

Abstract
A‐kinase anchoring proteins (AKAPs) have emerged as a converging point of diverse signals to achieve spatiotemporal resolution of directed cellular regulation. With the extensive studies of AKAP79/150 in regulation of ion channel activity, the major questions to be posed centre on the mechanism and functional role of synergistic regulation of ion channels by such signalling proteins. In this review, we summarize recent discoveries of AKAP79/150‐mediated modulation of voltage‐gated neuronal M‐type (KCNQ, Kv7) K+ channels and L‐type CaV1 Ca2+ channels, on both short‐ and longer‐term time scales, highlighting the dynamics of the macromolecular signalling complexes in brain and peripheral nerve We also discuss several models for the possible mechanisms of these multi‐protein assemblies and how they serve the agenda of the neurons in which they occur.
Abbreviations
- AKAP
A‐kinase anchoring protein
- CaN
calcineurin
- CaM
calmodulin
- DAG
diacylglycerol
- DN
dominant negative
- IP3
inositol trisphosphate
- L‐channels
L‐type Ca2+ channels
- M‐channels
KCNQ (M‐type) K+ channels
- NFAT
nuclear factor of activated T‐cells
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PKA
protein kinase A
- PKC
protein kinase C
- STORM
stochastic optical reconstruction microscopy
- WT
wild type
Introduction
A‐kinase anchoring proteins (AKAPs) are a diverse scaffold protein family that orchestrates enzymes with their substrates into protein complexes, which ensures specificity and optimal facilitation of signal transduction (Bauman et al. 2006). Over 70 AKAPs have been identified in different cell types. They all share the common ability to bind protein kinase A (PKA), as well as roles in organizing multi‐protein signalling complexes in specific subcellular locations (Wong & Scott, 2004). The well‐studied isoforms AKAP79/150 (human AKAP79/rodent AKAP150) are widely expressed in the nervous system, and regulate diverse neuronal ion channels through phosphorylation or dephosphorylation by signalling proteins such as PKA, PKC, calmodulin (CaM), calcineurin (CaN), and phosphatidylinositol 4,5‐bisphosphate (PIP2). The channels modulated include AMPA‐type glutamate receptors, the inwardly rectifying potassium channel Kir 2.1, L‐type Ca2+ channels (L‐channels), KCNQ (M‐type) K+ channels, TRPV cation channels, and various G protein‐coupled receptors, making it a point of convergence for integration of diverse signals, as discussed in previous reviews (Wong & Scott, 2004; McConnachie et al. 2006; Esseltine & Scott, 2013). In this mini‐review, we focus on the novel discoveries of AKAP79/150 in directing multi‐protein signalling complexes with reference to M‐type (Kv7) K+ channels and L‐type (CaV1) Ca2+ channels, and the mechanisms and dynamics of these complexes in the nervous system.
AKAP79/150 actions on M‐type K+ channels: altered PIP2 sensitivity, and effects on CaM interactions
Voltage‐gated M‐type (KCNQ, Kv7) K+ channels, expressed in a wide variety of neurons, play critical roles in modulation of excitability and action potential firing (Brown & Adams, 1980; Constanti & Brown, 1981; Delmas & Brown, 2005; Hernandez et al. 2008). Heteromers of KCNQ2 and KCNQ3, which were identified from an inherited human neonatal form of epilepsy called benign familial neonatal convulsions, underlie neuronal M‐current. Given their critical role in regulation of neuronal excitability, M‐channels are closely regulated by various neurotransmitters through three mechanisms from four related second messenger signalling events in neurons: First, M‐channels are highly sensitive to the binding of membrane PIP2; thus, depletion of membrane [PIP2] or change in the PIP2 sensitivity of the channels strongly depress M‐current by reducing the open probability of the channel (Suh & Hille, 2008; Logothetis et al. 2010). Such unbinding of PIP2 comes from stimulation of Gq/11‐coupled neurotransmitter receptors that activate phospholipase C (PLC), which hydrolyses PIP2 to diacylglycerol (DAG) and inositol trisphosphate (IP3). Secondly, DAG and IP3 generated by PIP2 breakdown potentially activate PKC and generate rises in intracellular Ca2+ () from internal Ca2+ stores. Third, Ca2+–CaM binds to and suppresses M‐channels (Gamper & Shapiro, 2003; Delmas & Brown, 2005). Fourth, AKAP79/150 physically associates with KCNQ2‐5 subtypes, recruiting PKC and facilitating subsequent phosphorylation and enhancing suppression of KCNQ channels (Hoshi et al. 2003; Bal et al. 2010; Zhang et al. 2011). These related, but seemingly complex and redundant, signalling pathways underlying M‐channel modulation made us inquire as to the physiological function of AKAP‐PKC on M‐channels, synergistic with PIP2 reduction and CaM action.
One attractive model that synergizes PIP2, Ca2+, CaM and PKC actions involves allosteric effects of one signalling molecule on the affinities of the others for KCNQ channels, with the focus on AKAP79/150 (Kosenko et al. 2012; Kosenko & Hoshi, 2013; Fig. 1). For the case of PKC, association of apoCaM with the A and B helices of the C‐terminus of the channels is obligatory to maintain tonic PIP2 affinity (Kosenko et al. 2012). AKAP79/150‐mediated PKC phosphorylation of KCNQ channels is suggested to induce rearrangement of CaM on the C‐terminus of the channels, resulting in lowered PIP2 affinity and sensitization to muscarinic depression of M‐current. At the same time, Ca2+ binding to CaM is suggested to likewise induce a conformational change in CaM bound to the channels, again reducing the channel–PIP2 affinity (Kosenko & Hoshi, 2013). Such a conformational change is consistent with the crystal structure of Ca2+–CaM bound solely to the KCNQ channel B helix (Xu et al. 2013 b), whereas apoCaM likely cross‐links the A and B helices (Wen & Levitan, 2002; Yus‐Najera et al. 2002; Gamper & Shapiro, 2003; Xu et al. 2013 b). Interestingly, the Hoshi group has suggested that AKAP79/150 acts as an acceptor for CaM dissociated from the channels after their PKC phosphorylation or increases (Kosenko et al. 2012; Kosenko & Hoshi, 2013), a hypothesis that would seem at odds with maintained CaM binding to the channels regardless of Ca2+ binding (Bal et al. 2008).
Figure 1. Model of 3‐step rearrangement of AKAP–KCNQ channel complex .
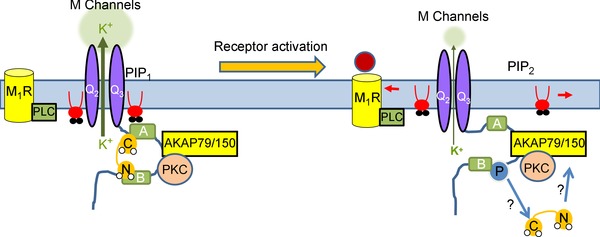
See Kosenko et al. (2012) and Kosenko & Hoshi (2013). AKAP79/150 organizes a signalling complex at the carboxyl terminus of KCNQ channels, including PKC and CaM. Stable KCNQ–CaM association is required for maintaining tonic PIP2 affinity. Stimulation of Gq/11‐coupled muscarinic receptors depletes PIP2 and activates AKAP79/150‐anchored PKC, which phosphorylates KCNQ subunits near the CaM binding sites of KCNQ channels. PKC phosphorylation, or signals, triggers CaM conformational changes and/or dissociation from the cytoplasmic tail of KCNQ channels, resulting in lowered PIP2 affinity and suppressed channel activity. AKAP79/150 may act as an acceptor to bind to dissociated CaM. Thus, together with PIP2 depletion, receptor stimulation suppresses KCNQ channel activity by inducing CaM rearrangement and dissociation from the channel complex. Note that the depression of M‐current by reducing channel opening is schematically indicated here by a squeezing together of the two halves of the channel, and a thinning of the directional arrow of potassium flux. See the text for issues concerning this model.
We recently suggested a dual mode of mixed convergence–competition model to explain AKAP79/150 involvement in M‐current suppression by only one group of neurotransmitter receptors that are localized away from ER Ca2+ stores and do not induce IP3‐mediated rises (Fig. 2 A; Zhang et al. 2011). Our thinking is based on the following observations: (1) these receptors physically associate with AKAP79 (Hoshi et al. 2003; Bal et al. 2010), (2) AKAP79/150 and CaM share overlapping binding sites on the C‐terminus of KCNQ channels, and Ca2+–CaM disrupts AKAP79/150‐channel interactions (Bal et al. 2010), and (3) over‐expression of WT, but not DN, CaM prevents AKAP79/150‐mediated channel sensitization (Bal et al. 2010). In this model, rearrangement of CaM induced by Ca2+ binding prevents AKAP79/150‐mediated PKC phosphorylation by interfering with the association of AKAP79/150 with KCNQ complexes (Fig. 2 B). Association of receptors with those complexes is required to co‐localize M‐channels with putative transient, local depletions of PIP2 and activation of PKC that may be more physiologically relevant (Fig. 2 A). Thus, M‐channels reduce their activity in response to two types of concurrently sensed responses: either PIP2 depletion + AKAP79/150‐mediated PKC phosphorylation (Fig. 2 A), or IP3‐mediated Ca2+ release + Ca2+–CaM actions on M‐channels (Fig. 2 B). Such convergent dual mechanisms may serve as a coincidence‐detector to fine tune the spatiotemporal resolution of directed signals.
Figure 2. Modified concurrent dual mode of KCNQ channel inhibition .
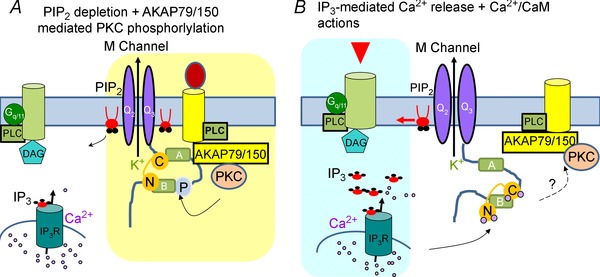
In this model, phosphorylation of KCNQ channels functions to reduce their PIP2 affinity, priming them to small changes in PIP2 abundance induced by physiological stimulation of receptors. A, activation of receptors that are part of AKAP79150–KCNQ complexes rapidly depletes PIP2 and activates AKAP79/150‐anchored PKC, which phosphorylates KCNQ subunits near the CaM binding site on the B helix, altering the configuration of CaM and KCNQ channels. KCNQ channels thus sense both PIP2 depletion and phosphorylation‐mediated PIP2‐affinity reduction. B, activation of receptors located close to IP3 receptors, but outside of the AKAP–KCNQ complexes, induces intracellular Ca2+ rises and Ca2+ binding to CaM, which undergoes a conformational change on the overlapping binding sites with AKAP150 on the carboxy‐terminus of KCNQ channels. This induces a lowered interaction between KCNQ and AKAP79/150, thus preventing PKC phosphorylation.
Crucial questions arising from the two models are (1) whether CaM constitutively binds to KCNQ channels in native neurons, (2) whether CaM–KCNQ association is obligatory for channel activity, and (3) whether expression of functional channels or ‐sensing is the more critical role of CaM. At this moment, it is hard to answer question (3), but biochemical studies pointing out both a changed conformation and affinity of the CaM–KCNQ complex after Ca2+ binds (Black & Persechini, 2011; Wang et al. 2013; Alaimo et al. 2014) suggest it is likely that these two mechanisms are not exclusive. Recently, it has been shown that the N‐lobe of apoCaM binds to helix B due to its greater affinity, whereas the C‐lobe moves from helix A to helix B upon Ca2+ binding (Alaimo et al. 2014). Our current thinking has thus been modified accordingly (Fig. 2): (1) cross‐linking of apoCaM with helices A and B is important to maintain their helical structure, which we suppose is critical to stabilize channel interactions with PIP2; (2) PKC phosphorylation on helix B (Fig. 2 A) weakens the interaction between helix B and the N‐lobe of CaM, resulting in reduced PIP2 affinity and suppressed M‐current (consistent with both models), and (3) maximal Ca2+ binding to CaM (Fig. 2 B) induces the C‐lobe to move from helix A to helix B and a subsequent conformational change of both CaM and KCNQ channels which causes reduced PIP2 affinity, weaker AKAP79/150 association with (and perhaps AKAP79/150 dissociation from) the channel complex and prevention of PKC phosphorylation (Fig. 2 B). Further direct biochemical analysis in native neurons of KCNQ channels, AKAP79/150 and CaM would be very helpful, and direct observation of native KCNQ channel–AKAP150–CaM dynamics upon receptor stimulation would be exciting.
AKAP79/150 interactions on L‐type Ca2+ channels: priming by PKA and Ca2+ sensing by CaN
Another ion channel family whose activity is under close regulation is voltage‐gated Ca2+channels (VGCCs), especially L‐type (CaV1) channels which are critical for synaptic plasticity, axon growth and neuronal development. In the hippocampus, AKAP79/150 bound to the modified leucine zipper (LZ) motif of the distal C‐terminus of CaV1.2 channels anchors PKA to phosphorylate and up‐regulate channel activity, whereas the Ca2+–CaM‐regulated phosphatase, calcineurin (CaN), which is also recruited by AKAP79/150, counterbalances PKA actions, serving as an activity‐dependent negative feedback mechanism (Hall et al. 2007; Oliveria et al. 2007). Moreover, AKAP79/150 intimately regulates gene transcription via Ca2+–CaN, inducing CaN‐mediated NFAT (nuclear factor of activated T‐cells) dephosphorylation and translocation to the nucleus, where NFAT interacts with regulatory elements of NFAT‐sensitive genes (Graef et al. 1999, 2003; Hudry et al. 2012; Wu et al. 2012; Murphy et al. 2014). We have recently identified similar L‐channel–AKAP–CaN protein complexes in sympathetic ganglia that initiate NFAT signalling to up‐regulate M‐channel expression, serving as a longer‐term feedback mechanism to counter increased neuronal hyperexcitability (Zhang & Shapiro, 2012). We suggest that upregulating M‐channels is a critical ‘anti‐epileptogenic’ mechanism in brain regions such as the hippocampus to prevent development of epilepsy. Unlike in hippocampus, the L‐channel–NFAT pathway is slightly more complex in sympathetic neurons, in which L‐channels underlie less than 15% of the total Ca2+ current, revealing both N‐ and L‐type Ca2+ channel activity to be necessary for NFAT activation (Zhang & Shapiro, 2012). We have suggested a dual requirement model in these cells: L‐channels serve as the critical initiating ‘sensor’ of activity and depolarization, and an elevated microdomain signal, which activates only AKAP–L‐channel complex‐bound CaN, despite the high abundance of CaN in neurons. Upon Ca2+ binding, CaN rapidly dissociates from AKAP79/150 complexes to dephosphorylate NFAT, because CaN affinity for AKAP79/150 is relatively low (K D ≈ 0.5 μm; Li et al. 2012). However, globally elevated [Ca2+]i (mostly mediated by N‐type Ca2+ channels in peripheral ganglia) is also required to keep CaN–NFAT activated during its import into the nucleus, This global elevation could probably come from various sources, not specifically from N‐channels (Zhang & Shapiro, 2012).
Besides M‐channels, large‐conductance Ca2+‐activated BKCa channels have been discovered as a downstream target of AKAP‐organized transcriptional signalling (Zhang & Shapiro, 2012; Nystoriak et al. 2014). In arterial myocytes, the same L‐channel–AKAP–CaN complex as described above mediates down‐regulation of BKCa channel β1 subunits, contributing to BKCa channel remodelling and enhanced vasoconstriction during type II diabetes mellitus (Nystoriak et al. 2014). The critical questions are (1) whether NFATs pre‐associate with the AKAP79/150–CaN–L‐channel complex at the membrane and (2) whether CaN stays associated with NFAT during its import into the nucleus, perhaps thus explaining its delayed time to be transported into the nuclear compartment.
An additional layer of flexibility and complexity of AKAP signalling pathways comes from the recent discovery of the regulatory property of AKAP15, which binds competitively to the same LZ motif on the distal C‐terminal domain of CaV1.2 channels as does AKAP79/150 and also seems to mediate PKA phosphorylation on L‐channels (Fuller et al. 2014). AKAP15 and AKAP79/150 actions were shown to have different effects, dependent on their differential associations with regulatory proteins, notably CaN (Fuller et al. 2014). However, previous studies involving AKAP79/150 knockdown or knockouts that disrupt L‐channel–AKAP–PKA–CaN complexes prevented speci‐fic PKA‐, or CaN‐mediated effects on L‐channel function. Exciting studies from new transgenic mice with a specific deletion of the PKA binding segment or the CaN‐binding PXIXIT motif on AKAP150, AKAP150ΔPKA or AKAP150ΔPIX, allow more specific probing of the physiological roles of AKAP‐anchored PKA or CaN on L‐channels. Surprisingly, removal of PKA from the CaV1.2–AKAP150 complex not only reduced basal L‐channel phosphorylation and L‐current density, but also impaired CaN‐mediated NFAT activation and translocation to the nucleus (Dittmer et al. 2014; Murphy et al. 2014).
Interplay between AKAP79/150 anchored ion channels in super multi‐channel complexes
Whether AKAP79/150‐anchored proteins are physically coupled as larger super‐complexes is an intriguing question to investigate, especially involving distinct ion channel complexes that shape neuronal activity (Fig. 3). The gating of individual CaV1.2 channels seems to be coupled (Navedo et al. 2010), probably via physical protein–protein interactions of CaV1.2 channels with one another at their carboxyl‐tails by AKAP79/150 (Cheng et al. 2011), resulting in amplification of Ca2+ influx and increased excitation–contraction coupling in ventricular myocytes (Dixon et al. 2012). Recent structural and biochemical studies also suggest such a possibility, as AKAP79/150 is reported to express as homodimers coupled with a PKA homodimer and two CaN heterodimers (Gold et al. 2011). Since each AKAP79/150 protomer in such putative homodimers binds one PKA (Gold et al. 2011), they could also physically couple two distinct ion channel complexes to form one macro‐molecular multi‐channel complex. Recent use of super‐resolution fluorescence imaging offering nanometer scale resolution has successfully revealed the molecular architecture of synapses, including organization of protein components of the presynaptic active zone and the postsynaptic density (Dani et al. 2010), and of actin–sepctrin–Na+ channels organization in axons (Xu et al. 2012, 2013 a). The next important step would be to elucidate the detailed mechanism of these actions in different excitable cells.
Figure 3. Model of putative AKAP79/150‐orchestrated multi‐channel protein complexes .
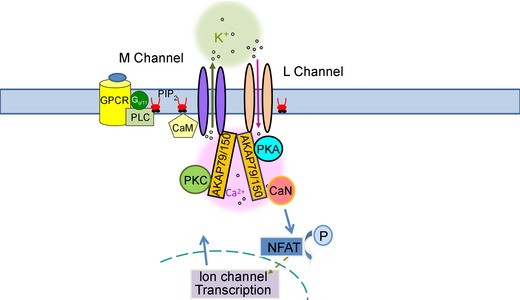
AKAP79/150 expresses as a homodimer, and each protomer of AKAP79/150 binds to one PKA molecule, and could physically couple two distinct ion channel complexes together to form one large macro‐molecular super‐complex, such as containing the M‐channel and L‐channel we illustrate here. In this large multi‐channel protein complex, channels are also likely to be functionally linked by AKAP79/150 (Dixon et al. 2012), via direct coupling of their gating, indirect coupling via Ca2+ or changes in [PIP2], or more slowly via transcriptional regulation.
Conclusions
AKAP79/ is particularly crucial in its ability to form focally compartmentalized multi‐protein signalling complexes in localized neuron structures, such as regulating pre‐ and postsynaptic plasticity (Sanderson & Dell'Acqua, 2011; Sanderson et al. 2012). Local perturbations of neuronal ion channel function by disrupted AKAP79/150 signalling complexes modulate synaptic transmission, and have been suggested to play a role in seizures, mental retardation, Alzheimer's disease and schizophrenia (Sanderson & Dell'Acqua, 2011; Esseltine & Scott, 2013). In this mini‐review, we summarize the dual fast and slow regulation of ion channel activity by AKAP‐organized protein complexes which includes rapidly altering the activity of ion channels already functioning in the membrane, and more slowly altering the population of expressed channels by transcriptional regulation in the nucleus, dependent on the activity of VGCCs. The highly dynamic feature of AKAP79/150 complexes upon neuronal stimulation suggests their novel ability to further fine tune the spatiotemporal resolution of localized signals. We also suggest that AKAP79/150 probably clusters together different ion channels that functionally interact, as a mechanism of feed‐back inhibition, amplification or focusing of neuronal signals.
Additional information
Competing interests
None declared.
Funding
This work was supported by NIH grants R01 NS43394 and R56 NS065138 (M.S.S), and NIH training grant T32 HL007446 (J.Z.).
Acknowledgements
We thank Pamela Reed for expert technical assistance and Frank S. Choveau, Crystal R. Archer, Clinton Taylor and Sonya M. Bierbower for scientific discussions.
Biography
Jie Zhang PhD received her Bachelor degree at SunYat‐sen University in 2007, and her PhD in the Department of Physiology at the University of Texas Health Science Centre in San Antonio (UTHSCSA) in 2011. During her PhD and post‐doctoral work under Mark S. Shapiro, she studied G‐protein and transcriptional modulation of neuronal ion channels by AKAP79/150. As a post‐doc under Paul A. Slesinger at the Icahn School of Medicine at Mount Sinai, she learned to generate cell‐based neurotransmitter in vivo reporters. Mark S. Shapiro PhD received his Bachelor degree in Physics at the University of Chicago, and his doctorate in Physiology at Rush University Medical Centre in Chicago, under Tom DeCoursey, where he studied the biophysics of K+ channels in lymphocytes. Dr Shapiro did his post‐doctoral work at the University of Washington in Seattle under Bertil Hille and William Zagotta, studying the mechanisms of G protein‐mediated regulation of Ca2+ channels and KCNQ (‘M‐type’) K+ channels, and the biophysics and molecular structure of cyclic nucleotide‐gated channels. In 2000, he joined the faculty of the University of Texas Health Science Centre in San Antonio, attaining tenure in 2005 and the rank of Professor in 2010. His research programme spans the physiology, modulation and functional role of voltage‐gated K+, Ca2+ channels, TRPV cation channels, and others, in neurons, smooth muscle and non‐excitable cells using patch‐clamp electrophysiology, biochemistry, confocal and total internal reflection fluorescence (TIRF) microscopy, molecular biology and live single‐cell and whole‐animal imaging.

This review was presented at the symposium Localised intracellular signalling in neurons, which took place at Physiology 2014, the annual meeting of The Physiological Society, London, UK on 1 July 2014.
References
- Alaimo A, Alberdi A, Gomis‐Perez C, Fernandez‐Orth J, Bernardo‐Seisdedos G, Malo C, Millet O, Areso P & Villarroel A (2014). Pivoting between calmodulin lobes triggered by calcium in the Kv7.2/calmodulin complex. PLoS One 9, e86711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Zaika O, Martin P & Shapiro MS (2008). Calmodulin binding to M‐type K+ channels assayed by TIRF/FRET in living cells. J Physiol 586, 2307–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Zhang J, Hernandez CC, Zaika O & Shapiro MS (2010). Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M‐Type) K+ channels. J Neurosci 30, 2311–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW & Scott JD (2006). Dynamic regulation of cAMP synthesis through anchored PKA–adenylyl cyclase V/VI complexes. Mol Cell 23, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DJ & Persechini A (2011). In calmodulin–IQ domain complexes, the Ca2+‐free and Ca2+‐bound forms of the calmodulin C‐lobe direct the N‐lobe to different binding sites. Biochemistry 50, 10061–10068. [DOI] [PubMed] [Google Scholar]
- Brown DA & Adams PR (1980). Muscarinic suppression of a novel voltage‐sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. [DOI] [PubMed] [Google Scholar]
- Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves‐Cintron M, Scott JD & Santana LF (2011). Restoration of normal L‐type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res 109, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constanti A & Brown DA (1981). M‐Currents in voltage‐clamped mammalian sympathetic neurones. Neurosci Lett 24, 289–294. [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C & Zhuang X (2010). Superresolution imaging of chemical synapses in the brain. Neuron 68, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P & Brown DA (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6, 850–862. [DOI] [PubMed] [Google Scholar]
- Dittmer PJ, Dell'Acqua ML & Sather WA (2014). Ca2+/calcineurin‐dependent inactivation of neuronal L‐type Ca2+ channels requires priming by AKAP‐anchored protein kinase A. Cell Rep 7, 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Yuan C, Cheng EP, Navedo MF & Santana LF (2012). Ca2+ signaling amplification by oligomerization of L‐type Cav1.2 channels. Proc Natl Acad Sci U S A 109, 1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine JL & Scott JD (2013). AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol Sci 34, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Fu Y, Scheuer T & Catterall WA (2014). Differential regulation of CaV1.2 channels by cAMP‐dependent protein kinase bound to A‐kinase anchoring proteins 15 and 79/150. J Gen Physiol 143, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N & Shapiro MS (2003). Calmodulin mediates Ca2+‐dependent modulation of M‐type K+ channels. J Gen Physiol 122, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Stengel F, Nygren PJ, Weisbrod CR, Bruce JE, Robinson CV, Barford D & Scott JD (2011). Architecture and dynamics of an A‐kinase anchoring protein 79 (AKAP79) signaling complex. Proc Natl Acad Sci U S A 108, 6426–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW & Crabtree GR (1999). L‐Type calcium channels and GSK‐3 regulate the activity of NF‐ATc4 in hippocampal neurons. Nature 401, 703–708. [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier‐Lavigne M & Crabtree GR (2003). Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113, 657–670. [DOI] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS & Hell JW (2007). Critical role of cAMP‐dependent protein kinase anchoring to the L‐type calcium channel CaV1.2 via A‐kinase anchor protein 150 in neurons. Biochemistry 46, 1635–1646. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP & Shapiro MS (2008). Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol 586, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA & Higashida H (2003). AKAP150 signaling complex promotes suppression of the M‐current by muscarinic agonists. Nat Neurosci 6, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Wu HY, Arbel‐Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires‐Jones TL, Betensky RA, Bacskai BJ & Hyman BT (2012). Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer's disease. J Neurosci 32, 3176–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko A & Hoshi N (2013). A change in configuration of the calmodulin–KCNQ channel complex underlies Ca2+‐dependent modulation of KCNQ channel activity. PLoS One 8, e82290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko A, Kang S, Smith IM, Greene DL, Langeberg LK, Scott JD & Hoshi N (2012). Coordinated signal integration at the M‐type potassium channel upon muscarinic stimulation. EMBO J 31, 3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML & Hogan PG (2012). Balanced interactions of calcineurin with AKAP79 regulate Ca2+–calcineurin–NFAT signaling. Nat Struct Mol Biol 19, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Petrou VI, Adney SK & Mahajan R (2010). Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch 460, 321–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnachie G, Langeberg LK & Scott JD (2006). AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12, 317–323. [DOI] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA & Dell'Acqua ML (2014). AKAP‐anchored PKA maintains neuronal L‐type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7, 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD & Santana LF (2010). Increased coupled gating of L‐type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystoriak MA, Nieves‐Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell'acqua ML et al (2014). AKAP150 contributes to enhanced vascular tone by facilitating large‐conductance Ca2+‐activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res 114, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML & Sather WA (2007). AKAP79/150 anchoring of calcineurin controls neuronal L‐type Ca2+ channel activity and nuclear signaling. Neuron 55, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL & Dell'Acqua ML (2011). AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist 17, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS & Dell'Acqua ML (2012). AKAP150‐anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+‐permeable AMPA receptors. J Neurosci 32, 15036–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC & Hille B (2008). PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37, 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang P, Hoffman L, Tripathi S, Homouz D, Liu Y, Waxham MN & Cheung MS (2013). Protein recognition and selection through conformational and mutually induced fit. Proc Natl Acad Sci U S A 110, 20545–20550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H & Levitan IB (2002). Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22, 7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W & Scott JD (2004). AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5, 959–970. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ & Hyman BT (2012). Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid‐β revealed by a novel fluorescence resonance energy transfer assay. J Neurosci 32, 5298–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Babcock HP & Zhuang X (2012). Dual‐objective STORM reveals three‐dimensional filament organization in the actin cytoskeleton. Nat Methods 9, 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G & Zhuang X (2013. a). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Chang A, Tolia A & Minor DL, Jr (2013. b). Structure of a Ca2+/CaM:Kv7.4 (KCNQ4) B‐helix complex provides insight into M current modulation. J Mol Biol 425, 378–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus‐Najera E, Santana‐Castro I & Villarroel A (2002). The identification and characterization of a non‐continuous calmodulin binding site in non‐inactivating voltage‐dependent KCNQ potassium channels. J Biol Chem 277, 28545–28553. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bal M, Bierbower S, Zaika O & Shapiro MS (2011). AKAP79/150 signal complexes in G‐protein modulation of neuronal ion channels. J Neurosci 31, 7199–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J & Shapiro MS (2012). Activity‐dependent transcriptional regulation of M‐Type (Kv7) K+ channels by AKAP79/150‐mediated NFAT actions. Neuron 76, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]