

Key points
Single‐channel recordings in CA3 pyramidal neurons revealed that large‐conductance calcium‐activated K+ (BK) channel open probability was reduced by loss of fragile X mental retardation protein (FMRP) and that FMRP acts on BK channels by modulating the channel's gating kinetics.
Fmr1/BKβ4 double knockout mice were generated to genetically upregulate BK channel activity in the absence of FMRP.
Deletion of the BKβ4 subunit alleviated reduced BK channel open probability via increasing BK channel open frequency, but not through prolonging its open duration.
Genetic upregulation of BK channel activity via deletion of BKβ4 normalized action potential duration, excessive glutamate release and short‐term synaptic plasticity during naturalistic stimulus trains in excitatory hippocampal neurons in the absence of FMRP.
Genetic upregulation of BK channel activity via deletion of BKβ4 was sufficient to normalize excessive epileptiform activity in an in vitro model of seizure activity in the hippocampal circuit in the absence of FMRP.
Abstract
Loss of fragile X mental retardation protein (FMRP) causes fragile X syndrome (FXS), yet the mechanisms underlying the pathophysiology of FXS are incompletely understood. Recent studies identified important new functions of FMRP in regulating neural excitability and synaptic transmission via both translation‐dependent mechanisms and direct interactions of FMRP with a number of ion channels in the axons and presynaptic terminals. Among these presynaptic FMRP functions, FMRP interaction with large‐conductance calcium‐activated K+ (BK) channels, specifically their auxiliary β4 subunit, regulates action potential waveform and glutamate release in hippocampal and cortical pyramidal neurons. Given the multitude of ion channels and mechanisms that mediate presynaptic FMRP actions, it remains unclear, however, to what extent FMRP–BK channel interactions contribute to synaptic and circuit defects in FXS. To examine this question, we generated Fmr1/β4 double knockout (dKO) mice to genetically upregulate BK channel activity in the absence of FMRP and determine its ability to normalize multilevel defects caused by FMRP loss. Single‐channel analyses revealed that FMRP loss reduced BK channel open probability, and this defect was compensated in dKO mice. Furthermore, dKO mice exhibited normalized action potential duration, glutamate release and short‐term dynamics during naturalistic stimulus trains in hippocampal pyramidal neurons. BK channel upregulation was also sufficient to correct excessive seizure susceptibility in an in vitro model of seizure activity in hippocampal slices. Our studies thus suggest that upregulation of BK channel activity normalizes multi‐level deficits caused by FMRP loss.
Key points
Single‐channel recordings in CA3 pyramidal neurons revealed that large‐conductance calcium‐activated K+ (BK) channel open probability was reduced by loss of fragile X mental retardation protein (FMRP) and that FMRP acts on BK channels by modulating the channel's gating kinetics.
Fmr1/BKβ4 double knockout mice were generated to genetically upregulate BK channel activity in the absence of FMRP.
Deletion of the BKβ4 subunit alleviated reduced BK channel open probability via increasing BK channel open frequency, but not through prolonging its open duration.
Genetic upregulation of BK channel activity via deletion of BKβ4 normalized action potential duration, excessive glutamate release and short‐term synaptic plasticity during naturalistic stimulus trains in excitatory hippocampal neurons in the absence of FMRP.
Genetic upregulation of BK channel activity via deletion of BKβ4 was sufficient to normalize excessive epileptiform activity in an in vitro model of seizure activity in the hippocampal circuit in the absence of FMRP.
Abbreviations
- AHP
afterhyperpolarization
- AP
action potential
- APV
2‐amino‐5‐phosphonopentanoic acid
- BK channel
large‐conductance calcium‐activated K+ channel
- dKO
Fmr1/β4 double knockout (mice)
- DNQX
6,7‐dinitroquinoxaline‐2,3‐dione
- EPSC
excitatory postsynaptic current
- Fmr1
fragile X mental retardation 1 gene
- FMRP
fragile X mental retardation protein
- FXS
fragile X syndrome
- ISI
inter‐stimulus interval
- KO
knock out
- mGluR
metabotropic glutamate receptor
- PC
pyramidal cell
- PPR
paired‐pulse ratio
- SC
Schaffer collaterals
- SR
stratum radiatum
- VGCCs
voltage‐gated calcium channels
- WT
wild‐type
Introduction
Transcriptional silencing of the Fmr1 gene encoding fragile X mental retardation protein (FMRP) is the most common inheritable cause of intellectual disability, known as FXS. This disorder is also characterized by high co‐morbidity with autism and epilepsy (Pfeiffer & Huber, 2009). FMRP is widely recognized to play important roles in synaptic function, yet how FMRP loss leads to synaptic and circuit defects in FXS remains incompletely understood (Brager & Johnston, 2014; Contractor et al. 2015).
While FMRP has been widely recognized to play a major role as a regulator of local translation in axons and dendrites, recent studies indicate that a number of FMRP actions, particularly its regulation of neuronal excitability and neurotransmitter release, are, at least in part, translational‐independent and mediated by FMRP interactions with a number of ion channels (Brown et al. 2010; Zhang et al. 2012; Deng et al. 2013 a; Myrick et al. 2015). FMRP directly modulates gating of a sodium‐activated potassium (K+) channel Slack to regulate neural excitability (Brown et al. 2010; Zhang et al. 2012), while FMRP binding to presynaptic N‐type voltage‐gated calcium channels (VGCCs) regulates their surface expression, presynaptic calcium influx and neurotransmitter release in dorsal root ganglion (DRG) neurons (Ferron et al. 2014). We recently identified FMRP function in regulating action potential (AP) duration and neurotransmitter release in excitatory central neurons mediated by FMRP's interaction with large‐conductance calcium‐activated K+ (BK) channels, particularly their regulatory β4 subunits (Deng et al. 2013 a). Evidence that FMRP modulates AP duration both in hippocampal and cortical pyramidal neurons suggests that this BK channel‐dependent regulation of presynaptic function by FMRP may be a widespread phenomenon contributing to the synaptic deficits in FXS (Deng et al. 2013 a; Zhang et al. 2014). Indeed, contribution of BK channel dysfunctions to FXS behavioural and cognitive phenotypes has been suggested by several recent studies (Laumonnier et al. 2006; Higgins et al. 2008; Skafidas et al. 2012; Zhang et al. 2014) and FMRP–BK channel interactions have been implicated in a specific subset of fragile X phenotypes found in a patient with an Fmr1 missense mutation (Myrick et al. 2015). However, given the multitude of FMRP interacting partners and mechanisms mediating FMRP presynaptic functions, the contributions of BK channel deficits to the synaptic and circuit pathophysiology of FXS remains poorly understood.
To examine the role of BK channels in multi‐level deficits in FXS mouse model, we generated Fmr1/β4 double KO (dKO) mice. Within a range of physiological calcium concentrations below several tens of micromolar, β4 serves as a negative regulator of BK channel activity (Brenner et al. 2000; Ha et al. 2004). Therefore our working model predicts that genetic deletion of β4 should increase BK channel activity and lead to improvements in BK channel‐dependent FMRP functions. Our analyses of dKO mice revealed that genetic deletion of BKβ4 normalized a number of defects associated with FMRP loss spanning from single‐channel to synaptic and neuronal levels, and also significantly reduced seizure susceptibility in hippocampal circuits, which is abnormally elevated in Fmr1 KO mice. These results suggest that BK channel dysfunction plays a major role in synaptic and circuit deficits in the FXS mouse model.
Methods
Ethical approval
All animal procedures conformed to the National Institutes of Health guidelines and were approved by the Washington University Animal Studies Committee.
Animals
Fmr1 KO and control strain mice on FVB background were obtained from the Jackson Laboratory. Sloβ4 KO mice on C57/BL/6 background were generously provided by Dr Robert Brenner (University of Texas, San Antonio, TX USA). Mice were fed ad libitum. The dKO mice were generated in a two‐level breeding scheme using Fmr1 KO females and β4 KO males and thus have a mixed FVB/C57BL/6 genetic background. To control for the background variation, we backcrossed wild‐type (WT) FVB and Fmr1 KO FVB mice to C57BL/6 WT mice to match the mixed FVB/C57BL/6 background of the dKO mice. In most recordings, WT, Fmr1 KO and dKO mice on the same mixed FVB/C57BL/6 background were used. Analysis of AP duration in WT and Fmr1 KO mice on FVB, C57/BL/6 or mixed FVB/C57/BL/6 backgrounds (Fig. 2 C and D), indicated that genetic background did not have any measurable effects in our recordings. Similarly, we did not observe any measureable effects of genetic background in recordings of synaptic transmission (Fig. 4 D) or in recordings of circuit excitability (see text for details). Data on FVB and mixed backgrounds were therefore pooled in all measurements.
Figure 2. BKβ4 deletion normalizes excessive AP broadening defects in excitatory CA3 neurons caused by loss of FMRP .
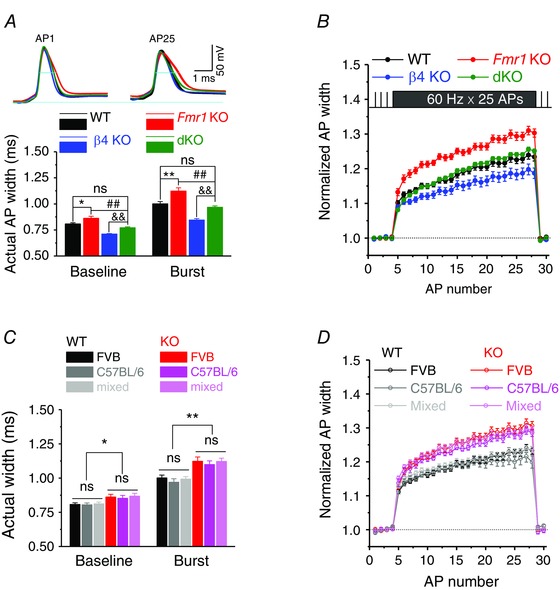
A, upper panel, sample traces of the 1st and last APs of the 25‐AP train at 60 Hz. Long line indicates the baseline of the first APs, short lines AP duration measured at −10 mV level. A, lower panel, summarized data of actual AP widths for baseline and burst. B, time course of normalized AP width during 25‐AP train at 60 Hz. AP duration was normalized to its own basal values. C, grouped data of actual AP widths of baseline and burst for different background mice. D, time course of normalized AP width during 25‐AP train at 60 Hz among different background mice. *P < 0.05, **P < 0.01 vs. WT; ## P < 0.01 vs. Fmr1 KO; && P < 0.01 vs. β4 KO; ns, not significant. Error bars represent SEM.
Figure 4. BKβ4 deletion corrects neurotransmission and short‐term dynamics defects at excitatory hippocampal CA3–CA1 synapses in the absence of FMRP .
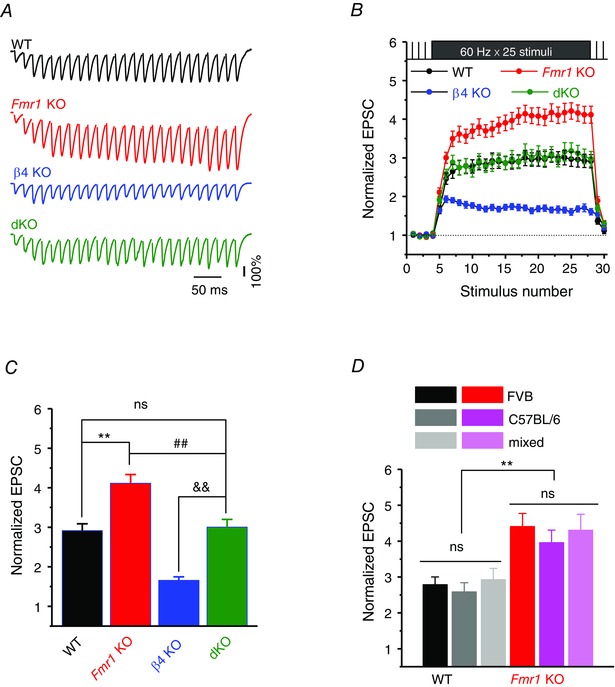
A, normalized EPSC traces recorded in CA1 PCs in response to SC stimulation with a train of 25 stimuli at 60 Hz. Note that EPSC traces were normalized to their own baseline amplitude measured at 0.2 Hz of the same train for better visual comparison (the first EPSCs of the burst in each condition were scaled to the same value, representing 100% shown as a bar). EPSC stimulus artifacts were removed for clarity. B, summarized data of EPSC amplitude during the train. Data were normalized to their own baseline. C, normalized EPSC amplitudes averaged from the last two EPSCs of the burst. D, the normalized EPSC amplitudes at the end of burst for WT and KO mice among different backgrounds. **P < 0.01, ## P < 0.01, && P < 0.01 vs. β4 KO; ns, not significant. Error bars represent SEM.
Slice preparation
Slices were prepared as previously described (Deng et al. 2011). In brief, both male and female 16‐ to 20‐day‐old mice were used. Animals were killed with carbon dioxide narcosis followed by decapitation. Carbon dioxide was administered by inhalation and delivered at a flow rate of 20% chamber volume displacement per minute, using a flow meter to control delivery. Brains were dissected out in ice‐cold saline containing the following (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 5.0 MgCl2, and 10 glucose, pH 7.4 (saturated with 95% O2 and 5% CO2). Horizontal brain slices (350 μm) including the hippocampi were cut using a vibrating microtome (Leica VT1100S). Slices were initially incubated in the above solution at 35°C for 1 h for recovery and then kept at room temperature (∼23°C) until use.
Single‐channel recordings
Single‐channel recordings were performed in inside‐out configuration at 33–34°C using an Axopatch 700B (Molecular Devices) in CA3 pyramidal cells (PCs). The pipette solution contained (in mm): 140 KCl, 10 HEPES, 10 4‐aminopyridine, 1 MgCl2, and 0.001 TTX; pH 7.35. Once the presence of an active BK channel in the patch was determined, the bath was replaced with a HEPES‐based solution containing (in mm): 140 KCl, 10 HEPES, 2 MgCl2, 2 EGTA and Ca2+ in appropriate concentrations to yield the desired free Ca2+ concentrations, estimated using MaxChelator (http://maxchelator.stanford.edu/). BK channels were identified based on the large amplitude of unitary currents and, in a subset of recordings, by their paxilline sensitivity. Only patches containing single BK channels were included in analysis.
Action potential recordings
APs were recorded from current‐clamped CA3 PCs at 33–34°C. The recording electrodes were filled with the following (in mm): 130 potassium gluconate, 0.5 EGTA, 2 MgCl2, 5 NaCl, 2 ATPNa2, 0.4 GTPNa, and 10 Hepes, pH 7.3. The extracellular solution contained (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 and 10 glucose, with APV (50 μm), DNQX (10 μm), gabazine (5 μm), pH 7.4 (saturated with 95% O2 and 5% CO2). For evoked APs recordings, APs were evoked by repetitive injection of a 1‐ms current to evoke a 25‐AP train at 60 Hz. Membrane potential was set at −65 mV by automatic slow current injection to ensure stability of the resting potential and to prevent spontaneous AP firing. Each AP train was evoked four to six times in each neuron, and each train was separated by a 2‐min rest period. AP duration during bursts was normalized to an average duration of four low‐frequency APs (0.2 Hz) evoked before each train. Trains at 60 Hz have fractional 16.67 ms inter‐stimulus intervals (ISIs). Because of software rounding of submillisecond timing to a whole millisecond, every third ISI is 1 ms shorter in our 60 Hz stimulus trains, causing apparent periodicity in the data (Deng et al. 2013 b). This periodicity was no longer present in 58.8 Hz trains (constant 17 ms ISIs) (Deng et al. 2013 b). This effect is very small (<2%) and is present in all recordings at 60 Hz.
For spontaneous APs recordings, the resting membrane potential was depolarized to ∼−51 mV via automatic slow current injection. AP duration was determined as the time interval between AP upstroke and downstroke at −10 mV level, to avoid potential errors introduced by variations in the baseline during AP bursts. The amplitude of fAHP was defined as the voltage difference between resting membrane potential before an AP and the lowest point of corresponding fAHP.
Whole‐cell EPSC recordings
EPSCs were recorded from CA1 pyramidal neurons in whole‐cell configuration at 33–34°C at a holding potential of −65 mV by stimulating Schaffer collaterals (SCs) with a monopolar electrode in the presence of gabazine (5 μm) and APV (50 μm). EPCSs during the stimulus trains were normalized to an average of four low‐frequency (0.2 Hz) control stimuli preceding each train to produce changes in synaptic strength. Each stimulus train was presented four to six times in each cell, and each presentation was separated by 2 min. The overlap of EPSCs at short ISIs was corrected for using a normalized scaled template of EPSC waveform as described previously (Deng et al. 2013 a).
Recordings of spontaneous epileptiform activity
Spontaneous epileptiform activity was induced in hippocampal slices as previously described (Perreault & Avoli, 1991; Gonzalez‐Sulser et al. 2012) by adding 4‐aminopyridine (4‐AP, 100 μm) to the standard extracellular solution described above. In addition, extracellular KCl concentration was elevated to 5 mm. Simultaneous field potential recording and whole‐cell current‐clamp recording were made in the CA3 region at 33–34°C. Briefly, a field recording pipette containing 2 m NaCl was placed in CA3 pyramidal layer to record epileptiform activity as described previously (Perreault & Avoli, 1991; Gonzalez‐Sulser et al. 2012). Whole‐cell AP recordings were made from CA3 PCs situated 50—100 μm away from field recording pipettes. Latency of epileptiform activity was determined by the time interval between initiation of 4‐AP perfusion and the first epileptiform discharge. Frequency and duration of epileptiform events were determined from a 4‐min time period following stabilization of epileptiform activity. For the number of APs per epileptiform event, only APs with a peak potential >0 mV were counted. The epileptiform events were analysed in MatLab or Mini Analysis.
Statistical analysis
Data are presented as means ± SEM. Student's unpaired t test, or one‐way ANOVA with post hoc Bonferroni multiple comparison procedures were used for statistical analysis as appropriate; significance was set as P < 0.05; n was number of neurons tested.
Results
Genetic deletion of BKβ4 compensates the decreased BK channel open probability caused by FMRP loss
Our recent studies indicate that loss of FMRP decreases whole‐cell BK currents in CA3 pyramidal cells (Deng et al. 2013 a). However, how FMRP modulates BK channel activity remains poorly understood. Therefore, we examined the single channel properties of BK channels at ∼33°C in CA3 pyramidal cells in acute brain slices from Fmr1 KO and WT mice. Single channel recordings were performed in inside‐out configuration at the holding potential of −80 mV and free Ca2+ concentration of 10 μm. We found that loss of FMRP caused a significant decrease in BK channel open probability (n = 6 (WT); 6 (KO): P < 0.0001; Fig. 1 A, B and D), without detectable changes in single channel conductance (P = 0.45; Fig. 1 C). These results suggest that FMRP modulates gating of BK channels in excitatory hippocampal neurons. These findings also provide support for a non‐canonical function of FMRP in regulation of ion channel activity previously only described for SlackB channels (Brown et al. 2010; Zhang et al. 2012).
Figure 1. BKβ4 deletion alleviates the decreased BK channel open probability in the absence of FMRP .
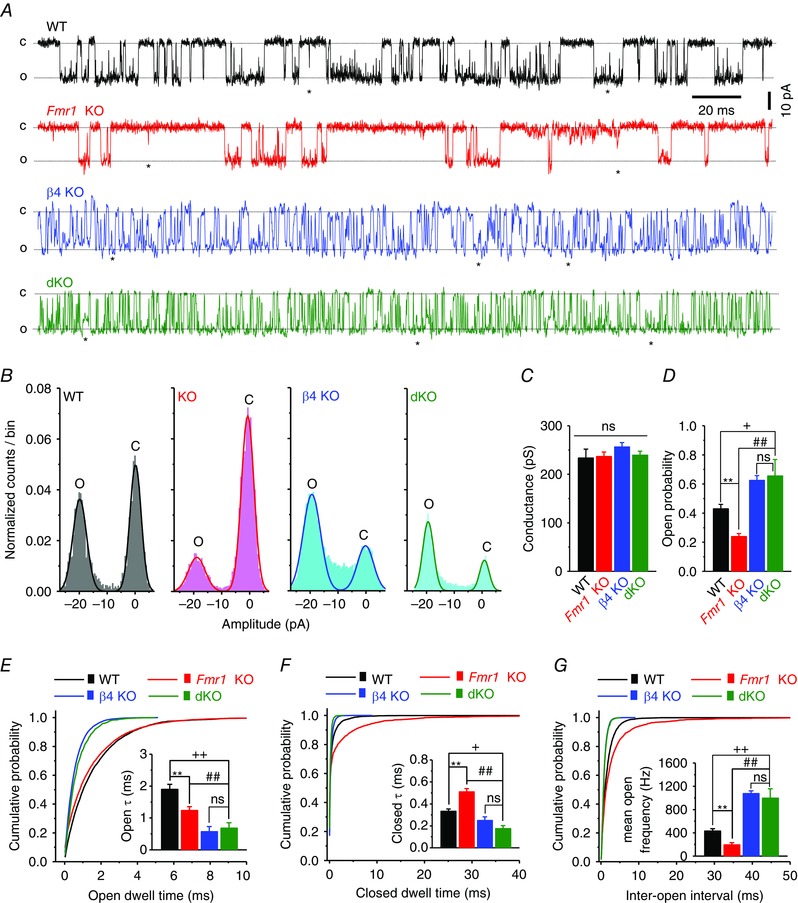
A, traces of BK single channel recordings at −80 mV, 10 μm free Ca2+, 33°C, from CA3 PCs of acute hippocampal slices. C, closed state; O, open state; ★, subconductance state. B–D, all‐point amplitude histograms (B) were fitted by two Gaussian fits, representing the open (peak around −20 pA) and closed (peak around 0 pA) states. BK unitary current was determined by the amplitude difference between two peaks of Gaussian fits, which then was used to estimate BK single channel conductance (C). Open probabilities were calculated by the area under the Gaussian fits [open area/(open area + closed area)], and summarized in D. E–G, representative cumulative probability of open dwell time (E), closed dwell time (F) and inter‐open intervals (G). Insets represent the mean open dwell time constant (E), the mean closed dwell time constant (F), and the mean open frequency (G). **P < 0.01 Fmr1 KO vs. WT; ## P < 0.01 dKO vs. KO; + P < 0.01, ++ P < 0.01 dKO vs. WT; ns, not significant. Error bars represent SEM.
Our current model suggests that FMRP regulates BK channel activity predominately via interactions with the BKβ4 subunit, and that in normal neurons FMRP acts to counter‐balance the β4's negative regulation of BK channel activity. This β4‐suppressing action of FMRP is lost in Fmr1 KO neurons, leading to reduced BK channel activity, excessive AP broadening and exaggerated glutamate release in the absence of FMRP. We therefore hypothesized that interference with BKβ4 should reduce or alleviate these defects in the absence of FMRP. To test this idea and start to determine the role of BK channels in synaptic abnormalities in FXS, we created Fmr1 and β4 double knockout mice (dKO) using Fmr1 KO females and β4 KO males in a two‐stage breeding scheme. In agreement with the role of β4 as a negative regulator of BK channel activity (Brenner et al. 2000; Ha et al. 2004), BK channel open probability was significantly increased in dKO mice compared with Fmr1 KO mice (n = 6 (KO), 6 (dKO); P < 0.001; Fig. 1 A, B and D), while the single‐channel conductance was unaffected (P = 0.93; ANOVA; Fig. 1 C).
We also observed that loss of FMRP significantly shortened the BK channel open dwell time constant (P = 0.0052; Fig. 1 E), prolonged the channel's closed dwell time constant (P = 0.0011; Fig. 1 F), and reduced the channel open frequency compared with WT controls (P < 0.0001; Fig. 1 G). As expected from the characteristic effect of β4 on slowing the channel gating, BK channels in β4 KO or dKO neurons exhibited much more frequent switching between open and closed states, as can be seen by the increased channel open frequency (n = 7 (β4 KO), 6 (dKO); Fig. 1 G). As a result of this rapid gating, both open and closed dwell times were much shorter in dKO neurons than either in WT or Fmr1 KOs (Fig. 1 E and F). These results indicate that FMRP regulates BK channel activity by modulating the channel gating kinetics and that deletion of BKβ4 corrects BK channel open probability via increasing BK channel open frequency, but not through prolonging its open duration (i.e. open dwell time).
Genetic deletion of BKβ4 normalizes defects in AP duration caused by FMRP loss in excitatory hippocampal neurons
Our previous studies suggested that BK channel‐mediated modulation of AP duration is a major mechanism by which FMRP regulates neurotransmitter release in hippocampal excitatory neurons. We thus first assessed the effects of β4 deletion on AP waveform in dKO mice during high‐frequency AP trains or spontaneous AP firing. APs were recorded in current‐clamped CA3 pyramidal cells
(PCs) in 16‐ to 20‐day‐old dKO mice and compared to age‐matched Fmr1 KO, b4 KO and WT mice. Recordings were performed at near‐physiological temperature (∼33°C) in the presence of APV (50 μm), DNQX (10 μm) and gabazine (5 μm).
To examine the effects of β4 deletion on evoked APs, trains of 25 stimuli at 60 Hz were used and the resting membrane potential (RMP) was set at −65 mV via automatic slow current injection to avoid spontaneous AP firing. As we reported previously (Deng et al. 2013 a; Myrick et al. 2015), AP duration was significantly prolonged in Fmr1 KO relative to WT mice for both single APs and APs during the trains (Fig. 2 A and B). In contrast, AP duration was significantly shorter in β4 KO compared to WT mice (Fig. 2 A and B), as expected from the increased BK channel activity in β4 KO animals. Most importantly, we found that excessive AP broadening defect observed in Fmr1 KO mice was normalized in dKO mice and was not significantly different from WT for both single evoked APs at 0.2 Hz (n = 15 (WT), 18 (Fmr1 KO), 15 (β4 KO) 26 (dKO); dKO v.s KO, P = 0.009; dKO vs. WT, P = 0.396; Fig. 2 A and B), as well as for APs during high‐frequency trains (dKO vs. KO, P = 0.0095; dKO vs. WT, P = 0.46; Fig. 2 A and B). The AP amplitude was not significantly different in KO and WT mice (Deng et al. 2013 a) and was not altered in dKO mice (baseline: P = 0.69, ANOVA; at the end of burst: P = 0.78, ANOVA). Together, these results indicate that genetic deletion of β4 normalizes the AP broadening defect observed in Fmr1 KO mice. These data support a critical role of BK channels as the major mechanism mediating effects of FMRP loss on AP waveform.
We note that in our earlier studies of the AP broadening defects in Fmr1 KO mice (Deng et al. 2013 a) we used Fmr1 KO and WT mice on FVB genetic background, while in the current study we used WT, Fmr1 KO and dKO mice on a mixed FVB/C57BL/6 background. We therefore tested whether genetic background may contribute to our measurements of AP duration. The AP widths were measured in WT and Fmr1 KO mice on FVB, C57BL/6, and mixed FVB/C57BL/6 backgrounds. We found that the differences in AP duration between KO and WT mice were conserved among all backgrounds for both baseline APs and APs during high‐frequency trains (Fig. 2 C and D). These results suggest that the effects of FMRP loss on AP duration and its normalization in dKO mice are independent of the mice genetic background.
We then assessed the effects of β4 deletion on spontaneous AP firing in dKO mice. Spontaneous APs were recorded in CA3 pyramidal neurons using the approach described previously (Deng et al. 2013 a) at a membrane potential of ∼−51 mV set via automatic slow current injection. As we reported previously, the duration of spontaneous APs was excessively prolonged in Fmr1 KO mice relative to WT at all firing frequencies (Deng et al. 2013 a). This excessive broadening of spontaneous APs was normalized in dKO mice and was indistinguishable from WT at all firing frequencies (Fig. 3 A and B). The RMP and AP amplitude were not different significantly among groups (RMP: WT −52.0 ± 1.9 mV, n = 19; Fmr1 KO −51.6 ± 1.7 mV, n = 20; β4 KO 51.1 ± 2.1 mV, n = 15; dKO −51.9 ± 1.5 mV, n = 26, P = 0.563, ANOVA; AP amplitude: WT 100.2 ± 1.15 mV; Fmr1 KO 99.6 ± 1.29 mV; β4 KO 97.8 ± 2.8 mV; dKO 98.5 ± 1.5 mV, P = 0.625, ANOVA). We further examined another aspect of AP waveform that was abnormal in Fmr1 KO mice, the fast after‐hyperpolarization (fAHP), which depends in a large part on BK channel activity in CA3 PCs and was strongly reduced in Fmr1 KOs (Deng et al. 2013 a). We observed a significant increase of fAHP in dKO mice (KO vs. WT, P < 0.001; dKO vs. KO, P < 0.001; dKO vs. WT, P = 0.015; Fig. 3 C and D). It is noteworthy that fAHP in dKO failed to reach the WT levels, suggesting that additional mechanisms may contribute to fAHP reduction in the absence of FMRP.
Figure 3. BKβ4 deletion ameliorates spontaneous AP defects in excitatory CA3 neurons caused by loss of FMRP .
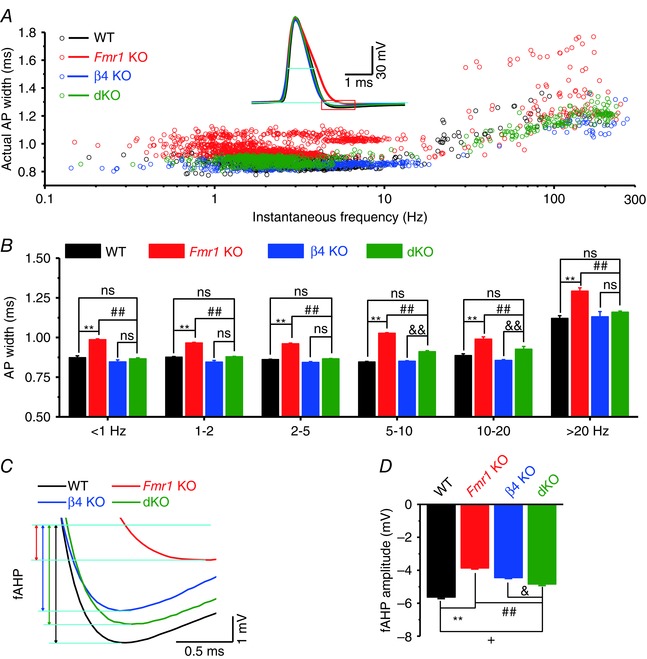
A, spontaneous AP (sAP) duration measured at −10 mV level. Inset, sample sAP traces showing the level of AP duration measured (short cyan line). Red‐boxed area is enlarged in C. B, data from A averaged by frequency bands shown on x‐axis. C, enlargement of the red box in A indicates the fAHP measurements. fAHP is defined by the potential difference between the baseline and the lowest point within 5 ms after AP peak. Baseline was determined as an average of a 0.5‐ms time period in the recording trace immediately before AP initiation (2–2.5 ms before individual AP threshold). D, summarized data of fAHP amplitude. **P < 0.01, + P < 0.05 vs. WT; ## P < 0.01 vs. KO; & P < 0.05, && P < 0.01 vs. β4 KO; ns, not significant. Error bars represent SEM.
Genetic deletion of BKβ4 normalizes synaptic transmission defects in excitatory hippocampal synapses caused by loss of FMRP
AP duration is a major determinant of presynaptic calcium influx and neurotransmitter release. Given that the AP broadening defect is normalized in hippocampal CA3 neurons of dKO mice, we next tested whether genetic deletion of β4 also normalizes the defects in synaptic transmission caused by FMRP loss. Synaptic transmission was assessed using whole‐cell recordings of EPSCs evoked in CA1 PCs by Schafer collateral stimulation. As we reported previously, loss of FMRP leads to excessive enhancement of neurotransmitter release during repetitive activity (Deng et al. 2011, 2013 a; Fig. 4 A–C). In contrast, deletion of β4 alone led to a significant reduction in release relative to WT mice during trains (n = 15 (WT), 8 (β4 KO); P < 0.001, Fig. 4 A–C), which is consistent with the narrower APs in β4 KO animals (Fig. 2 A–B). Most importantly, we found that synaptic transmission during constant‐frequency stimulation (25 stimuli, 60 Hz) was normalized in dKO mice and was indistinguishable from WT (n = 14 (WT), 14 (Fmr1 KO), 8 (β4 KO), 10 (dKO); KO vs. WT, P < 0.001; dKO vs. KO, P < 0.001; dKO vs. WT, P = 0.397; Fig. 4 A–C).
As in measurements of AP duration, we note that in our earlier studies we used Fmr1 KO and WT mice on FVB genetic background, while in the current study WT, Fmr1 KO and dKO mice have a mixed FVB/C57BL/6 background. We therefore tested whether mice genetic background may affect our measurements of synaptic transmission. EPSCs were evoked by trains of Schafer collateral stimuli and recorded as described above in WT and Fmr1 KO mice on FVB, C57BL/6, and mixed FVB/C57BL/6 backgrounds. We found that synaptic transmission during trains and the differences between Fmr1 KO and WT mice were conserved among all backgrounds (Fig. 4 D). These results suggest that the effects of FMRP loss on synaptic transmission and its normalization in dKO mice are independent of the mice genetic background. The data on FVB and mixed FVB/C57BL/6 backgrounds were therefore pooled.
We further examined synaptic transmission during naturalistic stimulus patterns recorded in vivo from hippocampal place cells in exploring rodents (provided by Drs A. Fenton and R. Muller, SUNY; Fenton & Muller, 1998), as a more realistic representation of the spike frequencies and inter‐spike intervals that hippocampal synapses may experience in vivo. Here, we used a long segment of such naturalistic spike trains, consisting of 128 spikes with a mean burst frequency of 38 ± 27 Hz (mean ± SD) and duration of 15 ± 6 spikes burst−1 (mean ± SD; Fig. 5 A). We have previously shown that, similarly to constant‐frequency trains, synaptic transmission was markedly elevated in Fmr1 KO mice during naturalistic spike trains at all frequencies above 5 Hz (Fig. 5 A–C). Here we found that this defect was normalized in dKO mice and was not different from WT levels at all frequencies (n = 12 (WT), 14 (Fmr1 KO), 7 (β4 KO), 8 (dKO); average gain during bursts: KO vs. WT, P < 0.001; dKO vs. WT, P = 0.355; Fig. 5 A–C). Taken together, these results indicate that genetic deletion of BKβ4 normalized defects in synaptic transmission in excitatory hippocampal synapses caused by the loss of FMRP.
Figure 5. BKβ4 deletion corrects abnormal short‐term dynamics during natural stimulus patterns at excitatory hippocampal CA3–CA1 synapses in the absence of FMRP .
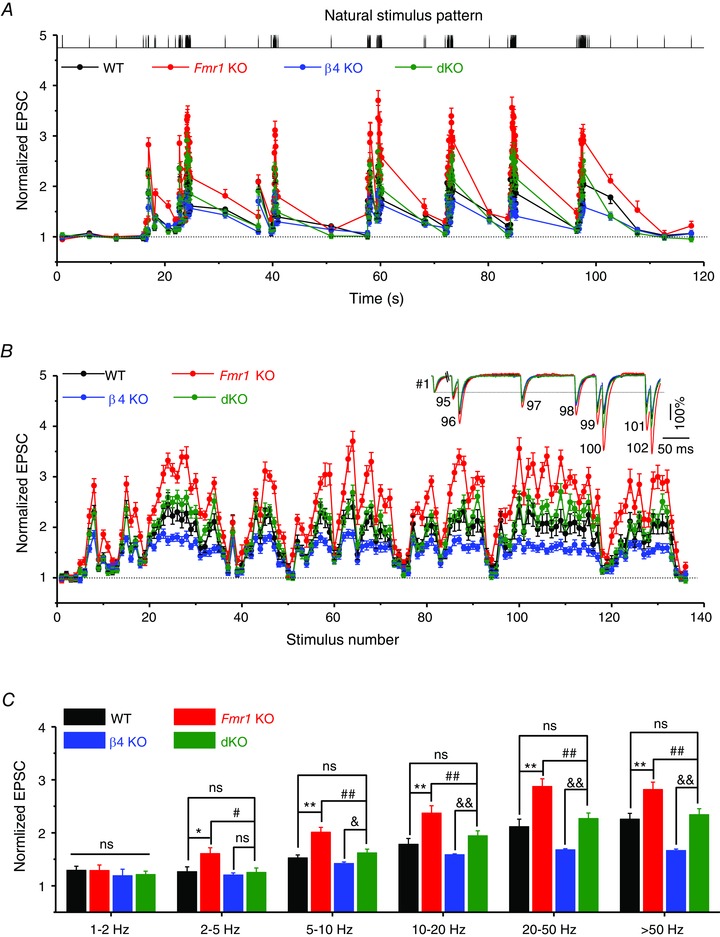
A, changes in EPSC amplitude during natural stimulus train plotted as a function of time. The top shows the natural stimulus pattern used (Fenton & Muller, 1998), preceded and followed by four control stimuli at 0.2 Hz. B, same data as in A but plotted as a function of stimulus number for better visual comparison. Inset shows EPSCs 95–102 during the natural stimulus train, scaled to their own controls. C, average synaptic gain during natural stimulus trains plotted as a function of frequency bands. *P < 0.05, # P < 0.05, **P < 0.01, ## P < 0.01, & P < 0.05, && P < 0.01 vs. β4 KO; ns, not significant. Error bars represent SEM.
Genetic deletion of BKβ4 alleviates increased seizure susceptibility caused by FMRP loss in vitro
Excitation–inhibition (E–I) imbalance and seizure activity are common in many circuits of Fmr1 KO mice and have been proposed to play major roles in the neuropathology of FXS (Contractor et al. 2015). Because BK channels have been reported to preferentially regulate glutamate over GABA release in hippocampal and cortical circuits (Martire et al. 2010) and considering the above evidence that glutamate release is normalized in dKO mice, we examined whether β4 deletion also alters circuit hyperexcitability and seizures in the absence of FMRP. We used a common in vitro model of seizure activity (Perreault & Avoli, 1991; Gonzalez‐Sulser et al. 2012) induced in the CA3 area of hippocampal slices by bath application of a broad‐spectrum K+ channel blocker 4‐AP (to which BK channels are insensitive). This treatment induces epileptiform discharges in WT hippocampal circuits, which we recorded using simultaneous whole‐cell and field potential recordings to distinguish single‐cell firing events from the circuit activity (Fig. 6). As expected from circuit hyperexcitability in Fmr1 KO mice, the latency to the first discharge, a widely used measure of circuit excitability, was markedly reduced in the absence of FMRP (n = 7 (WT); 8 (KO); P < 0.001; Fig. 6 E), and the frequency of discharges was significantly increased (P < 0.001; Fig. 6 A, B and F). The duration of epileptiform discharges and the number of APs per discharge were not significantly different between Fmr1 KO and WT slices (duration: P = 0.62; AP number per event: P = 0.55; Fig. 6 G and H). Importantly, both the latency and discharge frequency were normalized in dKO mice to WT levels (n = 8 (dKO); latency: P = 0.26 vs. WT; frequency: P = 0.60 vs. WT; Fig. 6 E and F), while the discharge duration and the number of APs per discharge remained unaffected (Fig. 6 G and H).
Figure 6. BKβ4 deletion normalizes elevated epileptiform activity in the hippocampal slices caused by FMRP loss in vitro .
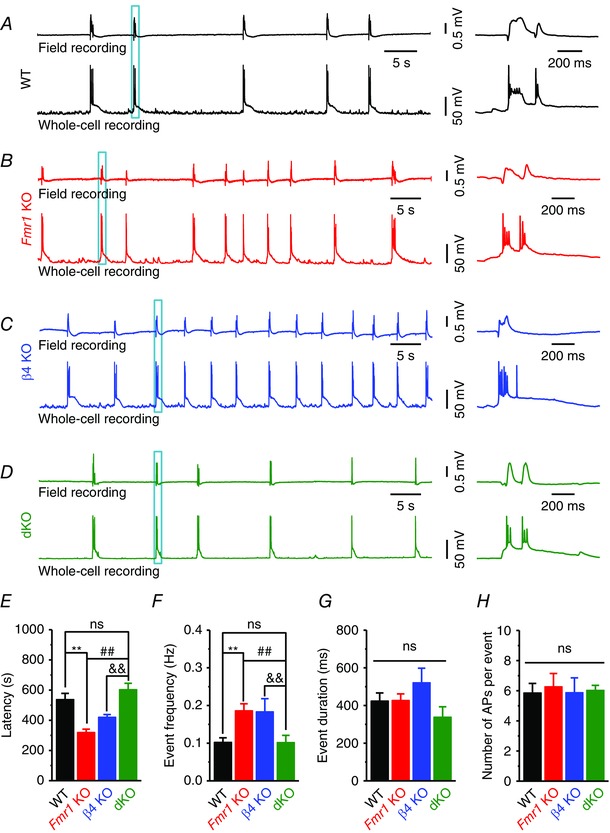
Epileptiform activity in slices was induced by bath application of 4‐AP (100 μm). Field recording electrodes were put at CA3 PC layer; APs recordings were made by whole‐cell configuration of CA3 PCs 50–100 μm apart from field recording electrodes. A–D, simultaneous field recordings of epileptiform activity from CA3 PC layer and whole‐cell recording of APs from CA3 PCs. Right panels are enlarged plots of the boxed area in the left traces. E, pooled data showing the epileptiform activity latency, i.e. time interval from 4‐AP application to the occurrence of first event. F, frequencies of epileptiform activity measured upon even stabilization. G, duration of epileptiform events. H, number of APs per epileptiform event. Only the APs with a peak potential above 0 mV were counted. **P < 0.01 vs. WT; ## P < 0.01 vs. KO; && P < 0.01 vs. β4 KO; ns, not significant. Error bars represent SEM.
As in measurements on AP duration and synaptic transmission, we also tested whether genetic background may contribute to measurements of circuit excitability. Recordings of 4‐AP evoked seizure activity were performed as described above in WT and Fmr1 KO mice on FVB, C57BL/6 and mixed FVB/C57BL/6 backgrounds. We found that all main parameters of discharges and the differences between Fmr1 KO and WT mice were conserved among the backgrounds (data not shown; briefly, latency and frequency among backgrounds within genotypes: P > 0.05 for both WT and KO, ANOVA;
latency and frequency between genotypes within backgrounds, P < 0.01 for all genotypes, paired t test within genotypes), suggesting that the effects of FMRP loss on circuit excitability and its normalization in dKO mice are independent of the mice genetic background.
We also note that analysis of epileptiform discharges in β4 KO mice revealed increased seizure susceptibility (n = 7, Fig. 6 C, E–H), supporting the complex bi‐directional role of BK channels in circuit excitability (Brenner et al. 2005; Gu et al. 2007; N'Gouemo, 2011; Zhang et al. 2014) and suggesting that additional BK channel‐dependent mechanisms, beyond regulation of AP duration in CA3 excitatory neurons contribute to hippocampal circuit hyperexcitability in the absence of FMRP.
Taken together, these results indicate that genetic deletion of BKβ4 normalizes the hippocampal circuit hyperexcitability caused by FMRP loss in vitro and point to a major contribution of BK channels to hyperexcitability defects in neural circuits of Fmr1 KO mice.
Discussion
Here we demonstrate that FMRP loss reduces activity of BK channels by modulating the channel gating kinetics and that abnormalities in AP waveform, glutamate release and short‐term synaptic plasticity caused by FMRP loss in hippocampal excitatory neurons can be normalized by genetic upregulation of BK channel activity. These results suggest a central role of FMRP–BK channel interactions in the defects of these presynaptic processes in a FXS mouse model. Moreover, analysis of circuit hyperexcitability using an in vitro seizure model indicates that genetic upregulation of BK channel activity is also sufficient to normalize increased seizure susceptibility in the hippocampal circuit in the absence of FMRP. These results suggest that BK channel dysfunction plays a major role in synaptic and circuit defects caused by loss of FMRP.
Mechanisms of FMRP–BK channel interactions
FMRP is generally thought to affect neural excitability via translational regulation mechanisms and recent studies in Fmr1 KO mice have identified a number of ion channels which abnormal expression has been linked to altered excitability in the absence of FMRP (Strumbos et al. 2010; Gross et al. 2011; Lee et al. 2011; Brager et al. 2012; Routh et al. 2013). In contrast, our single‐channel analyses indicate that FMRP acts on BK channels by regulating their gating kinetics, and provide extensive evidence that FMRP can modulate activity of an ion channel in mammalian central neurons. To the best of our knowledge, this non‐canonical function of FMRP has been demonstrated previously only for Slack channels studied in heterologous expression systems and Aplysia bag cell neurons (Brown et al. 2010; Zhang et al. 2012). This result supports the emerging view that FMRP role in regulating neural excitability is not limited to translational regulation, but FMRP also acts in part by directly modulating activity of a subset of ion channels (Contractor et al. 2015).
Our approach to upregulate BK channel activity in the absence of FMRP was based on our earlier observations that FMRP actions on BK channel activity are mediated in large part by interactions with the channel's β4 subunit. Because no specific pharmacological inhibitor of β4 has been developed thus far, we sought to develop a genetic, proof‐of‐principle approach to increase BK channel activity via β4 deletion. Here we found that genetic deletion of β4 upregulates BK channel open probability in dKO neurons as would be expected from the known role of β4 as a negative regulator of BK channel activity (Brenner et al. 2000; Ha et al. 2004). This genetic manipulation thus provided us with a useful tool to study, as a proof‐of‐principle approach, the extent to which upregulation of BK currents restores synaptic deficits caused by loss of FMRP.
We note, however, that this genetic approach does not represent a rescue of BK channel function, but rather a compensatory effect in which β4 deletion acts largely by facilitating channel gating kinetics and increasing channel open frequency, rather than open dwell time, as would be expected from previous studies of β4 (Brenner et al. 2000; Ha et al. 2004). The precise biophysical mechanisms by which β4 modulates BK channel activity remains, however, poorly understood, limiting further advances towards elucidating how FMRP impacts on BK channel gating. One possible mechanism involves FMRP acting as a part of the BK channel protein complex to weaken BKα–β4 interactions, thus altering the effects of β4 on channel kinetics. Moreover, each BK channel can bind up to four β subunits, and at least two other β subunits (β2 and β3) have also been described in neurons (Faber & Sah, 2003). Different channel kinetics in the WT and dKO neurons could thus also arise in part from FMRP altering the number of β4 subunits associated with each channel, or affecting the relative abundance of β4 and other β subunits acting on the same channel. In addition, we noted that β4 deletion over‐compensated BK channel open probability in dKO neurons beyond WT levels, while the cellular properties were indistinguishable in dKO neurons from that in WT. These differences could arise from a reduced functional channel surface expression in the absence of β4 that could partially compensate the increased channel open probability, a possibility which is in line with our observation of somewhat reduced chances of obtaining single‐channel recordings in β4 and dKO animals. Future studies will be needed to determine the molecular mechanisms by which FMRP regulates BK channel activity and to examine whether FMRP actions are mediated in part by altering the functional channel surface expression and/or balance of different β subunits.
It is important to emphasize that the normalization of synaptic and circuit functions we observed in dKO mice is not primarily driven by the deletion of β4 simply superseding effects of Fmr1 loss. Indeed β4 KO and dKO mice are significantly different in almost all of our measurements (except for BK open probability), including spontaneous and evoked AP duration, synaptic transmission and short‐term plasticity. Moreover, while Fmr1 KO and β4 KO exhibit opposite effects in all of these measurements relative to WT, deletion of Fmr1 or β4 has similar effects in reducing AHP amplitude and in circuit excitability measurements by increasing epileptiform activity. Still, dKO animals exhibit fully corrected epileptiform activity, and almost fully corrected AHP. These results provide strong evidence that the corrective effects in dKO animals are not simply driven by the dominate effects of deletion of β4.
BK channels and synaptic transmission defects in FXS neurons
BK channels, which are both voltage and calcium regulated, are unique in their ability to translate activity‐dependent changes into modulation of spike duration at synapses, and thus play key roles in regulating neurotransmitter release during neural activity (Faber & Sah, 2003). BK channels are ubiquitously expressed but found preferentially in axons and presynaptic terminals (Knaus et al. 1996; Martire et al. 2010), and their role as a negative‐feedback regulator of neurotransmitter release during neural activity is well established (Faber & Sah, 2003). Our previous studies established the role of BK channels in mediating FMRP regulation of AP duration in hippocampal and cortical excitatory neurons. However, given the multitude of FMRP targets and interacting partners (including a large number of other voltage‐gated K+ and Ca2+ channels), it remained unknown to what extent FMRP–BK channel interactions contribute to defects in synaptic transmission and plasticity in the FXS mouse model beyond AP modulation. Our results suggest that BK channels are both necessary and sufficient to mediate the effects of FMRP loss not only on AP duration but also on glutamate release and short‐term plasticity in excitatory hippocampal neurons. In particular, we demonstrate here that upregulation of BK channel activity is sufficient to fully normalize glutamate release and short‐term plasticity during naturalistic spike trains in these neurons. These results are unexpected given that FMRP has been recently found to regulate neurotransmitter release in DRG neurons by directly interacting with and regulating surface levels of presynaptic VGCCs (Ferron et al. 2014). While we did not observe significant changes in calcium currents in the soma of CA3 neurons (Deng et al. 2013 a), we cannot exclude a possibility that FMRP effects on these channels are different at the synaptic terminals. We note, however, that our previous mimicking experiments using BK channel blockers (Deng et al. 2013 a) together with the current experiments (Figs 4 and 5) indicate that no significant additional dysregulation of neurotransmitter release is present in hippocampal excitatory neurons beyond what is caused by BK channel deficits. Because neurotransmitter release is a very sensitive measure of presynaptic calcium influx due to the 4th‐power dependence of release on calcium levels, these findings suggest that in hippocampal neurons presynaptic VGCCs are not likely to be strongly affected by FMRP loss. It thus remains to be determined whether calcium channel regulation by FMRP is specific to peripheral sensory neurons or whether this mechanism also contributes to abnormal release in other types of central neurons.
BK channels and circuit excitability defects in FXS
FMRP loss affects excitability and neurotransmitter release in both excitatory and inhibitory neurons in several circuits, yet the variable extent and often opposing directions of these changes complicates understanding of the causes of hyperexcitability in FXS (Contractor et al. 2015). Highly diverse pharmacological treatments targeting dendritic spine structure (Dolan et al. 2013), protein synthesis (Osterweil et al. 2013), or the endocanabinoid system (Busquets‐Garcia et al. 2013) improve or ameliorate the seizure activity in Fmr1 KO mice. Recent evidence also suggested that hyperexcitability of cortical circuits in Fmr1 KO mice is mGluR5 dependent (Gibson et al. 2008; Hays et al. 2011). Our analyses of circuit hyperexcitability in the hippocampus revealed that genetic upregulation of BK channel activity is sufficient to correct increased epileptiform activity in an in vitro hippocampal seizure model in the absence of FMRP (Fig. 6). This finding suggests a major contribution of abnormal BK channel activity to circuit hyperexcitability in the hippocampus of FXS mice.
BK channels have been found to play complex roles in neuronal excitability: they typically act to repolarize the membrane, shorten the AP duration and reduce excitability (Faber & Sah, 2003); yet in some neurons, increased BK channel activation has been found to facilitate high‐frequency firing (Brenner et al. 2005; Gu et al. 2007; Shruti et al. 2008). This effect could arise from enhanced fAHP, which promotes the recovery of Na+ channels from inactivation (Klyachko et al. 2001) and may explain the otherwise paradoxical findings that both loss‐of‐function and gain‐of‐function mutations in the BK channel α subunit have been associated with seizures (N'Gouemo, 2011). In the present study, we also observed that β4 KO mice per se exhibited faster BK channel gating activity and increased seizure susceptibility, as reported previously (Brenner et al. 2005; Gu et al. 2007). This dual role of BK channels is not unique, however, since gain‐of‐function mutations in Slack K+ channels can lead to epileptic seizures in patients (Kim et al. 2014). These findings that both loss‐ and gain‐of‐function in K+ channels cause circuit hyperexcitability suggests that a delicate balance of various ion channel functions within and across neuronal populations is required to maintain circuit functionality.
In addition, the effects of altered BK channel activity in inhibitory interneurons could contribute to circuit‐level effects observed here. Indeed, our findings on the cellular effects of BK channel upregulation concerns the excitatory neurons only, while circuit‐level activity is determined by interplay of both excitatory and inhibitory neurons. Our recent studies indicate that GABA release is abnormal in the subset of local circuits in the hippocampus of Fmr1 KO mice (Wahlstrom‐Helgren & Klyachko, 2015), but whether inhibitory synapse dysfunction is present in the CA3 area and has a significant contribution from BK channel dysregulation remains to be determined. However, the emerging evidence supports the notion that reduced BK channel activity is associated with increased neural and circuit excitability in the FXS mouse model (Zhang et al. 2014; Myrick et al. 2015). Future work will determine the cell‐type and circuit specificity of the BK channel's functions in neurotransmission and its contribution to circuit dysfunction in FXS. The contribution of FMRP–BK channel interactions to circuit deficits in FXS may also depend, in part, on the degree of overlap and compensation of different β subunit expression. Thus, further studies of the BK channel subunit distribution and functions are needed to better understand their contributions to circuit excitability defects in different brain areas.
Additional information
Competing interests
The authors declare no competing financial interest.
Author contributions
P.Y.D. and V.A.K. conceptualized and designed the experiments. P.Y.D. collected and analysed the data. P.Y.D. and V.A.K. interpreted the data, drafted and revised the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported in part by grant to V.A.K. from the NINDS R01 NS089449.
Acknowledgements
We thank Dr Brenner (University of Texas‐San Antonio, TX USA) for providing sloβ4 KO mice, and Dr Cavalli and Ms Owyoung for their constructive comments.
References
- Brager DH, Akhavan AR & Johnston D (2012). Impaired dendritic expression and plasticity of h‐channels in the fmr1−/y mouse model of fragile X syndrome. Cell Rep 1, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH & Johnston D (2014). Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull 103, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL & Aldrich RW (2005). BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci 8, 1752–1759. [DOI] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y & Aldrich RW (2000). Cloning and functional characterization of novel large conductance calcium‐activated potassium channel β subunits, hKCNMB3 and hKCNMB4. J Biol Chem 275, 6453–6461. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D & Kaczmarek LK (2010). Fragile X mental retardation protein controls gating of the sodium‐activated potassium channel Slack. Nat Neurosci 13, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets‐Garcia A, Gomis‐Gonzalez M, Guegan T, Agustin‐Pavon C, Pastor A, Mato S, Perez‐Samartin A, Matute C, de la Torre R, Dierssen M, Maldonado R & Ozaita A (2013). Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19, 603–607. [DOI] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA & Portera‐Cailliau C (2015). Neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS & Klyachko VA (2013. a). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS & Klyachko VA (2013. b). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 78, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Sojka D & Klyachko VA (2011). Abnormal presynaptic short‐term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci 31, 10971–10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY & Tonegawa S (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small‐molecule PAK inhibitor FRAX486. Proc Natl Acad Sci USA 110, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES & Sah P (2003). Calcium‐activated potassium channels: multiple contributions to neuronal function. Neuroscientist 9, 181–194. [DOI] [PubMed] [Google Scholar]
- Fenton AA & Muller RU (1998). Place cell discharge is extremely variable during individual passes of the rat through the firing field. Proc Natl Acad Sci USA 95, 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron L, Nieto‐Rostro M, Cassidy JS & Dolphin AC (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N‐type calcium channel density. Nat Commun 5, 3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA & Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol 100, 2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Sulser A, Wang J, Queenan BN, Avoli M, Vicini S & Dzakpasu R (2012). Hippocampal neuron firing and local field potentials in the in vitro 4‐aminopyridine epilepsy model. J Neurophysiol 108, 2568–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A & Bassell GJ (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31, 5693–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K & Storm JF (2007). BK potassium channels facilitate high‐frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol 580, 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TS, Heo MS & Park CS (2004). Functional effects of auxiliary β4‐subunit on rat large‐conductance Ca2+‐activated K+ channel. Biophys J 86, 2871–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Huber KM & Gibson JR (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci 31, 14223–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JJ, Hao J, Kosofsky BE & Rajadhyaksha AM (2008). Dysregulation of large‐conductance Ca2+‐activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p.R419X mutation. Neurogenetics 9, 219–223. [DOI] [PubMed] [Google Scholar]
- Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R & Kaczmarek LK (2014). Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko VA, Ahern GP & Jackson MB (2001). cGMP‐mediated facilitation in nerve terminals by enhancement of the spike afterhyperpolarization. Neuron 31, 1015–1025. [DOI] [PubMed] [Google Scholar]
- Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML & Sperk G (1996). Distribution of high‐conductance Ca2+‐activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci 16, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Roger S, Guerin P, Molinari F, M'Rad R, Cahard D, Belhadj A, Halayem M, Persico AM, Elia M, Romano V, Holbert S, Andres C, Chaabouni H, Colleaux L, Constant J, Le Guennec JY & Briault S (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry 163, 1622–1629. [DOI] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson‐Baldwin A, Smith SJ, Jan YN & Jan LY (2011). Bidirectional regulation of dendritic voltage‐gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire M, Barrese V, D'Amico M, Iannotti FA, Pizzarelli R, Samengo I, Viggiano D, Ruth P, Cherubini E & Taglialatela M (2010). Pre‐synaptic BK channels selectively control glutamate versus GABA release from cortical and hippocampal nerve terminals. J Neurochem 115, 411–422. [DOI] [PubMed] [Google Scholar]
- Myrick LK, Deng PY, Hashimoto H, Oh YM, Cho Y, Poidevin MJ, Suhl JA, Visootsak J, Cavalli V, Jin P, Cheng X, Warren ST & Klyachko VA (2015). Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci USA 112: 949–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N'Gouemo P ( 2011). Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets 15, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK & Bear MF (2013). Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault P & Avoli M (1991). Physiology and pharmacology of epileptiform activity induced by 4‐aminopyridine in rat hippocampal slices. J Neurophysiol 65, 771–785. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE & Huber KM (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh BN, Johnston D & Brager DH (2013). Loss of functional A‐type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J Neurosci 33, 19442–19450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shruti S, Clem RL & Barth AL (2008). A seizure‐induced gain‐of‐function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol Dis 30, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skafidas E, Testa R, Zantomio D, Chana G, Everall IP & Pantelis C (2012). Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry 19, 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB & Kaczmarek LK (2010). Fragile X mental retardation protein is required for rapid experience‐dependent regulation of the potassium channel Kv3.1b. J Neurosci 30, 10263–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom‐Helgren S & Klyachko VA (2015). GABAB receptor‐mediated feed‐forward circuit dysfunction in the mouse model of fragile X syndrome. J Physiol 593, DOI: 10.1113/JP271190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, Sans N, Rossier J, Oostra B, LeMasson G & Frick A (2014). Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1−/y mice. Nat Neurosci 17, 1701–1709. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Brown MR, Hyland C, Chen Y, Kronengold J, Fleming MR, Kohn AB, Moroz LL & Kaczmarek LK (2012). Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci 32, 15318–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]