

Key points
Excessive production of reactive oxygen species (ROS) is implicated in many central nervous system disorders; however, the physiological role of ROS in spinal ventral horn (VH) neurons remains poorly understood.
We investigated how pathological levels of H2O2, an abundant ROS, regulate synaptic transmission in VH neurons of rats using a whole‐cell patch clamp approach.
H2O2 increased the release of glutamate and GABA from presynaptic terminals.
The increase in glutamate release involved N‐type voltage‐gated calcium channels (VGCCs), ryanodine receptors (RyRs), and inositol trisphosphate receptors (IP3Rs); the increase in GABA release, which inhibited glutamatergic transmission, involved IP3R.
Inhibiting N‐type VGCCs and RyRs attenuates excitotoxicity resulting from increased glutamatergic activity while preserving the neuroprotective effects of GABA, and may represent a novel strategy for treating H2O2‐induced motor neuron disorders resulting from trauma or ischaemia–reperfusion injury.
Abstract
Excessive production of reactive oxygen species (ROS) is a critical component of the cellular and molecular pathophysiology of many central nervous system (CNS) disorders, including trauma, ischaemia–reperfusion injury, and neurodegenerative diseases. Hydrogen peroxide (H2O2), an abundant ROS, modulates synaptic transmission and contributes to neuronal damage in the CNS; however, the pathophysiological role of H2O2 in spinal cord ventral horn (VH) neurons remains poorly understood, despite reports that these neurons are highly vulnerable to oxidative stress and ischaemia. This was investigated in the present study using a whole‐cell patch clamp approach in rats. We found that exogenous application of H2O2 increased the release of glutamate from excitatory presynaptic terminals and γ‐aminobutyric acid (GABA) from inhibitory presynaptic terminals. The increase of glutamate release was induced in part by an increase in Ca2+ influx through N‐type voltage‐gated calcium channels (VGCCs) as well as by ryanodine receptor (RyR)‐ and inositol trisphosphate receptor‐mediated Ca2+ release from the endoplasmic reticulum (ER). In inhibitory presynaptic neurons, increased IP3R‐mediated Ca2+ release from the ER increased GABAergic transmission, which served to rescue VH neurons from excessive release of glutamate from presynaptic terminals. These findings indicate that inhibiting N‐type VGCCs or RyRs may attenuate excitotoxicity resulting from increased glutamatergic activity while preserving the neuroprotective effects of GABA, and may therefore represent a novel and targeted strategy for preventing and treating H2O2‐induced motor neuron disorders.
Key points
Excessive production of reactive oxygen species (ROS) is implicated in many central nervous system disorders; however, the physiological role of ROS in spinal ventral horn (VH) neurons remains poorly understood.
We investigated how pathological levels of H2O2, an abundant ROS, regulate synaptic transmission in VH neurons of rats using a whole‐cell patch clamp approach.
H2O2 increased the release of glutamate and GABA from presynaptic terminals.
The increase in glutamate release involved N‐type voltage‐gated calcium channels (VGCCs), ryanodine receptors (RyRs), and inositol trisphosphate receptors (IP3Rs); the increase in GABA release, which inhibited glutamatergic transmission, involved IP3R.
Inhibiting N‐type VGCCs and RyRs attenuates excitotoxicity resulting from increased glutamatergic activity while preserving the neuroprotective effects of GABA, and may represent a novel strategy for treating H2O2‐induced motor neuron disorders resulting from trauma or ischaemia–reperfusion injury.
Abbreviations
- ADPR
adenosine diphosphate ribose
- 2‐APB
2‐aminoethoxydiphenil borate
- Bic
bicuculline
- BPB
4‐bromophenacyl bromide
- CaN
calcineurin
- CICR
Ca2+‐induced Ca2+‐release
- CSA
cyclosporine A
- Dant
dantrolene sodium
- ER
endoplasmic reticulum
- IP3R
inositol trisphosphate receptor
- ω‐Aga
ω‐agatoxin IVA
- MPT
mitochondrial permeability transition pore
- ω‐Ctx
ω‐conotoxin GVIA
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SCI
spinal cord injury
- Stry
strychnine
- VH
ventral horn
- VGCC
voltage‐gated calcium channel
Introduction
Reactive oxygen species (ROS) are generated naturally during cell metabolism, and their levels are tightly regulated by the intracellular antioxidant network in the central nervous system (CNS) and other organs (Giorgio et al. 2007; Rice, 2011). However, excessive ROS production is deleterious since it contributes to neuronal damage and degeneration, a hallmark of many pathological conditions of the CNS including trauma, ischaemia–reperfusion injury, and neurodegenerative diseases like amyotrophic lateral sclerosis (ALS) (Halliwell, 1992; Coyle & Puttfarcken, 1993).
Hydrogen peroxide (H2O2) is a diffusible and relatively stable ROS that is present in low micromolar concentrations under physiological conditions (Lei et al. 1998). H2O2 levels are significantly increased in ischaemia–reperfusion lesions of the brain (Hyslop et al. 1995) and in spinal cord injury (SCI) (Liu et al. 1999). Although the mechanisms of neuronal damage induced by H2O2 are not entirely understood, it is known that elevated H2O2 levels result in elevated glutamate levels (Mailly et al. 1999) and activation of the glutamate receptors including the N‐methyl‐d‐aspartate (NMDA) receptor (Mailly et al. 1999; Avshalumov & Rice, 2002). The consequent accumulation of high levels of intracellular Ca2+ leads to excitotoxicity, which is characterized by neuronal damage and death (Pellegrini‐Giampietro et al. 1990; Roettger & Lipton, 1996; Shaw & Ince, 1997). Attenuating excitotoxicity is therefore a potential approach for treating CNS injuries and diseases. For instance, riluzole, which reduces overall excitability and neurotransmitter release by inhibiting presynaptic voltage‐dependent sodium channels (Leach et al. 1986; Doble, 1996), is indicated for use in ALS (Shaw & Ince, 1997) and is being investigated for its therapeutic potential in SCI (Wilson & Fehlings, 2014). However, this class of drugs inhibits not only the release of glutamate but also that of other neurotransmitters including γ‐aminobutyric acid (GABA) (Leach et al. 1986; Martin et al. 1993; Jehle et al. 2000). Since GABA provides neuroprotection in cerebral ischaemia via its well‐established role in neural inhibition (Schwartz‐Bloom & Sah, 2001), selectively inhibiting glutamate release is a better strategy for neuroprotection against excitotoxicity. To date, there has been no study on the effect of H2O2 on synaptic transmission in spinal ventral horn (VH) neurons, despite the finding that these neurons are highly vulnerable to oxidative stress (Carri et al. 2003) and ischaemia (Sakurai et al. 1997; Nohda et al. 2007).
The present study investigated the mechanisms by which pathological levels of H2O2 affect synaptic transmission in VH neurons using a whole‐cell patch clamp approach. We demonstrate that elevated H2O2 levels increased glutamate and GABA release from excitatory and inhibitory presynaptic terminals, respectively. The former was partly due to increased presynaptic Ca2+ influx through N‐type voltage‐gated calcium channels (VGCCs) and Ca2+ release from the endoplasmic reticulum (ER) mediated by the ryanodine and inositol trisphosphate receptors (RyR and IP3R, respectively), whereas increased GABAergic transmission was due to IP3R‐mediated Ca2+ release from the ER, which inhibited excessive glutamate release from presynaptic terminals.
Methods
Study approval
All experimental procedures involving the use of animals were approved by the Animal Care and Use Committee at Niigata University Graduate School of Medical and Dental Sciences (Niigata, Japan).
Preparation of spinal cord slices
Neonatal Wistar rats of either sex (131 rats, 7–15 days old) were anaesthetized with urethane (1.5 g kg−1 by intraperitoneal injection). A dorsal laminectomy was performed and the lumbosacral segment (L1–S3) of the spinal cord was removed (Honda et al. 2011). The rats were immediately killed by exsanguination. Spinal cord specimens were placed in pre‐oxygenated ice‐cold (2–4°C) artificial cerebrospinal fluid (ACSF) containing 117 mm NaCl, 3.6 mm KCl, 2.5 mm CaCl2, 1.2 mm MgCl2, 1.2 mm NaH2PO4, 25 mm NaHCO3, and 11.5 mm d‐glucose. After cutting the ventral and dorsal roots, the pia‐arachnoid membrane was removed, and the spinal cord was mounted on the metal stage of a microslicer (Linear Slicer PRO 7; Dosaka, Kyoto, Japan) and cut into 500 μm transverse slices. Each of these was transferred to a recording chamber and placed on the stage of an upright microscope equipped with an infrared–differential interference contrast (IR‐DIC) system (E600FN; Nikon, Tokyo, Japan). The slice was fixed with an anchor and superfused at 5–6 ml min−1 with ACSF equilibrated with a gas mixture of 95% O2 and 5% CO2 (pH 7.4), with the temperature maintained at 36 ± 0.5°C using a temperature controller (TC‐324B; Warner Instruments, Hamden, CT, USA). Slices were used for recording after a recovery period of ≥ 40 min.
Patch clamp recordings from spinal VH neurons
Laminar regions were identified under low magnification (5× objective) and individual neurons were identified using the 40× objective lens of an IR‐DIC microscope and monitored with a charge‐coupled device camera (C2400‐79H; Hamamatsu Photonics, Hamamatsu, Japan). Whole‐cell patch clamp recordings were made from the large VH neurons of Rexed lamina IX (size: > 25 μm), which were most frequently seen in the ventrolateral and ventromedial areas. A somal size > 20 μm was used as the cut‐off in past studies of postnatal rat lumbar motor neurons (Takahashi, 1990); however, large interneurons (> 20 μm) have been detected in the ventral half of the rat lumbar spinal cord (Thurbon et al. 1998 a,b). Therefore, putative motor neurons were identified as the largest cells in the VH (somal diameter > 25 μm). After the whole‐cell configuration was established, neurons were held in voltage‐clamp at −70 mV to record excitatory postsynaptic currents (EPSCs) or at 0 mV for recording inhibitory postsynaptic currents (IPSCs). In spinal VH neurons, the reversal potentials of EPSCs are 0 mV, and those of IPSCs are −70 mV (Aoyama et al. 2010; Honda et al. 2012; Yamamoto et al. 2012). Therefore, EPSCs in the membrane current trace were specifically recorded as downward deflections at −70 mV, while IPSCs were specifically recorded as upward deflections at 0 mV. Cell input resistance (R in) was calculated by measuring the current response to 5 mV hyperpolarizing steps from a −70 mV holding potential (Pagnotta et al. 2005; Quitadamo et al. 2005). To investigate the effects of H2O2 on presynaptic terminals of fibres synaptically connected to the VH neuron being recorded, all patch clamp recordings were made in the presence of the Na+ channel blocker tetrodotoxin (TTX; 1μm). Since conduction of action potentials is blocked by TTX, the effects of H2O2 on presynaptic terminals could be isolated. In the presence of TTX, postsynaptic responses to spontaneously released neurotransmitters were detected as miniature (m) EPSCs and mIPSCs. Whole‐cell patch clamp pipettes were constructed from borosilicate glass capillaries (1.5 mm outer diameter; World Precision Instruments, Sarasota, FL, USA). The resistance of a typical pipette was 4–8 MΩ when filled with internal solution composed of 110 mm Cs2SO4, 5 mm tetraethylammonium, 0.5 mm CaCl2, 2 mm MgCl2, 5 mm EGTA, 5 mm Hepes and 5 mm ATP‐Mg.
Signals were amplified with an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA), filtered at 2 kHz, and digitized at 5 kHz. Data were stored and analysed using the pCLAMP 10.3 data acquisition program (Molecular Devices).
Drug application
The following pharmacological agents were purchased from Wako (Osaka, Japan): H2O2 (1 mm), TTX (1 μm), CGP35348 (20 μm), dantrolene sodium (Dant; 10 μm), 2‐aminoethoxydiphenyl borate (2‐APB; 200 μm), flufenamic acid (FFA; 100 μm), 4‐bromophenacyl bromide (BPB; 20 μm), cyclosporine A (CSA; 10 μm), nifedipine (10 μm), ω‐conotoxin GVIA (ω‐Ctx; 0.5 μm), and SNX‐482 (0.1 μm). Bicuculline (Bic; 20 μm), strychnine (Stry; 2 μm), and ω‐agatoxin IVA (ω‐Aga; 0.2 μm) were purchased from Sigma‐Aldrich (St Louis, MO, USA). H2O2 was freshly dissolved in ACSF immediately before the experiments. TTX, Stry, CGP35348, ω‐Aga and ω‐Ctx were first dissolved in distilled water at 1000 times the working concentration. Bic, Dant, 2‐APB, BPB, CSA, nifedipine and SNX‐482 were dissolved in dimethyl sulfoxide at a 1000 times the concentration for storage, and then diluted to the working concentration in ACSF immediately before use. The osmotic pressure of nominally Ca2+‐free, high‐Mg2+ (5 mm) ACSF was adjusted by lowering the Na+ concentration. Drugs were applied to the whole slice by perfusion via a three‐way stopcock without changing the perfusion rate or temperature. The volume of the recording chamber was approximately 1.0 ml. The drugs reached the recording chamber within 15 s of opening the stopcock and were completely washed out within 90 s of its being closed. Both the test inhibitor and TTX were superfused at least 4 min before starting H2O2 superfusion. Since the focus of the present study was on the acute toxicity of ROS, H2O2 was applied for 5 min at a concentration of 1 mm, which was previously shown to induce neurotoxicity in spinal neurons (Taccola et al. 2008) and hypoglossal motoneurons (Nani et al. 2010) in vitro. Slices were discarded after each H2O2 application experiment.
Data analysis
Changes in frequency of postsynaptic currents following bath application of drugs were analysed by determining the time course of postsynaptic current frequency and amplitude before and after drug application with a time bin of 30 s using the Mini Analysis program 6.0.7 (Synaptosoft, Decatur, GA, USA). The threshold for detection of postsynaptic currents was set at 2 times the root mean square of the background noise and each automatically identified event was visually confirmed to be free of artefacts.
Statistics
All numerical data are expressed as the mean ± SEM. Student's t test (paired or unpaired) or one‐way analysis of variance (ANOVA) with a Bonferroni post hoc test was used to determine the statistical significance between means. The Kolmogorov–Smirnov test was used to compare cumulative distributions of postsynaptic current parameters in the absence and presence of the drugs. P < 0.05 was considered significant for all tests. For electrophysiological data, the number of samples n refers to the number of neurons recorded.
Results
H2O2 acts presynaptically to modulate spontaneous glutamate release in VH neurons
To determine whether H2O2 directly modulates excitatory neuronal activity, we measured mEPSCs under whole‐cell patch clamp at a holding potential of −70 mV in the presence of TTX. The mEPSCs had an average frequency of 9.5 ± 1.0 Hz and an average amplitude of 11.3 ± 0.5 pA (n = 11). H2O2 superfusion for 5 min produced biphasic changes in the frequency from baseline, specifically, an increase (maximum: 163.1 ± 10.9 % of the control) followed by a depression (51.7 ± 6.5% of the control) that persisted for at least 10 min following washout (Fig. 1 A–C). The maximal increase in frequency was observed 2–5.5 min after the start of H2O2 superfusion (mean = 3.6 ± 0.3 min) (Fig. 1 B). In contrast, mEPSC amplitude was unaffected by H2O2 treatment (Fig. 1 C, right panel). Figure 1 D shows that H2O2 increased the proportion of mEPSCs with a shorter inter‐event interval relative to the control (Kolmogorov–Smirnov test), and those with longer inter‐event intervals were observed for at least 10 min after the washout (left panel). However, H2O2 had no effect on the cumulative distribution of mEPSC amplitude over the recording period (Fig. 1 D, right panel).
Figure 1. H2O2 induces an increase followed by depression in mEPSC frequency without affecting amplitude .
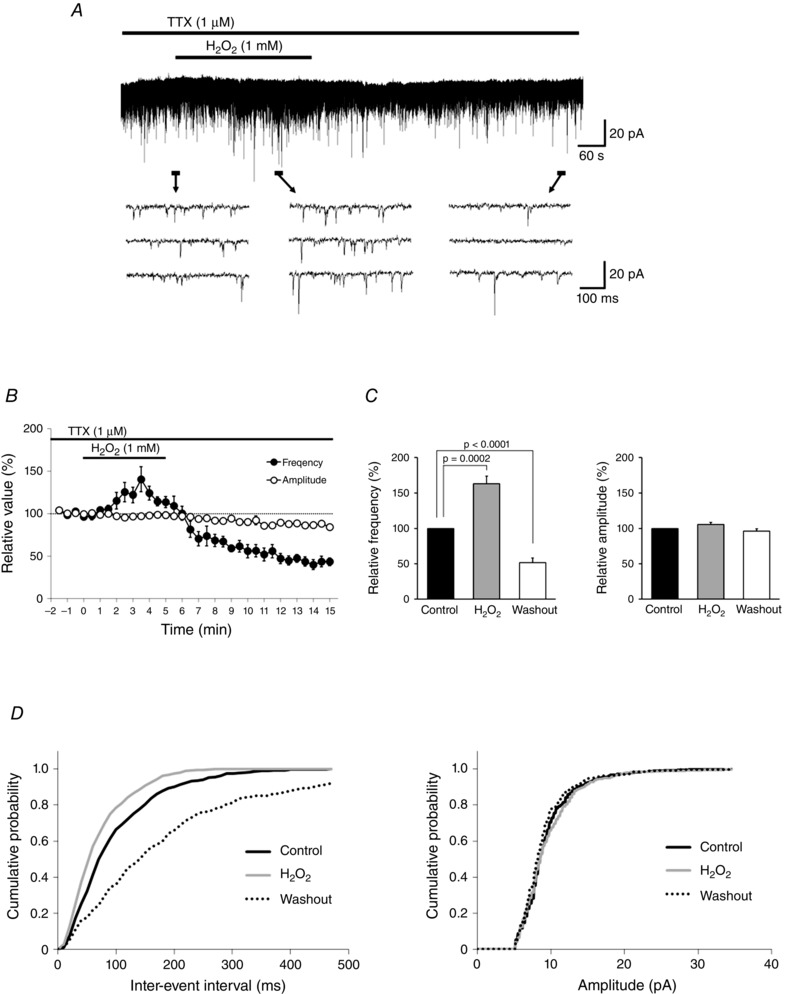
A, continuous chart recording of mEPSCs before, during and after H2O2 (1 mm) superfusion. B, average frequency (filled circles) and amplitude (open circles) of mEPSCs in the presence of H2O2 relative to the control, shown as a function of time (mean ± SEM, n = 11). C, summary of mEPSC frequency (left) and amplitude (right). Values are shown as percentage change in maximal values relative to the control during H2O2 superfusion and after washout (mean ± SEM, n = 11 each). P values were determined by a paired t test. D, cumulative distributions of the mEPSC inter‐event interval (left) and amplitude (right) before (continuous black line), during (grey line), and after (dotted line) H2O2 superfusion. Values were compared to the control with the Kolmogorov–Smirnov test. H2O2 had no effect on amplitude (during H2O2 superfusion: P = 0.13; after washout: P = 0.78), but inter‐event interval was shorter during H2O2 superfusion (P < 0.0001) and longer after washout (P < 0.0001). A and D were obtained from the same neuron. The holding potential was −70 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
We next examined the effect of H2O2 treatment on R in. Bath‐application of H2O2 (1 mm; 5 min) did not affect R in values, which were 144.4 ± 40.9 MΩ before H2O2 superfusion, 138.9 ± 38.0 MΩ after 5 min of H2O2 superfusion (98.3 ± 2.1% of the control, paired t test, P = 0.47, n = 6), and 147.5 ± 42.3 MΩ after 10 min of H2O2 washout (101.5 ± 1.8% of the control, paired t test, P = 0.44, n = 6), when measured from the same neurons before and after application. Moreover, to examine whether the depression phase of mEPSC frequency simply reflected damage to release machinery, we investigated the influence of a prolonged (15 min) application of H2O2 (Fig. 2 A). At 15 min after H2O2 superfusion, the increase of mEPSC frequency was sustained relative to control values, which are written in square brackets at their first mention (165.3 ± 21.4% of the control [12.8 ± 3.3 Hz], paired t test, P = 0.016, n = 7). This was followed by a depression that persisted for at least 10 min following washout (42.1 ± 8.4% of the control, paired t test, P = 0.001, n = 6; Fig. 2 B, left panel). In contrast, mEPSC amplitude was not affected significantly when compared with the control (Fig. 2 B, right panel).
Figure 2. Prolonged application of H2O2 results in an increase followed by depression in mEPSC frequency .
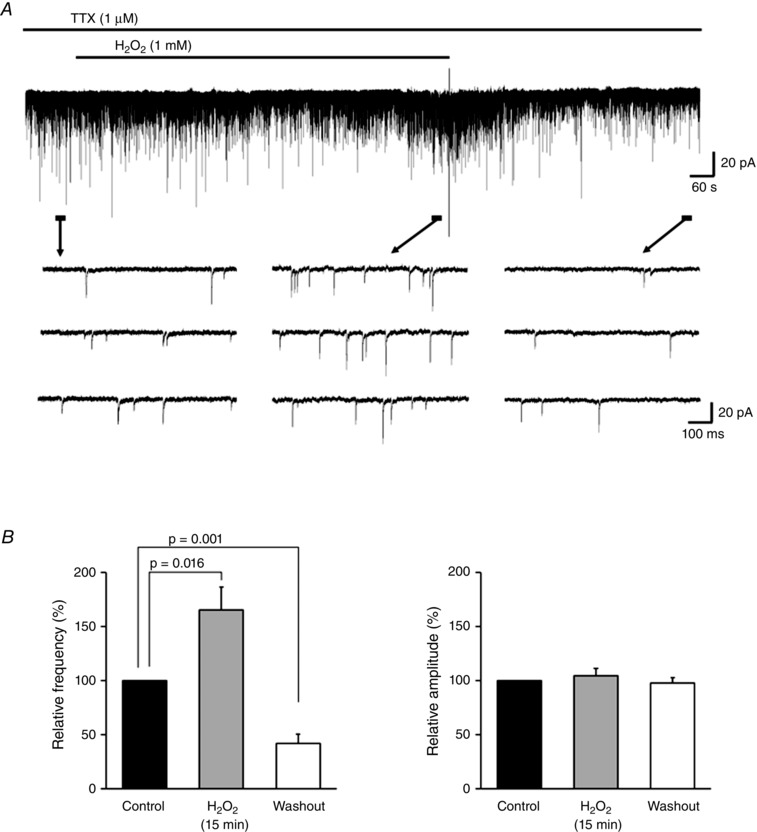
A, continuous chart recording of mEPSCs before, during and after H2O2 (1 mm, 15 min) superfusion. B, summary of mEPSC frequency data. Values are shown as percentage change relative to the control after 15 min of H2O2 superfusion (n = 7) and after washout (n = 6) (mean ± SEM). P values were determined by a paired t test. The holding potential was −70 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
H2O2 increases mEPSC frequency by causing an elevation in presynaptic Ca2+ concentration
We next investigated the mechanism underlying the H2O2‐induced increase in mEPSC frequency. Given that the frequency of spontaneous miniature synaptic events correlates with presynaptic Ca2+ concentration (Augustine et al. 2003), we investigated the source of presynaptic Ca2+. In general, these include influx through VGCCs in the plasma membrane (Catterall & Few, 2008) and release from the ER, which stores intracellular Ca2+. ER release is mediated by RyR or IP3R (Verkhratsky, 2002), whereas the mitochondria also play a role in regulating presynaptic Ca2+ concentration (Zenisek & Matthews, 2000; Billups & Forsythe, 2002).
Owing to the presence of exposed sulfhydryl groups, several ion channels and receptors including VGCCs (Nowicky AV & Duchen MR, 1998), RyR and IP3R (Gerich et al. 2009) are activated by changes in cytosolic redox balance and by the level of ROS such as H2O2. We therefore investigated the role of VGCC‐, mitochondria‐, RyR‐, and IP3R‐mediated Ca2+ signalling on H2O2‐induced excitatory activity by patch clamp combined with pharmacological manipulations. As a control, we recorded from cells exposed to both the inhibitor of interest and TTX but not H2O2. The values of the control are written in square brackets at their first mention. Given that the maximal increases in mEPSC frequency induced by H2O2 were observed between 2 and 5.5 min after the start of H2O2 superfusion, we evaluated maximal mEPSC frequency 1.5–6 min after superfusion.
We first examined whether H2O2‐induced increases in mEPSC frequency were dependent on extracellular Ca2+. In Ca2+‐free ACSF, H2O2 superfusion had little effect on mEPSC frequency (102.8 ± 6.1% of the control [8.4 ± 2.1 Hz]; paired t test, P = 0.66, n = 7; Fig. 3 A and B), suggesting that influx through VGCCs was partly responsible for the increase in intracellular Ca2+ level. In contrast, the subsequent depression in mEPSC frequency (48.9 ± 7.8% of the control; paired t test, P = 0.0006, n = 7) was not prevented in Ca2+‐free ASCF (i.e. was the same as observed in standard ACSF) (Fig. 3 A and B).
Figure 3. H2O2 induces an increase in mEPSC frequency by modulating Ca2+ influx via N‐type VGCCs .
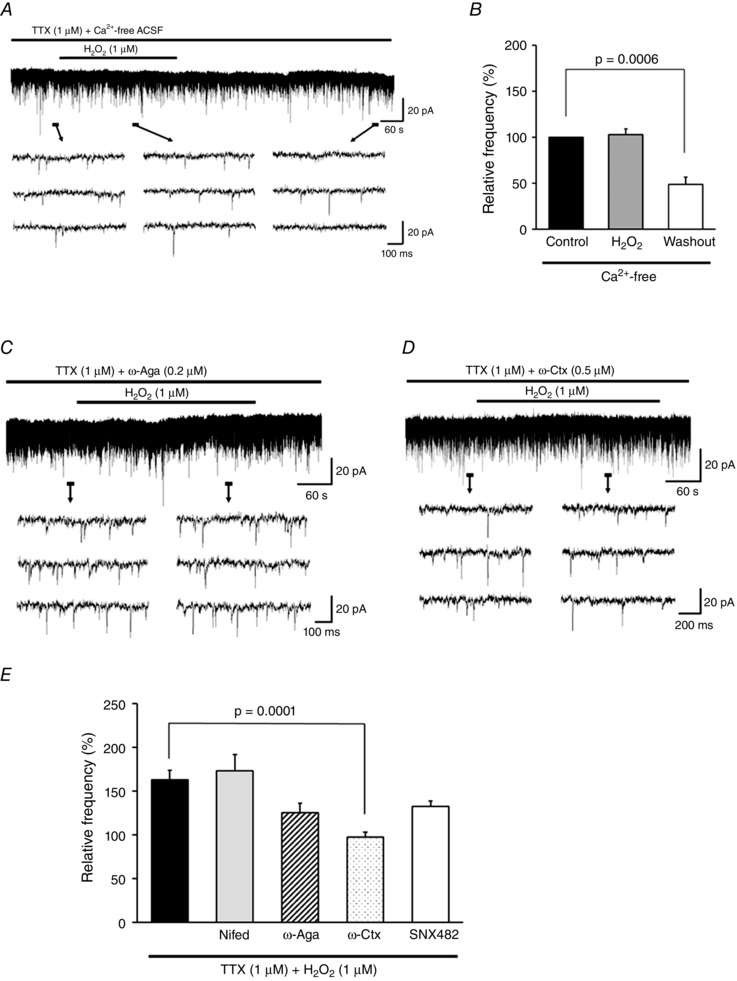
A, effect of H2O2 on mEPSCs in Ca2+‐free ACSF. Under this condition, the H2O2‐induced mEPSC increase but not its subsequent depression was completely blocked. B, summary of mEPSC frequency in Ca2+‐free ACSF (mean ± SEM, n = 7). Values in the charts correspond to percentage change in maximal values of mEPSC frequency relative to the control during H2O2 superfusion and after washout. The P value was determined by a paired t test. C, ω‐Aga (0.2 μm), a P/Q‐type VGCC blocker, partially suppressed the H2O2‐induced increase in mEPSC frequency. D, ω‐Ctx (0.5 μm), an N‐type VGCC blocker, completely suppressed the increase in frequency. E, summary of mEPSC frequencies observed during H2O2 superfusion with TTX alone (n = 11) or with nifedipine (L‐type VGCC blocker, 10 μm; n = 7), ω‐Aga (0.2 μm; n = 6), ω‐Ctx (0.5 μm; n = 6), or SNX‐482 (R‐type VGCC blocker, 0.1 μm; n = 6) relative to control (mean ± SEM). Maximal values of mEPSC frequency relative to the control 1.5–6 min after H2O2 superfusion are shown. P values were determined by one‐way ANOVA with a Bonferroni post hoc test. The holding potential was −70 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
Multiple types of VGCCs are expressed throughout the CNS (Frank, 2014); an immunocytochemical analysis revealed that rat spinal VH neurons express L‐, P/Q‐, N‐ and R‐type VGCCs (Westenbroek et al. 1998), although the presynaptic input to these cells presumably involves a different constellation of VGCCs. To determine the contribution of different presynaptic VGCC subtypes on the H2O2‐induced increase in spontaneous mEPSCs at excitatory synapses, we used VGCC inhibitors specific for each channel type during H2O2 superfusion of VH neurons.
Nifedipine, a dihydropyridine antagonist of L‐type VGCCs, had no effect on the H2O2‐induced increase in mEPSC frequency (173.3 ± 18.8% of the control [10.8 ± 1.8 Hz], n = 7). In contrast, inhibiting P/Q‐ or R‐type VGCCs with ω‐Aga and SNX‐482, respectively, partially blocked this increase (ω‐Aga, 125.5 ± 10.6% of the control [10.1 ± 1.0 Hz], n = 6, Fig. 3 C; SNX‐482, 132.6 ± 6.2% of the control [8.2 ± 1.2 Hz], n = 6), while a complete suppression was observed by blocking N‐type VGGCs with ω‐Ctx (97.7 ± 5.6% of the control [12.3 ± 2.5 Hz], n = 6; Fig. 3 D). A comparison between values obtained using VGCC inhibitors and TTX only by one‐way ANOVA revealed a significant effect of ω‐Ctx (F 5,38 = 7.73, P < 0.0001; Bonferroni post hoc test, TTX only vs. ω‐Ctx, P = 0.0001; Fig. 3 E). These VGCC blockers did not block the subsequent depression of mEPSC frequency observed after H2O2 washout (Nifedipine, 52.0 ± 10.6% of the control, n = 6; ω‐Aga, 52.8 ± 10.8% of the control, n = 5; ω‐Ctx, 58.5 ± 10.0% of the control, n = 6; SNX‐482, 61.3 ± 7.0% of the control, n = 6).
To determine whether intracellular Ca2+ stores are activated by H2O2, we targeted mitochondria and the ER using specific inhibitors. CSA, which blocks the opening of the mitochondrial permeability transition pore (MPT), had no effect on H2O2‐induced changes in mEPSC frequency (with H2O2 superfusion, 168.3 ± 15.3% of the control [7.4 ± 1.3 Hz]; after washout, 54.2 ± 6.2% of the control; n = 6 each). CSA also inhibits calcineurin (CaN). Therefore, these results further suggest that CaN does not mediate H2O2‐induced changes in mEPSC frequency in spinal VH neurons.
We then tested whether H2O2 affects intracellular Ca2+ release from the ER, which is mediated by RyR and IP3R. Dant, an RyR inhibitor, blocked the increase in mEPSC frequency induced by H2O2 (92.9 ± 8.3% of the control [8.1 ± 1.4 Hz], n = 8), but did not alter the subsequent decrease in frequency after H2O2 washout (42.7 ± 7.6% of the control, n = 5; Fig. 4 A). In the presence of 2‐APB, an IP3R antagonist, the H2O2‐induced increase in mEPSC frequency not only was blocked but was reduced after 5 min, reaching a maximum value between 1.5 and 6 min after the start of superfusion to 81.2 ± 11.0% of the control [12.7 ± 2.0 Hz] (n = 6; Fig. 4 B). These changes were accompanied by an outward current (> 5 pA) in five of six neurons (14.2 ± 1.6 pA, n = 5; Fig. 4 B). The decrease in mEPSC frequency was reversible, and subsequent depression of mEPSC frequency by H2O2 was completely blocked (99.5 ± 7.0% of the control, n = 6; Fig. 4 B). H2O2 treatment in the presence of 2‐APB had no effect in recordings of another five neurons: the R in values were 146.9 ± 31.9 MΩ before H2O2 superfusion, 150.24 ± 32.5 MΩ after 5 min of H2O2 superfusion (102.3 ± 2.5% of the control, paired t test, P = 0.42, n = 5), and 142.9 ± 29.2 MΩ after 10 min of H2O2 washout (98.5 ± 3.6% of the control, paired t test, P = 0.69, n = 5).
Figure 4. H2O2‐induced increases in mEPSC frequency are due to intracellular Ca2+ release via RyR and IP3R .
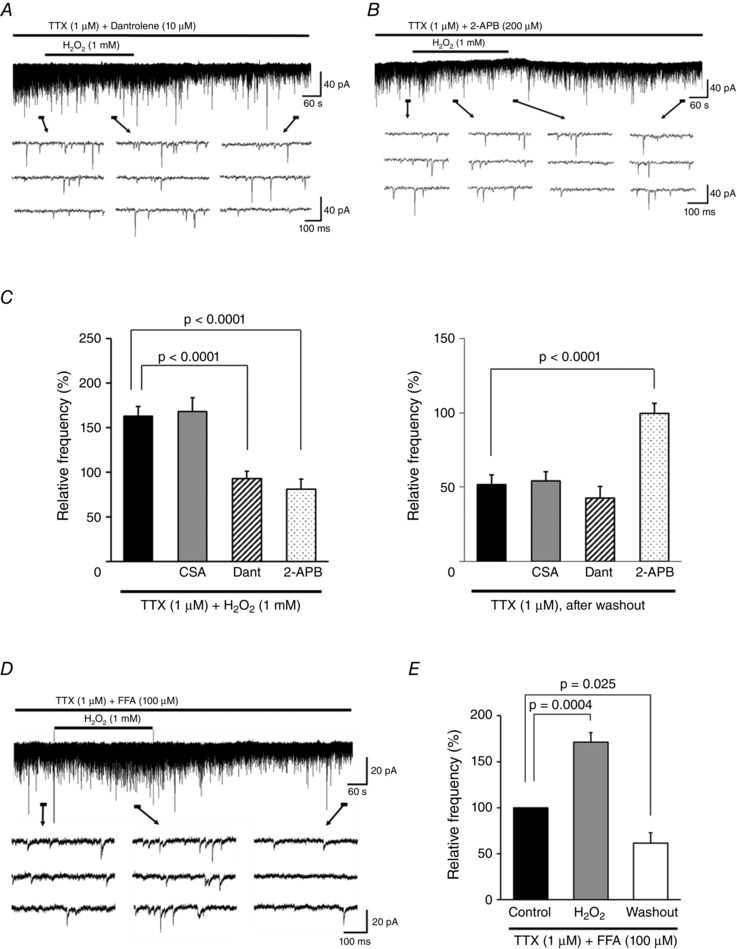
A, Dant (10 μm), an RyR antagonist, completely blocked the H2O2‐induced increase in mEPSC frequency but not the subsequent depression. B, the IP3R antagonist 2‐APB (200 μm) completely blocked both the increase and depression in frequency. Note that the increase not only was blocked but was reduced with respect to the control after 5 min of H2O2 superfusion (38.9 ± 3.2% of control, n = 6). C, summary of mEPSC frequency during (left) and after (right) H2O2 superfusion with TTX alone (n = 11) or with CSA (a MPT blocker/calcineurin inhibitor, 10 μm; n = 6), Dant (10 μm; n = 5–8), or 2‐APB (200 μm; n = 6) relative to the control (mean ± SEM). Values on the left indicate maximal values of mEPSC frequency relative to the control 1.5–6 min after H2O2 superfusion. P values were determined by one‐way ANOVA with a Bonferroni post hoc test. D, FFA (100 μm), a TRPM2 channel blocker, did not block the H2O2‐induced increase and subsequent depression in mEPSC frequency. E, summary of mEPSC frequency recordings in the presence of FFA (mean ± SEM, n = 7). Values in the charts correspond to percentage change in maximal values of mEPSC frequency relative to the control during H2O2 superfusion and after washout. The P value was determined by a paired t test. The holding potential was −70 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
A comparison between values obtained with CSA, Dant, or 2‐APB and TTX only by one‐way ANOVA showed significant effects of Dant and 2‐APB on the H2O2‐induced increase in mEPSC frequency (F 4,31 = 12.5, P < 0.0001; Bonferroni post hoc test, TTX only vs. Dant or 2‐APB, P < 0.0001 for each; Fig. 4 C, left panel), and a significant effect of 2‐APB on the subsequent depression (F 3,23 = 11.8, P < 0.0001; Bonferroni post hoc test, TTX only vs. 2‐APB, P < 0.0001; Fig. 4 C, right panel).
Although we used 2‐APB as an IP3 antagonist in this experiment, 2‐APB also blocks Na+ and Ca2+‐permeable melastatin‐related transient receptor potential 2 (TRPM2) channels (Xu et al. 2005; Togashi et al. 2008). We therefore examined the effect of another TRPM2 channel blocker, flufenamic acid (FFA) (Hill et al. 2004; Naziroğlu et al. 2011; Lee et al. 2011, 2013), on the H2O2‐induced changes of mEPSC frequency (Fig. 4 D). FFA (100 μm) did not block the initial H2O2‐induced increased of mEPSC frequency (171.5 ± 10.1% of control [13.4 ± 2.5 Hz], paired t test, P = 0.0004, n = 7) or the subsequent depression after H2O2 washout (61.5 ± 11.0% of the control, paired t test, P = 0.025, n = 7) (Fig. 4 E).
IP3, the natural ligand for IP3R, arises from the enzymatic activity of phospholipase C (PLC)‐β, which is downstream of G protein‐coupled receptor activation, as detailed in Fig. 8 (Rhee & Bae, 1997). We therefore examined the effect of the PLC/A2 inhibitor BPB on the H2O2‐induced increase and subsequent depression in mEPSC frequency. Unlike the IP3R antagonist 2‐APB, BPB did not block this increase (167.8 ± 15.4% of the control [13.6 ± 3.5 Hz], paired t test, P = 0.0071, n = 6) and subsequent depression (59.5 ± 6.5% of the control, paired t test, P = 0.0034, n = 5), indicating that H2O2‐mediated IP3R‐related signalling is downstream of PLC‐β.
Figure 8. Model of H2O2‐induced modulation of presynaptic activity in spinal VH neurons .
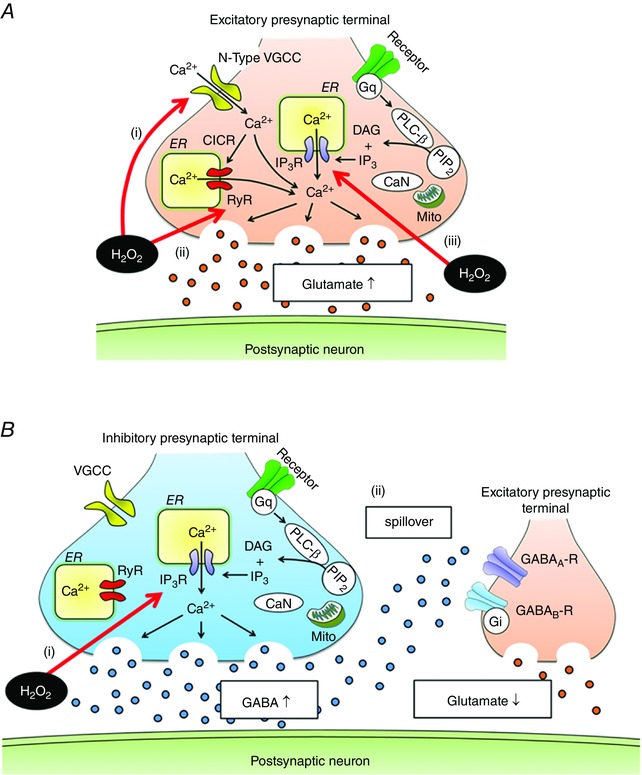
A, H2O2 stimulates glutamate release by Ca2+ influx via N‐type VGCC (i) and Ca2+ release from the ER via RyR (ii) and IP3R (iii). Oxidation of RyR cysteine residues increases RyR sensitivity to Ca2+, resulting in excessive activation of RyR‐mediated CICR. The effects of H2O2 on IP3R‐related signalling are downstream of PLC‐β. B, H2O2 increases GABAergic transmission by stimulating Ca2+ release from the ER via IP3R; its site of action is downstream of PLC‐β (i). Moreover, diffusion of GABA away from the release site activates presynaptic GABAA and to a lesser degree GABAB receptors (ii), thereby inhibiting glutamate release from excitatory presynaptic terminals. These effects by GABA may protect postsynaptic neurons from excitotoxicity resulting from excessive glutamate. Mito, mitochondria; CaN, calcineurin; PIP2, phosphatidylinositol 4,5‐bisphosphate; DAG, diacylglycerol.
H2O2‐induced enhancement of GABAergic activity reduces mEPSC frequency following the initial increase
In spinal dorsal horn neurons, ischaemia leads to an increase in the release of GABA and glycine (Kawasaki et al. 2004) and a decrease in glutamate release from nerve terminals via activation of GABA receptors (Matsumoto et al. 2003). H2O2‐induced increases in GABA release have also been reported in the spinal dorsal horn (Takahashi et al. 2007). Based on these findings, we investigated whether GABA and/or glycine receptors at presynaptic terminals are involved in the depression of mEPSC frequency by H2O2 using specific receptor antagonists.
GABA receptors are ionotropic A‐type (GABAA) or metabotropic G protein‐coupled B‐type (GABAB) (Kumamoto, 1997), and both receptors have been detected in nerve terminals of spinal motor neurons (Rekling et al. 2000). The addition of Bic or CGP35348 – GABAA and GABAB receptor antagonists, respectively – led to a robust enhancement of the H2O2‐mediated increase in mEPSC frequency (Bic, 241.1 ± 37.3% of the control [7.3 ± 1.8 Hz], n = 7; CGP35348, 282.3 ± 69.4% of the control [7.1 ± 0.7 Hz], n = 6; Fig. 5 A and B). The subsequent depression in mEPSC frequency was blocked by Bic (108.7 ± 10.7% of the control, n = 7; Fig. 5 A) but not by CGP35348 (48.2 ± 12.2% of the control, n = 6; Fig. 5 B). Treating neurons with the glycine receptor antagonist Stry had no effect on H2O2‐mediated changes in mEPSC (during H2O2 superfusion, 162.6 ± 19.5% of the control [8.6 ± 1.1 Hz]; after washout, 44.9 ± 7.3% of the control; n = 6 for each; Fig. 5 C). A comparison between values obtained with Bic, CGP35348, or Stry and TTX only revealed a trend of enhanced H2O2‐induced increases in mEPSC frequency in the presence of Bic or CGP35348; however, the differences were not statistically significant (one‐way ANOVA, F 3,26 = 2.86, P = 0.056; Fig. 5 D, left panel). In contrast, Bic had a significant effect on the subsequent depression of mEPSC frequency (one‐way ANOVA, F 3,26 = 11.05, P < 0.0001; Bonferroni post hoc test, TTX only vs. Bic, P < 0.0001; Fig. 5 D, right panel). These data suggest that GABAA and GABAB receptor activation in terminals that innervate VH neurons inhibits the excessive presynaptic release of glutamate induced by H2O2 superfusion, and that GABAA receptor activation is involved in the subsequent depression in mEPSC frequency.
Figure 5. Effects of GABA and glycine receptor antagonists on H2O2‐induced changes in mEPSC frequency .
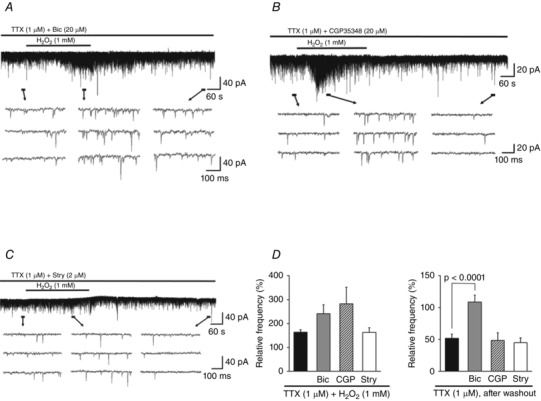
A, Bic (20 μm), a GABAA receptor antagonist, enhanced H2O2‐induced increases in mEPSC frequency and blocked its subsequent depression. B, CGP35348 (20 μm), a GABAB receptor antagonist, enhanced the increase in frequency but had no effect on the depression. C, Stry (2 μm), a glycine receptor antagonist, had no effect on H2O2‐induced changes in mEPSC frequency. D, summary of mEPSC frequency data during (left) and after (right) H2O2 superfusion with TTX alone (n = 11) or with Bic (20 μm; n = 7), CGP35348 (CGP, 20 μm; n = 6), or Stry (2 μm; n = 6) relative to the control (mean ± SEM). Values on the left indicate maximal values of mEPSC frequency relative to the control 1.5–6 min after H2O2 superfusion. P values were determined by one‐way ANOVA with a Bonferroni post hoc test. The holding potential was −70 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
H2O2 increases the frequency of GABAergic miniature inhibitory postsynaptic currents (mIPSCs)
The above findings prompted us to examine the effects of H2O2 on GABA release from presynaptic terminals by recording GABAergic mIPSCs in VH neurons. Since GABA and glycine are both released from axon terminals that synapse on spinal motor neurons (Ornung et al. 1994; Taal & Holstege, 1994; Jonas et al. 1998; Baer et al. 2003), GABAergic mIPSCs were isolated by adding TTX and Stry to the ACSF.
GABAergic mIPSCs were measured at a holding potential of 0 mV; the frequency and amplitude were 1.9 ± 0.4 Hz and 10.0 ± 0.7 pA, respectively (n = 8). H2O2 superfusion resulted in a marked increase in frequency (647.8 ± 201.4% of the control) but only a minimal change in amplitude (120.0 ± 8.3% of the control; paired t test, P = 0.057; Fig. 6 A and C). These changes were accompanied by a more than 5 pA of outward current in seven of eight neurons examined (25.0 ± 10.1 pA, n = 7; Fig. 6 A). Figure 6 B shows the time course of the average change in GABAergic mIPSC frequency and amplitude in response to H2O2. The maximal increase in frequency was delayed, appearing 4.5–7 min (mean = 5.9 ± 0.4 min) after initiating H2O2 superfusion or approximately 2 min after initiating the washout (Fig. 6 C). The effect was reversible, but persisted for 10 min after the washout (Fig. 6 C); it also coincided with the decrease in mEPSC frequency observed after H2O2 washout (Fig. 1 B). These results suggest that changes in mEPSCs induced by H2O2 are mediated by GABA. Figure 6 D shows that H2O2 application shortened the average inter‐event interval without causing a significant change in amplitude relative to the control (during H2O2 superfusion: frequency, P < 0.0001; amplitude, P = 0.051; after washout: frequency, P < 0.0001; amplitude, P = 0.061; Kolmogorov–Smirnov test).
Figure 6. H2O2 increases the frequency of GABAergic mIPSCs .
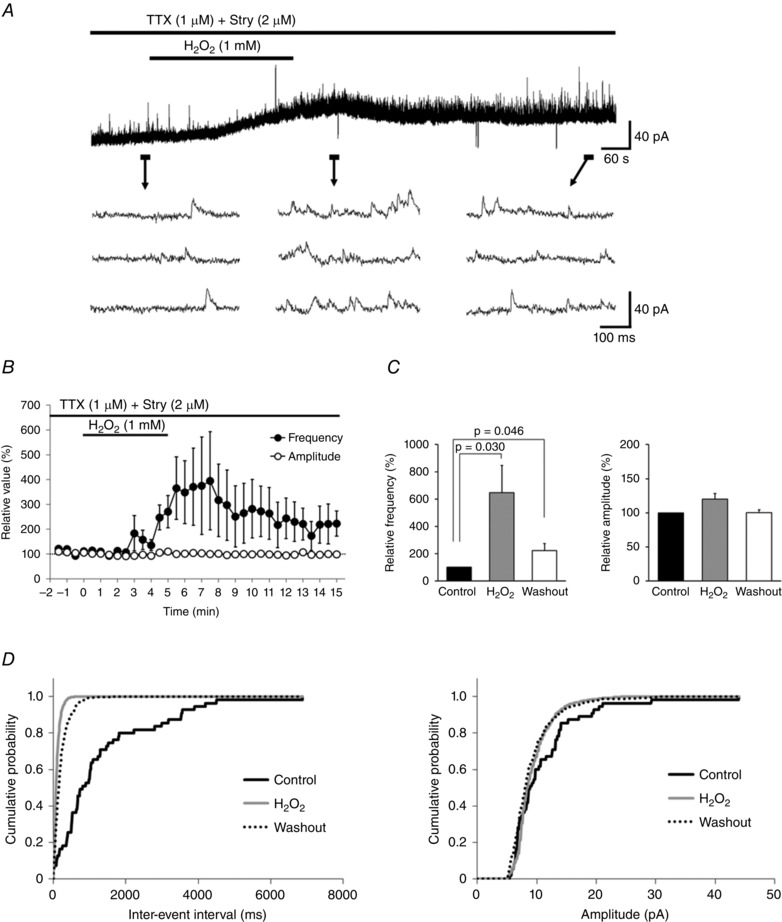
A, continuous chart recording of GABAergic mIPSCs before during, and after H2O2 superfusion. H2O2‐induced increases in GABAergic mIPSC frequency were often accompanied by an outward current. B, average frequency (filled circles) and amplitude (open circles) of GABAergic mIPSCs after H2O2 superfusion relative to the control as a function of time (mean ± SEM, n = 8). C, summary of GABAergic mIPSC frequency (left) and amplitude (right) relative to the control (mean ± SEM, n = 8 each) showing an increase in frequency but not amplitude induced by H2O2. Middle columns of charts indicate maximal values of GABAergic mIPSC frequency and amplitude relative to the control following H2O2 superfusion. P values were determined by a paired t test. D, cumulative distributions of inter‐event interval (left) and amplitude (right) of GABAergic mIPSCs before (continuous black line), during (grey line), and after (dotted line) H2O2 superfusion. H2O2 did not significantly affect the distribution of amplitude (during H2O2 superfusion: P = 0.051; after washout: P = 0.061; Kolmogorov–Smirnov test) relative to the control, but shortened the inter‐event interval during H2O2 superfusion and increased the interval after washout (P < 0.0001 for each). A and D were obtained from the same neuron. The holding potential was 0 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
H2O2‐induced increase in GABAergic mIPSC frequency involves intracellular Ca2+ release via IP3R
We next examined the role of Ca2+ signalling in the modulation of GABAergic mIPSC frequency by H2O2. In Ca2+‐free ACSF, H2O2 superfusion increased the frequency of GABAergic mIPSCs (413.0 ± 94.2% of the control [1.6 ± 0.2 Hz], n = 6; Fig. 7 A and B) to a level similar to that observed in normal ACSF (P = 0.36, unpaired t test), indicating that Ca2+ influx is not involved in the H2O2‐induced increase in GABA release. The effect was reversible, but persisted for 10 min after the washout (181.2 ± 29.4% of the control, n = 6). In addition, the H2O2‐induced increase of GABAergic mIPSC frequency was accompanied by an outward current (> 5pA) in three of six neurons examined (18.1 ± 10.9 pA, n = 3).
Figure 7. H2O2‐induced increase in GABAergic mIPSC frequency is due to intracellular Ca2+ release via IP3R .
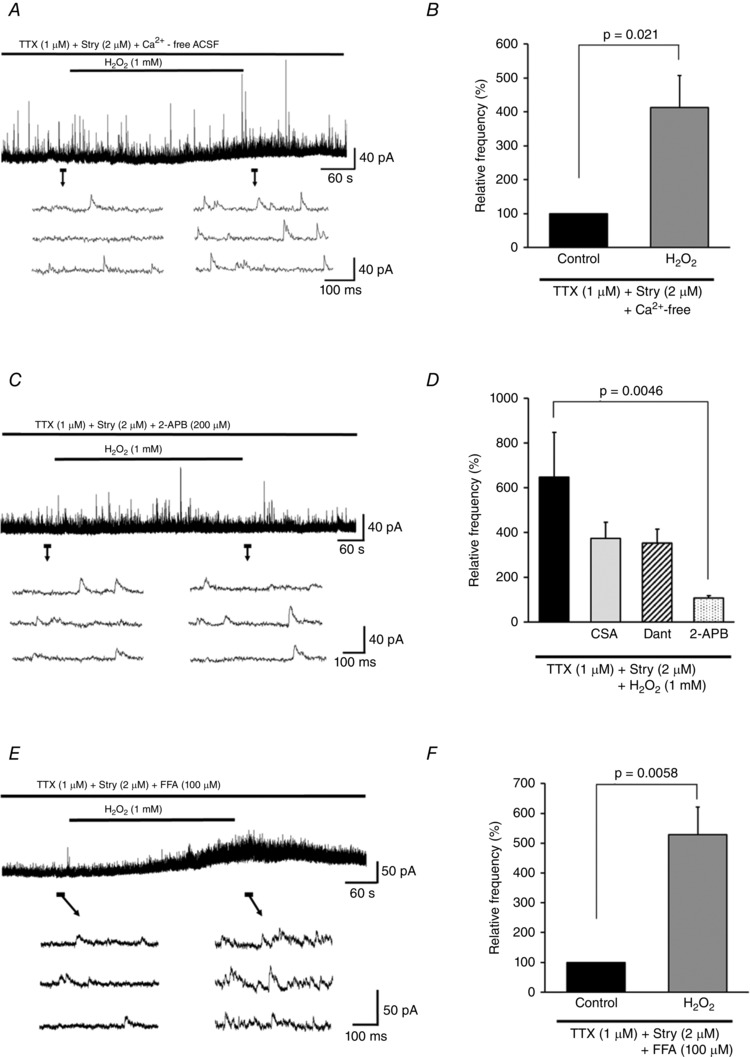
A, H2O2‐induced increase in GABAergic mIPSC frequency was unaffected by Ca2+‐free ACSF. B, summary of GABAergic mIPSC frequency relative to the control in Ca2+‐free ACSF (mean ± SEM, n = 6). Values in charts correspond to percentage change in maximal values of GABAergic mIPSC frequency relative to the control following H2O2 superfusion. P values were determined by a paired t test. C, H2O2‐induced increase in GABAergic mIPSC frequency and outward current were completely blocked by 2‐APB (200 μm). D, summary of GABAergic mIPSC frequencies observed during H2O2 superfusion with TTX combined with either Stry (2 μm; n = 8), CSA (10 μm; n = 6), Dant (10 μm; n = 6), or 2‐APB (200 μm; n = 7) relative to the control (mean ± SEM). Maximal values of GABAergic mIPSC frequency relative to the control following H2O2 superfusion are shown. P values were determined by one‐way ANOVA with a Bonferroni post hoc test. E, FFA (100 μm) did not block the H2O2‐induced increase of GABAergic mIPSC frequency. F, summary of GABAergic mIPSC frequency data during FFA treatment (mean ± SEM, n = 6). Values in the charts correspond to the percentage change in maximal values of GABAergic mIPSC frequency relative to the control following H2O2 superfusion. The P value was determined by a paired t test. The holding potential was 0 mV for all recordings. Washout indicates the 10 min period after initiating H2O2 washout.
We then pharmacologically targeted mitochondria and the ER to determine whether intracellular Ca2+ stores are activated by H2O2 at GABAergic inhibitory presynaptic terminals. CSA (an MPT blocker/calcineurin inhibitor) and Dant (an RyR antagonist) had no effect on H2O2‐induced increases in mIPSC frequency during H2O2 superfusion (CSA, 373.6 ± 72.5% of the control [1.8 ± 0.4 Hz], n = 6; Dant, 352.0 ± 63.3% of the control [2.1 ± 0.7 Hz], n = 6) and after H2O2 washout (CSA, 202.3 ± 34.1% of the control, n = 6; Dant, 184.6 ± 30.2% of the control, n = 6). Moreover, H2O2‐induced outward currents (> 5 pA) were seen in three of six neurons treated with CSA (17.4 ± 9.5 pA, n = 3) and in four of six neurons treated with Dant (28.5 ± 12.7 pA, n = 4). In contrast, 2‐APB (an IP3R antagonist) completely blocked H2O2‐induced increases in mIPSC frequency during H2O2 superfusion (106.6 ± 11.8% of the control [2.1 ± 0.4 Hz], n = 7) and after H2O2 washout (101.0 ± 12.8% of the control, n = 5) as well as the outward current (Fig. 7 C). A comparison between values during H2O2 superfusion obtained for CSA, Dant, or 2‐APB and TTX + Stry only showed a significant effect of 2‐APB (F 3,23 = 3.31, P = 0.038; one‐way ANOVA with Bonferroni post hoc test, vs. TTX + Stry, P = 0.0046; Fig. 7 D). Moreover, the H2O2‐induced increase of GABAergic mIPSC frequency was not blocked by the application of the TRPM2 channel blocker FFA (during H2O2 superfusion, 528.1 ± 92.8% of the control [2.2 ± 0.7 Hz], paired t test, P = 0.0058, n = 6, Fig. 7 E and F; after H2O2 washout, 188.4 ± 20.5% of the control, paired t test, P = 0.0076, n = 6; accompanied by an outward current (> 5 pA) in four of six neurons (22.0 ± 6.5 pA, n = 4; Fig. 7 E)). These results suggest that the IP3R‐sensitive pool in the ER is the source of the Ca2+ responsible for the increase in GABAergic mIPSC frequency. However, this was not blocked by the application of the PLC‐β inhibitor BPB (during H2O2 superfusion, 503.7 ± 95.6% of the control [1.9 ± 1.1 Hz], paired t test, P = 0.0083, n = 6; after H2O2 washout, 224.7 ± 40.4% of the control, paired t test, P = 0.027, n = 6), indicating that H2O2 acts downstream of PLC‐β, as in the case of mEPSCs.
Discussion
The present study investigated the cellular mechanisms underlying the effects of H2O2 on synaptic transmission in spinal VH neurons. We demonstrated that acute H2O2 administration induces biphasic changes in mEPSC frequency, characterized by an initial increase followed by a prolonged depression. On the other hand, prolonged H2O2 administration resulted in the high frequency of mEPSC that persisted before H2O2 washout, suggesting that the subsequent depression of mEPSC frequency observed upon acute H2O2 administration is associated with the removal of H2O2 and does not simply reflect damage to glutamate release machinery. In addition, H2O2 markedly increased GABA release from presynaptic terminals at a time point coinciding with the depression in mEPSC frequency, suggesting that the latter was caused by GABA‐mediated inhibitory activity. These results also indicate that H2O2‐induced changes in excitatory and GABAergic inhibitory activities have a presynaptic origin, given that increases in mEPSC and mIPSC frequency occurred in the absence of significant changes in amplitude.
H2O2 increases mEPSC frequency by Ca2+ influx via N‐type VGCCs
The results of VGCC inhibition suggested that the H2O2‐induced increase in mEPSC frequency was due to Ca2+ influx mainly through N‐ and, to a lesser extent, P/Q‐ and R‐type VGCCs (Fig. 8 Aa (i)). In contrast, in hippocampal neurons, H2O2‐induced increases in Ca2+ flux occur through L‐type VGCCs (Akaishi et al. 2004) whereas in Xenopus oocytes, P/Q‐type VGCCs are mainly involved (Li et al. 1998). These differences may be due to the variable expression of VGCC subtypes across cell types and/or parts of the cell. P/Q‐ and N‐type Ca2+ channels are predominant in synaptic nerve terminals, but their relative expression levels vary across the nervous system given the distinct physiological and pathological roles of VGCCs (Evans & Zamponi, 2006). In hippocampal neurons, the function and regulation of subtypes depends on whether they are located on excitatory or inhibitory terminals (Potier et al. 1993). In inhibitory spinal neurons, P/Q‐type VGCCs are primarily responsible for presynaptic Ca2+ influx (Takahashi & Momiyama, 1993), whereas our findings indicate that this function is attributable to N‐type VGCCs at excitatory presynaptic terminals of spinal VH neurons under oxidative conditions.
H2O2‐induced increases in mEPSC frequency are associated with intracellular Ca2+ release via RyR and IP3R
Similar to the effects of several VGCC blockers, the H2O2‐induced increase in mEPSC frequency was suppressed by antagonists of either RyR or IP3R, both of which mediate Ca2+ release from the ER. These data suggest that the mechanism underlying H2O2‐induced increases in mEPSC frequency involves Ca2+ release from the intracellular pool via RyR and IP3R. Intracellular Ca2+‐induced Ca2+‐release (CICR) in response to activity is dependent on RyR and, to a lesser extent, on IP3R. The oxidation of cysteine residues on RyR increases Ca2+ sensitivity, leading to excessive RyR‐mediated CICR (Marengo et al. 1998). Our data suggest that the increase in Ca2+ sensitivity of RyR is responsible for the observed H2O2‐induced increase in mEPSC frequency (Fig. 8 A (ii)).
On the other hand, treatment with the IP3R antagonist 2‐APB blocked the H2O2‐induced increase in mEPSC frequency, which was often accompanied by an outward current. However, blocking G‐coupled protein receptor signalling, which is upstream of IP3R, had no effect on mEPSC frequency. It was recently reported that ROS can activate IP3 signalling and IP3Rs by modifying cysteinyl residues, possibly in an IP3R isoform‐dependent manner (Lencesova & Krizanova, 2012; Ivanova et al. 2014). Thus, H2O2 may act directly on IP3Rs either as a ligand or by modifying the IP3R protein in a manner that alters Ca2+ release in excitatory presynaptic terminals (Fig. 8 A (iii)). It is worth noting, however, that IP3R antagonism did not simply inhibit H2O2‐induced increases in mEPSC frequency but instead caused a significant reduction from the baseline value for reasons that are unclear. We speculate that activation of IP3R‐related signalling is fundamental to the increase in mEPSC frequency induced by H2O2, and that blocking IP3R activated the release of neurotransmitters such as adenosine, which induced further decreases in mEPSC frequency. This is supported by previous reports that H2O2 activates rat hippocampal neuron adenosine A1 receptors (Masino et al. 1999), leading to presynaptic inhibition of excitatory transmission and eliciting an outward current in spinal VH neurons (Miyazaki et al. 2008). However, it is unclear why H2O2 treatment in the presence of 2‐APB had no effect on R in even though an outward current and decrease of mEPSCs frequency were observed. The 5 mV hyperpolarizing steps we applied from a −70 mV holding potential might not be sufficient to evaluate the changes of R in induced by H2O2 treatment in the presence of 2‐APB. The precise mechanisms by which IP3R regulates excitatory activity in spinal VH neurons, as well as the possible roles of adenosine signalling and the voltage dependency of the outward current by H2O2 treatment in the presence of 2‐APB, remain to be elucidated.
Increases in H2O2‐induced GABA release from presynaptic terminals is attributable to Ca2+ release from the IP3R‐sensitive intracellular pool
We demonstrated that presynaptic IP3Rs have an essential role in modulating H2O2‐induced increases in GABA release from presynaptic terminals of spinal VH neurons (Fig. 8 B (i)). In the rat hippocampus, H2O2 induces Ca2+ release from the ER via RyR and IP3R by acting on the redox‐sensing elements of their hyper‐reactive sulfhydryl groups (Gerich et al. 2009). In contrast, in mouse spinal dorsal horn neurons this process depends on IP3R but not RyR (Takahashi et al. 2007). Thus, the two receptors have distinct roles depending on the CNS region, likely to be due to differences in expression and neuronal architecture. In addition, as observed for excitatory presynaptic terminals, blocking PLA2/C had no effect on the H2O2‐induced increase in GABA release, suggesting that H2O2 acts downstream of PLC‐β (Fig. 8). However, an alternative IP3 generating pathway may also be involved.
The H2O2‐induced increase in GABAergic mIPSC frequency was often accompanied by an outward current. Although the underlying mechanism is unknown, this may involve hyperpolarization of the resting membrane potential by GABAA activation, which would lead to Cl− influx into the cell body (Sivilotti & Nistri, 1991; Laube et al. 2002). Another possibility is that H2O2 increases the GABAA receptor‐mediated tonic currents in hippocampal neurons (Penna et al. 2014). Moreover, as we used neonatal rats, an exploration of the possibility of a postsynaptic uncovering of ‘silent synapses’ is needed. It has been reported that silent synapses reflect the functional presence of NMDA receptors but not α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole‐4‐propionic acid (AMPA) receptors, and that they are involved in a postsynaptic mechanism of long‐term potentiation (Kerchner & Nicoll, 2008). Recently, it has been reported that there is a similar form of GABA synapse plasticity (Inoue et al. 2013) and that mitochondrial ROS regulates the strength of GABA‐mediated synaptic transmission through a mechanism that recruits α3‐containig GABAA receptors into the synapse (Accardi et al. 2014). Additional insight may be provided by future studies examining the postsynaptic effects of H2O2 including the possible involvement of silent synapses.
H2O2‐induced increases in the frequency of mEPSC and GABAergic mIPSCs do not involve Ca2+ influx via TRPM2 channels
TRPM2 channels are widely expressed in the brain and have been implicated in neuronal damage induced by oxidative stress (Halliwell, 2006; Hara et al. 2002). Moreover, it has been reported that TRPM2 channels are also expressed in spinal VH of neonatal rats (Bianchetti et al. 2013) and can be activated by H2O2 in neuronal cells (Naziroğlu, 2011; Lee et al. 2011, 2013). In our acute H2O2 administration experiments, the TRPM2 channel blocker FFA did not block H2O2‐induced changes in mEPSCs and GABAergic mIPSCs, suggesting that the effects of H2O2 do not involve Ca2+ influx via TRPM2 channels in spinal VH neurons.
Although the mechanism of TRPM2 channel activation by H2O2 is not entirely resolved, it appears that the gating mechanism relies primarily on its ability to release adenosine diphosphate ribose (ADPR) from mitochondria (Ayub & Hallett, 2004) and the nucleus (Caiafa et al. 2009; Esposito & Cuzzocrea, 2009; Fauzee et al. 2010). This suggests that exogenous H2O2 may be needed to cross the plasma membrane and generate ADPR from mitochondria and/or the nucleus to activate TRPM2 channels. TRPM2 activation by exogenous H2O2 shows slow kinetics (Sumoza‐Toledo & Penner, 2011) and may therefore occur at a time point beyond our experimental range. Moreover, applying FFA for only 4 min before starting H2O2 superfusion might not have been sufficient to antagonize TRPM2 channels completely. Further study is needed to clarify the time dependence of FFA and H2O2 effects on TRPM2 channels in rat VH neurons.
H2O2‐induced increases in GABA release inhibit glutamatergic transmission
When H2O2 superfusion was combined with GABAA or GABAB receptor antagonist, a robust increase in mEPSC frequency was observed. In addition, GABAA receptor antagonism blocked the depression in mEPSC frequency observed after H2O2 washout, indicating that this was caused by GABAA receptor activation at presynaptic terminals. These results suggest that excess GABA released from terminals may activate presynaptic GABAA and GABAB receptors of glutamatergic interneurons, thereby suppressing glutamate release and excitatory activity (Fig. 8 B (ii)). In support of this possibility, a similar form of synaptic modulation in the cerebellar glomerulus is thought to result from GABA in single inhibitory axons (Mitchell & Silver, 2000). Nonetheless, it is unclear why subsequent depression of mEPSC frequency was blocked only by activation of GABAA receptors. One explanation is the differential distribution and physiological roles of GABA receptor subtypes in presynaptic terminals of spinal VH neurons. For example, in group Ia dorsal root afferent axons that make monosynaptic connections to motor neurons in the spinal cord, the GABAA but not the GABAB receptor, which is also present at Ia terminals, plays a predominant role in presynaptic inhibition (Stuart & Redman, 1992). Finally, as predicted by the absence of presynaptic glycine receptors in the spinal VH (Rekling et al. 2000), application of a glycine receptor antagonist had no effect on glutamate release induced by H2O2. However, as glycine is a key inhibitory transmitters in the spinal cord, the effect of H2O2 in glycinergic synaptic transmission should be studied.
Physiological significance of H2O2‐induced modulation of synaptic transmission
Our results demonstrate that both glutamatergic excitatory and GABAergic inhibitory synaptic transmission were enhanced by H2O2‐activated increases in Ca2+‐dependent transmitter release, albeit by different mechanisms. We do note that the apparent lack of an effect of some inhibitors might reflect the limited time (4 min) of exposure to them prior to H2O2 application. Notably, N‐type VGCC, RyR, and IP3R were fundamental to H2O2‐induced increases in glutamate release, while only IP3R was implicated in the increase in GABA release induced by H2O2. These results suggest a novel strategy for preventing and/or treating H2O2‐induced motor neuron disorders resulting from trauma or ischaemia–reperfusion injury by selectively blocking N‐type VGCCs or RyR but not IP3R, which would specifically suppress excitotoxicity caused by the H2O2‐induced increase in glutamate release without blocking the neuroprotective effects of GABA.
Additional information
Competing interests
The authors have declared that no conflicts of interest exist.
Author contributions
M.O. and N.O. conducted experiments and analysed data. M.O., T.H., K.W., K.K, N.E., H.B. and T.K conceived and designed the project. M.O. and T.K. wrote the manuscript. T.H., K.W., K.K., N.O., H.B. and N.O. revised the manuscript critically for important intellectual content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by a Grant for Promotion of Niigata University Research Projects (25C032), a Tsukada Grant for Niigata University Medical Research, and a Grant‐in‐Aid for Exploratory Research (grant number 25670668 and 15K19989) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Tokyo, Japan).
Acknowledgements
The authors would like to thank Yukio Sato for his technical assistance (Division of Anesthesiology, Niigata University Graduate School of Medical and Dental Sciences).
References
- Accardig MV, Daniels BA, Brown PM, Fritschy JM, Tyagarajan SK & Bowie D (2014). Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA‐mediated synaptic transmission. Nat Commun 5, 3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaishi T, Nakazawa K, Sato K, Saito H, Ohno Y & Ito Y (2004). Hydrogen peroxide modulates whole cell Ca2+ currents through L‐type channels in cultured rat dentate granule cells. Neurosci Lett 356, 25–28. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Koga S, Nakatsuka T, Fujita T, Goto M & Kumamoto E (2010). Excitation of rat spinal ventral horn neurons by purinergic P2X and P2Y receptor activation. Brain Res 1340, 10–17. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F & Tanaka K (2003). Local calcium signaling in neurons. Neuron 40, 331–346. [DOI] [PubMed] [Google Scholar]
- Avshalumov MV & Rice ME (2002). NMDA receptor activation mediates hydrogen peroxide‐induced pathophysiology in rat hippocampal slices. J Neurophysiol 87, 2896–2903. [DOI] [PubMed] [Google Scholar]
- Ayub K & Hallett MB (2004). The mitochondrial ADPR link between Ca2+ store release and Ca2+ influx channel opening in immune cells. FASEB J 18, 1335–1338. [DOI] [PubMed] [Google Scholar]
- Baer K, Waldvogel HJ, During MJ, Snell RG, Faull RL & Rees MI (2003). Association of gephyrin and glycine receptors in the human brainstem and spinal cord: an immunohistochemical analysis. Neuroscience 122, 773–784. [DOI] [PubMed] [Google Scholar]
- Bianchetti E, Mladinic M & Nistri A (2013). Mechanisms underlying cell death in ischemia‐like damage to the rat spinal cord in vitro. Cell Death Dis 4, e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B & Forsythe ID (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22, 5840–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caifa P, Guastafierro T & Zampieri M (2009). Epigenetics: poly(ADP‐ribosyl)ation of PARP‐1 regulates genomic methylation patterns. FASEB J 23, 672–678. [DOI] [PubMed] [Google Scholar]
- Carri MT, Ferri A, Cozzolino M, Calabrese L & Rotilio G (2003). Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull 61, 365–374. [DOI] [PubMed] [Google Scholar]
- Catterall WA & Few AP (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. [DOI] [PubMed] [Google Scholar]
- Coyle JT & Puttfarcken P (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695. [DOI] [PubMed] [Google Scholar]
- Doble A (1996). The pharmacology and mechanism of action of riluzole. Neurology 47, S233–241. [DOI] [PubMed] [Google Scholar]
- Esposito E & Cuzzocrea S (2009). Superoxide, NO, peroxynitrite and PARP in circulatory shock and inflammation. Front Biosci 14, 263–296. [DOI] [PubMed] [Google Scholar]
- Evans RM & Zamponi GW (2006). Presynaptic Ca2+ channels–integration centers for neuronal signaling pathways. Trends Neurosci 29, 617–624. [DOI] [PubMed] [Google Scholar]
- Fauzee NJ, Pan J & Wang YL (2010). PARP and PARG inhibitors: new therapeutic targets in cancer treatment. Pathol Oncol Res 16, 469–478. [DOI] [PubMed] [Google Scholar]
- Frank CA (2014). How voltage‐gated calcium channels gate forms of homeostatic synaptic plasticity. Front Cell Neurosci 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich FJ, Funke F, Hildebrandt B, Fasshauer M & Muller M (2009). H2O2‐mediated modulation of cytosolic signaling and organelle function in rat hippocampus. Pflugers Arch 458, 937–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E & Pelicci PG (2007). Hydrogen peroxide: a metabolic by‐product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8, 722–728. [DOI] [PubMed] [Google Scholar]
- Halliwell B (1992). Reactive oxygen species and the central nervous system. J Neurochem 59, 1609–1623. [DOI] [PubMed] [Google Scholar]
- Halliwell B (2006). Oxidative stress and neurodegeneration: where are we now? J Neurochem 97, 1634–1658. [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K & Mori Y (2002). LTRPC2 Ca2+‐permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Hill K, Benham CD, McNulty S & Randall AD (2004). Flufenamic acid is a pH‐dependent antagonist of TRPM2 channels. Neuropharmacology 47, 450–460. [DOI] [PubMed] [Google Scholar]
- Honda H, Baba H & Kohno T (2011). Electrophysiological analysis of vulnerability to experimental ischemia in neonatal rat spinal ventral horn neurons. Neurosci Lett 494, 161–164. [DOI] [PubMed] [Google Scholar]
- Honda H, Kawasaki Y, Baba H & Kohno T (2012). The mu opioid receptor modulates neurotransmission in the rat spinal ventral horn. Anesth Analg 115, 703–712. [DOI] [PubMed] [Google Scholar]
- Hyslop PA, Zhang Z, Pearson DV & Phebus LA (1995). Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 671, 181–186. [DOI] [PubMed] [Google Scholar]
- Inoue W, Baimoukhametova DV, Füzesi T, Wamsteeker JI, Koblinger K, Whelan PJ, Pittman QJ & Bains JS (2013). Noradrenaline is a stress‐associated metaplastic signal at GABA synapses. Nat Neurosci 16, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H & Bultynck G (2014). Inositol 1,4,5‐trisphosphate receptor‐isoform diversity in cell death and survival. Biochim Biophys Acta 1843, 2164–2183. [DOI] [PubMed] [Google Scholar]
- Jehle T, Bauer J, Blauth E, Hummel A, Darstein M, Freiman TM & Feuerstein TJ (2000). Effects of riluzole on electrically evoked neurotransmitter release. Br J Pharmacol 130, 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J & Sandkuhler J (1998). Corelease of two fast neurotransmitters at a central synapse. Science 281, 419–424. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Fujita T & Kumamoto E (2004). Enhancement of the releases of GABA and glycine during ischemia in rat spinal dorsal horn. Biochem Biophys Res Commun 316, 553–558. [DOI] [PubMed] [Google Scholar]
- Kerchner GA & Nicoll RA (2008). Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci 9, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto E (1997). The pharmacology of amino‐acid responses in septal neurons. Prog Neurobiol 52, 197–259. [DOI] [PubMed] [Google Scholar]
- Laube B, Maksay G, Schemm R & Betz H (2002). Modulation of glycine receptor function: a novel approach for therapeutic intervention at inhibitory synapses? Trends Pharmacol Sci 23, 519–527. [DOI] [PubMed] [Google Scholar]
- Leach MJ, Marden CM & Miller AA (1986). Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of action. Epilepsia 27, 490–497. [DOI] [PubMed] [Google Scholar]
- Lee CR, Witkovsky P & Rice ME (2011). Regulation of substantia nigra pars reticulate GABAergic neuron activity by H2O2 via flufenamic acid‐sensitive channels and KATP channels. Front Syst Neurosci 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Machold RP, Witkovsky P & Rice ME (2013). TRPM2 channels are required for NMDA‐induced burst firing and contribute to H2O2‐dependent modulation in substantia nigra pars reticulate GABAergic Neurons. J Neurosci 33, 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei B, Adachi N & Arai T (1998). Measurement of the extracellular H2O2 in the brain by microdialysis. Brain Res Brain Res Protoc 3, 33–36. [DOI] [PubMed] [Google Scholar]
- Lencesova L & Krizanova O (2012). IP3 receptors, stress and apoptosis. Gen Physiol Biophys 31, 119–130. [DOI] [PubMed] [Google Scholar]
- Li A, Segui J, Heinemann SH & Hoshi T (1998). Oxidation regulates cloned neuronal voltage‐dependent Ca2+ channels expressed in Xenopus oocytes. J Neurosci 18, 6740–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Liu J & Wen J (1999). Elevation of hydrogen peroxide after spinal cord injury detected by using the Fenton reaction. Free Radic Biol Med 27, 478–482. [DOI] [PubMed] [Google Scholar]
- Mailly F, Marin P, Israel M, Glowinski J & Premont J (1999). Increase in external glutamate and NMDA receptor activation contribute to H2O2‐induced neuronal apoptosis. J Neurochem 73, 1181–1188. [DOI] [PubMed] [Google Scholar]
- Marengo JJ, Hidalgo C & Bull R (1998). Sulfhydryl oxidation modifies the calcium dependence of ryanodine‐sensitive calcium channels of excitable cells. Biophys J 74, 1263–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Thompson MA & Nadler JV (1993). The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol 250, 473–476. [DOI] [PubMed] [Google Scholar]
- Masino SA, Mesches MH, Bickford PC & Dunwiddie TV (1999). Acute peroxide treatment of rat hippocampal slices induces adenosine‐mediated inhibition of excitatory transmission in area CA1. Neurosci Lett 274, 91–94. [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Kumamoto E, Furue H & Yoshimura M (2003). GABA‐mediated inhibition of glutamate release during ischemia in substantia gelatinosa of the adult rat. J Neurophysiol 89, 257–264. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ & Silver RA (2000). GABA spillover from single inhibitory axons suppresses low‐frequency excitatory transmission at the cerebellar glomerulus. J Neurosci 20, 8651–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki N, Nakatsuka T, Takeda D, Nohda K, Inoue K & Yoshida M (2008). Adenosine modulates excitatory synaptic transmission and suppresses neuronal death induced by ischaemia in rat spinal motoneurones. Pflugers Arch 457, 441–451. [DOI] [PubMed] [Google Scholar]
- Nani F, Cifra A & Nistri A (2010). Transient oxidative stress evokes early changes in the functional properties of neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci 31, 951–966. [DOI] [PubMed] [Google Scholar]
- Naziroğlu M (2011). TRPM2 cation channels, oxidative stress and neurological diseases: where are we now? Neurochem Res 36, 355–366. [DOI] [PubMed] [Google Scholar]
- Naziroğlu M, Özgül C, Ciğ B & Sözbir E (2011). Aminoethoxydiphenil borate and flufenamic acid inhibit Ca2+ influx through TRPM2 channels in rat dorsal root ganglion neurons activated by ADP‐ribose and rotenone. J Membr Biol 241, 69–75. [DOI] [PubMed] [Google Scholar]
- Nohda K, Nakatsuka T, Takeda D, Miyazaki N, Nishi H, Sonobe H & Yoshida M (2007). Selective vulnerability to ischemia in the rat spinal cord: a comparison between ventral and dorsal horn neurons. Spine 32, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Nowicky AV & Duchen MR (1998). Changes in [Ca2+]i and membrane currents during impaired mitochondrial metabolism in dissociated rat hippocampal neurons. J Physiol 507, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornung G, Shupliakov O, Ottersen OP, Storm‐Mathisen J & Cullheim S (1994). Immunohistochemical evidence for coexistence of glycine and GABA in nerve terminals on cat spinal motoneurones: an ultrastructural study. Neuroreport 5, 889–892. [DOI] [PubMed] [Google Scholar]
- Pagnotta SE, Lape R, Quitadamo C & Nistri A (2005). Pre‐ and postsynaptic modulation of glycinergic and GABAergic transmission by muscarinic receptors on rat hypoglossal motoneurons in vitro. Neuroscience 130, 783–795. [DOI] [PubMed] [Google Scholar]
- Pellegrini‐Giampietro DE, Cherici G, Alesiani M, Carla V & Moroni F (1990). Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia‐induced neuronal damage. J Neurosci 10, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A, Wang DS, Yu J, Lecker I, Brown PM, Bowie D & Orser BA (2014). Hydrogen peroxide increases GABAA receptor‐mediated tonic current in hippocampal neurons. J Neurosci 34, 10624–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier B, Dutar P & Lamour Y (1993). Different effects of omega‐conotoxin GVIA at excitatory and inhibitory synapses in rat CA1 hippocampal neurons. Brain Res 616, 236–241. [DOI] [PubMed] [Google Scholar]
- Quitadamo C, Fabbretti E, Lamanauskas N & Nistri A (2005). Activation and desensitization of neuronal nicotinic receptors modulate glutamatergic transmission on neonatal rat hypoglossal motoneurons. Eur J Neurosci 22, 2723–2734. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Funk GD, Bayliss DA, Dong XW & Feldman JL (2000). Synaptic control of motoneuronal excitability. Physiol Rev 80, 767–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG & Bae YS (1997). Regulation of phosphoinositide‐specific phospholipase C isozymes. J Biol Chem 272, 15045–15048. [DOI] [PubMed] [Google Scholar]
- Rice ME (2011). H2O2: a dynamic neuromodulator. Neuroscientist 17, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roettger V & Lipton P (1996). Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience 75, 677–685. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Aoki M, Abe K, Sadahiro M & Tabayashi K (1997). Selective motor neuron death and heat shock protein induction after spinal cord ischemia in rabbits. J Thorac Cardiovasc Surg 113, 159–164. [DOI] [PubMed] [Google Scholar]
- Schwartz‐Bloom RD & Sah R (2001). γ‐Aminobutyric acidA neurotransmission and cerebral ischemia. J Neurochem 77, 353–371. [DOI] [PubMed] [Google Scholar]
- Shaw PJ & Ince PG (1997). Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J Neurol 244, S3–14. [DOI] [PubMed] [Google Scholar]
- Sivilotti L & Nistri A (1991). GABA receptor mechanisms in the central nervous system. Prog Neurobiol 36, 35–92. [DOI] [PubMed] [Google Scholar]
- Stuart GJ & Redman SJ (1992). The role of GABAA and GABAB receptors in presynaptic inhibition of Ia EPSPs in cat spinal motoneurones. J Physiol 447, 675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumoza‐Toledo A & Penner R (2011). TRPM2: a multifunctional ion channel for calcium signaling. J Physiol 589, 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal W & Holstege JC (1994). GABA and glycine frequently colocalize in terminals on cat spinal motoneurons. Neuroreport 5, 2225–2228. [DOI] [PubMed] [Google Scholar]
- Taccola G, Margaryan G, Mladinic M & Nistri A (2008). Kainate and metabolic perturbation mimicking spinal injury differentially contribute to early damage of locomotor networks in the in vitro neonatal rat spinal cord. Neuroscience 155, 538–555. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Mikami M & Yang J (2007). Hydrogen peroxide increases GABAergic mIPSC through presynaptic release of calcium from IP3 receptor‐sensitive stores in spinal cord substantia gelatinosa neurons. Eur J Neurosci 25, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T (1990). Membrane currents in visually identified motoneurones of neonatal rat spinal cord. J Physiol 423, 27–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T & Momiyama A (1993). Different types of calcium channels mediate central synaptic transmission. Nature 366, 156–158. [DOI] [PubMed] [Google Scholar]
- Thurbon D, Luscher HR, Hofstetter T & Redman SJ (1998. a). Passive electrical properties of ventral horn neurons in rat spinal cord slices. J Neurophysiol 79, 2485–2502. [DOI] [PubMed] [Google Scholar]
- Thurbon D, Luscher HR, Hofstetter T & Redman SJ (1998. b). Passive electrical properties of ventral horn neurons in rat spinal cord slices. J Neurophysiol 80, 2485–2502. [DOI] [PubMed] [Google Scholar]
- Togashi K, Inada H & Tominaga M (2008). Inhibition of the transient receptor potential cation channel TRPM2 by 2‐aminoethoxydiphenyl borate (2‐APB). Br J Pharmacol 153, 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A (2002). The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium 32, 393–404. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L & Catterall WA (1998). Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci 18, 6319–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JR & Fehlings MG (2014). Riluzole for acute traumatic spinal cord injury: a promising neuroprotective treatment strategy. World Neurosurg 81, 825–829. [DOI] [PubMed] [Google Scholar]
- Xu SZ, Zeng F, Boulay G, Grimm C, Harteneck C & Beech DJ (2005). Block of TRPC5 channels by 2‐aminoethoxydiphenyl borate: a differential, extracellular and voltage‐dependent effect. Br J Pharmacol 145, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Honda H, Baba H & Kohno T (2012). Effect of xenon on excitatory and inhibitory transmission in rat spinal ventral horn neurons. Anesthesiology 116, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Zenisek D & Matthews G (2000). The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25, 229–237. [DOI] [PubMed] [Google Scholar]