanipulator‐attached glass rod. The total length of the nerve (∼5 mm) was increased by ∼20% (1 mm) within 30 s, after which the tension on the nerve was released. This is considered to produce a mild, slow strain with little or no damage to the axolemma (Shi & Whitebone, 2006). The stretching procedure usually moved the imaged axonal region out of the microscope field; thus, before acquiring the post‐stretch images, the original field was restored by moving the microscope stage.
Drugs and reagents
ω‐Agatoxin‐IVA and ω‐conotoxin GVIA were from Alomone Labs (Jerusalem, Israel); OG‐1 and FM4‐64 were from Life Technologies (Carlsbad, CA, USA); KB‐R7943 and SN‐6 were from Tocris (Bristol, UK). Other reagents were from Sigma‐Aldrich (St Louis, MO, USA).
Statistics
Statistical comparison among means was done using Student's two‐tailed t test for two means, or by ANOVA followed by Dunnett's multiple comparison test when multiple means were compared to control (Prism).
Results
Action potential stimulation elevates [Ca2+] around nodes of Ranvier in phrenic nerve
Figure 1 illustrates representative findings from a single OG‐1‐filled phrenic axon stimulated with trains of action potentials (50 Hz, 5 s). The low magnification field in Fig. 1 B shows a fluorescence image of the injected axon (green) superimposed on a phase image (grey) of the phrenic nerve trunk. Examination of the labelled axon at higher magnification (Fig. 1 A) revealed two nodes of Ranvier (phase and fluorescence images of node 1 and node 2 in Fig. 1 A) separated by a 700 μm internode. Each node shows the characteristic narrowing of the axonal diameter. In some experiments (e.g. Fig. 2 A) the identification of nodes was also indicated by identification of paranodal loops in myelin labelled with FM4‐64. The pseudocolour images of Fig. 1 C show OG‐1 fluorescence, normalized to resting values before stimulation (F/F rest). During stimulation, axoplasmic [Ca2+] became elevated (red‐yellow) at both nodal regions, but remained close to resting levels (blue) along most of the intervening internode. At both nodes, [Ca2+] recovered to low pre‐stimulation levels within 5 s after the end of the stimulation train.
The distinctly localized [Ca2+] elevations in Fig. 1 C suggest that the [Ca2+] elevation originates at or near nodes. If action potentials activate local Ca2+ influx (or release) at nodal regions, then the [Ca2+] increase is expected to be large and rapid at the Ca2+ source, but become smaller and slower at more distant regions. Figure 1 D–G shows measurements testing this possibility in a nodal region (node no. 1 in Fig. 1 A) stimulated at 50 Hz for 5 s. The colour image in Fig. 1 D depicts stimulation‐induced [Ca2+] changes (F/F rest) as a pseudocolour spatiotemporal map. During stimulation (between dashed lines) a high‐[Ca2+] zone (red‐yellow) spread from the node towards the internodal regions on both sides of the node. Traces in Fig. 1 E show the time course of the [Ca2+] elevation measured at the node and at various distances from the node. Consistent with the hypothesis that [Ca2+] elevations originate at the node, the [Ca2+] increase at the node had a greater magnitude and developed more rapidly than the [Ca2+] increase at more distant internodal regions (compare red nodal with black internodal curves).
Figure 1 F and G plots the spatial extent of [Ca2+] elevations at different times during (Fig. 1 F) and after stopping the stimulus train (Fig. 1 G). During the first 0.2 s of stimulation (red plot in Fig. 1 F), the elevated‐[Ca2+] region was sharply localized to an ∼7 μm region, centred on the node. During continued stimulation this high‐[Ca2+] region expanded rapidly to include an ∼40 μm region, still centred on the node (1.0 s, magenta trace). Expansion of the high‐[Ca2+] region proceeded more slowly with continued stimulation, establishing a node‐centred ‘standing gradient’ after ∼4 s (4.2–5.0 s, black traces). After stimulation ended [Ca2+] decreased faster at regions closer to the node (Fig. 1 E); spatial profiles in Fig. 1 G show that [Ca2+] at the node decreased below the levels in the flanking para/juxtaparanodal regions (see e.g. trace at 1.0 s after stimulation ended). This finding suggests that Ca2+ extrusion mechanisms are most active at the node.
The single node findings plotted in Fig. 1 D–G are representative of those measured at other phrenic axon nodes. Figure 2 A and B summarizes results (mean ± SEM) from 11 phrenic nerve axons stimulated at 50 Hz for 10 s. Spatial profiles in Fig. 2 A plot both F/F rest and the corresponding ∆[Ca2+] estimates (see Methods) from axonal locations that start at a node (N) and progress ∼30 μm towards the internode (IN). This segment is likely to encompass the paranode and juxtaparanode, and subsequent text uses the term peri‐nodal to refer to these structures collectively. The appearances of these structures in phase and fluorescence micrographs are illustrated under the abscissa for one of these axons in which myelin was labelled with FM4‐64. At all stimulation times [Ca2+] elevations (∆[Ca2+]) were largest at the node (e.g. compare plots at 1 and 10 s), decreasing to half‐amplitude at distances of 7–11 μm from the node after 1 s of stimulation and 16–20 μm after 10 s. A crude estimate for the rate of longitudinal progression of Ca2+ during stimulation was obtained by observing that the site at which ∆[Ca2+] reached 50 nm progressed laterally by ∼10 μm in 9 s (time elapsed between the 1 and 10 s curves in Fig. 2 A). For one‐dimensional diffusion with diffusion coefficient (D), an estimate of the average travel distance (x) in time (t) is given by Einstein's approximation equation: . For unbound Ca2+ in cytoplasm, D = 0.5–0.6 × 10−5 cm2 s−1 (Hodgkin & Keynes, 1957; Donahue & Abercrombie, 1987) and the distance travelled in 9 s would be ∼100 μm. The ∼10‐fold shorter travel distance observed experimentally is compatible with restrictions on Ca2+ diffusion imposed by binding to intracellular buffers and by possible active Ca2+ extrusion (and/or sequestration) at internodes. Averaged time course records in Fig. 2 B show that during stimulation at 50 Hz, ∆[Ca2+] reached a plateau within ∼1–2 s of stimulation, with a mean value of 0.096 ± 0.019 μm (SEM) at the node, 0.064 ± 0.010 μm at a region 10 μm distant from the node (likely to be juxtaparanodal), and 0.036 ± 0.007 μm (SEM) at a region 20 μm distant from the node (likely to be internodal).
The averaged records of Fig. 2 B show that intra‐axonal [Ca2+] at the node decreased to pre‐stimulation levels 2 s after stimulation stopped, and then decreased to below pre‐stimulation levels with a small ‘undershoot’. The maximal duration of this undershoot (after stimulus trains of 10–20 s at 50 Hz) was ∼40–80 s; due to its small size it was difficult to determine the exact time course of recovery.
Figure 3 investigates the frequency dependence of the stimulation‐induced [Ca2+] changes. In these experiments all stimulus trains consisted of 500 stimuli, but stimulation frequency varied from 12.5 to 100 Hz, in the physiological range for mammalian motor axons (Hennig & Lømo, 1985). Figure 3 A shows responses to 100 Hz and 25 Hz stimulation at a node and at regions on either side of the node, demonstrating that the peak [Ca2+] elevation at all locations was greater for the higher stimulation frequency. Figure 3 B plots peak F/F rest responses for four different frequencies of stimulation as a function of distance from the node, both during and after stimulation, and Fig. 3 C plots peak F/F rest at the node as a function of stimulus frequency. Interestingly, while the peak [Ca2+] elevation increased with increasing stimulation frequency, the peak reduction in [Ca2+] after stimulation did not change. One possible explanation for this finding is that the increased Ca2+ extrusion rate suggested by the reduced post‐stimulation [Ca2+] (see Discussion), is already maximally activated by the lowest frequency trains used here.
Figure 3. Frequency dependence of responses before and after stimulation in a phrenic axon .
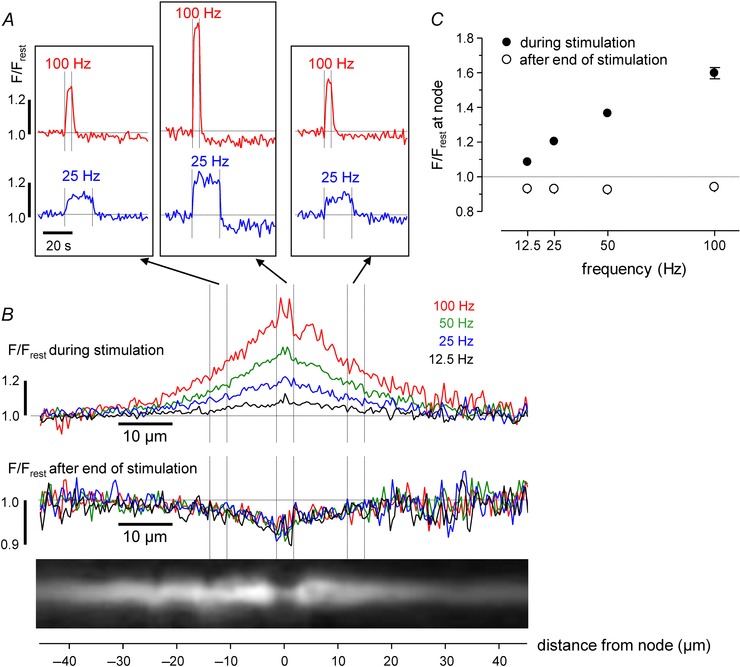
Each stimulus train consisted of 500 stimuli administered at the indicated frequency (12.5 Hz for 40 s, 25 Hz for 20 s, 50 Hz for 10 s and 100 Hz for 5 s). A, time course of F/F rest changes for 100 Hz and 25 Hz stimulation measured at the node and flanking regions of the axon illustrated in the fluorescence image in B. B, superimposed spatial profiles of F/F rest changes measured during (upper) and after (lower) stimulation at the indicated frequencies. Traces in A and B are averages of 3–4 repetitions at each frequency (repeated cycles of 12.5, 25, 50, 100 Hz). C, mean (± SEM) change in nodal F/F rest during (filled circles) and after (open circles) stimulation at the indicated frequencies (SEM was usually smaller than point diameter). In B, each trace labelled ‘during stimulation’ was generated by averaging spatial profiles (acquired each second) at times when nodal F/F rest stabilized at a plateau level. These times are the last 38 s of the 12.5 Hz train; last 18 s of 25 Hz train; last 8 s of 50 Hz train; last 3 s of 100 Hz train. Each trace labelled ‘after end of stimulation’ was generated by averaging spatial profiles over a period during which nodal F/F rest was relatively stable, 5–20 s after the end of stimulation, for all trains.
Stimulation‐induced nodal [Ca2+] elevations also occur in ventral root and intramuscular regions of motor axons
Figure 4 shows [Ca2+] responses to 50 Hz stimulation in myelinated motor axons in a ventral root (Fig. 4 A) and in an intramuscular axon ending in a motor terminal in the levator auris muscle (Fig. 4 B). Figure 4 Aa‐d shows that the ventral root node of Ranvier displayed a stimulation‐induced increase in OG‐1 fluorescence, with a spatial distribution and time course similar to those recorded from the phrenic nerve axons illustrated in Figs 1, 2, 3. Figure 4 Ba and b compares stimulation‐induced [Ca2+] elevations imaged simultaneously in a motor terminal and in nodes (N1, N2) at two branch points of the same intramuscular axon. At the nodes, ∆[Ca2+] was smaller than that in the terminal (∼0.1 vs. ∼1.0 μm) and lacked the prominent post‐stimulation tail of elevated residual Ca2+ observed at the terminal (Fig. 4 Bc and d; terminal responses were previously described in David (1999); David & Barrett (2000) and García‐Chacón et al. (2006)). These data suggest that modest elevations of [Ca2+] localized around nodes of Ranvier constitute a common response to repetitive nerve stimulation all along the peripheral extent of large‐diameter motor axons, i.e. from the ventral root all the way to the innervated muscle.
Peri‐nodal [Ca2+] elevations are mediated by Ca2+ influx via T‐type Ca2+ channels and reverse‐mode Na+/Ca2+ exchange
Figure 5 illustrates results of experiments testing various possible mechanisms underlying the stimulation‐induced nodal [Ca2+] elevations. Figure 5 A and F demonstrates that [Ca2+] responses were eliminated reversibly by removal of bath Ca2+, suggesting a major role for Ca2+ influx as distinct from release of intra‐axonal Ca2+. This suggestion is supported by the finding (Fig. 5 B and F) that stimulation‐induced [Ca2+] elevations were not reduced by a 30–60 min exposure to cyclopiazonic acid (CPA, 10 μm), a blocker of the endoplasmic reticulum (ER) Ca2+‐ATPase that is used to deplete ER Ca2+ stores. Ca2+ release from ER stores is thought to contribute to pathological [Ca2+] elevations in metabolically compromised CNS axons (Stirling & Stys, 2010).
We next tested blockers of various depolarization‐activated Ca2+ channels. Figure 5 C and F demonstrates that blockers of high voltage‐activated (HVA) Ca2+ channels, including L‐type (Cav1.x, nimodipine, 4 μm), P/Q‐type (Cav2.1, ω‐agatoxin IVA, 1 μm) and N‐type (Cav2.2, ω‐conotoxin GVIA, 3 μm), did not reduce the nodal [Ca2+] elevations. In contrast, the large stimulation‐induced [Ca2+] elevations in motor nerve terminals were dramatically reduced by the P/Q‐type blocker ω‐agatoxin IVA (mean block of 89.1 ± 2.4% (SEM), n = 6 levator auris terminals, not illustrated). Nodal [Ca2+] elevations were significantly diminished by blockers of low voltage‐activated (LVA) T‐type channels (Cav3.x), mibefradil (2 μm) and ML218 (6 μm, Xiang et al. 2011), which reduced the response by ∼40–60% (Fig. 5 D and F). Thus the stimulation‐induced [Ca2+] elevations recorded in motor axons and motor terminals involve activation of different Ca2+ channels, LVA T‐type at nodes vs. HVA P/Q‐type at motor terminals.
Another pathway for Ca2+ entry into axons is reverse‐mode operation of the plasma membrane Na+/Ca2+ exchanger (NCX), thought to mediate ischaemia‐induced [Ca2+] elevations in rat dorsal root myelinated axons (Petrescu et al. 2007). At physiological intra‐axonal [Ca2+] (50–100 nm) and [Na+] (10 mm; Stys et al. 1997; David et al. 1997 a), the reversal potential of NCX (E NCX, −60 to −40 mV) is more positive than the resting potential (−70 to −80 mV) and the exchanger operates in forward mode, extruding Ca2+ and importing Na+. However, the direction of net transport at nodes can reverse during repetitive action potentials, due to both membrane potential depolarization and elevation of intra‐axonal [Na+]. Figure 5 E and F shows that pharmacological blockers of NCX, KB‐R7943 and SN‐6 (10 μm; Török, 2007), reduced stimulation‐induced nodal [Ca2+] elevations by 30–40%. A combination of NCX and T‐type Ca2+ channel blockers (KB‐R7943 and mibefradil) blocked 82% of the [Ca2+] response (Fig. 5 E and F).
The NCX inhibitors used here are not entirely specific (Török, 2007), so the experiments of Fig. 6 used an additional strategy for investigating the importance of Ca2+ entry via reverse‐mode NCX. These experiments tested the prediction that increasing intra‐axonal [Na+] (via various mechanisms listed in Fig. 6 A, right) would increase the stimulation‐induced nodal [Ca2+] elevations. Figure 6 B shows that prolonging action potential duration, and thus increasing Na+ accumulation, using veratridine (which inhibits Na+ channel inactivation) produced an irreversible increase in the Ca2+ response. Figure 6 C shows that elevating intra‐axonal [Na+] (and [Ca2+]) using a long intervening conditioning train of action potentials also increased the [Ca2+] response (compare the response to trains no. 1 and no. 3; note also the increase in response amplitude during conditioning train no. 2). Figure 6 D shows results of experiments designed to increase intra‐axonal [Na+] by inhibiting Na+ extrusion via the Na+/K+‐ATPase pump. Inhibiting pump activity with ouabain (0.2 mm) increased the [Ca2+] response at first, but then gradually abolished the response altogether, probably due to blockade of action potential conduction via axonal depolarization. Inhibiting pump activity by decreasing extracellular [K+] produced a long‐lasting increase in the [Ca2+] response without blocking conduction. (NCX might also be implicated in the decrease in resting [Ca2+], since low extracellular [K+] hyperpolarizes axons (Morita et al. 1993), which would favour Ca2+ extrusion via forward‐mode NCX activity.) Thus a variety of techniques for increasing intra‐axonal [Na+] all increased the stimulation‐induced nodal [Ca2+] elevation, supporting the hypothesis that Ca2+ entry via reverse mode NCX makes a significant contribution to this response.
Figure 6. Treatments that elevate intra‐axonal [Na+] increase the stimulation‐induced nodal [Ca2+] elevations in phrenic axons .
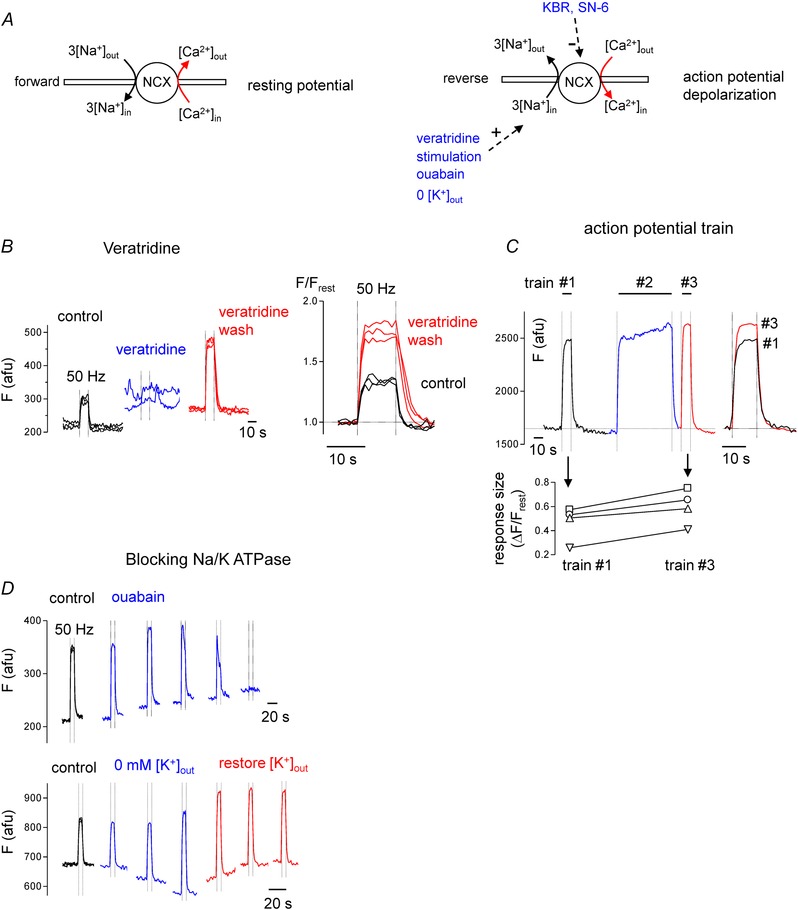
A, diagrams illustrate forward operation of NCX at the resting potential (left), and reverse‐mode NCX operation during action potential depolarization combined with 1 of the 4 listed strategies for elevating intra‐axonal [Na+] (right). B (left to right), control response to stimulation, abolition of response in veratridine (10 μm), enhanced response during veratridine washout, and comparison of control and veratridine wash responses. C, control response to 10 s train (no. 1), response during prolonged conditioning train (no. 2), response to repeated 10 s train (no. 3), and comparison of responses to train no. 1 and no. 3. In the graph below, each line connects the response to train no. 1 and no. 3 measured in each of 4 separate axons; points labelled with upright triangles (∆) correspond to the axon depicted in C. D, effects of blocking Na+/K+‐ATPase activity with ouabain (0.2 mm, upper traces) or by removing and later restoring extracellular [K+] (lower traces). Each record plots sequential responses to 50 Hz stimulation trains repeated at 10 min intervals. In B–D fluorescence (F) is plotted in arbitrary fluorescence units (afu) to indicate changes in both resting and stimulated fluorescence.
Peri‐nodal axolemma contains L‐type Ca2+ channels that are normally silent, but can be activated following BAYK‐8644 or paranodal demyelination
The lack of effect of nimodipine on stimulation‐induced [Ca2+] elevations (Fig. 5 C and F) suggests that Ca2+ influx via L‐type Ca2+ channels makes little or no contribution to the [Ca2+] increase. This finding (also reported for frog peripheral axons by Zhang et al. (2010)) could result from at least two possible scenarios: (1) L‐type channels might be absent, or (2) L‐type channels might be present, but in a sub‐myelin location that precludes their activation by action potentials. L‐type channels require large depolarizations to activate (half‐activation voltage −36 to −18 mV; Tuckwell, 2012). Depolarizations of this magnitude occur in nodal axolemma, but not in peri‐nodal/internodal axolemma covered by myelin. In these sub‐myelin regions most of the action potential depolarization occurs across the myelin, with only ∼10–20 mV depolarization across the sub‐myelin axolemma (Blight, 1985; David et al. 1995). Thus, if L‐type channels are present in sub‐myelin axolemma, they would be silent under normal conditions. We used two strategies to try to unmask any sub‐myelin L‐type channels: (1) treatment that shifts channel activation to more negative voltages and (2) acute paranodal demyelination, thereby increasing the magnitude of the action potential depolarization experienced by sub‐myelin axolemma (Blight, 1985).
BAY‐K8644 shifts the activation of L‐type channels towards more negative potentials, prolongs the open‐time by ∼10‐fold and causes a ∼5‐fold increase in Ca2+ current elicited by a 20 mV depolarization from rest (Nowycky et al. 1985; Scroggs & Fox, 1991). Figure 7 shows that BAY‐K8644 (2 μm) increased the magnitude of the stimulation‐induced nodal Ca2+ elevation by ∼130% (Fig. 7 A and C) and expanded the spatial extent of the high‐[Ca2+] region (Fig. 7 B). L‐type Ca2+ channels modified by BAY‐K8644 retain their sensitivity to nimodipine block (McCarthy & TanPiengco, 1992). Figure 7 shows that most of the BAY‐K8644‐induced augmentation of the [Ca2+] response was suppressed by nimodipine. Nimodipine's effect confirms that the enhancement of the [Ca2+] elevation by BAY‐K8644 was indeed attributable to activation of L‐type Ca2+ channels, and that nimodipine has access to these channels. Thus, nimodipine's lack of effect on [Ca2+] elevations in naïve axons (without BAYK‐K8644 enhancement, Fig. 5 C and F) is not due to limited drug access or to absence of L‐type channels. Rather, the nimodipine‐resistance in intact axons indicates that Ca2+ influx via (existing) L‐type channels is normally small relative to the influx mediated by T‐type Ca2+ channels and reverse NCX (Fig. 5 D and E).
Figure 7. BAYK‐8644, which enhances activation of and influx through L‐type Ca2+ channels, increases stimulation‐induced [Ca2+] elevations in phrenic axons .
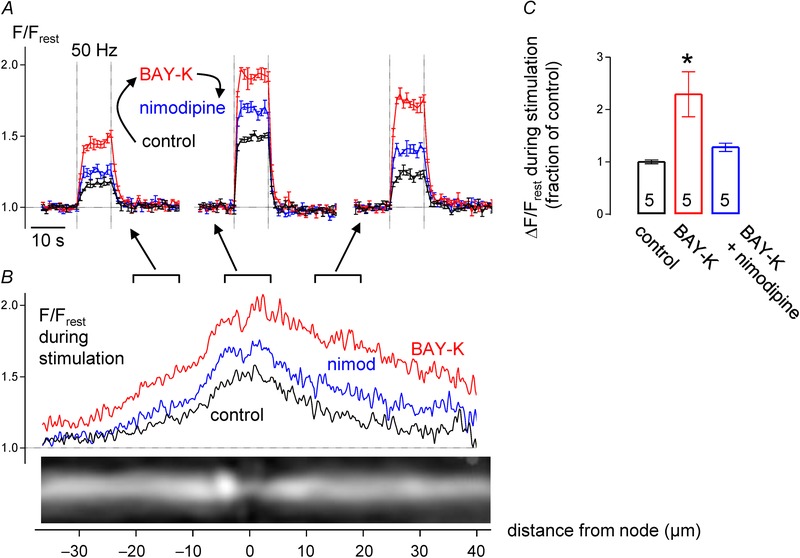
A, superimposed time courses of F/F rest responses to 50 Hz stimulus train, recorded in node (centre) and flanking internodal regions in control solution, after addition of BAYK‐8644 (BAY‐K, 2 μm), and after addition of nimodipine to the BAYK‐8644 solution. B, spatial extent of F/F rest elevation in control, BAY‐K and Bay‐K + nimodipine. Records in A and B came from the axon illustrated in B; records in A plot mean ± SEM of 4 responses. C, ΔF/F rest (mean ± SEM) of peak nodal responses to stimulation in the indicated solutions (n = 5 axons, BAY‐K response significantly different from control at P < 0.01). Spatial profile curves in B were generated by averaging spatial profiles (acquired each second) at times when nodal F/F rest stabilized at a plateau level, between the 5th and 10th second of stimulation at 50 Hz.
Figure 8 demonstrates that acute disruption of paranodal axon–Schwann cell junctions, by application of detergent or by mechanical stretch, enhances stimulation‐induced [Ca2+] elevations. Figure 8 A illustrates effects of the detergent lysophosphatidylcholine (LPC, 1 mg mL−1). A 5–15 min exposure to LPC causes retraction of paranodal Schwann cell loops, there by exposing the underlying axolemma, a procedure used to permit activation of K+ channels normally masked by their location in sub‐myelin axolemma (Hall & Gregson, 1971; Gregson & Hall 1973; Chiu & Ritchie, 1980, 1981). OG‐1 fluorescence images show the peri‐nodal morphology of a phrenic nerve axon before and after a 10 min exposure to LPC. The control image shows a 4 μm‐long constricted segment formed by the normal node and its flanking paranodes. The apparent elongation of this constricted segment after LPC is likely to be due to swollen Schwann cell paranodal loops impinging on the axon (Ouyang et al. 2010; Sotnikov et al. 2011). The illustrated F/F rest recordings show the time course (upper traces) and spatial extent (lower traces) of stimulation‐induced [Ca2+] elevations in this axon before and after LPC exposure. LPC increased both the magnitude and spatial extent of the [Ca2+] elevations. Data summarized from five axons show that LPC increased the magnitude of nodal [Ca2+] responses by 101%. All five axons treated with LPC displayed increased peri‐nodal [Ca2+] responses, but in two of these axons the effect was much larger on one side of the node (e.g. left side of node in
Figure 8. Paranodal demyelination increases stimulation‐induced [Ca2+] responses by enabling activation of L‐type Ca2+ channels .
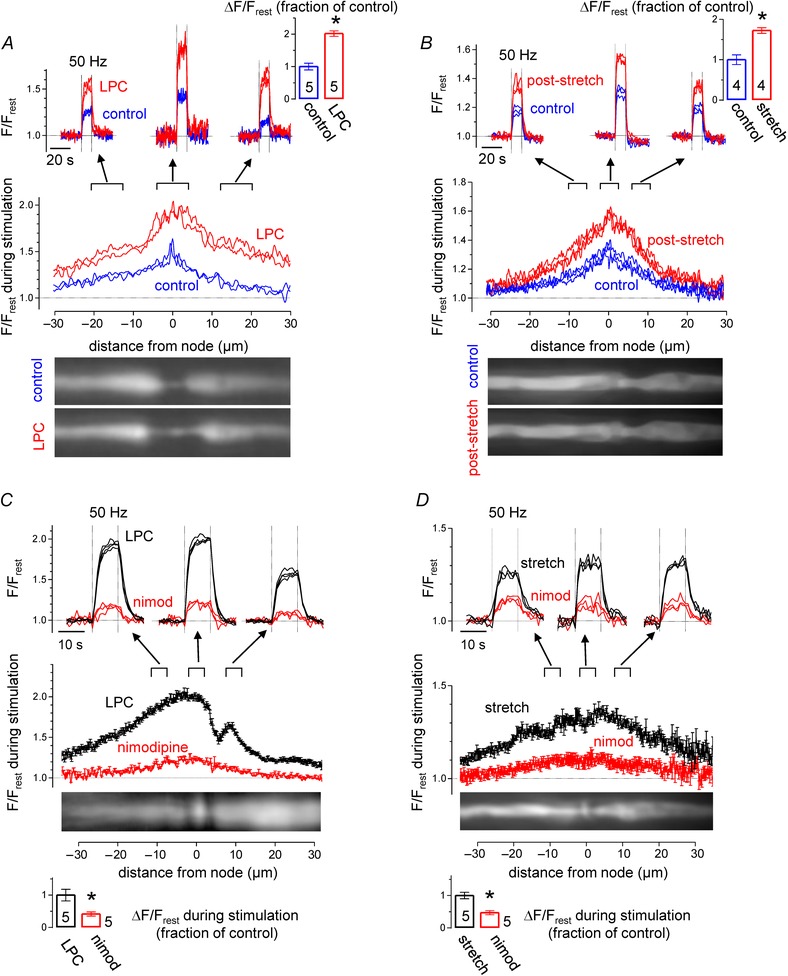
A, effects of brief exposure to LPC. Traces plot time courses and spatial extents of F/F rest elevations recorded around the illustrated node before and after exposure to LPC. Bar graph summarizes results of nodal responses in 5 LPC‐treated axons. B, similar recordings in axons subjected to brief mechanical stretch. C and D, time courses and spatial extents of the F/F rest responses recorded in an LPC‐treated axon (C) or a stretched axon (D) before and following application of nimodipine. Other aspects of the format are similar to that in A. P < 0.01 in A, P < 0.05 in B, P < 0.05 in C, P < 0.01 in D. Spatial profile curves were generated by averaging spatial profiles (acquired each second) at times when nodal F/F rest stabilized at a plateau level, between the 5th and 10th second of stimulation at 50 Hz.
Fig. 8 C). This asymmetry might be due to different degrees of LPC‐induced myelin detachment at the two paranodes.
Mechanical stretch of mammalian myelinated axons also produces acute paranodal demyelination (Chiu & Ritchie, 1980; 1981). After obtaining control records of stimulation‐induced [Ca2+] responses, the phrenic nerve containing the imaged axon was subjected to a brief longitudinal stretch, as described in Methods. Figure 8 B compares control and post‐stretch stimulation‐induced [Ca2+] elevations in the illustrated phrenic axon, demonstrating that stretch increased the magnitude of nodal and peri‐nodal [Ca2+] elevations. Results from four axons indicate an average increase of 72% in the nodal response.
Figure 8 C and D shows that the large, stimulation‐induced [Ca2+] elevations recorded after LPC and stretch were markedly suppressed by nimodipine. This blocker of L‐type Ca2+ channels decreased the post‐LPC and post‐stretch responses by 58 and 53%, respectively.
The findings that acute paranodal demyelination increases the magnitude and spatial extent of stimulation‐induced [Ca2+] elevations, and that this effect is greatly reduced by nimodipine, supports the conclusion that peri‐nodal axolemma contains L‐type Ca2+ channels that are not normally activated by action potentials, but can be activated following disruption of the myelin that normally covers them.
Discussion
Differential utilization of Ca2+ entry pathways in motor neuron somata, axons and terminals
Work presented here demonstrates that repetitive stimulation of mouse motor axons produces [Ca2+] elevations of ∼0.1 μm (at 50 Hz) localized around nodes of Ranvier. This finding appears to be a general feature of large‐diameter motor axons, since it was observed in ventral roots, peripheral nerve and intramuscular nerve branches, in axons whose somata are located in a cranial nucleus and in cervical and lumbar spinal cord. [Ca2+] elevations localized to nodes were also reported in some frog sciatic axons by Zhang et al. (2010); they presented evidence that Ca2+ entry contributed to these elevations, but did not identify the underlying influx pathways. We present evidence that in mammalian motor axons the stimulation‐induced nodal [Ca2+] elevations are mediated by Ca2+ entry with roughly equal contributions from T‐type Ca2+ channels and NCX (reverse mode).
Previous work has demonstrated that mammalian motor neuron somata express multiple types of depolarization‐activated Ca2+ channels, including both high voltage‐activated (HVA) P/Q‐, N‐, L‐ and R‐type channels (Westenbroek et al. 1998) and low voltage‐activated (LVA) channels (Palecek et al. 1999). The differential utilization of these channels in various parts of the motor neuron is diagrammed in Fig. 9. Fluorescence imaging and voltage clamp studies indicate that in somata HVA channels contribute much more than LVA channels to stimulation‐induced [Ca2+] elevations (Palecek et al. 1999). For example, in hypoglossal motor neurons 85% of the Ca2+ current elicited by depolarization from −70 to 0 mV entered via HVA Ca2+ channels (P/Q 50%, N 29% and L 6%), with the remaining 15% via LVA Ca2+ channels (Umemiya & Berger, 1994). Both pharmacological and immunohistochemical studies demonstrate that in mammalian motor nerve terminals the major Ca2+ channel responsible for triggering transmitter release is the P/Q‐type (Day et al. 1997; Urbano et al. 2002; and this paper). Our data from motor axons indicate the presence of T‐type channels and NCXs near nodes, but cannot distinguish whether these Ca2+ entry pathways are nodal and/or peri‐nodal. Thus the pathways for peri‐nodal Ca2+ entry in motor axons (T‐type LVA channels, reverse‐mode NCX) differ from those utilized in somata and motor nerve terminals. Both HVA and T‐type channels are reported to play significant roles in the axon initial segment (Bender et al. 2012; reviewed by Kole & Stuart, 2012).
Figure 9. Diagrams summarizing the dominant pathways used for stimulation‐induced Ca2+ entry into motor neuron soma, motor axon and motor terminal (A) and the appearance of Ca2+ entry via L‐type channels in axons following acute paranodal demyelination (B) .
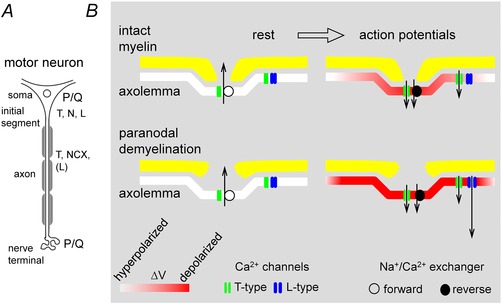
Diagrams in B show nodes at rest (left) and during repetitive stimulation (right), with a normal node at the top (intact myelin) and a disrupted node at the bottom (paranodal demyelination). In all cases T‐type channels in nodal/paranodal axolemma are activated during stimulation, and the direction of NCX‐mediated Ca2+ transport shifts from extrusion in the resting axon to entry during stimulation. With intact myelin the depolarization of the axolemma during action potentials (shades of red) is largely restricted to the node; the much smaller depolarization in peri‐nodal regions (under myelin) is sufficient to activate LVA T‐type channels but not HVA L‐type channels. Following acute paranodal demyelination, action potentials produce greater depolarization of peri‐nodal axolemma, sufficient to activate L‐type channels. For simplicity, peri‐nodal channels were drawn only on the right side of each node. Information in A concerning soma and initial segment comes from published studies (see Discussion). The exact distribution of T‐type channels and NCX between nodal and peri‐nodal axolemma is not known, and possible contributions of reverse‐mode NCX‐mediated transport to Ca2+ entry into soma and terminal have (to our knowledge) not been determined.
While HVA Ca2+ channels appear to play little role in normally myelinated motor axons, acute paranodal demyelination produces large increases in stimulation‐induced [Ca2+] elevations, due mostly to L‐type channels (nimodipine‐sensitive). Thus the axolemma of myelinated axons contains L‐type channels, but they appear to be located in regions that are covered by myelin and therefore normally experience only a small portion of the action potential depolarization, not sufficient to activate these HVA channels (see intact myelin in Fig. 9 B). Consistent with this idea, in intact axons L‐type channels made significant contributions to peri‐nodal [Ca2+] elevations only after the activation threshold of L‐type channels was reduced by BAY‐K8644 (Fig. 7). After myelin disruption by LPC or stretch, channels in peri‐nodal axolemma experience more of the action potential depolarization, sufficient to activate L‐type channels (Fig. 8, paranodal demyelination in Fig. 9 B).
Thus it appears that motor neurons normally transport T‐, L‐ and P/Q‐type channels down their axons, but insert these channels in different locations, T‐ and L‐types near nodes, with P/Q reserved for terminals. Different circumstances can alter this channel sorting: for example, in transgenic mice lacking P/Q‐type channels, N‐ and R‐type channels appear in terminals (Urbano et al. 2002), and L‐ and N‐type channels are transiently present in motor terminals regenerating after botulinum toxin‐induced damage (Santafé et al. 2000).
Note that in the diagram in Fig. 9 the direction of Ca2+ transport via NCX reverses from extruding Ca2+ at rest, to importing Ca2+ during repetitive nerve stimulation. As discussed in Results, this reversal is consistent with changes in the electrochemical gradients governing NCX transport expected during the conversion from rest to repetitive activation. Evidence for Ca2+ import during stimulation (reverse‐mode NCX) includes the findings that stimulation‐induced [Ca2+] elevations in axons with intact myelin were reduced to about half by NCX inhibitors (Fig. 5), and increased by strategies that increase Na+ influx (Fig. 6). NCX transcripts are expressed in spinal motor neurons (Papa et al. 2003), and immunohistochemical evidence suggests that they are expressed throughout the axolemma (nodal/peri‐nodal/internodal, Steffensen et al. 1997). But electrochemical considerations indicate that the NCX contribution to Ca2+ entry during stimulation would be greatest near nodes, where both axolemmal depolarization and axoplasmic [Na+] would be maximal.
Measurements of the spatial profile of [Ca2+] decay during recovery from stimulation (Fig. 3) suggest powerful mechanisms for Ca2+ extrusion in the nodal region, likely to be due to both forward‐mode NCX transport and plasma membrane Ca2+‐ATPase (PMCA), which is localized near nodes in peripheral myelinated axons (Mata et al. 1988; Mizutani et al. 1995). Interestingly, Na+/K+‐ATPase‐mediated Na+ efflux is also thought to occur at higher rates at nodal vs. internodal regions (Connelly, 1959), yet additional Na+ extrusion also occurs along internodes (David et al. 1997 a). The post‐stimulation undershoot of [Ca2+] in nodal/peri‐nodal regions (e.g. Figs 2 B and 3) might occur because the Ca2+‐initiated activation of NCX and PMCA during the stimulation train decays quite slowly and thus allows higher Ca2+ extrusion rates to persist after the stimulation train (NCX, Scheff et al. 2014; PMCA, Caride et al. 2001)
Functional roles of stimulation‐induced nodal/peri‐nodal [Ca2+] elevations
The nodal [Ca2+] elevations that accompany stimulation in normally myelinated axons are modest, about 10‐fold lower than the elevations recorded in motor terminals (though in domains around open channels local [Ca2+] will of course be higher than the spatially averaged estimate). The role(s) of these nodal [Ca2+] elevations are as yet unknown, but the literature suggests several possibilities, which we present as speculations for future study.
First, nodal [Ca2+] elevations might stimulate exocytosis of membrane vesicles located in nodes of Ranvier. These vesicles are reported to contain proteins associated with Ca2+‐dependent transmitter release at synapses (e.g. SNARE complex proteins in developing rodent optic nerve – Alix et al. (2008); SV2 and synaptophysin in rat peripheral axons – Zimmermann (1996)). Exocytosis of membrane vesicles might participate in local membrane turnover and/or release some messenger substance(s) signalling axonal activation to the paranodal loops of overlying Schwann cells. One possible role for this hypothesized axon–glial communication is maintenance of the integrity and modulation of the thickness of the myelin sheath; evidence from zebra fish CNS suggests that axonal electrical activity promotes myelination (Hines et al. 2015; Mensch et al. 2015). In adult rat peripheral nerves whose activity is experimentally reduced (soleus nerve) or increased (radial nerve) by 14 days of hind limb unloading, myelin thickness (relative to axon diameter) decreased in the less active nerves and increased in the more active nerves (Canu et al. 2009; reviewed in Bucher & Goaillard, 2011).
Second, nodal [Ca2+] elevations might stimulate respiration, thereby increasing the supply of ATP needed to restore transmembrane ion gradients following repetitive stimulation. Ca2+ in the mitochondrial matrix can activate citric acid cycle enzymes, electron transport chain dehydrogenases and ATP synthase (reviewed in Glancy & Balban, 2012; Rueda et al. 2014), and elevations of cytosolic [Ca2+] can increase transport of energy substrates into the mitochondrial matrix (Gellerich et al. 2013; Llorente‐Folch et al. 2013).
Third, nodal [Ca2+] elevations might also influence axoplasmic mitochondrial transport. Zhang et al. (2010) present evidence that stimulation increased the proportion of stationary mitochondria in frog sciatic axons, an effect they attributed to the combination of (1) elevated [Ca2+] and (2) increased competition for ATP due to the increased Na+/K+‐ATPase activity needed to restore transmembrane ion gradients. However, investigators using mouse saphenous nerves reported evidence for the opposite effect, i.e. that axonal stimulation speeds mitochondrial transport (Sajic et al. 2013).
Fourth, nodal [Ca2+] elevations might modulate axonal excitability. Increased [Ca2+] in the physiological range (10–400 nm) suppresses current through KCNQ channels (Kv7.2 and 7.3) (Kosenko & Hoshi, 2013), which are present at peripheral nodes of Ranvier (Devaux et al. 2004; Schwarz et al. 2006). These channels mediate a slowly activating nodal K+ current thought to contribute to the axonal resting membrane potential (reviewed by Cooper, 2011), so Ca2+‐mediated inhibition of channel activity would be expected to depolarize the resting potential. Interaction with Ca2+‐activated K+ channels would be expected to hyperpolarize the axon during and after stimulation trains. Mi et al. (1999) found no immunohistochemical evidence for the presence of these channels in the axolemma of rat sciatic nerve, but these channels (BK) have been detected in amphibian patch‐clamped peripheral axon nodes at a relatively low density (Jonas et al. 1991), which might have precluded immunohistochemical detection.
The much larger nodal/peri‐nodal [Ca2+] elevations recorded in acutely (paranodally) demyelinated axons (∼0.5 μm during 50 Hz stimulation) might signal Schwann cells to repair injured myelin, perhaps via an axon‐to‐glia signalling pathway like that hypothesized above. But if demyelination becomes extensive and widespread, the large elevations of axoplasmic [Ca2+] accompanying axonal activity might become sufficient to activate intracellular proteases like μ‐calpain, thereby initiating axonal degeneration. Arroyo et al. (2004) report that the molecular organization of peripheral axon nodes becomes disrupted 5–7 days following brief exposure to LPC. Axonal degeneration occurs in some demyelinating diseases including Charcot–Marie–Tooth type 1A and multiple sclerosis (Krajewski et al. 2000; Friese et al. 2014). One hypothesized contributing factor is pathological influx of Ca2+. The literature suggests that during chronic demyelination this increased Ca2+ influx could occur via at least two mechanisms: (1) aberrant insertion of voltage‐gated Ca2+ channels into demyelinated axons (e.g. N‐type channels in multiple sclerosis), and (2) increased reverse mode NCX activity, due to the co‐localization of Na+ channels and NCX that develops along demyelinated plaques (Friese et al. 2014). Our findings with acutely demyelinated motor axons suggest that another contributor to pathological Ca2+ influx is unmasking of L‐type Ca2+ channels that are normally present in peri‐nodal axolemma. Such channels would increase activity‐associated Ca2+ influx immediately after myelin disruption.
In summary, measurements using a fluorescent [Ca2+] indicator injected into large‐diameter mouse motor axons demonstrate that repetitive action potential stimulation produces [Ca2+] elevations localized around nodes. Pharmacological evidence indicates that in normally myelinated axons these elevations are mediated mainly by Ca2+ influx through (1) T‐type (low voltage‐activated) Ca2+ channels and (2) reverse mode Na+/Ca2+ exchange. Stimulation‐induced nodal [Ca2+] elevations become much greater following acute myelin disruption, due largely to activation of L‐type (nimodipine‐sensitive) high voltage‐activated Ca2+ channels. The fact that L‐type channel activity becomes apparent immediately following myelin disruption suggests that these channels are normally present in peri‐nodal (sub‐myelin) axolemma. The physiological and pathophysiological functions of these stimulation‐induced peri‐nodal [Ca2+] elevations merit further study.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
G.D. designed the study, Z.Z. and G.D. performed the experiments, and G.D. analysed the data and wrote the manuscript. All authors are accountable for all aspects of the work and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. All authors read and approved the final version of the manuscript. Work was done at the University of Miami Miller School of Medicine.
Funding
This work was supported by a grant from the Muscular Dystrophy Association.
Acknowledgements
We thank Drs. Ellen Barrett and John Barrett for valuable discussions and help with the manuscript.
References
- Alix JJP, Dolphin AC & Fern R (2008). Vesicular apparatus, including functional calcium channels, are present in developing rodent optic nerve axons and are required for normal node of Ranvier formation. J Physiol 586, 4069–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angaut‐Petit D, Molgo J, Connold AL & Faille L (1987). The levator auris longus muscle of the mouse: a convenient preparation for studies of short‐ and long‐term presynaptic effects of drugs or toxins. Neurosci Lett 82, 83–88. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Sirkowski EE, Chitale R & Scherer SS (2004). Acute demyelination disrupts the molecular organization of peripheral nervous system nodes. J Comp Neurol 479, 424–434. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Uebele VN, Renger JJ & Trussell LO (2012). Control of firing patterns through modulation of axon initial segment T‐type calcium channels. J Physiol 590, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight AR (1985). Computer simulation of action potentials and afterpotentials in mammalian myelinated axons: the case for a lower resistance myelin sheath. Neuroscience 15, 13–31. [DOI] [PubMed] [Google Scholar]
- Bucher D & Goaillard J‐M (2011). Beyond faithful conduction: short‐term dynamics, neuromodulation, and long‐term regulation of spike propagation in the axon. Prog Neurobiol 94, 307–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canu M‐H, Carnaud M, Picquet F & Goutebroze L (2009). Activity‐dependent regulation of myelin maintenance in the adult rat. Brain Res 1252, 45–51. [DOI] [PubMed] [Google Scholar]
- Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi A & Penniston JT (2001). The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J Biol Chem 276, 39797–39804. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez‐Reyes E, Snutch TP & Striessnig J (2005). International Union of Pharmacology. XLVIII. Nomenclature and structure‐function relationships of voltage‐gated calcium channels. Pharmacol Rev 57, 411–425. [DOI] [PubMed] [Google Scholar]
- Chiu SY & Ritchie JM (1980). Potassium channels in nodal and internodal axonal membrane of mammalian myelinated fibres. Nature 284, 170–171. [DOI] [PubMed] [Google Scholar]
- Chiu SY & Ritchie JM (1981). Evidence for the presence of potassium channels in the paranodal region of acutely demyelinated mammalian single nerve fibres. J Physiol 313, 415–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CM (1959). Recovery processes and metabolism of nerve. Rev Mod Phys 31, 475–484. [Google Scholar]
- Cooper EC (2011). Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin Cell Dev Biol 22, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier F, Decker L, Funalot B, Vallat J‐M, Garcia‐Bragado F, Charnay P & Topilko P (2010). CNS/PNS boundary transgression by central glia in the absence of Schwann cells or Krox20/Egr2 function. J Neurosci 30, 5958–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G (1999). Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+]. J Neurosci 19, 7495–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G & Barrett EF (2000). Stimulation‐evoked increases in cytosolic [Ca2+] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature‐dependent. J Neurosci 20, 7290–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN & Barrett EF (1997. a). Spatiotemporal gradients of intra‐axonal [Na+] after transection and resealing in lizard peripheral myelinated axons. J Physiol 498, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN & Barrett EF (1997. b). Stimulation‐induced changes in [Ca2+] in lizard motor nerve terminals. J Physiol 504, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Modney B, Scappaticci KA, Barrett JN & Barrett EF (1995). Electrical and morphological factors influencing the depolarizing after‐potential in rat and lizard myelinated axons. J Physiol 489, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day NC, Wood SJ, Ince PG, Volsen SG, Smith W, Slater CR & Shaw PJ (1997). Differential localization of voltage‐dependent calcium channel α1 subunits at the human and rat neuromuscular junction. J Neurosci 17, 6226–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC & Scherer SS (2004). KCNQ2 is a nodal K+ channel. J Neurosci 24, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue BS & Abercrombie RF (1987). Free diffusion coefficient of ionic calcium in cytoplasm. Cell Calcium 8, 437–448. [DOI] [PubMed] [Google Scholar]
- Fazan VPS, Rodrigues Filho OA, Jordão CER & Moore KC (2009). Ultrastructural morphology and morphometry of phrenic nerve in rats. Anat Rec (Hoboken) 292, 513–517. [DOI] [PubMed] [Google Scholar]
- Friede RL, Benda M, Dewitz A & Stoll P (1984). Relations between axon length and axon caliber. “Is maximum conduction velocity the factor controlling the evolution of nerve structure”? J Neurol Sci 63, 369–380. [DOI] [PubMed] [Google Scholar]
- Friese MA, Schattling B & Fugger L (2014). Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol 10, 225–238. [DOI] [PubMed] [Google Scholar]
- García‐Chacón LE, Nguyen KT, David G & Barrett EF (2006). Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+–Ca2+ exchanger increases post‐tetanic evoked release. J Physiol 574, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z & Vielhaber S (2013). The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life 65, 180–190. [DOI] [PubMed] [Google Scholar]
- Glancy B & Balaban RS (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschall J (1981). The diaphragm of the rat and its innervation. Muscle fiber composition; perikarya and axons of efferent and afferent neurons. Anat Embryol (Berl) 161, 405–417. [DOI] [PubMed] [Google Scholar]
- Gregson NA & Hall SM (1973). A quantitative analysis of the effects of the intraneural injection of lysophosphatidyl choline. J Cell Sci 13, 257–277. [DOI] [PubMed] [Google Scholar]
- Hall SM & Gregson NA (1971). The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibres of the adult mouse. J Cell Sci 9, 769–789. [DOI] [PubMed] [Google Scholar]
- Hennig R & Lømo T (1985). Firing patterns of motor units in normal rats. Nature 314, 164–166. [DOI] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK & Appel B (2015). Neuronal activity biases axon selection for myelination in vivo . Nat Neurosci 18, 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL & Keynes RD (1957). Movements of labelled calcium in squid giant axons. J Physiol 138, 253–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Koh DS, Kampe K, Hermsteiner M & Vogel W. (1991). ATP‐sensitive and Ca‐activated K channels in vertebrate axons: novel links between metabolism and excitability. Pflugers Arch 418, 68–73. [DOI] [PubMed] [Google Scholar]
- Kole MHP & Stuart GJ (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. [DOI] [PubMed] [Google Scholar]
- Kosenko A & Hoshi N (2013). A change in configuration of the calmodulin‐KCNQ channel complex underlies Ca2+‐dependent modulation of KCNQ channel activity. PLoS One 8, e82290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J & Shy ME (2000). Neurological dysfunction and axonal degeneration in Charcot‐Marie‐Tooth disease type 1A. Brain 123, 1516–1527. [DOI] [PubMed] [Google Scholar]
- Lehning EJ, Doshi R, Isaksson N, Stys PK & LoPachin RM (1996). Mechanisms of injury‐induced calcium entry into peripheral nerve myelinated axons: role of reverse sodium‐calcium exchange. J Neurochem 66, 493–500. [DOI] [PubMed] [Google Scholar]
- Lev‐Ram V & Ellisman MH (1995). Axonal activation‐induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J Neurosci 15, 2628–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente‐Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B & Satrústegui J (2013). Calcium‐regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1‐malate aspartate shuttle in intact cortical neurons. J Neurosci 33, 13957–13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL & Svoboda K (2000). Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J 78, 2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Staple J & Fink DJ (1988). Cytochemical localization of Ca2+‐ATPase activity in peripheral nerve. Brain Res 445, 47–54. [DOI] [PubMed] [Google Scholar]
- Mattei C, Elmorjani K, Molgó J, Marion D & Benoit E (1998). The wheat proteins puroindoline‐a and α1‐purothionin induce nodal swelling in myelinated axons. Neuroreport 9, 3803–3807. [DOI] [PubMed] [Google Scholar]
- McCarthy RT & TanPiengco PE (1992). Multiple types of high‐threshold calcium channels in rabbit sensory neurons: high‐affinity block of neuronal L‐type by nimodipine. J Neurosci 12, 2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A & Lyons DA (2015). Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 18, 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Harris‐Warrick RM, Deerinck TJ, Inman I, Ellisman MH & Schwarz TL (1999). Identification and localization of Ca2+‐activated K+ channels in rat sciatic nerve. Glia 26, 166–175. [DOI] [PubMed] [Google Scholar]
- Mizutani Y, Hirai K, Wang GY, Toda N & Yamazaki Y (1995). Fine localization of low and high calcium dependent ATPase activities in the rat sciatic nerve. Brain Res 693, 95–100. [DOI] [PubMed] [Google Scholar]
- Morita K, David G, Barrett JN & Barrett EF (1993). Posttetanic hyperpolarization produced by electrogenic Na+‐K+ pump in lizard axons impaled near their motor terminals. J Neurophysiol 70, 1874–1884. [DOI] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP & Tsien RW (1985). Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443. [DOI] [PubMed] [Google Scholar]
- Ouyang H, Sun W, Fu Y, Li J, Cheng J‐X, Nauman E & Shi R (2010). Compression induces acute demyelination and potassium channel exposure in spinal cord. J Neurotrauma 27, 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek J, Lips MB & Keller BU (1999). Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J Physiol 520, 485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa M, Canitano A, Boscia F, Castaldo P, Sellitti S, Porzig H, Taglialatela M & Annunziato L (2003). Differential expression of the Na+‐Ca2+ exchanger transcripts and proteins in rat brain regions. J Comp Neurol 461, 31–48. [DOI] [PubMed] [Google Scholar]
- Petrescu N, Micu I, Malek S, Ouardouz M & Stys PK (2007). Sources of axonal calcium loading during in vitro ischemia of rat dorsal roots. Muscle Nerve 35, 451–457. [DOI] [PubMed] [Google Scholar]
- Rueda CB, Llorente‐Folch I, Amigo I, Contreras L, González‐Sánchez P, Martínez‐Valero P, Juaristi I, Pardo B, del Arco A & Satrústegui J (2014). Ca2+ regulation of mitochondrial function in neurons. Biochim Biophys Acta 1837, 1617–1624. [DOI] [PubMed] [Google Scholar]
- Sajic M, Mastrolia V, Lee CY, Trigo D, Sadeghian M, Mosley AJ, Gregson NA, Duchen MR & Smith KJ (2013). Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biol 11, e1001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santafé MM, Urbano FJ, Lanuza MA & Uchitel OD (2000). Multiple types of calcium channels mediate transmitter release during functional recovery of botulinum toxin type A‐poisoned mouse motor nerve terminals. Neuroscience 95, 227–234. [DOI] [PubMed] [Google Scholar]
- Scheff NN, Yilmaz E & Gold MS (2014). The properties, distribution and function of Na+‐Ca2+ exchanger isoforms in rat cutaneous sensory neurons. J Physiol 592, 4969–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H, Jensen HJ, Bjaerg M, Kamieniecka Z & Kurland L (1991). A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol 50, 192–204. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JR, Glassmeier G, Cooper EC, Kao T‐C, Nodera H, Tabuena D, Kaji R & Bostock H (2006). KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol 573, 17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS & Fox AP (1991). Distribution of dihydropyridine and ω‐conotoxin‐sensitive calcium currents in acutely isolated rat and frog sensory neuron somata: diameter‐dependent L channel expression in frog. J Neurosci 11, 1334–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R & Whitebone J (2006). Conduction deficits and membrane disruption of spinal cord axons as a function of magnitude and rate of strain. J Neurophysiol 95, 3384–3390. [DOI] [PubMed] [Google Scholar]
- Sotnikov OS, Kokurina TN, Solov'eva IA & Sergeeva SS (2011). Early reactive changes of myelin sheath in the area of myelin sheath gaps (nodes of Ranvier) in nerve fibers (a supravital study). Morfol 139, 46–50. [PubMed] [Google Scholar]
- Stathopoulos P, Alexopoulos H & Dalakas MC (2015). Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol 11, 143–156. [DOI] [PubMed] [Google Scholar]
- Steffensen I, Waxman SG, Mills L & Stys PK (1997). Immunolocalization of the Na+‐Ca2+ exchanger in mammalian myelinated axons. Brain Res 776, 1–9. [DOI] [PubMed] [Google Scholar]
- Stirling DP & Stys PK (2010). Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol Med 16, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Lehning E, Saubermann AJ & LoPachin RM (1997). Intracellular concentrations of major ions in rat myelinated axons and glia: calculations based on electron probe X‐ray microanalyses. J Neurochem 68, 1920–1928. [DOI] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE & Unser M (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7, 27–41. [DOI] [PubMed] [Google Scholar]
- Thomas D, Tovey SC, Collins TJ, Bootman MD, Berridge MJ & Lipp P (2000). A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 28, 213–223. [DOI] [PubMed] [Google Scholar]
- Török TL (2007). Electrogenic Na+/Ca2+‐exchange of nerve and muscle cells. Prog Neurobiol 82, 287–347. [DOI] [PubMed] [Google Scholar]
- Tuckwell HC (2012). Quantitative aspects of L‐type Ca2+ currents. Prog Neurobiol 96, 1–31. [DOI] [PubMed] [Google Scholar]
- Umemiya M & Berger AJ (1994). Properties and function of low‐ and high‐voltage‐activated Ca2+ channels in hypoglossal motoneurons. J Neurosci 14, 5652–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Rosato‐Siri MD & Uchitel OD (2002). Calcium channels involved in neurotransmitter release at adult, neonatal and P/Q‐type deficient neuromuscular junctions (Review). Mol Membr Biol 19, 293–300. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L & Catterall WA (1998). Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci 18, 6319–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Hopkins CR, Xie XS, Conn PJ, Weaver CD, Lindsley CW. (2011). The discovery and characterization of ML218: A novel, centrally active T‐Type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson's disease. ACS Chem Neurosci 2, 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wilson JA, Williams J & Chiu SY (2006). Action potentials induce uniform calcium influx in mammalian myelinated optic nerves. J Neurophysiol 96, 695–709. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Ho PL, Kintner DB, Sun D & Chiu SY (2010). Activity‐dependent regulation of mitochondrial motility by calcium and Na/K‐ATPase at nodes of Ranvier of myelinated nerves. J Neurosci 30, 3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H (1996). Accumulation of synaptic vesicle proteins and cytoskeletal specializations at the peripheral node of Ranvier. Microsc Res Tech 34, 462–473. [DOI] [PubMed] [Google Scholar]