

Abstract
Nitric oxide (NO) functions widely as a transmitter/diffusible second messenger in the central nervous system, exerting physiological effects in target cells by binding to specialized guanylyl cyclase‐coupled receptors, resulting in cGMP generation. Despite having many context‐dependent physiological roles and being implicated in numerous disease states, there has been a lack of clarity about the ways that NO operates at the cellular and subcellular levels. Recently, several approaches have been used to try to gain a more concrete, quantitative understanding of this unique signalling pathway. These approaches have included analysing the kinetics of NO receptor function, real‐time imaging of cellular NO signal transduction in target cells, and the use of ultrasensitive detector cells to record NO as it is being generated from native sources in brain tissue. The current picture is that, when formed in a synapse, NO is likely to act only very locally, probably mostly within the confines of that synapse, and to exist only in picomolar concentrations. Nevertheless, closely neighbouring synapses may also be within reach, raising the possibility of synaptic crosstalk. By engaging its enzyme‐coupled receptors, the low NO concentrations are able to stimulate physiological (submicromolar) increases in cGMP concentration in an activity‐dependent manner. When many NO‐emitting neurones or synapses are active simultaneously in a tissue region, NO can act more like a volume transmitter to influence, and perhaps coordinate, the behaviour of cells within that region, irrespective of their identity and anatomical connectivity.
Introduction
Nitric oxide (NO) is an evolutionary ancient but unique type of intercellular chemical messenger that operates widely within the central nervous system (and elsewhere) where it participates in many behaviours, including learning and memory, pain, feeding, sleeping, reproductive activity and anxiety (Garthwaite, 2008; Steinert et al. 2010). Like conventional second messengers, NO is synthesized intracellularly in response to external stimuli. In the brain, the best characterized stimulus is the neurotransmitter glutamate acting postsynaptically on NMDA receptors (Garthwaite et al. 1988). The coupling is aided by the neuronal subtype of NO synthase (nNOS) being physically tethered to these receptors through postsynaptic density‐95 protein (Brenman et al. 1996), an arrangement that localizes the synthetic enzyme to just 18 nm inside the postsynaptic cell membrane (Valtschanoff & Weinberg, 2001). The influx of Ca2+ through open NMDA receptor channels and their subsequent binding to calmodulin leads to nNOS becoming catalytically active, consuming the amino acid l‐arginine and synthesizing NO along with the co‐product l‐citrulline which, in turn, is recycled back to l‐arginine.
While the positioning of nNOS close to the mouth of NMDA receptor channels facilitates NO synthesis (Sattler et al. 1999; d'Anglemont de Tassigny et al. 2007), more global increases in Ca2+, as brought about, for example, by administering elevated K+ (Alagarsamy et al. 1994) can also be effective. When operating as a nitrergic transmitter in autonomic nerves, voltage‐dependent Ca2+ channels (primarily N‐type channels) provide the Ca2+ influx that initiates presynaptic NO formation (Toda & Okamura, 2003; Toda & Herman, 2005) and there is evidence that other transmitters, for example acetylcholine (de Vente, 2004), and other glutamate receptors (Southam et al. 1991), can be associated with NO synthesis in the brain.
NO sources and targets in the brain
What distinguishes NO from traditional second messengers are its low molecular weight (30 g mol−1), which enables a large aqueous diffusion coefficient (3.3 μm2 ms−1) and a correspondingly rapid rate of aqueous diffusion (about 2.5 μm in 1 ms), paired with its high lipid solubility, allowing rapid membrane permeation (Lancaster, 1997). In respect of the latter, NO concentrates about 4‐fold in model biological membranes where it has only a 10‐fold (rather than the expected 100‐fold) lower diffusion coefficient than in aqueous buffer (Moller et al. 2005), so that NO would cross the 3 nm hydrophobic membrane interior in about 3 ns. These physicochemical properties imply that NO will diffuse uniformly in all directions away from its site of synthesis and so can indulge in forms of cellular communication that bypass anatomical connectivity. The prototypic example of this type of signalling is in blood vessels where NO produced in endothelial cells diffuses to the underlying smooth muscle to cause relaxation (Furchgott, 1999). In the context of the central nervous system, with its closely packed, heterogeneous mixture of neuronal and glial elements, a host of intercellular signalling pathways could exist. One constraint is imposed by the cellular location of nNOS, which is exclusively neuronal and is usually concentrated in discrete neuronal subtypes, for example in interneurones and their processes in the striatum and cerebral cortex, although in other areas, such as the cerebellum, it is more widely distributed among the constituent neurones despite being absent in Purkinje cells, the sole output neurones of this brain region (Bredt et al. 1991; Vincent & Kimura, 1992).
But what of the cellular targets for NO and how do they relate anatomically to the sites of NO formation? Unlike conventional transmitters, the NO molecule lacks the chemical specializations normally exploited for binding to receptor proteins. Although a radical species, NO also exhibits only low chemical reactivity at the concentrations likely to be relevant physiologically (nm and below; see later) but there are two notable exceptions. One is its reactivity with other radicals (e.g. superoxide ions and lipid radicals) and the other is its high affinity for binding to transition metals, particularly iron (Hill et al. 2010). This latter property is exploited by the only known NO receptors, which consist of a prosthetic ferrous (Fe2+) haem group coordinated to a protein possessing intrinsic guanylyl cyclase activity. Binding of NO to the vacant coordination site on the metal results in a pivoting of the haem group, leading to strain on the bond between the haem and an underlying histidine group of the protein; breakage of this bond triggers a conformational change in the protein that propagates to the catalytic site, prompting the formation of cGMP from GTP (Waldman & Murad, 1987; Ma et al. 2007).
Other haemoproteins with a vacant coordination site can also react with NO, notably haemoglobin (forming nitrate and methaemoglobin in the presence of O2) and mitochondrial cytochrome c oxidase where binding of NO is in competition with O2, unlike with NO‐activated guanylyl cyclase whose haem excludes O2. Reaction with haemoglobin in red blood cells inactivates endothelium‐derived NO and is also likely to contribute a slow, basal component to the inactivation of NO formed within blood‐perfused tissues, imposing on it a half‐life of about 1 s (Santos et al. 2011). Binding of NO to cytochrome c oxidase leads to inhibition of mitochondrial respiration and remains of uncertain physiological significance, in part because in intact cells under normal levels of O2 (about 30 μm), half‐maximal inhibition of respiration requires more than 10‐fold higher NO concentrations than the 10 nm needed for half‐maximal activation of guanylyl cyclase (Bellamy et al. 2002; Rodriguez‐Juarez et al. 2007), with endogenous NO concentrations probably being well below this 10 nm value under physiological conditions (Hall & Garthwaite, 2009).
The NO receptor proteins are heterodimers of two main types: α1β1, which predominates in the cardiovascular system, and α2β1, which predominates in brain and possesses a PDZ domain enabling it to bind to synaptic scaffold proteins, including postsynaptic density‐95 protein (Friebe & Koesling, 2009), implying a possible location on either side of the synapse. One approach for identifying NO‐responsive cells has been to find out where NO raises cGMP levels, a method that became feasible using immunohistochemistry and a special antibody raised against formaldehyde‐fixed cGMP (de Vente & Steinbusch, 1992). Studies in vivo and in vitro indicated that responsiveness to NO was widespread in the brain and spinal cord, with neurones, nerve fibres and glial cells variously labelled, and in a pattern that was grossly complementary to the distribution of NO synthase (Southam & Garthwaite, 1993; de Vente et al. 1998). A limitation of this technique, however, is its relatively low sensitivity, so that cellular cGMP needs to rise above about 10 μm to be detected. In the hippocampus, pharmacological manipulations that boost cGMP beyond this threshold allow a much more extensive distribution than was evident beforehand to be visualized, with astrocytes, interneurones, axons and pyramidal cells all showing strongly positive NO‐evoked cGMP accumulation, results that resonate with the location of the NO receptor subunit mRNA by in situ hybridization and the protein by immunohistochemistry (Bartus et al. 2013). Clearly, a more precise, high‐resolution anatomical picture is needed to help understand more clearly the potential lines of communication between different NO‐generating neurones and their targets. In one of the few studies of this type (Burette et al. 2002) evidence from the hippocampus indicated that nNOS in pyramidal neurones is concentrated postsynaptically whereas NO‐activated guanylyl cyclase protein is mainly found presynaptically in excitatory axon terminals, giving anatomical support to the idea that NO can act as a retrograde trans‐synaptic messenger of the type deemed of importance for NMDA receptor‐dependent synaptic plasticity in this brain area and elsewhere. In another investigation, also in the hippocampus, nNOS was, surprisingly, also present postsynaptic to GABAergic nerve terminals, with the α1β1 subtype of NO‐activated guanylyl cyclase, and NMDA‐evoked cGMP accumulation, being found presynaptically, pointing to a similar retrograde signalling by NO at these inhibitory synapses, albeit with fewer NMDA receptors available than at excitatory synapses (Szabadits et al. 2007, 2011). These examples raise the possibility that NO has evolved to signal within discrete synaptic domains in the brain.
NO signalling at individual synapses
The feasibility of this type of point‐to‐point, or ‘wired’ (Agnati et al. 2010) transmission can be tested by modelling the production of NO at synapses, its spread by diffusion, and the resultant stimulation of cGMP accumulation in immediately adjacent structures. The spread of NO generated by an array of 49 postsynaptic nNOS molecules (1 per NMDA receptor) simultaneously producing 20 NO molecules per second each, had suggested that NO concentration gradients around the source would be very steep, reaching 1 nm at a distance of 60 nm away (equivalent to just inside the presynaptic terminal), falling to 250 pm, 1 μm away (Hall & Garthwaite, 2009). Whilst this extreme degree of NMDA receptor activation helps set an upper limit on the size of the NO gradients established around a synaptic source, many fewer receptors appear to become active following the normal synaptic release of glutamate, possibly only three to five (Silver et al. 1992; Nimchinsky et al. 2004). If a single active NMDA receptor couples to one nNOS protein producing 10 NO molecules per second (Salerno, 2008) and there are 4 active NMDA receptors, the total number of NO molecules generated within a synapse each second would only be about 40, rather than the nearly 1000 considered previously (Hall & Garthwaite, 2009).
These considerations prompt a re‐examination of the potential biological significance of the release of NO at single synapses. If the zone of NO production in the postsynaptic compartment is modelled as a disc of appropriate diameter (400 nm; Fig. 1) that produces NO steadily at 40 s−1, the NO concentration is predicted to peak (at the source) at about 60 pm and fall to 5 pm a distance of 1 μm away (Fig. 1 A). Superimposing this concentration profile on an image of a synapse (Fig. 1 B) emphasizes the very local spread of NO away from such a source, suggesting a high degree of synapse specificity.
Figure 1. Synaptic spread of NO .
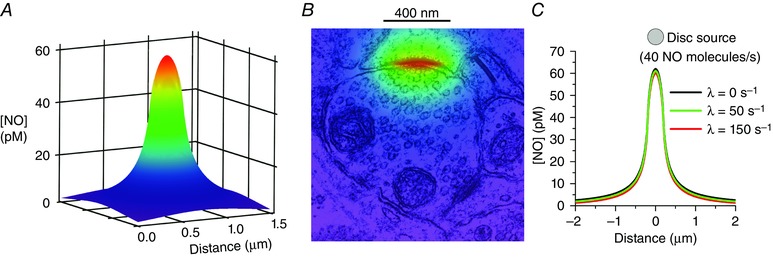
The zone of NO formation was modelled as a disc (0.4 μm diameter) that constantly emits 40 NO molecules per second, using the equation for an instantaneous disc source (Carslaw & Jaeger, 1986, eqn (10.3.9), p. 260) modified to include inactivation of NO as a first‐order decay term, and differentiated with respect to time. The profiles are taken 40 ms after the start, when the concentrations are at steady state. The plume of NO above a 1.5 μm × 1.5 μm grid (A) is shown compressed into 2‐dimensions in B and superimposed on an electron micrograph of an excitatory synapse with the plane of the disc centred on the synaptic cleft. In C, the profile in the plane of the disc surface is shown with rate constants for NO decay of 0, 50 and 150 s−1, corresponding to half‐lives of approximately 0, 14 and 5 ms, respectively.
NO is subject to a high rate of inactivation in brain tissue which, in cerebellum, appears to be through a saturable, enzymatic mechanism having an apparent Michaelis constant (K m) of 10 nm and a maximal velocity (V max) of 1.5 μm s−1 (Hall & Garthwaite, 2006). At NO concentrations well below the K m, NO consumption would correspond to a first‐order reaction governed by a rate constant (V max /K m) of 150 s−1, which is equivalent to an NO half‐life of 4.6 ms. The mechanism remains unclear, although cytochrome P450 oxidoreductase is a putative participant (Hall et al. 2009). At the level of the single synapse, an NO half‐life even as short as 5 ms would have very little effect on the local distribution of NO (Fig. 1 C) because diffusion over these dimensions is so rapid.
As the NO concentrations under consideration are so much lower than either the affinity of NO for binding to its receptor (K d = 20–50 nm) or the 10 nm NO concentration required to give 50% activation of guanylyl cyclase in cells (Griffiths et al. 2003; Roy et al. 2008; Tsai et al. 2012), it might be thought intuitively that such a tiny NO plume would be irrelevant. But NO‐activated guanylyl cyclase is an enzyme‐linked receptor whose transduction mechanism is most efficient when the agonist concentrations are low compared with the binding affinity (Batchelor et al. 2010). In this situation, and given a guanylyl cyclase activity found in rat platelets or cerebellar astrocytes (maximally, about 100 μm s−1), each NO molecule would stimulate the synthesis of 5000 molecules of cGMP each second at steady state, so 10 pm NO translates into 50 nm cGMP being formed per second (Batchelor et al. 2010), an astonishing feat for a simple, one‐component biological transducer. Attesting to these theoretical considerations, experiments in which cGMP was charted in real time using a fluorescent biosensor have documented the picomolar sensitivity of cells to perfusion of NO in constant concentration, or to sub‐second NO ‘puffs’ delivered by a local pipette (Batchelor et al. 2010).
Having in hand a quantitative kinetic description of NO‐activated guanylyl cyclase in cells (Roy et al. 2008; Halvey et al. 2009), modelling synaptic NO diffusion and signal transduction is achievable in principle but, without specialized computing resources, the very small time and distance steps needed to handle both components accurately makes such an undertaking prohibitive. A good approximation, however, can be achieved economically by dividing up the synaptic space into multiple concentric hemispheres, with one set of hemispheres whose outer dimensions resemble those of a nerve terminal or dendritic spine head (radius = 0.6 μm) being designated the target structure. The NO transduction machinery is present in all the hemispheres constituting the target structure and NO is generated in a 400 nm diameter disc at its base (Fig. 2 A). The fluxes of NO in and around the target can be calculated in a manner similar to that done for Ca2+ (Nowycky & Pinter, 1993; McHugh & Kenyon, 2004) and the associated cGMP generation and breakdown (by phosphodiesterase enzymes) within the target structure hemispheres quantified. To provide a minimal stimulus, the input NO profile is set to approximate to the profile of Ca2+ concentration in individual dendritic spine heads seen on activation of synaptic NMDA receptors (Sabatini et al. 2002), peaking after 40 ms and then declining over several 100 ms, and the peak amplitude is set (as above) to correspond to the formation of 40 NO molecules per second (inset, Fig. 2 B). The resulting diffusional spread of NO in the compartmental model matches the spread derived from solving analytically the diffusion of NO from a disc source (Fig. 2 B), giving credibility to the compartmental approach. If the NO‐receptive compartments possess 3.3 μm NO‐activated guanylyl cyclase, a value similar to that estimated for rat platelets and cerebellar astrocytes (Batchelor et al. 2010), a single ‘synaptic’ NO pulse results in a cGMP response that is effectively uniform within the target structure (because of cGMP diffusion) but peaks at only 10 nm cGMP (Fig. 2 C), a concentration that is probably too low to have biological activity bearing in mind that its affinity for downstream cGMP‐dependent protein kinases is in the 100 nm range (Francis & Corbin, 1994; Vaandrager et al. 2005). On the other hand, with trains of NO pulses to simulate tetanic synaptic stimulation, cGMP accumulates into its presumed active concentration range (Fig. 2 D). The computed frequency dependence of cGMP accumulation, saturating at about 20 Hz (Fig. 2 E), resembles the frequency dependence of smooth muscle relaxation observed experimentally in many tissues when nitrergic nerves are stimulated (Gibson, 2001). Similarly, orthograde NO‐mediated transmission between pairs of snail neurones is not effected by single presynaptic action potentials but becomes readily visible when bursts of action potentials at frequencies of 10 Hz or more are delivered (Park et al. 1998). The model predicts and explains these biological observations and, more generally, implies that NO in synapses is likely to function activity‐dependently, a property likely to be important for its participation in synaptic plasticity and other phenomena.
Figure 2. Compartmental analysis of synaptic NO signal transduction .
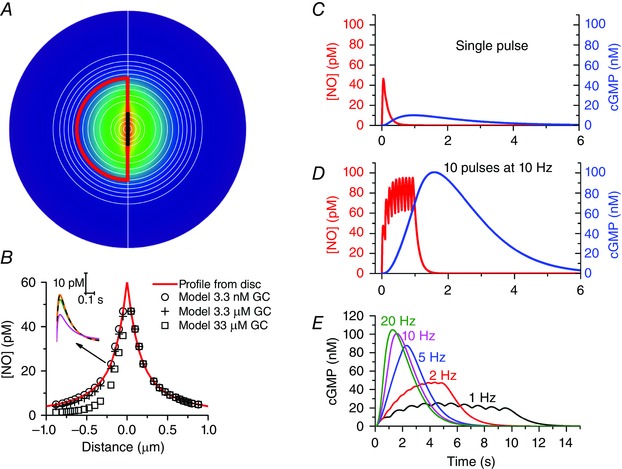
In the model A, which is an extension of one described previously (Wood et al. 2011), the synaptic space is divided into multiple concentric hemispheres, a group of which (outlined in red; radius = 0.6 μm) are designated the NO target structure. NO is generated in a 0.4 μm diameter zone (thick black line) at the base of the target structure and the colour‐coded NO profile from Fig. 1 B is shown centred on this zone. Distances correspond to the x‐axis in B. The target contains NO‐activated guanylyl cyclase (usually 3.3 μm) and cGMP‐stimulated phosphodiesterase‐5 having a maximal activity of 106 μm s−1 and a basal activity of 0.2% of this value. The kinetic schemes describing both these components were as published (Batchelor et al. 2010; Wood et al. 2011). NO is produced as a pulse having the shape depicted in the inset in B and peaking at a rate of 40 molecules s−1, with half the NO flowing each side of the emission zone through all available surfaces. The fluxes of NO in each hemisphere and of cGMP within the target hemispheres are calculated similarly to the method adopted for Ca2+ (Nowycky & Pinter, 1993; McHugh & Kenyon, 2004). The diffusion coefficients for NO and cGMP were those used previously (Wood et al. 2011) and NO was subject to first‐order decay (rate constant = 150 s−1) in each compartment. In B, the peak NO concentration in each hemisphere is plotted as a function of the concentration of NO‐activated guanylyl cyclase (GC) together with the concentrations obtained by solving analytically the equation for diffusion from a disc surface (Carslaw & Jaeger, 1986, eqn (10.3.10), p. 260) modified to include first‐order decay. The inset shows sample NO concentration profiles within the target structure, 0.2 μm from the emitting zone (orange line: profile from disc; dashed black line, green line and magenta line: profiles from model assuming NO‐activated guanylyl cyclase concentrations of 3.3 nm, 3.3 μm and 33 μm, respectively. The cGMP responses (blue lines; right‐hand ordinates) to either a single NO pulse (C) or to repeated pulses at 10 Hz (D) illustrate the likely activity dependence of NO‐mediated synaptic transmission effected largely by temporal summation at the level of guanylyl cyclase/cGMP. The sample NO traces (red lines; left‐hand ordinates) are from the central hemisphere. E shows the time‐courses of cGMP accumulation in response to 10 NO pulses delivered at different frequencies.
The model also addresses another outstanding question. It has been speculated that the low micromolar concentrations of NO‐activated guanylyl cyclase found in at least some cells may act as a sink for the (up to) million‐fold lower NO concentrations present in the vicinity (Batchelor et al. 2010). With the small size of the cellular compartments of interest here, however, NO gradients would be little affected by 3.3 μm NO‐activated guanylyl cyclase because diffusion is still rapid relative to the rate of NO binding to its receptors whereas a 10‐fold higher concentration of receptor more effectively hinders NO diffusion without affecting NO concentrations outside the target structure (Fig. 2 B).
Synaptic crosstalk through NO
The foregoing analysis indicates that NO could indeed transmit physiologically relevant signals in an activity‐dependent manner at the level of a single synapse. Indeed, at first glance, the limited spread of NO expected outside a synapse (Fig. 1) suggests that the principal way it functions would be with effective synapse specificity. This picture, however, ignores the anatomical realities of the arrangement of synapses in the brain. In the cerebral cortex, hippocampus and dentate gyrus, for example, the mean distance from one synapse to another in the neuropil is only about 0.5 μm and some synapses would even be located side‐by‐side (Rusakov et al. 1999; Merchan‐Perez et al. 2014). Perhaps even more pertinently, inhibitory GABAergic terminals synapsing onto pyramidal cell bodies in the hippocampus are less than 0.2 μm away from excitatory glutamatergic axo‐dendritic synapses (Merchan‐Perez et al. 2009). With these small distances, it must at least be plausible for NO generated in one synapse to signal to immediately neighbouring synapses. From the high levels of nNOS found in populations of GABAergic nerve terminals in the cerebral cortex and hippocampus (Aoki et al. 1997; Fuentealba et al. 2008), it is tempting to suggest that NO formed therein could signal inter‐synaptically to influence glutamatergic synapses and possibly contribute to plastic changes in those synapses.
Volume transmission by NO in the central nervous system
Because of its physicochemical nature, NO has long been a candidate volume transmitter (Agnati et al. 2010), i.e. one able to influence the activity of cells located within an active area of brain tissue, irrespective of synaptic connectivity. Several studies have invoked NO acting in such a way, for example, in insect brain (Ott et al. 2007), and in the mammalian optic nerve (Garthwaite et al. 2006), hypothalamus (Bellefontaine et al. 2014), auditory brainstem (Steinert et al. 2008) and, for targeting oligodendrocytes, the cerebellum (Garthwaite et al. 2015).
A recent example of presumed volume transmission is in the preoptic area of the hypothalamus in which the hormone leptin acts to stimulate NO formation from the abundant nNOS‐expressing neurones in this region, ultimately to cause release of luteinizing hormone from the pituitary gland which then acts on the gonads to affect fertility (Bellefontaine et al. 2014). The proposed mechanism here is that when nNOS activity is coordinated across the population of neurones, the NO concentration in between those neurones builds up to levels capable of stimulating neurones residing there to release gonadotrophin‐releasing hormone, which then acts on the pituitary gland. Realistic modelling of the size and distribution of the nNOS neurones in this brain area fully supports the proposed mechanism provided that two conditions are met: firstly, that the numbers of active nNOS neurones prior to leptin administration are half or fewer than those active in the presence of leptin, and secondly, that the rate of inactivation of NO is high enough to permit the NO‐responsive cells to discriminate between a sparsely activated and a more fully activated population of nNOS neurones. The computed NO inactivation rate allowing this mode of signalling is equivalent to a half‐life of 5 ms, a value neatly coinciding with the rate estimated for rat cerebellum (Hall & Garthwaite, 2006).
In addition to populations of large cells generating a volume signal, modelling of the coordinated activity in thin NO synthase‐expressing nerve fibres running in parallel or as a branched plexus also predict a build‐up of NO in the intervening space to an extent dependent on the fibre density (Philippides et al. 2005). The conditions under which NO is produced along the whole length of a nerve fibre, if any, remain to be defined. The plexus‐type anatomical model is probably more relevant to volume transmission involving endothelial NO synthase in the dense networks of capillaries, whose activity can be sustained by phosphorylation mechanisms and which, in the optic nerve, provides signals that target axons throughout the nerve to modify their excitability (Garthwaite et al. 2006).
Where axons express nNOS, it is more likely that NO synthesis is confined to nerve terminals where voltage‐sensitive Ca2+channels are found, and as occurs in the case of peripheral nitrergic nerves (Toda & Okamura, 2003; Toda & Herman, 2005). Is there scope for volume transmission when NO is formed in such small structures? The limited spread of NO beyond the boundaries of individual synapses (Fig. 1) might suggest otherwise but it can be tested by modelling arrays of these small sources at varying densities. With the sources taken as 0.4 μm diameter spheres each generating 40 NO molecules per second (as above) and located in a planar array, there is little build‐up of NO in between the sources when they are relatively well separated (4 μm apart), but as they get closer together, NO progressively accumulates in the intervening space (Fig. 3 A–C), even when it is subject to rapid inactivation (half‐life = 5 ms). When the sources are 2 μm apart, this space contains about half the peak NO concentration found at the source itself and, in a 3‐dimensional array with the same source separation, the build‐up is more pronounced (Fig. 3 C). In fact, this 3‐dimensional array approximates to a continuum of NO sources (Wood & Garthwaite, 1994) which, at the corresponding net rate of NO production per unit volume (10 nm s−1) and an NO inactivation rate constant of 150 s−1 (half‐life = 4.6 ms), predicts (from the ratio of the two) a steady‐state tissue NO concentration of 67 pm, a value close to the average found in the array (Fig. 3 C). Hence, should a field of bouton‐like structures (pre‐ or postsynaptic) simultaneously generate NO, and be close enough together, a volume‐type NO signal is the expected result. This scenario may explain how NO generated in cerebellar neurones signals to astrocytes (de Vente et al. 1990) or to oligodendrocytes, potentially to affect the myelination of local afferent and efferent axons (Garthwaite et al. 2015).
Figure 3. Conditions for volume transmission with multiple small NO emitters .
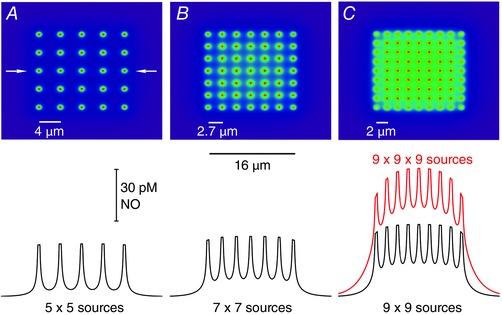
Spheres of radius = 0.2 μm and numbering 25 (A), 49 (B) and 81 (C) are arranged in 2‐dimensional arrays within a fixed area (16 μm × 16 μm). The spheres generate NO at their surfaces at the rate of 40 molecules s−1 and the resultant NO concentrations throughout the array at steady state are calculated as described (Bellefontaine et al. 2014). The upper panels illustrate the distributions of NO within and outside the area of emitters and the traces below (black lines) are sample concentrations taken through the centre of each array (marked by arrows in A, upper panel). The red trace (C) extends the array into 3 dimensions.
Concluding remarks
An initial attempt to gain understanding of the principles of NO signalling in the brain concluded that, when produced at a single point source, NO could diffuse in active concentrations within a sphere of 200 μm diameter, thereby influencing up to 2 million synapses (Wood & Garthwaite, 1994), a conclusion that is starkly different from the one made here, which is that NO produced at a single synapse is only likely to function very locally, specifically within submicrometre dimensions. The discrepancy arises from the previous reliance on the only estimate then available of the NO concentration found outside an NO‐producing cell, in this case an endothelial cell, which was reported using an electrochemical probe as 1 μm (Malinski & Taha, 1992). In the intervening years, such probes have given improbably large variations in estimates of NO concentration, ranging across six orders of magnitude (Hall & Garthwaite, 2009). The subnanomolar NO concentrations considered here, on the other hand, cohere quantitatively with the concentrations that NO receptors are tuned to detect, the concentrations expected from nNOS localization and enzymology, and the endogenous NO concentrations reported using highly sensitive NO‐detector cells when nNOS in brain slices is activated (Wood et al. 2011). Accordingly, the rate of NO formation at a single source assumed here (7 × 10−23 mol s−1) is more than five orders of magnitude lower than the one used initially (Wood & Garthwaite, 1994).
With the focus of synaptic NO signalling now narrowed down to the level of the single synapse, and with the elaboration of quantitative descriptions of NO signal transduction through guanylyl cyclase‐coupled receptors in hand (Batchelor et al. 2010; Garthwaite, 2010), it has become possible to model NO signalling between closely apposed partners (whether synaptic or not). The picture becomes one in which pre‐ or postsynaptically generated NO is likely to function in an activity‐dependent manner, a property that arises from the elementary NO pulse being of low amplitude (pm) and duration (sub‐second) balanced by NO receptor activity and cGMP in the target structure being able to undergo temporal summation, thereby allowing cGMP to accumulate into the submicromolar levels needed to engage downstream pathways.
At the same time, and as envisaged earlier (Wood & Garthwaite, 1994), NO could also subserve a more diffuse type of transmission in which NO from multiple sources in a volume of tissue, be they whole cells, blood capillaries or discrete pre‐ or postsynaptic sources, is able to summate spatially to provide a local cloud of NO able to engage recipient cells irrespective of anatomical connectivity. This diffuse type of signalling has been highlighted as a plausible mechanism for establishing synaptic connectivity during development (Gally et al. 1990; Montague & Sejnowski, 1994) and, more generally, allows NO to target structures located distantly from the individual sources, such as other neurones, astrocytes, oligodendrocytes and blood vessels, presumably to transmit information on the overall levels of neuronal activity taking place within a given brain subregion.
With the continuing development of fluorescent biosensors responsive to physiological levels of cGMP (Gorshkov & Zhang, 2014) it is becoming possible to visualize NO signalling through its receptors in real time and even in small synaptic dimensions (Bhargava et al. 2013). Being genetically encoded, these biosensors could be expressed in specific cell types in the brain, or in specific subcellular compartments, and are likely to prove invaluable in putting theory to the test and so further advance our understanding of NO‐mediated transmission.
Additional information
Competing interests
None declared.
Funding
This work is supported by The Wellcome Trust, grant number 081512/Z/06/Z.
Biography
Armed with a BSc in Biochemistry, John Garthwaite undertook research leading to a PhD in Pharmacology under the supervision of Prof. David A. Brown at the School of Pharmacy, London. Afterwards, he did postdoctoral research in the MRC Developmental Neurobiology Unit before becoming a Lecturer in Physiology in the University of Liverpool (1980), ultimately holding a personal chair there. In 1992 he was appointed Head of Neuroscience Research at the Wellcome Research Laboratories and in 1996 became a founder member of The Wolfson Institute for Biomedical Research in University College London. He is interested in the processing and storage of information in the brain, with particular emphasis on the role of nitric oxide.

This review was presented at the symposium “Localised intracellular signalling in neurons”, which took place at Physiology 2014 in London, UK, 30 June – 2 July 2014.
References
- Agnati LF, Guidolin D, Guescini M, Genedani S & Fuxe K (2010). Understanding wiring and volume transmission. Brain Res Rev 64, 137–159. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S, Lonart G & Johnson KM (1994). The role of P‐type calcium channels in the depolarization‐induced activation of nitric oxide synthase in frontal cortex. J Neurochem 62, 400–403. [DOI] [PubMed] [Google Scholar]
- Aoki C, Rhee J, Lubin M & Dawson TM (1997). NMDA‐R1 subunit of the cerebral cortex co‐localizes with neuronal nitric oxide synthase at pre‐ and postsynaptic sites and in spines. Brain Res 750, 25–40. [DOI] [PubMed] [Google Scholar]
- Bartus K, Pigott B & Garthwaite J (2013). Cellular targets of nitric oxide in the hippocampus. PLoS One 8, e57292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Bartus K, Reynell C, Constantinou S, Halvey EJ, Held KF, Dostmann WR, Vernon J & Garthwaite J (2010). Exquisite sensitivity to subsecond, picomolar nitric oxide transients conferred on cells by guanylyl cyclase‐coupled receptors. Proc Natl Acad Sci USA 107, 22060–22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy TC, Griffiths C & Garthwaite J (2002). Differential sensitivity of guanylyl cyclase and mitochondrial respiration to nitric oxide measured using clamped concentrations. J Biol Chem 277, 31801–31807. [DOI] [PubMed] [Google Scholar]
- Bellefontaine N, Chachlaki K, Parkash J, Vanacker C, Colledge W, d'Anglemont de Tassigny X, Garthwaite J, Bouret SG & Prevot V (2014). Leptin‐dependent neuronal NO signaling in the preoptic hypothalamus facilitates reproduction. J Clin Invest 124, 2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava Y, Hampden‐Smith K, Chachlaki K, Wood KC, Vernon J, Allerston CK, Batchelor AM & Garthwaite J (2013). Improved genetically‐encoded, FlincG‐type fluorescent biosensors for neural cGMP imaging. Front Mol Neurosci 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM & Snyder SH (1991). Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron 7, 615–624. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC & Bredt DS (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD‐95 and α1‐syntrophin mediated by PDZ domains. Cell 84, 757–767. [DOI] [PubMed] [Google Scholar]
- Burette A, Zabel U, Weinberg RJ, Schmidt HH & Valtschanoff JG (2002). Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J Neurosci 22, 8961–8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carslaw HS & Jaeger JC (1986). Conduction of Heat in Solids, 2nd edn Clarendon Press, Oxford. [Google Scholar]
- d'Anglemont de Tassigny X, Campagne C, Dehouck B, Leroy D, Holstein GR, Beauvillain JC, Buee‐Scherrer V & Prevot V (2007). Coupling of neuronal nitric oxide synthase to NMDA receptors via postsynaptic density‐95 depends on estrogen and contributes to the central control of adult female reproduction. J Neurosci 27, 6103–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vente J (2004). cGMP: a second messenger for acetylcholine in the brain? Neurochem Int 45, 799–812. [DOI] [PubMed] [Google Scholar]
- de Vente J, Bol JGJM, Berkelmans HS, Schipper J & Steinbusch HW (1990). Immunocytochemistry of cGMP in the cerebellum of the immature, adult, and aged rat: the involvement of nitric oxide. A micropharmacological study. Eur J Neurosci 2, 845–862. [DOI] [PubMed] [Google Scholar]
- de Vente J, Hopkins DA, Markerink‐Van Ittersum M, Emson PC, Schmidt HH & Steinbusch HW (1998). Distribution of nitric oxide synthase and nitric oxide‐receptive, cyclic GMP‐producing structures in the rat brain. Neuroscience 87, 207–241. [DOI] [PubMed] [Google Scholar]
- de Vente J & Steinbusch HW (1992). On the stimulation of soluble and particulate guanylate cyclase in the rat brain and the involvement of nitric oxide as studied by cGMP immunocytochemistry. Acta Histochem 92, 13–38. [DOI] [PubMed] [Google Scholar]
- Francis SH & Corbin JD (1994). Structure and function of cyclic nucleotide‐dependent protein kinases. Annu Rev Physiol 56, 237–272. [DOI] [PubMed] [Google Scholar]
- Friebe A & Koesling D (2009). The function of NO‐sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide 21, 149–156. [DOI] [PubMed] [Google Scholar]
- Fuentealba P, Begum R, Capogna M, Jinno S, Marton LF, Csicsvari J, Thomson A, Somogyi P & Klausberger T (2008). Ivy cells: a population of nitric‐oxide‐producing, slow‐spiking GABAergic neurons and their involvement in hippocampal network activity. Neuron 57, 917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF (1999). Endothelium‐derived relaxing factor: discovery, early studies, and identification as nitric oxide. Biosci Rep 19, 235–251. [DOI] [PubMed] [Google Scholar]
- Gally JA, Montague PR, Reeke GN Jr & Edelman GM (1990). The NO hypothesis: possible effects of a short‐lived, rapidly diffusible signal in the development and function of the nervous system. Proc Natl Acad Sci USA 87, 3547–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite G, Bartus K, Malcolm D, Goodwin DA, Kollb‐Sielecka M, Dooldeniya C & Garthwaite J (2006). Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci 26, 7730–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite G, Hampden‐Smith K, Wilson GW, Goodwin DA & Garthwaite J (2015). Nitric oxide targets oligodendrocytes and promotes their morphological differentiation. Glia 63, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J (2008). Concepts of neural nitric oxide‐mediated transmission. Eur J Neurosci 27, 2783–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J (2010). New insight into the functioning of nitric oxide‐receptive guanylyl cyclase: physiological and pharmacological implications. Mol Cell Biochem 334, 221–232. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL & Chess Williams R (1988). Endothelium‐derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336, 385–388. [DOI] [PubMed] [Google Scholar]
- Gibson A (2001). Phosphodiesterase 5 inhibitors and nitrergic transmission – from zaprinast to sildenafil. Eur J Pharmacol 411, 1–10. [DOI] [PubMed] [Google Scholar]
- Gorshkov K & Zhang J (2014). Visualization of cyclic nucleotide dynamics in neurons. Front Cell Neurosci 8, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths C, Wykes V, Bellamy TC & Garthwaite J (2003). A new and simple method for delivering clamped nitric oxide concentrations in the physiological range: application to activation of guanylyl cyclase‐coupled nitric oxide receptors. Mol Pharmacol 64, 1349–1356. [DOI] [PubMed] [Google Scholar]
- Hall CN & Garthwaite J (2006). Inactivation of nitric oxide by rat cerebellar slices. J Physiol 577, 549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN & Garthwaite J (2009). What is the real physiological NO concentration in vivo? Nitric Oxide 21, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Keynes RG & Garthwaite J (2009). Cytochrome P450 oxidoreductase participates in nitric oxide consumption by rat brain. Biochem J 419, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvey EJ, Vernon J, Roy B & Garthwaite J (2009). Mechanisms of activity‐dependent plasticity in cellular nitric oxide‐cGMP signaling. J Biol Chem 284, 25630–25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Dranka BP, Bailey SM, Lancaster JR Jr & Darley‐Usmar VM (2010). What part of NO don't you understand? Some answers to the cardinal questions in nitric oxide biology. J Biol Chem 285, 19699–19704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster JR Jr (1997). A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide 1, 18–30. [DOI] [PubMed] [Google Scholar]
- Ma X, Sayed N, Beuve A & van den Akker F (2007). NO and CO differentially activate soluble guanylyl cyclase via a heme pivot‐bend mechanism. EMBO J 26, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh JM & Kenyon JL (2004). An Excel‐based model of Ca2+ diffusion and fura 2 measurements in a spherical cell. Am J Physiol Cell Physiol 286, C342–C348. [DOI] [PubMed] [Google Scholar]
- Malinski T & Taha Z (1992). Nitric oxide release from a single cell measured in situ by a porphyrinic‐based microsensor. Nature 358, 676–678. [DOI] [PubMed] [Google Scholar]
- Merchan‐Perez A, Rodriguez JR, Gonzalez S, Robles V, Defelipe J, Larranaga P & Bielza C (2014). Three‐dimensional spatial distribution of synapses in the neocortex: a dual‐beam electron microscopy study. Cereb Cortex 24, 1579–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchan‐Perez A, Rodriguez JR, Ribak CE & Defelipe J (2009). Proximity of excitatory and inhibitory axon terminals adjacent to pyramidal cell bodies provides a putative basis for nonsynaptic interactions. Proc Natl Acad Sci USA 106, 9878–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller M, Botti H, Batthyany C, Rubbo H, Radi R & Denicola A (2005). Direct measurement of nitric oxide and oxygen partitioning into liposomes and low density lipoprotein. J Biol Chem 280, 8850–8854. [DOI] [PubMed] [Google Scholar]
- Montague PR & Sejnowski TJ (1994). The predictive brain: temporal coincidence and temporal order in synaptic learning mechanisms. Learn Mem 1, 1–33. [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG & Svoboda K (2004). The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci 24, 2054–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC & Pinter MJ (1993). Time courses of calcium and calcium‐bound buffers following calcium influx in a model cell. Biophys J 64, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott SR, Philippides A, Elphick MR & O'Shea M (2007). Enhanced fidelity of diffusive nitric oxide signalling by the spatial segregation of source and target neurones in the memory centre of an insect brain. Eur J Neurosci 25, 181–190. [DOI] [PubMed] [Google Scholar]
- Park JH, Straub VA & O'Shea M (1998). Anterograde signaling by nitric oxide: characterization and in vitro reconstitution of an identified nitrergic synapse. J Neurosci 18, 5463–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippides A, Ott SR, Husbands P, Lovick TA & O'Shea M (2005). Modeling cooperative volume signaling in a plexus of nitric‐oxide‐synthase‐expressing neurons. J Neurosci 25, 6520–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Juarez F, Aguirre E & Cadenas S (2007). Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration. Biochem J 405, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Halvey EJ & Garthwaite J (2008). An enzyme‐linked receptor mechanism for nitric oxide‐activated guanylyl cyclase. J Biol Chem 283, 18841–18851. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullman DM & Stewart MG (1999). Hippocampal synapses: do they talk to their neighbours? Trends Neurosci 22, 382–388. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG & Svoboda K (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–452. [DOI] [PubMed] [Google Scholar]
- Salerno JC (2008). Neuronal nitric oxide synthase: prototype for pulsed enzymology. FEBS Lett 582, 1395–1399. [DOI] [PubMed] [Google Scholar]
- Santos RM, Lourenco CF, Pomerleau F, Huettl P, Gerhardt GA, Laranjinha J & Barbosa RM (2011). Brain nitric oxide inactivation is governed by the vasculature. Antioxid Redox Signal 14, 1011–1021. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF & Tymianski M (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD‐95 protein. Science 284, 1845–1848. [DOI] [PubMed] [Google Scholar]
- Silver RA, Traynelis SF & Cull‐Candy SG (1992). Rapid‐time‐course miniature and evoked excitatory currents at cerebellar synapses in situ . Nature 355, 163–166. [DOI] [PubMed] [Google Scholar]
- Southam E, East SJ & Garthwaite J (1991). Excitatory amino acid receptors coupled to the nitric oxide/cyclic GMP pathway in rat cerebellum during development. J Neurochem 56, 2072–2081. [DOI] [PubMed] [Google Scholar]
- Southam E & Garthwaite J (1993). The nitric oxide‐cyclic GMP signalling pathway in rat brain. Neuropharmacology 32, 1267–1277. [DOI] [PubMed] [Google Scholar]
- Steinert JR, Chernova T & Forsythe ID (2010). Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16, 435–452. [DOI] [PubMed] [Google Scholar]
- Steinert JR, Kopp‐Scheinpflug C, Baker C, Challiss RA, Mistry R, Haustein MD, Griffin SJ, Tong H, Graham BP & Forsythe ID (2008). Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron 60, 642–656. [DOI] [PubMed] [Google Scholar]
- Szabadits E, Cserep C, Ludanyi A, Katona I, Gracia‐Llanes J, Freund TF & Nyiri G (2007). Hippocampal GABAergic synapses possess the molecular machinery for retrograde nitric oxide signaling. J Neurosci 27, 8101–8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadits E, Cserep C, Szonyi A, Fukazawa Y, Shigemoto R, Watanabe M, Itohara S, Freund TF & Nyiri G (2011). NMDA receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J Neurosci 31, 5893–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda N & Herman AG (2005). Gastrointestinal function regulation by nitrergic efferent nerves. Pharmacol Rev 57, 315–338. [DOI] [PubMed] [Google Scholar]
- Toda N & Okamura T (2003). The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev 55, 271–324. [DOI] [PubMed] [Google Scholar]
- Tsai AL, Berka V, Martin E & Olson JS (2012). A ‘sliding scale rule’ for selectivity among NO, CO, and O2 by heme protein sensors. Biochemistry 51, 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaandrager AB, Hogema BM & de Jonge HR (2005). Molecular properties and biological functions of cGMP‐dependent protein kinase II. Front Biosci 10, 2150–2164. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG & Weinberg RJ (2001). Laminar organization of the NMDA receptor complex within the postsynaptic density. J Neurosci 21, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SR & Kimura H (1992). Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience 46, 755–784. [DOI] [PubMed] [Google Scholar]
- Waldman SA & Murad F (1987). Cyclic GMP synthesis and function. Pharmacol Rev 39, 163–196. [PubMed] [Google Scholar]
- Wood J & Garthwaite J (1994). Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signalling and its pharmacological properties. Neuropharmacology 33, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Wood KC, Batchelor AM, Bartus K, Harris KL, Garthwaite G, Vernon J & Garthwaite J (2011). Picomolar nitric oxide signals from central neurons recorded using ultrasensitive detector cells. J Biol Chem 286, 43172–43181. [DOI] [PMC free article] [PubMed] [Google Scholar]