

Key points
Glucose regulates the density and function of 5‐HT3 receptors on gastric vagal afferent neurones.
Diet‐induced obesity compromises the excitability and responsiveness of vagal afferents.
In this study, we assessed whether exposure to a high fat diet (HFD) compromises the glucose‐dependent modulation of 5‐HT responses in gastric vagal afferents prior to the development of obesity.
We show that HFD does not alter the response of gastric vagal afferent nerves and neurones to 5‐HT but attenuates the ability of glucose to amplify 5‐HT3‐induced responses.
These results suggest that glucose‐dependent vagal afferent signalling is compromised by relatively short periods of exposure to HFD well in advance of the development of obesity or glycaemic dysregulation.
Abstract
Glucose regulates the density and function of 5‐HT3 receptors on gastric vagal afferent neurones. Since diet‐induced obesity attenuates the responsiveness of gastric vagal afferents to several neurohormones, the aim of the present study was to determine whether high fat diet (HFD) compromises the glucose‐dependent modulation of 5‐HT responses in gastric vagal afferents prior to the development of obesity. Rats were fed control or HFD (14% or 60% kilocalories from fat, respectively) for up to 8 weeks. Neurophysiological recordings assessed the ability of 5‐HT to increase anterior gastric vagal afferent nerve (VAN) activity in vivo before and after acute hyperglycaemia, while electrophysiological recordings from gastric‐projecting nodose neurones assessed the ability of glucose to modulate the 5‐HT response in vitro. Immunocytochemical studies determined alterations in the neuronal distribution of 5‐HT3 receptors. 5‐HT and cholecystokinin (CCK) induced dose‐dependent increases in VAN activity in all rats; HFD attenuated the response to CCK, but not 5‐HT. The 5‐HT‐induced response was amplified by acute hyperglycaemia in control, but not HFD, rats. Similarly, although 5‐HT induced an inward current in both control and HFD gastric nodose neurones in vitro, the 5‐HT response and receptor distribution was amplified by acute hyperglycaemia only in control rats. These data suggest that, while HFD does not affect the response of gastric‐projecting vagal afferents to 5‐HT, it attenuates the ability of glucose to amplify 5‐HT effects. This suggests that glucose‐dependent vagal afferent signalling is compromised by short periods of exposure to HFD well in advance of obesity or glycaemic dysregulation.
Key points
Glucose regulates the density and function of 5‐HT3 receptors on gastric vagal afferent neurones.
Diet‐induced obesity compromises the excitability and responsiveness of vagal afferents.
In this study, we assessed whether exposure to a high fat diet (HFD) compromises the glucose‐dependent modulation of 5‐HT responses in gastric vagal afferents prior to the development of obesity.
We show that HFD does not alter the response of gastric vagal afferent nerves and neurones to 5‐HT but attenuates the ability of glucose to amplify 5‐HT3‐induced responses.
These results suggest that glucose‐dependent vagal afferent signalling is compromised by relatively short periods of exposure to HFD well in advance of the development of obesity or glycaemic dysregulation.
Abbreviations
- CCK
cholecystokinin
- GI
gastrointestinal
- HFD
high fat diet
- VAN
vagal afferent nerve(s)
Introduction
Obesity, and its associated co‐morbidities, including hyperlipidaemia, type 2 diabetes, coronary heart disease, stroke and hypertension, is now recognized as the second leading cause of preventable death (Das, 2010). While multifactorial in origin, obesity is essentially a disorder of energy homeostasis. The vago‐vagal neurocircuit between the gastrointestinal (GI) tract and the brainstem is crucial in the regulation of energy homeostasis and food intake (Woods et al. 1998; Schwartz, 2000; Berthoud, 2008). Sensory information from the GI tract is transmitted through the afferent vagus nerve to the brainstem via a glutamatergic synapse at the level of the nucleus of the tractus solitarius (NTS) (Andresen & Kunze, 1994; Alywin et al. 1997). The NTS integrates these afferent signals with other inputs from CNS centres involved in regulation of autonomic homeostatic functions creating an integrated output response that is relayed to the nearby dorsal motor nucleus of the vagus (DMV), using glutamate, GABA or noradrenaline (norepinephrine) as neurotransmitters (Travagli et al. 1991; Sivarao et al. 1998; Browning & Travagli, 2014). The DMV contains the preganglionic parasympathetic neurones that provide the motor output to much of the upper GI tract via the efferent vagus nerve (Shapiro & Miselis, 1985; Altschuler et al. 1989; Berthoud et al. 1991; Browning & Travagli, 2014).
The GI tract is responsible for initiating homeostatic processes that regulate short‐term food intake and regulate blood glucose levels (Rayner et al. 2001; Horowitz et al. 2002). Nutrients within the intestinal lumen induce the release of a variety of neuroendocrine hormones that modulate GI functions, including inhibiting gastric emptying and modulation of gastrointestinal motility patterns and pancreatic secretion (Lloyd et al. 1993; Holzer et al. 1994; Raybould, 1999; Li et al. 2001; Zhou et al. 2008; Holmes et al. 2009 a; Babic et al. 2012 a,b). Glucose, in particular, induces the release of 5‐hydroxytryptamine (5‐HT; serotonin) from enterochromaffin cells within the intestine; the released 5‐HT activates 5‐HT3 receptors present on vagal afferent terminals and relays the afferent signal to the CNS (Li et al. 2001; Zhu et al. 2001; Raybould et al. 2003). Furthermore, circulating platelet‐free 5‐HT levels rise almost threefold following meal ingestion (Houghton et al. 2003) and blood‐borne substances have relatively free access to vagal afferent cell bodies contained within the paired nodose ganglia (Lacolley et al. 2006). Once absorbed from the GI tract, glucose itself is also able to act directly on vagal afferent neurones to modulate neuronal activity (Grabauskas et al. 2006, 2010) as well as modulate the response of GI vagal afferent neurones to 5‐HT by trafficking of 5‐HT3 receptors to the neuronal membrane (Babic et al. 2012 b).
Studies from many laboratories have demonstrated that the excitability and responsiveness of GI vagal afferent neurones and fibres is modulated by exposure to a high fat diet (HFD) or by diet‐induced obesity (Donovan et al. 2007; Daly et al. 2011; de Lartigue et al. 2011; Kentish et al. 2012). Few studies, however, have investigated either the effects of a high fat diet on the responsiveness of vagal afferents before obesity develops or whether the attenuated responsiveness of vagal afferents occurs in response to obesity. The aim of the present study, therefore, was to use neurophysiological, electrophysiological, and immunocytochemical techniques to investigate whether exposure to a HFD alters the ability of glucose to modulate 5‐HT3 receptor signalling in gastric vagal afferents, prior to the development of obesity or loss of glycaemic control.
Methods
Ethical approval
All experiments were approved by the Pennsylvania State University's Institutional Animal Care and Use Committee. Male Sprague–Dawley rats were used for the gastric vagal afferent nerve (VAN) recordings while rats of both sexes were used for electrophysiological and immunocytochemical studies. At 28 days of age, rats were divided into two diet groups and fed either a control diet (kilocalories: 14% from fat, 27% from protein and 59% from carbohydrate) or high fat diet (HFD; kilocalories: 60% from fat, of which 9% is from soybean oil and 91% from lard, 20% from protein and 20% from carbohydrate; both Harlan Teklad, Madison, WI, USA). Rats were maintained on these diets for periods of time ranging from 3 days to 8 weeks prior to experimentation. Briefly, in total the study used 13 rats (6 male, 7 female) after 3 days HFD exposure and 14 age‐matched control rats (11 female, 3 male). After 1 week HFD exposure, 14 rats were used (8 male, 6 female) and compared to 16 age‐matched control rats (7 male, 9 female). After 2 weeks HFD exposure, 16 rats were used (10 male, 6 female) and compared to 17 age‐matched control rats (13 male, 4 female). After 4 weeks HFD exposure, 28 rats were used (11 male, 17 female) and compared to 21 age‐matched control rats (9 male, 13 female). Finally, after 8 weeks HFD exposure, 22 rats were used (17 male, 5 female) and compared to 16 age‐matched control rats (9 male, 7 female). Body weight and peripheral blood glucose levels (OneTouch glucometer; LifeScan, Inc., Wayne, PA, USA) were measured at the time of experimentation in all rats.
Gastric vagal afferent nerve (VAN) recording
Male rats were fed either a control (n = 10) or HFD (n = 10) for 2 or 8 weeks prior to experimentation. Rats were anaesthetized with isoflurane (2–3% in 100% O2) and instrumented with catheters in the jugular vein (anaesthetic and fluids), femoral vein (glucose infusions) and brachial artery (blood pressure and blood glucose measurement). To deliver 5‐HT to the stomach and upper GI tract only, an arterial catheter (heat‐stretched microrenathane tubing MRE‐025, Braintree Scientific, Inc., Braintree, MA, USA) was introduced into the coeliac artery via the femoral artery and advanced 3–5 mm past the coeliac–aorta junction. After tracheal cannulation, rats were artificially ventilated and tidal volume (∼1 ml (100 g body weight)−1) or respiratory rate (60–80 breaths min−1) was adjusted to maintain end‐tidal CO2 between 3.5 and 4.5%. Rectal temperature was maintained at 37 ± 0.5°C with a water‐circulating blanket. At the end of the experiments, toluidine blue dye (0.5%, 100 μl) was injected through the intra‐coeliac artery to confirm the placement of the catheter.
Through a mid‐line laparotomy, the anterior gastric branch of the vagus nerve, which provides innervation to the anterior stomach and upper small intestine (Berthoud & Powley, 1991), was isolated, placed on a bipolar stainless steel electrode, crushed proximally, and insulated with KWIK‐SIL (World Precision Instruments, Sarasota, FL, USA). Gastric VAN activity was amplified (×10,000) and filtered (100–1000 Hz) using a differential AC amplifier (Model 1700, A‐M Systems, Sequim, WA, USA). Signals were digitized (2 kHz), rectified and integrated (1 s time constant) using Spike 2 software (Cambridge Electronic Design, Cambridge, UK). Baseline VAN was normalized to 100% after noise subtraction and assessed at the end of experiments by crushing the distal nerve. Following completion of all surgical procedures, isoflurane anaesthesia was replaced by thiobutabarbital (Inactin; 120 mg kg−1). Rats were allowed to stabilize for 60 min prior to experimentation. The response to intra‐coeliac administration of CCK (0.03–0.1 μg kg−1) or 5‐HT (0.03–10 μg kg−1) was assessed through 100 μl infusions over 15 s. In a second set of experiments, VAN responses to intra‐coeliac 5‐HT (1 μg kg−1) were assessed at 3 time‐points separated by 10 min before and after acute hyperglycaemia produced by i.v. infusion of 50% glucose for 10–30 min.
Retrograde tracing
Gastric‐projecting vagal afferent neurones were labelled with the fluorescent neuronal tracer 1,1‐dioctadecyl‐3,3,3,3‐tetramethyl‐indocarbocyanine perchlorate (DiI; Life Technologies, Grand Island, NY, USA) via application to the GI tract as described previously (Browning et al. 1999; Babic et al. 2012 b). Briefly, after induction of anaesthesia with isofluorane (2.5% in 100% O2), the abdominal area of 14‐day‐old rats (n = 51) was cleaned before exposing the stomach and proximal intestine via a laparotomy. After isolating the stomach from the surrounding viscera, crystals of DiI were placed onto the greater curvature of the fundus, corpus or antrum/pylorus area. Using a fast‐hardening epoxy resin, the DiI crystals were embedded in place; prior to closing the abdomen, the surgical area was washed with sterile saline. Rats were allowed a minimum of 17 days to recover before experimentation.
Dissociation of nodose ganglia
Rats were killed via anaesthetic overdose (isofluorane, 5%) followed by administration of a bilateral pneumothorax. Under microscopic guidance, nodose ganglia were removed and placed in chilled, oxygenated Hanks’ balanced salt solution (HBSS; Gibco Laboratories, Life Technologies, Grand Island, NY, USA) and dissociated as described previously (Babic et al. 2012 b). Dissociated neurones were plated on poly‐l‐lysine‐coated coverslips and incubated in a humid incubator (5% CO2) at 37°C. Cells were fed after 2 h and electrophysiological recordings and immunocytochemical studies were carried out within 24 h of dissociation.
Electrophysiological recordings
Cover glasses with plated dissociated nodose ganglion neurones were placed in a custom‐made perfusion chamber (volume, 500 μl; Michigan Precision Instruments, Parma, MI, USA) and maintained at 35°C by perfusion with warmed Krebs solution at a rate of 2.5–3.0 ml min−1. A Nikon E600FN microscope (Nikon Corporation, Tokyo, Japan) equipped with tetramethylrhodamine isothiocyanate (TRITC) epifluorescent filters was used to identify DiI labelled neurones. Once identified, DiI‐filled neurones were recorded electrophysiologically under bright‐field illumination.
Whole‐cell recordings were made with patch pipettes of resistance 2–4 MΩ when filled with potassium gluconate intracellular solution (see below). Data were acquired using an Axopatch 200A amplifier (Molecular Devices, Sunnyvale, CA, USA) at a rate of 10 kHz, filtered at 2 kHz, and digitized via a Digidata 1440 interface (Molecular Devices), before being stored and analysed on a personal computer using pCLAMP10 software (Molecular Devices). Only those neurones with a series resistance of <15 MΩ were considered acceptable. The basic membrane properties (action potential duration at threshold, after‐hyperpolarization amplitude from baseline, after‐hyperpolarization duration, firing frequency, input resistance) were assessed for recorded neurones as described previously (Browning et al. 1999). A separate patch pipette placed close to the recorded neurone was used to assess the response of neurones (voltage clamped at −50 mV) to picospritz application of 5‐HT (100 μm; 10–50 ms duration; Parker Hannifin Corp., Cleveland, OH, USA). Each neurone served as its own control and the response to 5‐HT was assessed before, during and after perfusion with equi‐osmolar Krebs solution containing different glucose concentrations (2.5–20 mm) using ANOVA with repeated measures. Results are expressed as means ± SEM with statistical significance set at P < 0.05.
Immunocytochemistry
Prior to fixation in 4% paraformaldehyde (15 min), dissociated nodose neurones were exposed to feeding medium with varying glucose concentrations (30 min; 1.25–20 mm). Cells were washed (5 min) in 50% ethanol, followed by 3 × 5 min washes in phosphate‐buffered saline (PBS; Babic et al. 2012 b) containing 0.1% bovine serum albumin (PBS‐BSA). Coverslips were then placed in rabbit anti‐5‐HT3B receptor antibody (1:500; ab39629, Abcam, Cambridge, MA, USA; diluted in PBS‐BSA) and incubated overnight at room temperature. Coverslips were washed (3 × 10 min) with PBS‐BSA before incubation in secondary antibody (AlexaFluor488‐conjugated goat anti‐rabbit IgG, 1:100; Life Technologies Corp., Grand Island, NY, USA) for 1 h at room temperature, washed 3 × 5 min in PBS‐BSA and mounted in Fluoromount‐G (Southern Biotech, Birmingham, AL, USA).
Confocal microscopy
Confocal photomicrographs of 5‐HT3 receptor‐immunoreactive neurones were taken on an Olympus IX81 microscope equipped with Olympus Fluoview 1000 software (Olympus America Inc., Center Valley, PA, USA) and were imported into ImageJ software (http://rsbweb.nih.gov/ij). Membrane‐associated 5‐HT3 receptor density was quantified as described previously (Babic et al. 2012 b) and the percentage of membrane‐associated 5‐HT3 receptors relative to total 5‐HT3 receptor density calculated. Data were analysed by comparing the percentage of membrane‐associated receptor density between different groups using an ANOVA followed by Student's t test. Results are expressed as means ± SEM and statistical significance was set at P < 0.05. Linear regression analysis was used to determine the correlation between the glucose concentration and the percentage of membrane‐associated 5‐HT3 receptor density.
Oral glucose tolerance testing
Oral glucose tolerance testing was performed on rats (n = 10 control and n = 10 HFD) at each experimental time point. Rats were fasted overnight (water ad libitum), and received a 1.25 mg kg−1 oral glucose gavage as previously described (Kawano et al. 1992). Peripheral blood glucose was measured via blood drawn from the tail vein at time 0, and then at 15, 30, 60 and 90 min post‐gavage. Results were averaged and plotted as glucose level (mg dl−1) over time.
Statistics
Neurophysiological data were analysed with Systat 10.2 software (Systat Software Inc., San Jose, CA, USA) using a two‐way ANOVA with repeated measures; post hoc comparisons were made using independent or paired t tests with a layered Bonferroni correction. Electrophysiological data were analysed with GraphPad Prism software using an unpaired t test. Immunohistochemical data were analysed with GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA) using an ANOVA followed by Student's t test. Linear regression analysis was used to determine the correlation between the glucose concentration and the percentage of membrane‐associated 5‐HT3 receptor density. All results are expressed as means ± SEM and statistical significance was set at P < 0.05.
Chemicals and drugs
Supplemented minimal essential medium (sMEM): minimal essential medium (Gibco, Life Technologies) supplemented with 0.15 g (100 ml)−1 NaHCO3 and 29.2 mg l‐glutamine, pH 7.4.
Plating medium: sMEM supplemented with fetal calf serum (10%), penicillin–streptomycin (1%), and CaCl2 (4 mm).
Feeding medium: sMEM supplemented with fetal calf serum (10%), and penicillin–streptomycin (1%).
Phosphate buffered saline (mm): 115 NaCl, 75 Na2HPO4, 7.5 KH2PO4.
Paraformaldehyde fixative: 4% paraformaldehyde in PBS.
Krebs solution (mm): 126 NaCl, 25 NaHCO3, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4 and 5 d‐glucose maintained at pH 7.4 by bubbling with 95% O2–5% CO2.
Intracellular pipette solution (mm): 128 potassium gluconate, 10 KCl, 0.3 CaCl2, 1 MgCl2, 10 Hepes, 1 EGTA, 2 NaATP and 0.25 NaGTP adjusted to pH 7.35 with KOH.
Dispase was purchased from Roche Diagnostics (Indianapolis, IN, USA); all other chemicals were purchased from Sigma Chemical Co. (St Louis, MO, USA).
Results
HFD exposure for up to 8 weeks does not induce obesity or glycaemic dysregulation
The weight and blood glucose levels did not differ between controls and HFD rats until 8 weeks exposure to HFD, when male rats were significantly heavier than their age‐matched control counterparts (604 ± 38 g vs. 488 ± 10 g, respectively, P < 0.05; Fig. 1 A), although not obese, defined as 20% increase in body weight relative to control. Additionally, after 8 weeks exposure to HFD, male rats had significantly elevated blood glucose levels compared to age‐matched control rats (248 ± 42 mg dl−1 vs. 149 ± 10 mg dl−1, respectively, measured between 09.00 and 10.00 h; P < 0.05; Fig. 1 B). To examine whether exposure to a HFD altered the ability of rats to release and respond to insulin, oral glucose tolerance testing was performed on rats (n = 10 control and n = 10 HFD). HFD did not alter the response of rats to oral glucose administration at any of the time points studied (Fig. 1 C).
Figure 1. HFD exposure for up to 8 weeks does not induce obesity or glycaemic dysregulation .
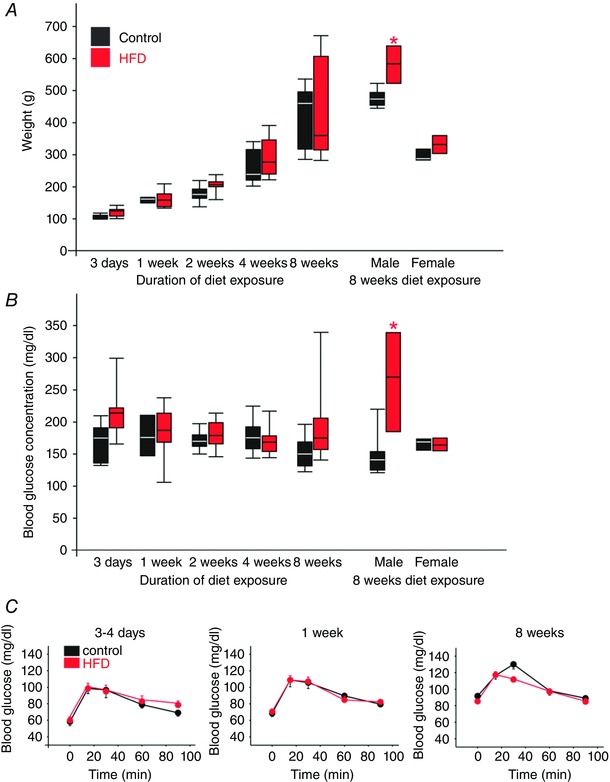
A, graphical representation of body weights of control (black) and HFD (red) rats at the experimental time‐points used in the present study illustrating the median, upper and lower quartile weights and, where appropriate, the 10th and 90th percentile weights. Note that the HFD‐fed rats were not significantly heavier than control rats until 8 weeks exposure to diet, when male HFD rats were significantly heavier than their age‐matched control counterparts. B, graphical representation of blood glucose levels of control (black) and HFD (red) rats at the experimental time‐points used in the present study illustrating the median, upper and lower quartile glycaemic levels and, where appropriate, the 10th and 90th percentile glycaemic levels. Note that blood glucose levels were unaffected by diet, until 8 weeks of exposure to diet, when male rats had significantly higher blood glucose levels compared to their age‐matched controls. C, oral glucose‐tolerance testing in control (black) and HFD (red) rats after 3 days, 1 week and 8 weeks HFD. Note that neither the peak response nor the time course of hyperglycaemia was altered by HFD.
These results suggest that after 8 weeks of exposure to HFD (i) rats are overweight but not obese, and (ii) in this pre‐obese state, HFD does not alter the ability of rats either to release insulin in response to oral glucose administration, or lower blood glucose in response to that released insulin.
The ability of CCK to increase gastric VAN activity is attenuated by HFD
Rats were fed either control diet (n = 10) or HFD (n = 10) for either 2 or 8 weeks prior to experimentation. There were no differences in baseline or evoked VAN responses to CCK or 5‐HT between age groups for either control or HFD animals, therefore, results were combined. Baseline VAN activity did not differ between control and HFD animals (0.86 ± 0.19 vs. 1.06 ± 0.27 μV, respectively; P > 0.05). Intra‐coeliac application of CCK induced a dose‐dependent increase in gastric VAN activity (Fig. 2). As described previously (Covasa & Ritter, 2000; Covasa et al. 2000 b; Daly et al. 2011), the gastric VAN response to CCK was attenuated by exposure to a HFD (Fig. 2 B and C).
Figure 2. HFD attenuates the response of gastric vagal afferent nerves to CCK .
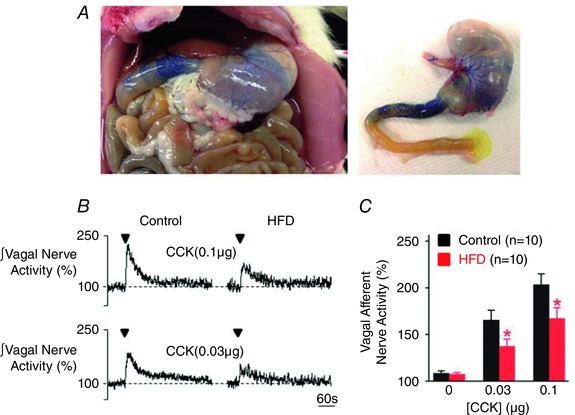
A, photographs of a rat following intra‐coeliac application of the dye toluidine blue at the conclusion of the experiment. Note that dye is restricted to the stomach and upper small intestine. B, representative rectified and integrated recordings of gastric vagal afferent nerve (VAN) activity in control (left) and HFD (right) rats following intra‐coeliac application of CCK (0.1 μg kg−1, upper; and 0.03 μg kg−1, lower). C, graphical summary of the attenuated increase in gastric VAN activity in response to intra‐coeliac application of CCK in HFD rats. *P < 0.05 vs. control.
The ability of 5‐HT to increase gastric vagal afferent nerve activity is not affected by HFD
Intra‐coeliac application of 5‐HT (0.3–10 μg kg−1) induced a dose‐dependent increase in gastric VAN activity in both control (n = 10) and HFD (n = 10) rats (Fig. 3 A–C; P > 0.05 vs. control). The responses were not different between control and HFD rats (P > 0.05 from overall ANOVA) but were attenuated significantly (>75%) by intra‐coeliac infusion of the 5‐HT3 antagonist ondansetron (1 mg kg−1, data not shown). To test whether glucose modulates the 5‐HT‐induced increases in gastric VAN activity, responses to intra‐coeliac application of 5‐HT (1 μg kg−1) were assessed before and during acute hyperglycaemia (>10 min). In control rats, acute hyperglycaemia (71 ± 4 to 498 ± 15 mg dl−1) enhanced the VAN response to 5‐HT (Fig. 3 D and F). In contrast, the 5‐HT response in HFD rats was not altered by acute hyperglycaemia (78 ± 6 to 510 ± 17 mg dl−1; Fig. 3 E and F). Acute hyperglycaemia itself did not significantly alter baseline gastric VAN nerve activity in control (baseline: 0.88 ± 0.17 μV vs. hyperglycaemia: 0.78 ± 0.12 μV) or HFD rats (baseline: 0.91 ± 0.27 μV vs. hyperglycaemia: 0.80 ± 0.12 μV).
Figure 3. HFD does not affect the response of gastric vagal afferent nerves to 5‐HT, but attenuates the ability of glucose to modulate the 5‐HT‐induced response .
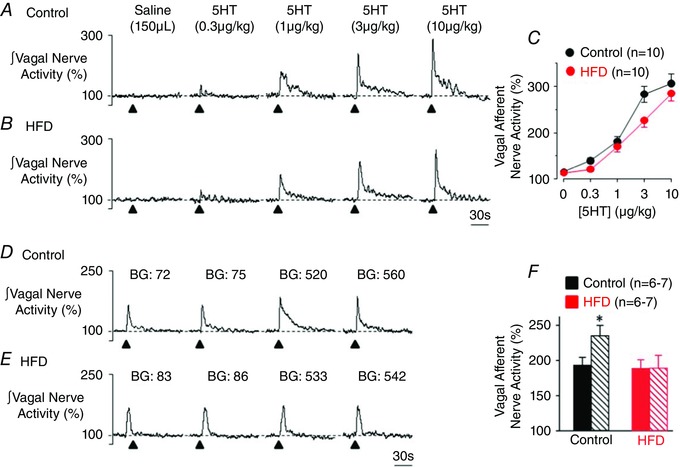
Representative rectified and integrated recordings illustrating the dose‐dependent increase in gastric VAN activity in response to intra‐coeliac application of 5‐HT in a control (A) and HFD (B) rat. C, graphical summary of the dose‐dependent increase in gastric VAN activity in control and HFD rats in response to 5‐HT. Representative rectified and integrated recordings of gastric VAN activity in a control (D) and HFD (E) rat in response to intra‐coeliac application of 5‐HT (1 μg kg−1) at normal blood glucose (BG) levels (control BG 72, 75 mg dl−1; HFD BG 83, 86 mg dl−1) and following acute elevation of blood glucose levels (control BG 520, 560 mg dl−1; HFD BG 533, 542 mg dl−1). Note the increased response to 5‐HT following hyperglycaemia in the control, but not HFD, rat. F, graphical summary of the increased gastric VAN activity in response to 5‐HT following hyperglycaemia in control (grouped data = 71 ± 4 to 498 ± 15 mg dl−1), but not HFD (grouped data = 78 ± 6 to 510 ± 17 mg dl−1), rats. *P < 0.05 vs. normal blood glucose level.
HFD does not alter the basic membrane properties of acutely dissociated gastric nodose neurones
As shown in Table 1, the basic membrane properties of gastric nodose neurones did not differ either between control and HFD groups at any time‐point studied or with increased duration of HFD exposure, suggesting that, in the pre‐obese state, HFD does not alter the membrane properties or excitability of gastric nodose neurones.
Table 1.
Basic membrane properties of gastric vagal afferent neurones
| 3 days diet | 1 week diet | 2 weeks diet | 4 weeks diet | 8 weeks diet | ||
|---|---|---|---|---|---|---|
| (n = 12) | (n = 12) | (n = 13) | (n = 17) | (n = 9) | ||
| (n = 15) | (n = 12) | (n = 12) | (n = 26) | (n = 23) | ||
| Input resistance (MΩ) | Control | 299 ± 45 | 310 ± 38 | 397 ± 39 | 295 ± 40 | 286 ± 50 |
| HFD | 287 ± 40 | 357 ± 36 | 299 ± 40 | 341 ± 29 | 343 ± 39 | |
| Action potential duration (ms) | Control | 4.5 ± 0.5 | 4.8 ± 0.4 | 3.9 ± 0.3 | 4.2 ± 0.3 | 4.6 ± 0.4 |
| HFD | 4.1 ± 0.2 | 4.4 ± 0.5 | 4.3 ± 0.3 | 4.4 ± 0.2 | 3.9 ± 0.2 | |
| After‐hyperpolarization amplitude (mV) | Control | 13.6 ± 1.4 | 12.6 ± 1.7 | 15.5 ± 1.3 | 15.6 ± 1.0 | 13.1 ± 1.4 |
| HFD | 15.0 ± 0.9 | 18.7 ± 1.4a | 14.3 ± 1.5 | 17.1 ± 0.9 | 13.6 ± 1.1 | |
| After‐hyperpolarization duration (ms) | Control | 64.4 ± 14.6 | 63.6 ± 16.3 | 62.4 ± 9.0 | 45.5 ± 6.7 | 47.4 ± 7.7 |
| HFD | 58.7 ± 11.5 | 51.6 ± 8.5 | 51.3 ± 11.1 | 98.1 ± 26.9 | 50.3 ± 7.5 |
P < 0.05 vs. control.
HFD does not alter the response of gastric nodose neurones to 5‐HT
To determine the effect of HFD on the inward current induced by picrospritz application of 5‐HT (100 μm), gastric nodose neurones were voltage clamped at −50 mV. In the presence of 5 mm extracellular glucose, 5‐HT induced an inward current in approximately 62% of control neurones (41 out of 67 neurons recorded from 30 rats); the proportion of responsive neurones did not change across time‐points studied (P > 0.05; Fig. 4 A and B). Additionally, in the presence of 5 mm extracellular glucose, the magnitude of the 5‐HT‐induced inward current in response to picospritz application of 5‐HT was not affected by age of rat or duration of HFD (55 out of 90 neurons recorded from 37 HFD rats; P > 0.05; Fig. 4 C).
Figure 4. HFD does not alter the response of gastric nodose neurones to 5‐HT .
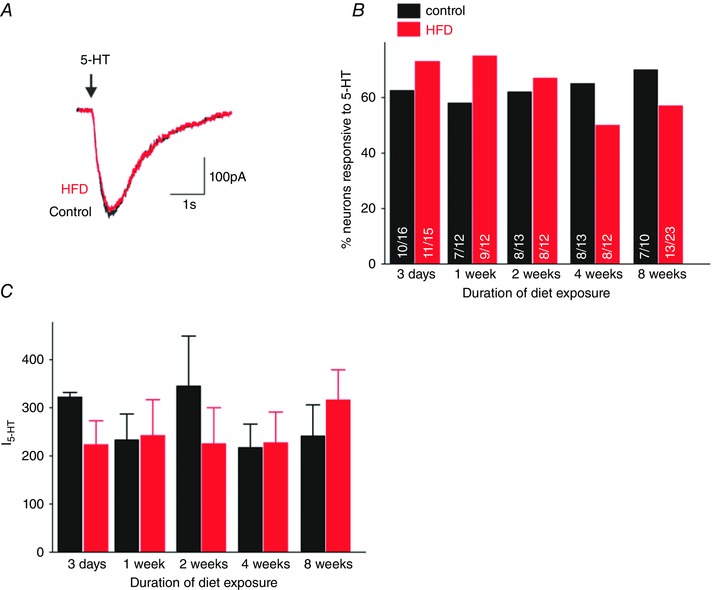
A, representative traces from control (black) and HFD (red) neurones voltage clamped at −50 mV prior to picospritz application of 5‐HT (100 μm; arrow). Note that HFD had no effect upon either the amplitude or duration of the 5‐HT‐induced inward current. B, graphical summary of the proportion of control (black) and HFD (red) gastric nodose neurones responding to 5‐HT with an inward current. Numbers within each bar describe the number of responsive/total number of neurones tested. C, graphical summary of the magnitude of the 5‐HT‐induced inward current in control (black) and HFD (red) gastric nodose neurones. *P < 0.05 vs. control 5 mm glucose; **P < 0.05 vs. HFD 5 mm glucose.
These results suggest that, in the absence of obesity, HFD does not affect either the magnitude of the 5‐HT‐induced inward current in gastric nodose neurones or the proportion of responsive neurones.
HFD attenuates the ability of glucose to modulate 5‐HT currents in gastric vagal afferent neurones
Previous studies have demonstrated that the 5‐HT‐induced inward current in gastric vagal afferent neurons is sensitive to extracellular glucose concentration. Specifically, elevating extracellular glucose levels increases, while lowering glucose levels decreases, the magnitude of the 5‐HT‐induced inward current (Babic et al. 2012 b). In the presence of 5 mm extracellular glucose, 5‐HT induced an inward current in control gastric nodose neurones in 43 neurons recorded from 32 rats across all the experimental time‐points (ranging in weight from 102 to 500 g). In 42 out of these 43 neurones (98%), the magnitude of the 5‐HT‐induced inward current was affected by the extracellular glucose concentration. Specifically, increasing extracellular glucose concentration from 5 to 10 mm increased the 5‐HT‐induced inward current from 274 ± 32 pA to 332 ± 50 pA, i.e. a 28 ± 4% increase in amplitude (P < 0.05), while decreasing extracellular glucose concentration to 2.5 mm decreased the amplitude of the 5‐HT‐induced inward current to 234 ± 37 pA, i.e. a 26 ± 4% decrease in amplitude (P < 0.05; Fig. 5 A and B). This ability of glucose to modulate the 5‐HT‐induced inward current was similar across all time‐points studied (Fig. 5 C).
Figure 5. HFD attenuates the ability of glucose to modulate the 5‐HT‐induced inward current .
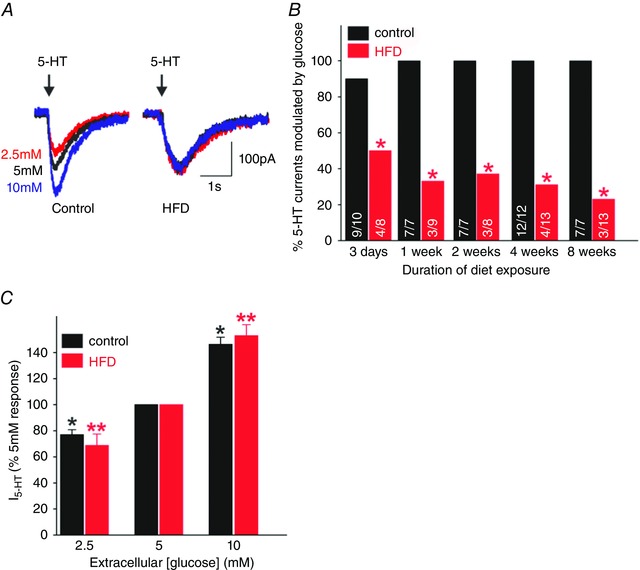
A, representative traces from control (left) and HFD (right) neurones voltage clamped at −50 mV prior to picospritz application of 5‐HT (100 μm; arrow). Note that in control, but not HFD, neurones, increasing the extracellular glucose concentration from 5 to 10 mm increased the magnitude of the 5‐HT‐induced inward current, while decreasing the extracellular glucose concentration to 2.5 mm decreased the magnitude of the current. B, graphical summary of the proportion of control (black) and HFD (red) gastric nodose neurones in which glucose modulated the amplitude of the 5‐HT‐induced inward current. Numbers within each bar describe the number of glucose‐modulated/total number of neurones tested. *P < 0.05 vs. age‐matched control. C, graphical summary of the changes in amplitude of 5‐HT‐induced inward current in control (black) and HFD (red) gastric nodose neurones responsive to extracellular glucose levels. Note that, in responsive neurones, the magnitude of the changes in 5‐HT current were similar between control and HFD neurones. F, graphical summary of the magnitude of the 5‐HT‐induced inward current in control (black) and HFD (red) gastric nodose neurones.
In contrast, the ability of extracellular glucose to regulate the 5‐HT‐induced inward current in gastric nodose neurones was attenuated by HFD at all time‐points studied (Fig. 5 A and C). Specifically, the magnitude of the 5‐HT response was unaffected by extracellular glucose concentration in 34 of the 51 gastric nodose neurones tested (P < 0.05 vs. control neurones; 260 ± 41 pA in 2.5 mm glucose, 266 ± 31 pA in 5 mm glucose and 270 ± 63 pA in 10 mm glucose, P > 0.05). In the remaining 17 neurones, extracellular glucose controlled the 5‐HT current to the same degree as in control neurones (Fig. 5 B).
Neither the duration of diet exposure nor the weight of the rat appeared to play a role in attenuating the actions of glucose to modulate the 5‐HT‐induced current in gastric nodose neurones in the current study. At the earliest time‐point of the study (3 days HFD exposure), for example, electrophysiological recordings were made from eight neurones from seven HFD rats (weights ranging from 111 to 130 g) as well as from 16 neurones from 10 control rats (weights ranging from 102 to 128 g). While 10/16 (62%) control neurones were considered ‘responsive’, in that glucose modulated the amplitude of the 5‐HT‐induced inward current, only four HFD neurones (50%; P ≤ 0.05 vs. control) were responsive and they were recorded from three rats weighing 111, 130 and 130 g. Similarly, at the 8 week time‐point of the study, electrophysiological recordings were made from seven neurones from five control rats (weights ranging from 329 to 500 g) and from 13 neurones from six HFD rats (weights ranging from 360 to 683 g). Whereas all seven control neurones were responsive, only three HFD neurones were responsive (one each from rats weighing 360 g, 465 g and 683 g) while the remaining 10 neurons were unresponsive. Furthermore, in every HFD rat in which a responsive neurone was recorded, non‐responsive neurones were also recorded from the same animal.
HFD attenuates the ability of glucose to traffic 5‐HT3 receptors in gastric vagal afferent neurones
The ability of glucose to traffic 5‐HT3 receptors was assessed in gastric‐projecting nodose ganglion neurons dissociated from control rats (n = 10, 10, 8, 11 and 6 rats at experimental time points 3 days, 1, 2, 4 and 8 weeks, respectively) and HFD rats (n = 7, 9, 4, 16 and 10 at experimental time points points 3 days, 1, 2, 4 and 8 weeks, respectively). Briefly, the proportion of membrane‐associated 5‐HT3 receptors, expressed as a percentage of total 5‐HT3 receptor density, was assessed following 30 min exposure to concentrations of extracellular glucose ranging from 1.25 to 20 mm (Fig. 6 A). As described previously (Babic et al. 2012 b), in control gastric vagal afferent neurones the proportion of membrane‐associated 5‐HT3 receptors was modulated by extracellular glucose levels, across all time‐points studied, increasing from 27.1 ± 0.5% at 1.25 mm glucose to 31.7 ± 0.4% at 20 mm glucose (Table 2; Fig. 6 B). In contrast, even after 3 days HFD exposure, the proportion of membrane‐associated 5‐HT3 receptors was no longer modulated by extracellular glucose levels, ranging from 28.8 ± 0.5% at 1.25 mm to 29.2 ± 0.6% at 20 mm glucose (P > 0.05; Table 2; Fig. 6 A and B).
Figure 6. HFD attenuates the ability of extracellular glucose to modulate the density of 5‐HT3 receptors associated with the membrane of gastric nodose neurones .
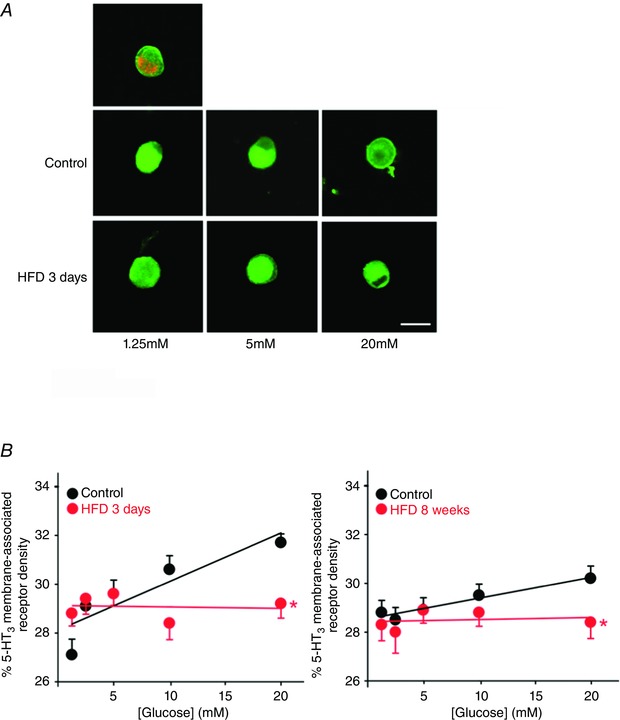
A, upper row, representative image of a gastric nodose neurone containing DiI (red) and 5‐HT3 receptor‐IR (green). Middle row, representative images of control gastric nodose neurones following incubation with 1.25 mm (left), 5 mm (middle) and 20 mm (right) glucose prior to fixation. Note that, as extracellular glucose levels increase, a greater proportion of 5‐HT3 receptors are associated with the neuronal membrane. Lower row, representative images of HFD gastric nodose neurones. Note that the proportion of membrane‐associated 5‐HT3 receptors is unaffected by extracellular glucose concentration. B, graphical summary of the proportion of membrane‐associated 5‐HT3 receptors in control (black) and HFD (red) gastric nodose neurones following 3 days (left) and 8 weeks (right) dietary exposure. Note that, following HFD, the proportion of membrane‐associated 5‐HT3 receptors is no longer regulated by extracellular glucose concentration. *P < 0.05 vs. control.
Table 2.
Effects of extracellular glucose to alter the proportion of membrane‐associated 5‐HT3 receptors in gastric‐projecting vagal afferent neurones
| 3 days diet | 1 week diet | 2 weeks diet | 4 weeks diet | 8 weeks diet | ||
|---|---|---|---|---|---|---|
| (n = 34–44) | (n = 26–40) | (n = 27–41) | (n = 36–43) | (n = 39–51) | ||
| [Glucose]ex | Treatment group | (n = 23–55) | (n = 24–43) | (n = 37–43) | (n = 33–40) | (n = 23–38) |
| 1.25 mm | Control | 27.0 ± 0.6 | 26.9 ± 0.5 | 27.9 ± 0.6 | 27.0 ± 0.6 | 28.8 ± 0.5 |
| HFD | 28.7 ± 0.5 | 26.8 ± 0.6 | 28.3 ± 0.7 | 25.8 ± 0.2 | 28.3 ± 0.7 | |
| 2.5 mm | Control | 29.1 ± 0.4 | 27.9 ± 0.6 | 28.0 ± 0.5 | 28.3 ± 0.4 | 28.4 ± 0.5 |
| HFD | 29.4 ± 0.6 | 27.3 ± 0.5 | 26.1 ± 0.5 | 26.8 ± 0.5 | 28.0 ± 0.9 | |
| 5 mm | Control | 29.8 ± 0.4 | 29.5 ± 0.4 | 28.5 ± 0.5 | 29.1 ± 0.6 | 29.0 ± 0.4 |
| HFD | 29.6 ± 0.5 | 28.9 ± 0.6 | 27.8 ± 0.5 | 26.8 ± 0.5 | 28.9 ± 0.5 | |
| 10 mm | Control | 30.6 ± 0.6 | 28.4 ± 0.6 | 27.6 ± 0.6 | 29.9 ± 0.5 | 29.5 ± 0.5 |
| HFD | 28.4 ± 0.7 | 29.0 ± 0.7 | 25.2 ± 0.5 | 26.0 ± 0.5 | 28.8 ± 0.6 | |
| 20 mm | Control | 31.7 ± 0.4 | 29.8 ± 0.6 | 30.3 ± 0.6 | 30.3 ± 0.5 | 30.2 ± 0.5 |
| HFD | 29.2 ± 0.6 | 28.9 ± 0.5 | 26.8 ± 0.7 | 26.2 ± 0.5 | 28.4 ± 0.7 |
Taken together with the neurophysiological and electrophysiological data, these data suggest that HFD exposure diminishes the ability of extracellular glucose to modulate both the response to activation of 5‐HT3 receptors in gastric‐projecting vagal afferent neurones and the proportion of 5‐HT3 receptors present on the neuronal membrane.
Discussion
Our results indicate that brief periods of exposure to HFD are sufficient, even in the absence of obesity, to attenuate the ability of glucose to modulate (1) the response of gastric vagal afferents to 5‐HT, (2) the 5‐HT‐induced inward current in gastric nodose neurones, and (3) the density of 5‐HT3 receptors associated with gastric nodose neuronal membranes. Conversely, brief periods of HFD do not alter either the 5‐HT‐induced increase in gastric vagal afferent activity, the number of gastric nodose neurones responsive to 5‐HT or the magnitude of the 5‐HT‐induced inward current.
While the regulation of energy food intake, energy balance and energy homeostasis is generally thought of as involving the integration of ‘higher’ CNS centres with autonomic nuclei (Schwartz et al. 2000; Berthoud & Morrison, 2008), the role of vago‐vagal neurocircuits in the regulation of early satiation has recently received more attention and has been the focus of investigations by several laboratories (Berthoud, 2004; Dockray, 2004; de Lartigue, 2014). Previously assumed to be static relay systems incapable of modulation, vagal neurocircuits controlling the stomach and upper GI tract are now known to be capable of a surprisingly large degree of modulation and integration (Browning & Travagli, 2010). The type and density of neurotransmitter receptors and ion channels displayed by nodose neurones, for example, is modulated by insult and injury (Myers et al. 2002; Bielefeldt et al. 2002 a,b; Lamb et al. 2003; Kang et al. 2005; Kollarik et al. 2007) as well as by feeding and fasting (Burdyga et al. 2006, 2008; de Lartigue et al. 2007).
The ability of vagal afferent neurons to respond to GI‐related feeding signals appears to be dependent, therefore, on ongoing physiological and pathophysiological conditions. Several studies have demonstrated that diet‐induced obesity significantly compromises the excitability and responsiveness of GI vagal afferents (Woods et al. 1998; Schwartz et al. 2000; Berthoud, 2008; Berthoud & Morrison, 2008; Paulino et al. 2009; de Lartigue et al. 2011; Kentish et al. 2012; de Lartigue, 2014) suggesting that vagally dependent signalling of early satiety and energy homeostasis is compromised by obesity (Covasa et al. 2000 a; Daly et al. 2011; Kentish et al. 2012). The mechanism responsible for this attenuated responsiveness has not been elucidated fully, although electrophysiological studies have shown that nodose neurones from obese mice and vagal efferent motor neurones from obese rats have a decreased input resistance and increased membrane capacitance, suggestive of an increase in resting background conductances (Daly et al. 2011; Browning et al. 2013).
Few studies, however, have investigated the responses of vagal afferents prior to the development of obesity and whether the attenuated responsiveness of vagal afferents is a cause of, or occurs in response to, obesity. In the present study, rats were given ad libitum access to HFD from postnatal day 28 onwards, i.e. after vagal neurocircuits are known to be fully developed (Rinaman & Levitt, 1993; Rinaman et al. 2000; Rinaman, 2003, 2004). During the time period of this study (8 weeks), rats became overweight but did not develop obesity, defined as a 20% increase in body weight relative to control. Earlier studies by several groups have demonstrated the tendency for Sprague–Dawley rats to separate into obese‐prone and obese‐resistant when fed a diet high in fat and/or carbohydrates in adulthood. The duration of exposure to a calorie‐dense diet required to produce obesity varies between studies and appears to depend on both diet composition (high fat vs. high fat/high carbohydrate) as well as the age of the rats at the start of the exposure (juvenile vs. adult; Levin et al. 1986, 1987; Gao et al. 2002). It is likely, therefore, that the duration of HFD exposure in the current study was not sufficient to evoke obesity within the 8 week time frame due to the young age of the rats (4 weeks) when exposed initially to the diet. In the current study, however, the effects of HFD to attenuate the actions of glucose to modulate 5‐HT‐induced currents were independent of obesity or rat weight, since neurones in which glucose was unable to modulate 5‐HT currents were observed across the weight spectrum of rats at every experimental time‐point studied. The mechanism responsible for this HFD‐induced attenuation in vagal afferent modulation is not yet clear.
Previous studies have also shown a feeding‐ or obesity‐dependent alteration in the distribution of neurotransmitter receptors (Burdyga et al. 2006, 2008; Paulino et al. 2009; de Lartigue et al. 2011; de Lartigue, 2014) providing a potential mechanism for the observed loss of responsiveness to a variety of GI‐related neurohormones, including CCK (Covasa & Ritter, 2000; Covasa et al. 2000 a,b; Swartz et al. 2010; Daly et al. 2011) and 5‐HT (Daly et al. 2011). In the present study, HFD attenuated the response of rat gastric VAN to CCK; in contrast to previous reports, however, HFD did not decrease neuronal excitability or attenuate the responsiveness of gastric VAN or nodose neurones to 5‐HT. There are several potential reasons for the observed differences including species differences (mice vs. rats), examination of the effects of 5‐HT in an obese (Daly et al. 2011) vs. pre‐obese state as well as assay techniques used (calcium imaging vs. whole cell patch clamp recordings; Daly et al. 2011). While the doses and concentrations of 5‐HT used in the present study exceed those measured in the post‐prandial circulation (30–80 nm; Houghton et al. 2003), they are consistent with those used in previous in vivo (Hillsley & Grundy, 1998; Hillsley et al. 1998; Daly et al. 2011) as well as in vitro (Moore et al. 2002; Daly et al. 2011; Babic et al. 2012 b) studies. It should be noted that the rapid enzymatic breakdown of neuropeptides can lead to the underestimation of their endogenous levels (Stengel et al. 2009). Furthermore, it has long been recognized that neurophysiological studies frequently require exogenous application of drugs up to three orders of magnitude greater than those measured in vivo and that systemic measurements of neurotransmitter or neurohormone levels may not accurately predict those at the site of release or action (Katz & Miledi, 1968); thus the present study is likely to be measuring a physiological, rather than pharmacological, response.
It is clear from the present study, however, that even short periods of exposure to a HFD alter vagal afferent neuronal signalling, suggesting that factors other than weight gain itself are responsible for this attenuation, providing further support for the idea put forth recently that the effects of diet on vagal neurocircuits can be distinguished from those of obesity (Daly et al. 2011; Browning et al. 2013). While the underlying mechanisms responsible for the loss of glucose‐dependent modulation of 5‐HT signalling in gastric VAN or nodose neurons is not yet clear, it is known that chronic ingestion of HFD alters the gut microbiota and induces intestinal inflammation (Cani et al. 2008; Hildebrandt et al. 2009; de La Serre et al. 2010; Ding et al. 2010), insulin resistance in neurons (reviewed in Kim & Feldman, 2012) and alters neuronal glucose ‘sensing’ and transport (Levin et al. 1998, 1999; Parton et al. 2007). Of note, the trafficking of 5‐HT3 receptors in model cell systems has been shown to occur via actin‐ and protein kinase C (PKC)‐dependent pathways (Sun et al. 2003; Ilegems et al. 2004; Connolly, 2008). PKC has been implicated as a ‘metabolic sensor’ and its expression is increased by HFD (Newton, 2009), both peripherally, in white adipose tissue (Bansode et al. 2008; Huang et al. 2009), as well as centrally, in the hypothalamus (Benoit et al. 2009). It remains to be determined, however, whether the short periods of HFD exposure employed in the present study alter PKC levels in VAN and nodose neurons and whether this plays a role in the apparent loss of 5HT3 receptor trafficking by glucose.
We have demonstrated previously that the response of GI nodose neurones to 5‐HT is modulated rapidly and reversibly by extracellular glucose levels. Immunocytochemical studies showed that increasing extracellular glucose levels are accompanied by the trafficking of 5‐HT3 receptors to the neuronal membrane whereas lowering extracellular glucose levels induced receptor internalization (Babic et al. 2012 b). Since ingested glucose induces the release of 5‐HT from intestinal enterochromaffin cells, this suggests that, by modulating the ability of vagal afferent fibres to respond to released 5‐HT, glucose amplifies or prolongs its own visceral sensory signalling. The present study, however, demonstrated that the ability of glucose to modulate the response of gastric VAN and neurones to 5‐HT, as well as the trafficking of 5‐HT3 receptors, was compromised by HFD exposure. Postprandial glucose levels in humans typically range between 6 and 8 mm (108–144 mg dl−1) although glycaemic levels approaching 15–20 mm (270–360 mg dl−1) have been reported in poorly controlled diabetic patients (Erlinger & Brancati, 2001; Monnier et al. 2011) . While the level of hyperglycaemia employed in the current study is higher than those used previously to modulate VAN activity (Mei, 1978; Niijima, 1982) or to induce vagally dependent gastric relaxation (Shi et al. 2005; Zhou et al. 2008), those studies used either local application of glucose to VAN endings or much longer periods of hyperglycaemia to induce their observed responses (20–30 min). It is also important to note that, in the present study, VAN activity was recorded only from the anterior gastric branch, i.e. the subdiaphragmatic vagal branch that provides most of the innervation to the anterior stomach and proximal duodenum and, most likely, underestimates the magnitude and sensitivity of the total vagal nerve response, which would include activity within the other four branches of the subdiaphragmatic vagus. In contrast, the glucose concentrations used in the current in vitro studies (1.5–20 mm; 27–260 mg dl−1) are closer to physiological glycaemic levels demonstrated previously by ourselves and other groups to modulate the activity of gastric nodose neurons (Grabauskas et al. 2006, 2010; Babic et al. 2012 b).
While further studies will be required to assess the impact of the HFD‐induced loss in glucose‐dependent regulation of 5‐HT signalling, as well as the mechanism by which HFD induces this effect, it is clear that nodose neuronal responsiveness is compromised even after relatively short periods of HFD exposure, well in advance of the development of obesity or loss of glycaemic regulation. The present study also implies that vagal afferents are remarkably sensitive to the adverse effects of diet and, further, that altered vagal afferent responsiveness may precede, and even contribute to, the development of obesity and glycaemic dysregulation; loss of the ability of glucose to amplify and prolong 5‐HT‐dependent vagal afferent signalling would be expected to compromise the glucose‐induced gastric relaxation, altered gastric motility and delayed gastric emptying, which are critically important for early satiation and meal termination, leading to increased food ingestion
Summary and conclusion
The present study suggests that HFD exposure disrupts the actions of extracellular glucose to modulate 5‐HT‐dependent vagal signalling. Since glucose induces the release of 5‐HT from enterochromaffin cells by also regulating the number of functional 5‐HT3 receptors present on the membrane of gastric vagal afferent neurons, glucose is able to modulate its own ‘perception’, hence amplify or prolong vagal afferent signalling. However, even relatively short periods of HFD exposure disrupt this glucose‐dependent modulation of vagal afferent 5‐HT‐dependent signalling, suggesting that diet, independently of obesity, may profoundly affect vagal afferent responsiveness and, further, that altered vagal afferent responsiveness may precede, and even contribute to, the development of obesity and glycaemic dysregulation.
Additional information
Competing interests
None declared.
Author contributions
A.E.T., S.D.S. and K.N.B. were responsible for study design; A.E.T. and K.N.B. performed the in vitro electrophysiology and immunocytochemical experiments and analyses; S.S.S. and S.D.S. performed the in vivo neurophysiology experiments and analyses. All experiments were carried out at Penn State University College of Medicine, Hershey, PA, USA. All authors were involved in drafting and critically revising the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
These studies were supported by NIH (NIDDK) grant DK078364 to K.N.B., and NIH (NHBLI) grant HL113270 and American Heart Association Established Investigator Award to S.D.S.
Acknowledgements
The authors wish to thank F. Holly Coleman for performing the gastric labelling surgeries and Sam Fortna for assistance in confocal microscopy imaging. Special thanks are due to W. Nairn Browning for support and encouragement, Dr Arthur Berg for his assistance with statistical analyses and Dr R. Alberto Travagli for his critical input on previous manuscript versions.
References
- Altschuler SM, Bao X, Bieger D, Hopkins DA & Miselis RR (1989). Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol 283, 248–268. [DOI] [PubMed] [Google Scholar]
- Alywin ML, Horowitz JM & Bonham AC (1997). NMDA receptors contribute to primary visceral afferent transmission in the nucleus of the solitary tract. J Neurophysiol 77, 2539–2548. [DOI] [PubMed] [Google Scholar]
- Andresen MC & Kunze DL (1994). Nucleus tractus solitarius – gateway to neural circulatory control. Annu Rev Physiol 56, 93–116. [DOI] [PubMed] [Google Scholar]
- Babic T, Browning KN, Kawaguchi Y, Tang X & Travagli RA (2012. a). Pancreatic insulin and exocrine secretion are under the modulatory control of distinct subpopulations of vagal motoneurones in the rat. J Physiol 590, 3611–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic T, Troy AE, Fortna SR & Browning KN (2012. b). Glucose‐dependent trafficking of 5‐HT3 receptors in rat gastrointestinal vagal afferent neurons. Neurogastroenterol Motil 24, e476–e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansode RR, Huang W, Roy SK, Mehta M & Mehta KD (2008). Protein kinase C deficiency increases fatty acid oxidation and reduces fat storage. J Biol Chem 283, 231–236. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, Lefevre AL, Cruciani‐Guglielmacci C, Magnan C, Yu F, Niswender K, Irani BG, Holland WL & Clegg DJ (2009). Palmitic acid mediates hypothalamic insulin resistance by altering PKC‐θ subcellular localization in rodents. J Clin Invest 119, 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud H‐R (2004). The caudal brainstem and the control of food intake and energy balance In Neurobiology of Food and Fluid Intake, ed. Stricker E. & Woods S, pp. 195–240. Plenum, New York. [Google Scholar]
- Berthoud H‐R (2008). The vagus nerve, food intake and obesity. Regul Pept 149, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud H‐R, Carlson NR & Powley TL (1991). Topography of efferent vagal innervation of the rat gastrointestinal tract. Am J Physiol 260, R200–R207. [DOI] [PubMed] [Google Scholar]
- Berthoud H‐R & Morrison C (2008). The brain, appetite, and obesity. Annu Rev Psychol 59, 55–92. [DOI] [PubMed] [Google Scholar]
- Berthoud H‐R & Powley TL (1991). Morphology and distribution of efferent vagal innervation of rat pancreas as revealed with anterograde transport of Dil. Brain Res 553, 336–341. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N & Gebhart GF (2002. a). Experimental ulcers alter voltage‐sensitive sodium currents in rat gastric sensory neurons. Gastroenterology 122, 394–405. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N & Gebhart GF (2002. b). Mild gastritis alters voltage‐sensitive sodium currents in gastric sensory neurons in rats. Gastroenterology 122, 752–761. [DOI] [PubMed] [Google Scholar]
- Browning KN, Fortna SR & Hajnal A (2013). Roux‐en‐Y gastric bypass reverses the effects of diet‐induced obesity to inhibit the responsiveness of central vagal motoneurones. J Physiol 591, 2357–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Renehan WE & Travagli RA (1999). Electrophysiological and morphological heterogeneity of rat dorsal vagal neurones which project to specific areas of the gastrointestinal tract. J Physiol 517, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN & Travagli RA (2010). Plasticity of vagal brainstem circuits in the control of gastric function. Neurogastroenterol Motil 22, 1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN & Travagli RA (2014). Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr Physiol 4, 1339–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, de Lartigue G, Raybould HE, Morris R, Dimaline R, Varro A, Thompson DG & Dockray GJ (2008). Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci 28, 11583–11592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG & Dockray GJ (2006). Feeding‐dependent depression of melanin‐concentrating hormone and melanin‐concentrating hormone receptor‐1 expression in vagal afferent neurones. Neuroscience 137, 1405–1415. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM & Burcelin R (2008). Changes in gut microbiota control metabolic endotoxemia‐induced inflammation in high‐fat diet‐induced obesity and diabetes in mice. Diabetes 57, 1470–1481. [DOI] [PubMed] [Google Scholar]
- Connolly CN (2008). Trafficking of 5‐HT3 and GABAA receptors (Review). Mol Membr Biol 25, 293–301. [DOI] [PubMed] [Google Scholar]
- Covasa M, Grahn J & Ritter RC (2000. a). High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul Pept 86, 83–88. [DOI] [PubMed] [Google Scholar]
- Covasa M, Grahn J & Ritter RC (2000. b). Reduced hindbrain and enteric neuronal response to intestinal oleate in rats maintained on high‐fat diet. Auton Neurosci 84, 8–18. [DOI] [PubMed] [Google Scholar]
- Covasa M & Ritter RC (2000). Adaptation to high‐fat diet reduces inhibition of gastric emptying by CCK and intestinal oleate. Am J Physiol Regul Integr Comp Physiol 278, R166–R170. [DOI] [PubMed] [Google Scholar]
- Daly DM, Park SJ, Valinsky WC & Beyak MJ (2011). Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol 589, 2857–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das UN (2010). Obesity: genes, brain, gut, and environment. Nutrition 26, 459–473. [DOI] [PubMed] [Google Scholar]
- de Lartigue G (2014). Putative roles of neuropeptides in vagal afferent signaling. Physiol Behav 136, 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, de La Serre CB & Raybould HE (2011). Vagal afferent neurons in high fat diet‐induced obesity; intestinal microflora, gut inflammation and cholecystokinin. Physiol Behav 105, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Dimaline R, Varro A & Dockray GJ (2007). Cocaine‐ and amphetamine‐regulated transcript: stimulation of expression in rat vagal afferent neurons by cholecystokinin and suppression by ghrelin. J Neurosci 27, 2876–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC & Raybould HE (2010). Propensity to high‐fat diet‐induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299, G440–G448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C & Lund PK (2010). High‐fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5, e12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray G (2004). Gut endocrine secretions and their relevance to satiety. Curr Opin Pharmacol 4, 557–560. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Paulino G & Raybould HE (2007). CCK1 receptor is essential for normal meal patterning in mice fed high fat diet. Physiol Behav 92, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlinger TP & Brancati FL (2001). Postchallenge hyperglycemia in a national sample of U.S. adults with type 2 diabetes. Diabetes Care 24, 1734–1738. [DOI] [PubMed] [Google Scholar]
- Gao J, Ghibaudi L, van Heek M & Hwa JJ (2002). Characterization of diet‐induced obese rats that develop persistent obesity after 6 months of high‐fat followed by 1 month of low‐fat diet. Brain Res 936, 87–90. [DOI] [PubMed] [Google Scholar]
- Grabauskas G, Song I, Zhou SY & Owyang C (2010). Electrophysiological identifications of glucose‐sensing neurons in the rat nodose ganglia. J Physiol 588, 617–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabauskas G, Zhou SY, Song I & Owyang C (2006). Nodose ganglia neurons demonstrate gluco‐responsiveness mediated via voltage‐dependent potassium channels. Gastroenterology 130, 507 (A74).16472603 [Google Scholar]
- Hildebrandt MA, Hoffmann C, Sherrill‐Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F & Wu GD (2009). High‐fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137, 1716–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillsley K & Grundy D (1998). Sensitivity to 5‐hydroxytryptamine in different afferent subpopulations within mesenteric nerves supplying the rat jejunum. J Physiol 509, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillsley K, Kirkup AJ & Grundy D (1998). Direct and indirect actions of 5‐hydroxytryptamine on the discharge of mesenteric afferent fibres innervating the rat jejunum. J Physiol 506, 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GM, Browning K, Tong M, Qualls‐Creekmore E & Travagli RA (2009. a). Vagally‐mediated effects of glucagon‐like peptide 1 (GLP‐1): in vitro and in vivo gastric actions. J Physiol 587, 4749–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GM, Tong M & Travagli RA (2009. b). Effects of brain stem cholecystokinin‐8 s on gastric tone and esophageal‐gastric reflex. Am J Physiol Gastrointest Liver Physiol 296, G621–G631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer HH, Turkelson CM, Solomon TE & Raybould HE (1994). Intestinal lipid inhibits gastric emptying via CCK and a vagal capsaicin‐sensitive afferent pathway in rats. Am J Physiol 267, G625–G629. [DOI] [PubMed] [Google Scholar]
- Horowitz M, O'Donovan D, Jones KL, Feinle C, Rayner CK & Samsom M (2002). Gastric emptying in diabetes: clinical significance and treatment. Diabet Med 19, 177–194. [DOI] [PubMed] [Google Scholar]
- Houghton LA, Atkinson W, Whitaker RP, Whorwell PJ & Rimmer MJ (2003). Increased platelet depleted plasma 5‐hydroxytryptamine concentration following meal ingestion in symptomatic female subjects with diarrhoea predominant irritable bowel syndrome. Gut 52, 663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Bansode R, Mehta M & Mehta KD (2009). Loss of protein kinase Cβ function protects mice against diet‐induced obesity and development of hepatic steatosis and insulin resistance. Hepatology 49, 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilegems E, Pick HM, Deluz C, Kellenberger S & Vogel H (2004). Noninvasive imaging of 5‐HT3 receptor trafficking in live cells: from biosynthesis to endocytosis. J Biol Chem 279, 53346–53352. [DOI] [PubMed] [Google Scholar]
- Kang YM, Lamb K, Gebhart GF & Bielefeldt K (2005). Experimentally induced ulcers and gastric sensory‐motor function in rats. Am J Physiol Gastrointest Liver Physiol 288, G284–G291. [DOI] [PubMed] [Google Scholar]
- Katz B & Miledi R (1968). The role of calcium in neuromuscular facilitation. J Physiol 195, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano K, Hirashima S, Mori Y, Saitoh Y & Kurosumi M (1992). Spontaneous long‐term hyperglycemic rat with diabetic complications. Otsuka Long‐Evans Tokushima Fatty (OLETF) strain. Diabetes 41, 1422–1428. [DOI] [PubMed] [Google Scholar]
- Kentish S, Li H, Philp LK, O'Donnell TA, Isaacs NJ, Young RL, Wittert GA, Blackshaw LA & Page AJ (2012). Diet‐induced adaptation of vagal afferent function. J Physiol 590, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B & Feldman EL (2012). Insulin resistance in the nervous system. Trends Endocrinol Metab 23, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollarik M, Ru F & Undem BJ (2007). Acid‐sensitive vagal sensory pathways and cough. Pulm Pharmacol Ther 20, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacolley P, Owen JR, Sandock K, Lewis TH, Bates JN, Robertson TP & Lewis SJ (2006). Occipital artery injections of 5‐HT may directly activate the cell bodies of vagal and glossopharyngeal afferent cell bodies in the rat. Neuroscience 143, 289–308. [DOI] [PubMed] [Google Scholar]
- Lamb K, Kang YM, Gebhart GF & Bielefeldt K (2003). Gastric inflammation triggers hypersensitivity to acid in awake rats. Gastroenterology 125, 1410–1418. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn‐Meynell AA & Routh VH (1999). Brain glucose sensing and body energy homeostasis: role in obesity and diabetes. Am J Physiol 276, R1223–R1231. [DOI] [PubMed] [Google Scholar]
- Levin BE, Govek EK & Dunn‐Meynell AA (1998). Reduced glucose‐induced neuronal activation in the hypothalamus of diet‐induced obese rats. Brain Res 808, 317–319. [DOI] [PubMed] [Google Scholar]
- Levin BE, Triscari J, Hogan S & Sullivan AC (1987). Resistance to diet‐induced obesity: food intake, pancreatic sympathetic tone, and insulin. Am J Physiol 252, R471–R478. [DOI] [PubMed] [Google Scholar]
- Levin BE, Triscari J & Sullivan AC (1986). Metabolic features of diet‐induced obesity without hyperphagia in young rats. Am J Physiol 251, R433–R440. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu XY, Zhu JX & Owyang C (2001). Intestinal serotonin acts as paracrine substance to mediate pancreatic secretion stimulated by luminal factors. Am J Physiol Gastrointest Liver Physiol 281, G916–G923. [DOI] [PubMed] [Google Scholar]
- Lloyd KCK, Holzer HH, Zittel TT & Raybould HE (1993). Duodenal lipid inhibits gastric acid secretion by vagal, capsaicin‐sensitive pathways in rats. Am J Physiol 264, G659–G663. [DOI] [PubMed] [Google Scholar]
- Mei N (1978). Vagal glucoreceptors in the small intestine of the cat. J Physiol 282, 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier L, Colette C & Owens D (2011). Postprandial and basal glucose in type 2 diabetes: assessment and respective impacts. Diabetes Technol Ther 13 (Suppl. 1), S25–S32. [DOI] [PubMed] [Google Scholar]
- Moore KA, Oh EJ & Weinreich D (2002). 5‐HT3 receptors mediate inflammation‐induced unmasking of functional tachykinin responses in vitro. J Appl Physiol (1985) 92, 2529–2534. [DOI] [PubMed] [Google Scholar]
- Myers AC, Kajekar R & Undem BJ (2002). Allergic inflammation‐induced neuropeptide production in rapidly adapting afferent nerves in guinea pig airways. Am J Physiol Lung Cell Mol Physiol 282, L775–L781. [DOI] [PubMed] [Google Scholar]
- Newton AC (2009). Lipid activation of protein kinases. J Lipid Res 50 (Suppl.), S266–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima A (1982). Glucose‐sensitive afferent nerve fibres in the hepatic branch of the vagus nerve in the guinea‐pig. J Physiol 332, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA & Lowell BB (2007). Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449, 228–232. [DOI] [PubMed] [Google Scholar]
- Paulino G, Barbier de la Serre C, Knotts TA, Oort PJ, Newman JW, Adams SH & Raybould HE (2009). Increased expression of receptors for orexigenic factors in nodose ganglion of diet‐induced obese rats. Am J Physiol Endocrinol Metab 296, E898–E903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE (1999). Nutrient tasting and signaling mechanisms in the gut. I. Sensing of lipid by the intestinal mucosa. Am J Physiol 277, G751–G755. [DOI] [PubMed] [Google Scholar]
- Raybould HE, Glatzle J, Robin C, Meyer JH, Phan T, Wong H & Sternini C (2003). Expression of 5‐HT3 receptors by extrinsic duodenal afferents contribute to intestinal inhibition of gastric emptying. Am J Physiol Gastrointest Liver Physiol 284, G367–G372. [DOI] [PubMed] [Google Scholar]
- Rayner CK, Samsom M, Jones KL & Horowitz M (2001). Relationships of upper gastrointestinal motor and sensory function with glycemic control. Diabetes Care 24, 371–381. [DOI] [PubMed] [Google Scholar]
- Rinaman L (2003). Postnatal development of hypothalamic inputs to the dorsal vagal complex in rats. Physiol Behav 79, 65–70. [DOI] [PubMed] [Google Scholar]
- Rinaman L (2004). Postnatal development of central feeding circuits In Neurobiology of Food and Fluid Intake, ed. Stricker E. & Woods S, pp. 159–194. Plenum, New York. [Google Scholar]
- Rinaman L & Levitt P (1993). Establishment of vagal sensorimotor circuits during fetal development in rats. J Neurobiol 24, 641–659. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Levitt P & Card JP (2000). Progressive postnatal assembly of limbic‐autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J Neurosci 20, 2731–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ (2000). The role of gastrointestinal vagal afferents in the control of food intake: current prospects. Nutrition 16, 866–873. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Seeley RJ & Baskin DG (2000). Central nervous system control of food intake. Nature 404, 661–671. [DOI] [PubMed] [Google Scholar]
- Shapiro RE & Miselis RR (1985). The central organization of the vagus nerve innervating the stomach of the rat. J Comp Neurol 238, 473–488. [DOI] [PubMed] [Google Scholar]
- Shi M, Jones AR, Ferreira M Jr, Sahibzada N, Gillis RA & Verbalis JG (2005). Glucose does not activate nonadrenergic, noncholinergic inhibitory neurons in the rat stomach. Am J Physiol Regul Integr Comp Physiol 288, R742–R750. [DOI] [PubMed] [Google Scholar]
- Sivarao DV, Krowicki ZK & Hornby PJ (1998). Role of GABAA receptors in rat hindbrain nuclei controlling gastric motor function. Neurogastroenterol Motil 10, 305–313. [DOI] [PubMed] [Google Scholar]
- Stengel A, Keire D, Goebel M, Evilevitch L, Wiggins B, Tache Y & Reeve JR Jr (2009). The RAPID method for blood processing yields new insight in plasma concentrations and molecular forms of circulating gut peptides. Endocrinology 150, 5113–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Hu XQ, Moradel EM, Weight FF & Zhang L (2003). Modulation of 5‐HT3 receptor‐mediated response and trafficking by activation of protein kinase C. J Biol Chem 278, 34150–34157. [DOI] [PubMed] [Google Scholar]
- Swartz TD, Savastano DM & Covasa M (2010). Reduced sensitivity to cholecystokinin in male rats fed a high‐fat diet is reversible. J Nutr 140, 1698–1703. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD & Vicini S (1991). Glutamate and GABA‐mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol 260, G531–G536. [DOI] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Porte DJ & Schwartz MW (1998). Signals that regulate food intake and energy homeostasis. Science 280, 1378–1382. [DOI] [PubMed] [Google Scholar]
- Zhou SY, Lu YX & Owyang C (2008). Gastric relaxation induced by hyperglycemia is mediated by vagal afferent pathways in the rat. Am J Physiol Gastrointest Liver Physiol 294, G1158–G1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JX, Wu XY, Owyang C & Li Y (2001). Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J Physiol 530, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]