

Key points
The hD4.7 variant has been linked to attention‐deficit/hyperactivity disorder (ADHD); however, the underlying mechanism is unknown.
We found that activation of hD4.7 induced over‐suppression of glutamatergic excitatory network bursts and under‐suppression of GABAergic inhibitory network bursts in the prefrontal cortex (PFC) circuitry.
Methylphenidate, a psychostimulant drug used to treat ADHD, normalized the effects of hD4.7 on synchronous network bursts in PFC pyramidal neurons.
The findings of the present study suggest that the aberrant regulation of PFC synchronous network activity by hD4.7 may underlie its involvement in ADHD.
Abstract
A unique feature of the human D4 receptor (hD4R) gene is the existence of a large number of polymorphisms in exon 3 coding for the third intracellular loop, which consists of a variable number of tandem repeats. The hD4R variants with long repeats have been linked to attention‐deficit/hyperactivity disorder (ADHD); however, the underlying mechanism is unknown. Emerging evidence suggests that selective attention is controlled by the rhythmic synchronization in the prefrontal cortex (PFC) and its connected networks. In the present study, we examined the role of hD4R variants in regulating PFC synchronous network activity. D4R knockout mice with viral infection of hD4.4 or hD4.7 in the medial PFC were used. Whole‐cell patch‐clamp recordings were performed to examine the effects of activating hD4.x on the spontaneous large scale correlated activity in PFC pyramidal neurons. We found that, compared to the normal four‐repeat variant hD4.4, the ADHD‐linked variant hD4.7 induces more suppression of glutamatergic excitatory network bursts and less suppression of GABAergic inhibitory network bursts in the PFC circuitry. Methylphenidate, a psychostimulant drug used to treat ADHD, normalized the effects of hD4.7 on synchronous network bursts in PFC pyramidal neurons. These results reveal the differential effects of hD4R variants on the integrated excitability of PFC circuits. It is suggested that the aberrant regulation of PFC network activity by hD4.7 may underlie its involvement in ADHD. The methylphenidate‐induced normalization of synaptic circuitry regulation may contribute to its effectiveness in ADHD treatment.
Key points
The hD4.7 variant has been linked to attention‐deficit/hyperactivity disorder (ADHD); however, the underlying mechanism is unknown.
We found that activation of hD4.7 induced over‐suppression of glutamatergic excitatory network bursts and under‐suppression of GABAergic inhibitory network bursts in the prefrontal cortex (PFC) circuitry.
Methylphenidate, a psychostimulant drug used to treat ADHD, normalized the effects of hD4.7 on synchronous network bursts in PFC pyramidal neurons.
The findings of the present study suggest that the aberrant regulation of PFC synchronous network activity by hD4.7 may underlie its involvement in ADHD.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- ADHD
attention‐deficit/hyperactivity disorder
- AMPAR
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor
- APV
dl‐2‐amino‐5‐phosphonovaleric acid
- DNQX
6,7‐dinitroquinoxaline‐2,3‐dione
- GABAAR
GABAA receptor
- GFP
green fluorescent protein
- hD4R
human dopamine D4 receptor
- NMDAR
NMDA receptor
- PFC
prefrontal cortex
- PSD
postsynaptic density
Introduction
Attention‐deficit/hyperactivity disorder (ADHD) is a prevalent and debilitating disorder diagnosed on the basis of overactivity, inattention and impulsivity. A meta‐analysis suggests that ADHD is associated with significant weaknesses in several key executive functions, including response inhibition, vigilance, working memory and planning (Willcutt et al. 2005), all of which are controlled by the prefrontal cortex (PFC). Although the pathophysiological mechanisms underlying ADHD remain to be clarified, genetic studies have linked the dopamine D4 receptor, which is highly enriched in the PFC (Mrzljak et al. 1996; Wedzony et al. 2000), to ADHD (Bobb et al. 2005; Grady et al. 2003; Swanson et al. 2007).
The gene encoding human D4 receptor (hD4R) contains a large number of polymorphisms in the coding region for the third intracellular loop, which consists of a variable number (2–11) of 48 bp tandem repeats (Van Tol et al. 1992). The three most common variants contain two, four and seven repeats (D4.2, D4.4, D4.7), with a global frequency of 8%, 64% and 21%, respectively (Chang et al. 1996). These proline‐rich repeats provide the binding sites for other proteins containing the SH3 domain. So far, the molecular mechanism and functional significance of the remarkable variable number tandem repeat polymorphism of hD4R is poorly understood. Importantly, several studies have reported a significant association of ADHD with the D4R gene seven‐repeat allele (LaHoste et al. 1996; Rowe et al. 1998; Smalley et al. 1998; Faraone et al. 1999; El‐Faddagh et al. 2004; Li et al. 2006). However, the underlying mechanism is unknown.
Emerging evidence suggests that the control of attention arises from interactions between widespread cortical and subcortical networks regulated via their rhythmic synchronization (Buschman & Miller, 2007; Gregoriou et al. 2009. Womelsdorf et al. 2010; Miller and Buschman, 2013). The synchronized network activity is an important intrinsic feature of cortical circuits (Opitz et al. 2002; Buzsáki & Draguhn, 2004) and plays a significant role in controlling brain functions (Varela et al. 2001; von der Malsburg et al. 2010). Key factors involved in synchronous network activity include the strength of glutamatergic input from the recurrent excitatory connections between pyramidal neurons (Stoop et al. 2003; Panuccio et al. 2009) and the strength of GABAergic input from interneurons (Korn et al. 1987; Traub et al. 1989). Excitatory and inhibitory synaptic conductances may be exquisitely balanced in normal neuronal activity (Borg‐Graham et al. 1998; Haider et al. 2006), which is critical for complex behaviours (Klausberger & Somogyi 2008; Womelsdorf et al. 2010; Yizhar et al. 2011).
An understanding of how the synchronized cortical network activity is regulated in normal and ADHD conditions is lacking. In the present study, we examined the role of hD4R variants in regulating the integrated excitability of cortical circuits by measuring their impact on spontaneous large scale correlated activity in PFC pyramidal neurons. We show that the synchronized network activity in the PFC is more prominently suppressed by hD4.7. These results provide a potential pathophysiological basis for the frontal hypoactivity found in the diagnosis of ADHD (Dickstein et al. 2006; Fernández et al. 2009).
Methods
The generation of hD4.x viruses
The HA‐tagged human D4.4 and D4.7 plasmids (Rondou et al. 2008) were kindly provided by Dr Kathleen Van Craenenbroeck at the Laboratory of Eukaryotic Gene Expression and Signal Transduction (LEGEST), BELGIUM. The generation of Sindbis virus used the same procedure as that described previously (Liu et al. 2011). Briefly, the cDNAs encoding green fluorescent protein (GFP) or GFP‐hD4.x were subcloned to pSinRep5 vector (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. Recombinant GFP‐pSinRep5 or GFP‐hD4.x‐pSinRep5 was linearized with NotI. The DH26S plasmid was linearized using XhoI. The linearized templates were transcribed in vitro using the mMessage Machine SP6 kit (Ambion, Austin, TX, USA) and the RNAs were electroporated into baby hamster kidney cells. The extracellular medium containing the recombinant viruses was harvested after 24–48 h. The medium was concentrated on a discontinuous sucrose gradient (55% and 20% sucrose) using ultracentrifugation (160,000 g for 90 min at 4°C).
In vivo delivery of hD4.x viruses
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the State University of New York at Buffalo. D4R knockout mice (Rubinstein et al., 1997, 2001) were originally provided by Dr David Grandy at Oregon Health & Science University and later bred and maintained in our own laboratory for the experiments. In vivo virus‐based gene delivery into medial PFC was performed as described previously (Duffney et al. 2015). In brief, D4R knockout mice (2–3 months old) were anaesthetized by an i.p. injection of pentobarbital (50 mg kg−1) and placed on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The Sindbis viral suspension (0.5 μl) was injected with a Hamilton syringe (needle gauge 31) at a speed of ∼0.2 μl min−1, and the needle was kept in place for an additional 5 min. The virus was delivered bilaterally to the medial prelimbic area of mice using the co‐ordinates: 2.0 mm anterior to bregma and 0.5 mm lateral. The needle was extended to a depth of 1.3 mm below the tissue surface, and the virus was injected to each side. Animals were allowed to recover for 24–48 h after viral injection, and analgesia was provided postoperatively during the recovery.
Slice preparation
Mice (2–3 months old) were deeply anaesthetized using isoflurane and killed by decapitation. The brain was removed quickly and placed into the ice‐cold sucrose solution containing (in mm): 234 sucrose, 4 MgSO4, 2.5 KCl, 1 NaH2PO4, 0.1 CaCl2, 15 Hepes and 11 glucose (pH 7.4, 300 mOsm). Coronal slices (350 μm) were cut with a vibratome (VP1000S; Leica Microsystems, Wetzlar, Germany). Slices were incubated at 32–34°C for 1 h in artificial cerebrospinal fluid (aCSF) (in mm: 130 NaCl, 26 NaHCO3, 1 CaCl2, 2 MgCl2, 3 KCl, 10 glucose and 1.25 NaH2PO4) bubbled with 95% O2 and 5% CO2. Then slices were kept at room temperature (22–24°C) in a modified aCSF solution (in mm: 2 CaCl2, 0.1 MgCl2 and 3.5 KCl) (Aradi & Maccaferri, 2004; Juuri et al. 2010; Ziburkus et al. 2013) for 1–2 h before recordings.
Electrophysiological recordings
The whole‐cell voltage‐clamp technique (Zhong & Yan, 2004, 2014; Yuen et al. 2012) was used to record the neuronal network currents. Slices were placed in a recording chamber and superfused with the modified aCSF solution containing low (0.1 mm) MgCl2, high (2 mm) CaCl2 and slightly higher (3.5 mm) KCl to keep the slices in an active state. Neurons infected with GFP‐conjugated hD4.4 or hD4.7 Sindbis virus in layer V of the medial PFC were visualized with a fluorescence microscope and selected for recordings. A Multuclamp 700A amplifier (Molecular Devices, Sunnyvale, CA, USA) and a Digidata1322A (Molecular Devices) were used. Patch electrodes were filled with the internal solution containing (in mm): 130 Cs‐methanesulphonate, 4 NaCl, 10 Hepes, 0.5 EGTA, 2 QX‐314, 12 phosphocreatine, 4 ATP and 0.5 GTP (pH 7.2–7.3, 265–270 mOsm). Tight seals (2–10 GΩ) were obtained by applying negative pressure. The membrane was disrupted with additional suction and the whole‐cell configuration was obtained. Neurons were voltage clamped at −70 mV or −20 mV throughout the recordings. Series resistances were ∼12 MΩ and were not compensated.
Statistical analysis
Data acquisition was carried out using Clampex, version 9 (Molecular Devices). The sampling rate was 5 kHz and the low‐pass filtering frequency was 1 kHz. Each neuron was continuously recorded for 10 min, and the averaged amplitude and frequency of network bursts within a 2 min timeframe at the stable state before and after drug application were calculated. Data analyses were performed with Clampfit (Molecular Devices), MiniAnalysis (Synaptosoft Inc., Fort Lee, NJ, USA) and KaleidaGraph (Synergy Software, Reading, PA, USA) software. All data are expressed as the mean ± SEM. Mann–Whitney U tests were used to determine the significance of the effects of various agents on network bursts. Experiments with two animal groups were analysed statistically using unpaired Student's t tests. Experiments with more than two animal groups were subjected to one‐way ANOVA, followed by post hoc Bonferroni tests.
Results
The synchronous network activity in the PFC
Neurons in the PFC are not highly ordered like those in hippocampus, and the loss of the integrity of inhibitory and excitatory synaptic transmissions in in vitro preparations has often led to difficulty in uncovering synchronous network activity in PFC slices. To elevate the neuronal activity in slices to the in vivo level, we performed whole‐cell patch clamp recordings in the modified aCSF with a lower Mg2+ concentration (0.1 mm) and a higher Ca2+ concentration (2 mm), as established previously (Aradi & Maccaferri, 2004; Juuri et al. 2010; Ziburkus et al. 2013).
In cortical slices (350 μm), the slowly rhythmic synchronous bursting of larger numbers of neurons in the network gave rise to the spontaneous large scale correlated activity. As shown in Fig. 1 A, spontaneous network bursts occurred on a background of asynchronous unitary postsynaptic currents in layer V PFC pyramidal neurons (held at −70 mV). The network bursting‐associated inward current had a mean ± SEM amplitude of 1810 ± 172 pA (n = 8) and a mean ± SEM interval of 18.3 ± 2.0 s (n = 8).
Figure 1. The synchronous network activity in the PFC is dependent on AMPAR‐ and NMDAR‐mediated glutamatergic transmission .
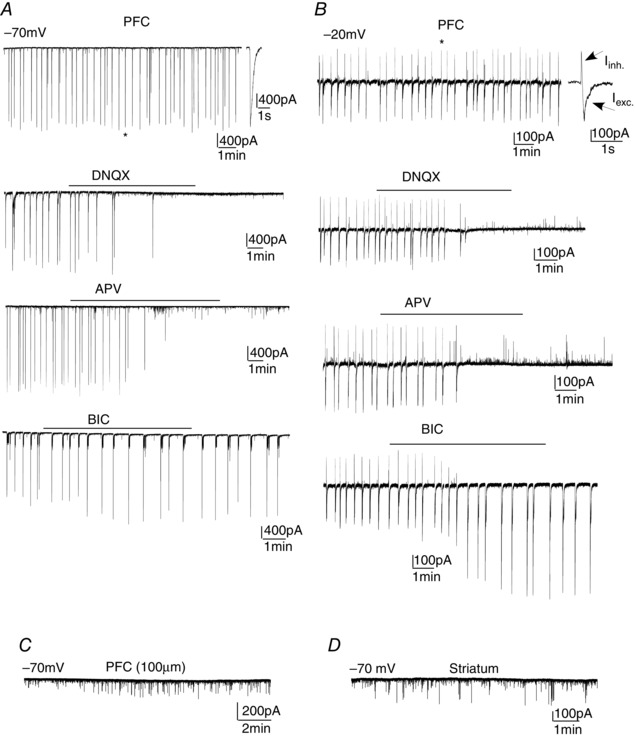
Network bursts recorded in PFC pyramidal neurons held at −70 mV (A) or −20 mV (B) in the absence or presence of DNQX (AMPAR antagonist), APV (NMDAR antagonist) or bicuculline (GABAAR antagonist). *Enlarged view of the network bursting‐associated currents. At −70 mV, the network burst is made of an inward excitatory current. At −20 mV, the network burst is composed of inward excitatory and outward inhibitory currents. C and D, synaptic currents recorded in PFC pyramidal neurons (held at −70 mV) from thin slices (100 μm) or medium spiny neurons (held at −70 mV) from striatal slices.
To examine the synaptic origin of synchronous network activity, we applied α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPAR), NMDA receptor (NMDAR) and GABAA receptor (GABAAR) antagonists, respectively. As shown in Fig. 1 A, the large‐scale network current was blocked by AMPAR antagonist 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX) (50 μm) (n = 5) or NMDAR antagonist dl‐2‐amino‐5‐phosphonovaleric acid (APV) (50 μm) (n = 6). Inhibition of GABAA receptors with bicuculline (20 μm) did not result in the blockade of synchronous network activity but led to the modulation of burst amplitude (14.2 ± 5.3% increase, n = 5, P < 0.05) and frequency (43 ± 4% decrease, n = 5, P < 0.01).
To reveal inhibitory synaptic currents in the measurement of network activity, we held the membrane potential at a more depolarized level (−20 mV). Under this condition, the rhythmic synchronous network bursting‐associated current was composed of two parts: an inhibitory outward current (amplitude: 218 ± 17 pA, n = 7) and an excitatory inward current (amplitude: 307 ± 21 pA, n = 7) (Fig. 1 B). The frequency of synchronous network activity was not influenced by holding potentials (interval: 19.4 ± 2.1 s, n = 12). Application of DNQX (50 μm) or APV (50 μm) blocked both excitatory inward network current and inhibitory outward network current (n = 6) (Fig. 1 B). Application of bicuculline blocked the inhibitory outward network current, and dramatically altered the excitatory inward network current (amplitude: 116 ± 14% increase; frequency: 46 ± 5% decrease, n = 8). This suggests that glutamatergic transmission, which is mediated by AMPA and NMDA receptors, is required for synchronous network activity, whereas GABAergic transmission modulates oscillatory network activity in the PFC.
Thin PFC slices (120 μm), which had severe loss of neuronal circuit integrity, failed to produce the large scale rhythmic synchronous network bursts (n = 8) (Fig. 1 C). No giant network bursts were observed in medium spiny neurons from striatal slices (n = 9) (Fig. 1 D). This suggests that the synchronized network activity requires intact neuronal connections in cortical circuits and occurs in a brain region‐specific manner.
The effect of hD4.7 and hD4.4 on synchronous network activity in PFC neurons
To clarify the role of human D4R variants in the cortical network, we examined the effects of the specific D4R agonist PD168077 on spontaneous network activity in the PFC of D4R knockout mice with viral infection of hD4.4 or hD4.7. The rodent D4R only contains two repeats, and wild‐type mice were also used for comparison. To avoid high overexpression of the hD4.x Sindbis virus, mice were killed within 2 days after viral delivery. PFC neurons expressing the GFP‐conjugated hD4.4 or hD4.7 exhibited normal morphological structures (Fig. 2 A). In hD4.7‐expressing PFC pyramidal neurons (held at −70 mV), PD168077 (20 μm) decreased the amplitude of network bursts by 40.7 ± 2.5% (n = 7), which was significantly (P < 0.01) larger than the reducing effect of PD168077 in hD4.4‐expressing PFC pyramidal neurons (20.8 ± 1.4%, n = 7) (Fig. 2 B, D and E) or in PFC pyramidal neurons from wild‐type mice that endogenously express D4.2 (20.4 ± 1.5%, n = 6) (Fig. 2 D and E). PD168077 reduced the frequency of network bursts to a similar level in D4.x‐expressing neurons (hD4.7: 31.9 ± 2.4%, n = 7; hD4.4: 30.3 ± 2.1%, n = 7) (Fig. 2 C, D and E) and neurons from wild‐type mice (30.7 ± 2.5%, n = 6) (Fig. 2 D and E). PD168077 had no effect on network activity in D4R knockout mice (amp: 3.7 ± 0.7%, freq: 3.2 ± 0.6%, n = 5) (Fig. 2 D and E), confirming the mediation by D4Rs. The baseline network bursts (held at −70 mV) were similar among different groups (wild‐type: 1760 ± 159 pA, 0.054 ± 0.005 Hz; D4KO: 1880 ± 181 pA, 0.061 ± 0.006 Hz; hD4.4: 1670 ± 145 pA, 0.052 ± 0.004 Hz; hD4.7: 1710 ± 152 pA, 0.051 ± 0.004 Hz) (Fig. 2 F). This suggests that hD4R activation results in a sustained decrease of burst occurrence without disrupting the patterned nature of activity, and activation of the ADHD‐linked variant hD4.7 results in more prominent hypoexcitability of the PFC network.
Figure 2. Activation of hD4.7 induces a stronger suppression of PFC synchronous network activity than hD4.4 .
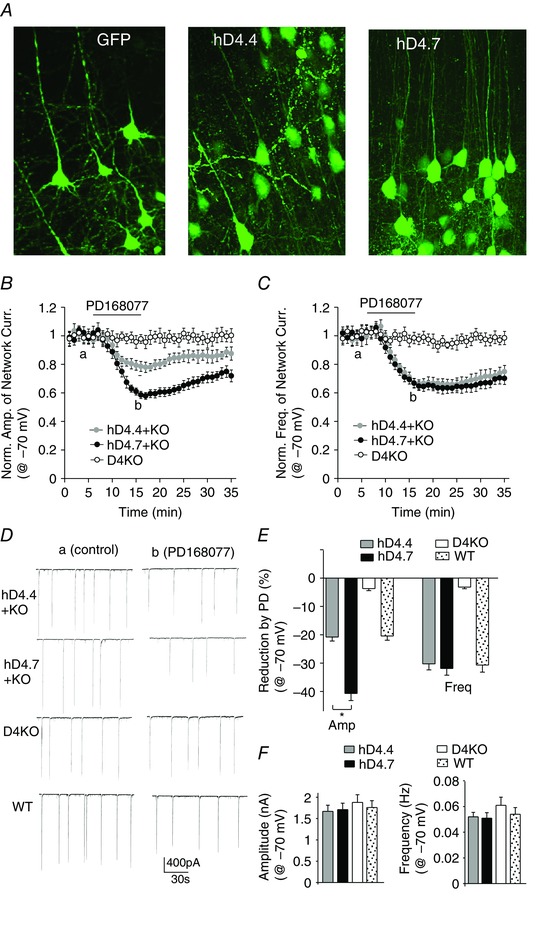
A, confocal images of PFC slices from D4R knockout mice with a stereotaxical injection of GFP or GFP‐conjugated hD4.4 or hD4.7 Sindbis virus. Network burst amplitudes (B) and frequencies (C) show the effect of bath applied D4R agonist PD168077 (20 μm) in PFC pyramidal neurons (held at −70 mV) of D4R knockout mice with or without viral infection of human D4.4 or D4.7 variants. D, representative network bursts in the absence or presence of PD168077 in PFC pyramidal neurons from different groups. E, percentage reduction of the amplitude and frequency of network bursts by PD168077 in different groups. *P < 0.01, ANOVA, hD4.4 vs. hD4.7. F, amplitude or frequency of baseline network bursting currents (held at −70 mV) in different groups.
The recordings described above were performed at room temperature (23°C). We also examined the network bursting current and its regulation by D4R activation at a more physiological temperature (32°C). We found that the rise in temperature increased the network current amplitude and frequency (amplitude: 28.9 ± 2.2%, frequency: 23.6 ± 2.7%, n = 5), although the reducing effect of PD168077 on network bursts was similar at different temperatures (23°C, amplitude: 20 ± 1.5%, frequency: 30.7 ± 2.5%, n = 6; 32°C, amplitude: 20.6 ± 1.9%, frequency: 28.8 ± 2.6%, n = 5).
Next, we compared the effect of hD4.x on excitatory and inhibitory network bursts in PFC pyramidal neurons held at a depolarized level (−20 mV). In hD4.7‐expressing neurons, PD168077 (20 μm) decreased the excitatory inward network current amplitude by 24.1 ± 1.9% (n = 8) (Fig. 3 A, C and D), whereas it had no effect on the inhibitory outward network current amplitude (1.8 ± 1.2%, n = 8) (Fig. 3 B to D). By contrast, in hD4.4‐expressing neurons, PD168077 (20 μm) had no effect on the excitatory inward network current amplitude (2.8 ± 1.4%, n = 7) (Fig. 3 A, C and D) but decreased the inhibitory outward network current amplitude by 22.8 ± 1.7% (n = 7) (Fig. 3 B to D), which was similar to the effect of PD168077 in the PFC pyramidal neurons from wild‐type mice (inward‐amplitude: 0.8 ± 1.6%, outward‐amplitude: 21.7 ± 2.1% reduction) (Fig. 3 C and D). The reducing effects of PD168077 on network burst frequency were similar in D4.x‐expressing neurons (hD4.7: 32.6 ± 3.5%, n = 8; hD4.4: 30.3 ± 2.8%, n = 7) and those from wild‐type mice (31.1 ± 2.9%, n = 5). The baseline network bursts (held at −20 mV) were similar among different groups (wild‐type, inward: 289 ± 19 pA, outward: 203 ± 15 pA, 0.055 ± 0.004 Hz; hD4.4, inward: 272 ± 13 pA, outward: 205 ± 16 pA, 0.053 ± 0.004 Hz; hD4.7, inward: 301 ± 21 pA, outward: 193 ± 17 pA, 0.050 ± 0.004 Hz) (Fig. 3 E). This suggests that, compared to the normal variant hD4.4, the ADHD‐linked variant hD4.7 induces more suppression of glutamatergic excitatory transmission and less suppression of GABAergic inhibitory transmission in the synaptic circuitries, which may collectively result in PFC network hypoactivity.
Figure 3. Activation of hD4.7 induces an over‐suppression of excitatory network activity and an under‐suppression of inhibitory network activity in the PFC .
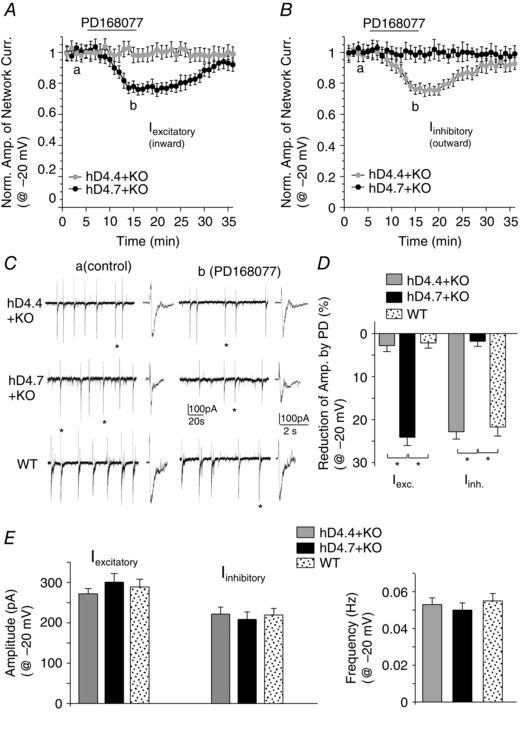
Excitatory or inhibitory network burst amplitudes (A) and frequencies (B) showing the effect of PD168077 (20 μm) in PFC pyramidal neurons (held at −20 mV) of D4R knockout mice with viral infection of human D4.4 or D4.7 variants. C, representative excitatory and inhibitory network bursts in the absence or presence of PD168077 in PFC pyramidal neurons from different groups. D, percentage reduction of the amplitude of excitatory or inhibitory network bursts by PD168077 in different groups. *P < 0.01, ANOVA. E, amplitude or frequency of baseline excitatory and inhibitory network bursts (held at −20 mV) in different groups.
To determine whether the virally expressed hD4R variants are activated by spontaneous dopamine release, we examined the effect of a D4R antagonist on network activity. As shown in Fig. 4 A and B, blocking D4R with the antagonist L‐745870 (20 μm) did not significantly alter network bursting currents in hD4.x‐expressing neurons (wild‐type, amplitude: 7.6 ± 1.8%, frequency: 8.2 ± 2.4%, n = 6; hD4.4, amplitude: 2.1 ± 1.3%, frequency: 5.1 ± 1.2%, n = 5; hD4.7, amplitude: 2.5 ± 1.4%, frequency: 4.2 ± 1.7%, n = 5, P > 0.05, ANOVA), suggesting that hD4R variants are not constitutively active.
Figure 4. Application of a D4R antagonist does not change network bursting currents .
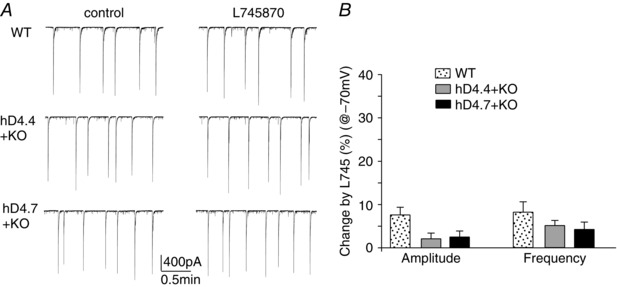
A, representative network bursts (−70 mV) in the absence or presence of the D4R antagonist L‐745870 (20 μm) in PFC pyramidal neurons from a wild‐type mouse or D4R knockout mice with viral infection of human D4.4 or D4.7 variants. B, effect of L‐745870 on the amplitude and frequency of network bursts.
The normalization of hD4.7 regulation of PFC network activity by methylphenidate (MPH)
To determine whether the aberrant regulation of PFC network activity by hD4.7 is related to its role in ADHD, we examined whether MPH, an effective agent for ADHD treatment, could normalize the effects of hD4.7 on synchronous network bursts in PFC pyramidal neurons. MPH (0.5 mg kg−1) or saline control was i.p. injected to hD4.7‐infected D4R knockout mice and, 1 h later, animals were killed before slicing (Cheng et al. 2014).
As shown in Fig. 5 A to C, in hD4.7‐expressing PFC pyramidal neurons (held at −70 mV) from MPH‐injected animals, PD168077 (20 μm) induced a significantly smaller reducing effect on network burst amplitude than in neurons from saline‐injected mice (MPH: 22.3 ± 1.7%, n = 6; saline: 38.4 ± 2.2%, n = 4, P < 0.01). Moreover, in hD4.7‐expressing PFC pyramidal neurons (held at −20 mV) from MPH‐injected animals (Fig. 5 D to G), PD168077 had no effect on the excitatory inward network current amplitude (MPH: 2.2 ± 1.2%, n = 5; saline: 22.3 ± 2.1%, n = 4), whereas it decreased the inhibitory outward network current amplitude (MPH: 22.1 ± 1.7%, n = 5; saline: 1.6 ± 1.1%, n = 4). The reducing effect of PD168077 on network current frequency was not affected by MPH (–70 mV, MPH: 30.7 ± 2.6%, n = 6; saline: 32.7 ± 3.2%, n = 4; −20 mV, MPH: 31.1 ± 2.9%, n = 5; saline: 29.4 ± 2.7%, n = 4). These data suggest that in vivo administration of MPH switches the regulatory effects of hD4.7 on PFC network activity to levels similar to those of hD4.4 (Figs 2 and 3). In vitro application of MPH (20 μm) decreased network burst amplitude (26.7 ± 2.9%, n = 6) but increased network burst frequency (31.3 ± 4.1%, n = 6) in hD4.7‐expressing neurons.
Figure 5. The hD4.7 regulation of PFC network activity is normalized by MPH injection .
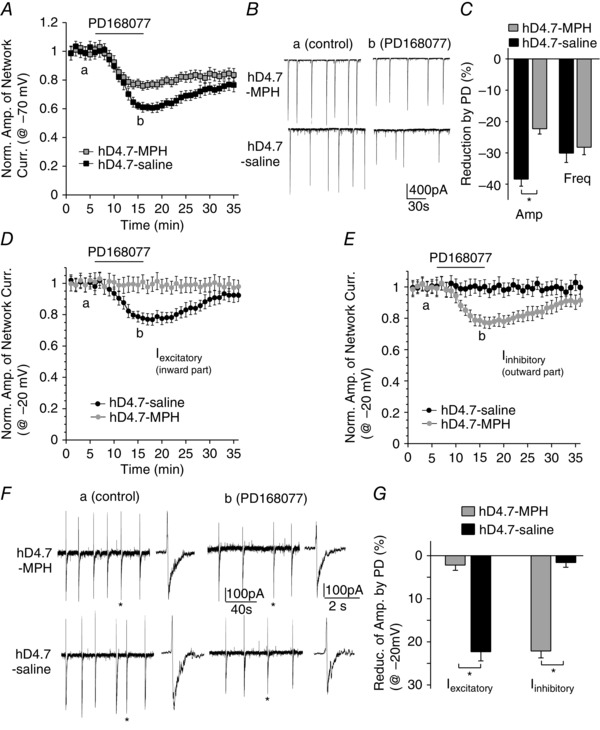
A, D and E, network burst amplitudes showing the effect of PD168077 (20 μm) in PFC pyramidal neurons of hD4.7‐infected, D4R knockout mice injected with MPH or saline control. B and F, representative network bursts in the absence or presence of PD168077 in hD4.7‐expressing neurons with MPH or saline injection. C and G, percentage reduction of the amplitude of network bursts by hD4.7 activation in PFC neurons from MPH‐ or saline‐injected mice. Cell membranes were held at −70 mV (A to C) or −20 mV (D to G). *P < 0.01, t test, hD4.7‐MPH vs. hD4.7‐saline.
We further examined the impact of MPH in hD4.4‐expressing neurons. Compared to saline‐injected mice, MPH injection (i.p.) did not alter the effect of PD168077 on network activity in D4.4‐expressing PFC pyramidal neurons recorded at −70 mV (MPH, amplitude: 19.8 ± 1.5% reduction, frequency: 29.3 ± 2.3% reduction, n = 6; saline, amplitude: 20.9 ± 1.6% reduction, frequency: 30.9 ± 2.4% reduction, n = 6, Fig. 6 A and C) or at −20 mV (MPH, inward‐amplitude: 2.8 ± 0.7%, outward‐amplitude: 22.9 ± 1.4% reduction, frequency, 34.5 ± 3.1% reduction, n = 6; saline, inward‐amplitude: −3.7 ± 1.5%, outward‐amplitude: 20.1 ± 1.7% reduction, frequency, 33.3 ± 2.3%, n = 5) (Fig. 6 B and D).
Figure 6. The hD4.4 regulation of PFC network activity is not altered by MPH injection .
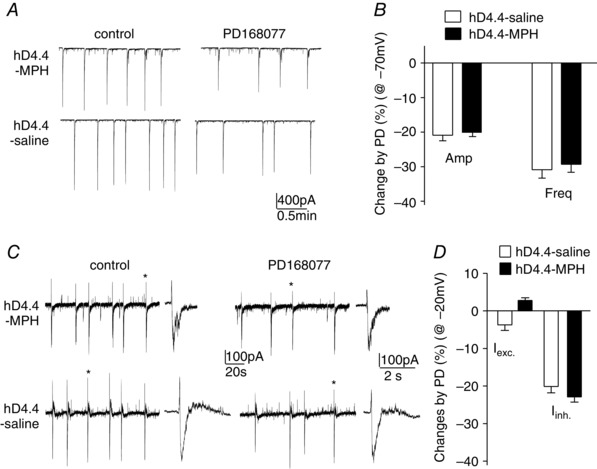
A and C, representative network bursts in the absence or presence of PD168077 in hD4.4‐expressing neurons with MPH or saline injection. B and D, percentage changes of the amplitude or frequency of network bursts by hD4.4 activation in PFC neurons from MPH‐ or saline‐injected mice. Cell membranes were held at −70 mV (A and B) or −20 mV (C and D).
Discussion
A unique primate‐specific feature of D4R is the additional 2–11 proline‐rich repeats located in the third intracellular loop (Wang et al. 2004), which allows more complex simultaneous interactions with other proteins containing the SH3 domain. The hD4R variants with long repeats have been linked to deficiencies in executive control processes in ADHD (LaHoste et al. 1996; Swanson et al. 1998; Talkowski et al. 2008; Gizer et al. 2009; Barnes et al. 2011). To understand the potential mechanism, we have examined their impact on synchronized network bursts originating from the large scale correlated activity of interconnected neurons, which controls PFC‐mediated cognitive function, such as attention (Buschman & Miller, 2007; Miller & Buschman, 2013).
Synchronized network activity in cortex has been suggested to co‐operatively support temporal representation and long‐term consolidation of information (Buzsáki & Draguhn, 2004). Local and long‐range rhythmic synchronization determines neuronal interactions and selective attention (Womelsdorf & Fries, 2007; Womelsdorf et al. 2007). Spontaneous synchrony in neural network is expected to depend on the finely tuned interplay of excitatory and inhibitory neuronal populations, which gives rise to the precisely timed and dynamically balanced excitatory and inhibitory conductances (Traub et al. 1989). The data obtained in the present study (Fig. 1) indicate that the synchronous network activity requires both AMPA and NMDA receptors, whereas GABAergic transmission mainly modulates oscillatory network activity.
Abnormal synchrony of spontaneous network activity has been associated with various neurological and psychiatric disorders, ranging from epileptic seizures (Steriade, 2003; Garcia Dominguez et al. 2005) and Parkinson's disease (Boraud et al. 2005; Uhlhaas & Singer, 2006) to schizophrenia (Spencer et al. 2003; Uhlhaas & Singer, 2010) and autism (Wilson et al. 2007; Yizhar et al. 2011). An imbalance favouring excitation may underlie the excessive synchrony in epilepsy (Dichter & Ayala, 1987). The data obtained in the present study (Figs 2 and 3) indicate that activation of hD4.4 reduced excitatory and inhibitory network activity, similar to the effects of D4 receptor activation in wild‐type mice. However, activation of the ADHD‐linked hD4.7 induces more suppression of the excitatory network bursts and less suppression of the inhibitory network bursts in the PFC circuitry, suggesting that it shifts the neuronal excitation–inhibition balance towards inhibition in postsynaptic neurons, which may explain the significant frontal hypoactivity detected in ADHD patients (Dickstein et al. 2006; Fernández et al. 2009).
Little is known about the mechanisms underlying the functional differences of hD4R variants. Using transfected cell lines, no major discrepancies in pharmacological profiles or the abilities to block cAMP production have been found among hD4.x isoforms (Asghari et al. 1995; Jovanovic et al. 1999). Using knock‐in mice that carry hD4.7 in the third intracellular loop of D4R, it was found that D4.7 does not form functional heteromers with the dopamine D2S receptor, which is assumed to affect presynaptic dopaminergic control of corticostriatal glutamate release (Gonzalez et al. 2012). Our previous studies in rodents have found that activation of D4 receptors in PFC pyramidal neurons produces a significant reduction of NMDAR‐mediated currents (Wang et al., 2003, 2006) and an activity‐dependent, homeostatic regulation of AMPAR‐mediated transmission (Yuen et al. 2010; Yuen & Yan, 2011) via mechanisms dependent on CaMKII and receptor channel trafficking. In addition, rodent D4R activation in PFC pyramidal neurons leads to a significant decrease of GABAAR currents (Wang et al. 2002) and GABAAR membrane trafficking via an actin/cofilin/myosin‐dependent mechanism (Graziane et al. 2009). These effects are similar to those of hD4.4 (normal variant) on excitatory and inhibitory network bursts. The additional proline‐rich repeats on hD4.7 (ADHD‐linked variant) enable the binding of more proteins containing the SH3 domain, such as many scaffolding proteins in the postsynaptic density (PSD). The binding of dopamine to hD4.7 may change the receptor conformation, disrupting the binding of hD4.7 to these SH3‐containing PSD proteins. Thus, the stronger suppression of glutamatergic transmission‐mediated excitatory network bursts by hD4.7 activation may be attributable to the larger inhibitory effect of hD4.7 on the membrane trafficking or maintenance of AMPA and NMDA receptors at PSD. Moreover, hD4.7 lost the regulation of GABAAR‐mediated inhibitory network bursts, which may be a result of the attenuated regulation of actin dynamics and the myosin‐based transport of GABAARs to the surface. The detailed mechanisms await further investigation.
To determine whether the aberrant regulation of PFC network activity by hD4.7 is related to its role in ADHD, we further examined whether MPH, a dopamine reuptake inhibitor approved for ADHD treatment, could normalize the effects of hD4.7 on synchronous network bursts in PFC pyramidal neurons. The data obtained in the present study (Fig. 5) indicate that MPH reduced the effect of hD4.7 on excitatory network bursts, and restored the effect of hD4.7 on inhibitory network bursts, bringing it close to hD4.4 in the regulation of PFC synchronized network activity. The mechanisms underlying MPH‐induced changes in hD4.7 regulation of network activity await clarification. In sum, the MPH‐induced normalization of synaptic circuitry regulation could contribute to its effectiveness in ADHD treatment.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
ZY and PZ conceived and designed the experiments. PZ and WL collected, analysed and interpreted the data. ZY drafted the article and revised it critically for important intellectual content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH grant DA037618 and VA merit award 1I01BX001633 to ZY, NSFC grant (31271124) and Guangdong Natural Science Foundation (S2013010016062) grant to WL.
Acknowledgements
We would like to thank Xiaoqing Chen for her excellent technical support.
References
- Aradi I & Maccaferri G (2004) Cell type‐specific synaptic dynamics of synchronized bursting in the juvenile CA3 rat hippocampus. J Neurosci 24, 9681–9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari V, Sanyal S, Buchwaldt S, Paterson A, Jovanovic V & Van Tol HH (1995) Modulation of intracellular cyclic AMP levels by different human dopamine D4 receptor variants. J Neurochem 65, 1157–1165. [DOI] [PubMed] [Google Scholar]
- Barnes JJ, Dean AJ, Nandam LS, O'Connell RG & Bellgrove MA (2011) The molecular genetics of executive function: role of monoamine system genes. Biol Psychiatry 69, e127–143. [DOI] [PubMed] [Google Scholar]
- Bobb AJ, Castellanos FX, Addington AM & Rapoport JL (2005) Molecular genetic studies of ADHD: 1991 to 2004. Am J Med Genet B Neuropsychiatr Genet 132B, 109–125. [PubMed] [Google Scholar]
- Boraud T, Brown P, Goldberg JA, Graybiel AM & Magill PJ (2005) Oscillations in the basal ganglia: the good, the bad, and the unexpected In The Basal Ganglia VIII, eds. Bolam JP, Ingham CA. & Magill PJ, pp. 3–24. Springer, New York, NY. [Google Scholar]
- Borg‐Graham LJ, Monier C & Frégnac Y (1998). Visual input evokes transient and strong shunting inhibition in visual cortical neurons. Nature 393, 369–373. [DOI] [PubMed] [Google Scholar]
- Buschman TJ & Miller EK (2007) Top‐down versus bottom‐up control of attention in the prefrontal and posterior parietal cortices. Science 315, 1860–1862. [DOI] [PubMed] [Google Scholar]
- Buzsáki G & Draguhn A (2004) Neuronal oscillations in cortical networks. Science 304, 1926–1929. [DOI] [PubMed] [Google Scholar]
- Chang FM, Kidd JR, Livak KJ, Pakstis AJ & Kidd KK (1996). The worldwide distribution of allele frequencies at the human dopamine D4 receptor locus. Hum Genet 98, 91–101. [DOI] [PubMed] [Google Scholar]
- Cheng J, Xiong Z, Duffney LJ, Wei J, Liu AY, Liu S, Chen GJ & Yan Z (2014). Methylphenidate exerts dose‐dependent effects on glutamate receptors and behaviors. Biol Psychiatry 76, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter M & Ayala GF (1987). Cellular mechanisms of epilepsy: a status report. Science 237, 157–164. [DOI] [PubMed] [Google Scholar]
- Dickstein SG, Bannon K, Castellanos FX & Milham MP (2006). The neural correlates of attention deficit hyperactivity disorder: an ALE meta‐analysis. J Child Psychol Psychiatry 47, 1051–1062. [DOI] [PubMed] [Google Scholar]
- Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, Ma K, Dietz DM, Kajiwara Y, Buxbaum JD & Yan Z (2015) Autism‐like deficits in Shank3‐deficient mice are rescued by targeting actin regulators. Cell Reports 11, 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Faddagh M, Laucht M, Maras A, Vöhringer L & Schmidt MH (2004). Association of dopamine D4 receptor (DRD4) gene with attention‐deficit/hyperactivity disorder (ADHD) in a high‐risk community sample: a longitudinal study from birth to 11 years of age. J Neural Transm 111, 883–889. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Weiffenbach B et al (1999). Dopamine D4 gene 7‐repeat allele and attention deficit hyperactivity disorder. Am J Psychiatry 156, 768–770. [DOI] [PubMed] [Google Scholar]
- Fernández A, Quintero J, Hornero R, Zuluaga P, Navas M, Gómez C, Escudero J, García‐Campos N, Biederman J & Ortiz T (2009). Complexity analysis of spontaneous brain activity in attention‐deficit/hyperactivity disorder: diagnostic implications. Biol Psychiatry 65, 571–577. [DOI] [PubMed] [Google Scholar]
- Garcia Dominguez L, Wennberg RA, Snead OC 3rd, Gaetz W, Perez Velazquez JL & Cheyne D. (2005). Enhanced synchrony in epileptiform activity? Local versus distant phase synchronization in generalized seizures. J Neurosci 25, 8077–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizer IR, Ficks C & Waldman ID (2009) Candidate gene studies of ADHD: a meta‐analytic review. Hum Genet 126, 51–90. [DOI] [PubMed] [Google Scholar]
- González S, Rangel‐Barajas C, Peper M, Lorenzo R, Moreno E, Ciruela F, Borycz J, Ortiz J, Lluís C, Franco R, McCormick PJ, Volkow ND, Rubinstein M, Floran B & Ferré S (2012). Dopamine D(4) receptor, but not the ADHD‐associated D(4.7) variant, forms functional heteromers with the dopamine D(2S) receptor in the brain. Mol Psychiatry 17, 650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady DL, Chi HC, Ding YC, Smith M, Wang E, Schuck S, Flodman P, Spence MA, Swanson JM & Moyzis RK (2003). High prevalence of rare dopamine receptor D4 alleles in children diagnosed with attention‐deficit hyperactivity disorder. Mol Psychiatry 8, 536–545. [DOI] [PubMed] [Google Scholar]
- Graziane NM, Yuen EY & Yan Z (2009). Dopamine D4 receptors regulate GABAA receptor trafficking via an actin/cofilin/myosin‐dependent mechanism. J Biol Chem 284, 8329–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoriou GG, Gotts SJ, Zhou H & Desimone R (2009). High‐frequency, long‐range coupling between prefrontal and visual cortex during attention. Science 324, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR & McCormick DA (2006) Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci 26, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic V, Guan HC & Van Tol HH (1999). Comparative pharmacological and functional analysis of the human dopamine D4.2 and D4.10 receptor variants. Pharmacogenetics 9, 561–568. [PubMed] [Google Scholar]
- Juuri J, Clarke VR, Lauri SE & Taira T (2010). Kainate receptor‐induced ectopic spiking of CA3 pyramidal neurons initiates network bursts in neonatal hippocampus. J Neurophysiol 104, 1696–1706. [DOI] [PubMed] [Google Scholar]
- Klausberger T & Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL & Dingledine R (1987). Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA‐mediated inhibition. J Neurophysiol 57, 325–3240. [DOI] [PubMed] [Google Scholar]
- LaHoste GJ, Swanson JM, Wigal SB, Glabe C, Wigal T, King N & Kennedy JL (1996). Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol Psychiatry 1, 121–124. [PubMed] [Google Scholar]
- Li D, Sham PC, Owen MJ & He L (2006). Meta‐analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum Mol Genet 15, 2276–2284. [DOI] [PubMed] [Google Scholar]
- Liu W, Dou F, Feng J & Yan Z (2011). RACK1 is involved in b‐amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiol Aging 32, 1818–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK & Buschman TJ (2013). Cortical circuits for the control of attention. Curr Opin Neurobiol 23, 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R & Goldman‐Rakic PS (1996). Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Nature 381, 245–248. [DOI] [PubMed] [Google Scholar]
- Opitz T, De Lima AD & Voigt T (2002). Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J Neurophysiol 88, 2196–2206. [DOI] [PubMed] [Google Scholar]
- Panuccio G, Curia G, Colosimo A, Cruccu G & Avoli M (2009). Epileptiform synchronization in the cingulate cortex. Epilepsia 50, 521–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondou P, Haegeman G, Vanhoenacker P & Van Craenenbroeck K (2008). BTB Protein KLHL12 targets the dopamine D4 receptor for ubiquitination by a Cul3‐based E3 ligase. J Biol Chem 283, 11083–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe DC, Stever C, Giedinghagen LN et al (1998). Dopamine DRD4 receptor polymorphism and attention deficit hyperactivity disorder. Mol Psychiatry 3, 419–426. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Cepeda C, Hurst RS, Flores‐Hernandez J, Ariano MA, Falzone TL, Kozell LB, Meshul CK, Bunzow JR, Low MJ, Levine MS & Grandy DK (2001). Dopamine D4 receptor‐deficient mice display cortical hyperexcitability. J Neurosci 21, 3756–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ & Grandy DK (1997). Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell 90, 991–1001. [DOI] [PubMed] [Google Scholar]
- Smalley SL, Bailey JN, Palmer CG et al (1998). Evidence that the dopamine D4 receptor is a susceptibility gene in attention deficit hyperactivity disorder. Mol Psychiatry 3, 427–430 [DOI] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME & McCarley RW (2003). Abnormal neural synchrony in schizophrenia. J Neurosci 23, 7407–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M (2003). Neuronal Substrates of Sleep and Epilepsy. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Stoop R, Conquet F, Zuber B, Voronin LL & Pralong E (2003). Activation of metabotropic glutamate 5 and NMDA receptors underlies the induction of persistent bursting and associated long‐lasting changes in CA3 recurrent connections. J Neurosci 23, 5634‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JM, Sunohara GA, Kennedy JL et al (1998). Association of the dopamine receptor D4 (DRD4) gene with a refined phenotype of attention deficit hyperactivity disorder (ADHD): a family‐based approach. Mol Psychiatry 3, 38–41. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, Taylor E, Casey BJ, Castellanos FX & Wadhwa PD (2007) Etiologic subtypes of attention‐deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev 17, 39–59. [DOI] [PubMed] [Google Scholar]
- Talkowski ME, Kirov G, Bamne M, Georgieva L, Torres G, Mansour H, Chowdari KV, Milanova V, Wood J, McClain L, Prasad K, Shirts B, Zhang J, O'Donovan MC, Owen MJ, Devlin B & Nimgaonkar VL (2008) A network of dopaminergic gene variations implicated as risk factors for schizophrenia. Hum Mol Genet 17, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Miles R & Wong RK (1989). Model of the origin of rhythmic population oscillations in the hippocampal slice. Science 243, 1319–1325. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ & Singer W (2006). Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52, 155–168. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ & Singer W (2010). Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11, 100–113. [DOI] [PubMed] [Google Scholar]
- Van Tol HH, Wu CM, Guan HC et al (1992). Multiple dopamine D4 receptor variants in the human population. Nature 358, 149–152. [DOI] [PubMed] [Google Scholar]
- Varela F, Lachaux JP, Rodriguez E & Martinerie J (2001). The brainweb: phase synchronization and large‐scale integration. Nat Rev Neurosci 2, 229–239. [DOI] [PubMed] [Google Scholar]
- von der Malsburg C, Phillips WA, Singer W. (2010). Dynamic Coordination in the Brain. MIT, Cambridge, MA. [Google Scholar]
- Wang E, Ding YC, Flodman P, Kidd JR, Kidd KK, Grady DL et al (2004). The genetic architecture of selection at the human dopamine receptor D4 (DRD4) gene locus. Am J Hum Genet 74, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gu Z, Zhong P, Chen G, Feng J & Yan Z (2006). Aberrant regulation of NMDA receptors by dopamine D4 signaling in rats after phencyclidine exposure. Mol Cell Neurosci 31, 15–25. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Gu Z & Yan Z (2003). Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci 23, 9852–9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhong P & Yan Z (2002). Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J Neurosci 22, 9185–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedzony K, Chocyk A, Mackowiak M, Fijal K & Czyrak A (2000). Cortical localization of dopamine D4 receptors in the rat brain–immunocytochemical study. J Physiol Pharmacol 51, 205–221. [PubMed] [Google Scholar]
- Willcutt EG, Doyle AE, Nigg JT, Faraone SV & Pennington BF (2005). Validity of the executive function theory of attention‐deficit/hyperactivity disorder: a meta‐analytic review. Biol Psychiatry 57, 1336–1346. [DOI] [PubMed] [Google Scholar]
- Wilson TW, Rojas DC, Reite ML, Teale PD & Rogers SJ (2007). Children and adolescents with autism exhibit reduced MEG steady‐state gamma responses. Biol Psychiatry 62, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womelsdorf T & Fries P (2007). The role of neuronal synchronization in selective attention. Curr Opin Neurobiol 17, 154–160. [DOI] [PubMed] [Google Scholar]
- Womelsdorf T, Schoffelen JM, Oostenveld R, Singer W, Desimone R, Engel AK & Fries P (2007). Modulation of neuronal interactions through neuronal synchronization. Science 316, 1609–1612. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P & Deisseroth K (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X & Yan Z (2012). Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73, 962–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY & Yan Z (2011). Cellular mechanisms for dopamine D4 receptor‐induced homeostatic regulation of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors. J Biol Chem 286, 24957–24965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Zhong P & Yan Z (2010). Homeostatic regulation of glutamatergic transmission by dopamine D4 receptors. Proc Natl Acad Sci USA 107, 22308–22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P & Yan Z (2004). Chronic antidepressant treatment alters the serotonergic regulation of GABA transmission in prefrontal cortical pyramidal neurons. Neurosci 129, 65–73. [DOI] [PubMed] [Google Scholar]
- Zhong P & Yan Z (2014). Distinct physiological effects of dopamine D4 receptors on prefrontal cortical pyramidal neurons and fast‐spiking interneurons. Cerebral Cortex doi: 10.1093/cercor/bhu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR & Schiff SJ (2013). Seizures as imbalance up states: excitatory and inhibitory conductances during seizure‐like evnets. J Neurophysiol 109, 1296–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]