

Key points
Interleukin‐13 (IL‐13) causes intestinal epithelial barrier dysfunction, and is implicated in the pathogenesis of Th2‐driven intestinal inflammation (e.g. ulcerative colitis). However, it is unclear whether the epithelial sodium channel (ENaC) – the main limiting factor for sodium absorption in the distal colon – is also influenced by IL‐13 and if so, by what mechanism(s).
We demonstrate in an intestinal cell model as well as in mouse distal colon that IL‐13 causes reduced ENaC activity.
We show that IL‐13 impairs ENaC‐dependent sodium transport by activating the JAK1/2–STAT6 signalling pathway.
These results improve our understanding of the mechanisms through which IL‐13 functions as a key effector cytokine in ulcerative colitis, thereby contributing to the distinct pathology of this disease.
Abstract
Interleukin‐13 (IL‐13) has been strongly implicated in the pathogenesis of ulcerative colitis, possibly by disrupting epithelial integrity. In the distal colon, the epithelial sodium channel (ENaC) is an important factor in the regulation of sodium absorption, and therefore plays a critical role in minimizing intestinal sodium and water losses. In the present study, we investigated whether IL‐13 also acts as a potent modulator of epithelial sodium transport via ENaC, and the signalling components involved. The effect of IL‐13 on ENaC was examined in HT‐29/B6‐GR/MR human colon cells, as well as in mouse distal colon, by measuring amiloride‐sensitive short‐circuit current (I SC) in Ussing chambers. The expression levels of ENaC subunits and the cellular components that contribute to ENaC activity were analysed by qRT‐PCR and promoter gene assay. We show that IL‐13, in both the cell model and in native intestinal tissue, impaired epithelial sodium absorption via ENaC (J Na) as a result of decreased transcription levels of β‐ and γ‐ENaC subunits and SGK1, a post‐translational regulator of ENaC activity, due to impaired promoter activity. The reduction in J Na was prevented by inhibition of JAK1/2–STAT6 signalling. This inhibition also affected the IL‐13‐induced decrease in p38 MAPK phosphorylation. The contribution of STAT6 to IL‐13‐mediated ENaC inactivation was confirmed in a STAT6−/− mouse model. In conclusion, these results indicate that IL‐13, the levels of which are elevated in ulcerative colitis, contributes to impaired ENaC activity via modulation of the STAT6/p38 MAPK pathways.
Key points
Interleukin‐13 (IL‐13) causes intestinal epithelial barrier dysfunction, and is implicated in the pathogenesis of Th2‐driven intestinal inflammation (e.g. ulcerative colitis). However, it is unclear whether the epithelial sodium channel (ENaC) – the main limiting factor for sodium absorption in the distal colon – is also influenced by IL‐13 and if so, by what mechanism(s).
We demonstrate in an intestinal cell model as well as in mouse distal colon that IL‐13 causes reduced ENaC activity.
We show that IL‐13 impairs ENaC‐dependent sodium transport by activating the JAK1/2–STAT6 signalling pathway.
These results improve our understanding of the mechanisms through which IL‐13 functions as a key effector cytokine in ulcerative colitis, thereby contributing to the distinct pathology of this disease.
Abbreviations
- DBA
dexamethasone plus butyrate and aldosterone
- ENaC
epithelial sodium channel
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- HRE
hormone response element
- IL‐13
interleukin‐13
- ISC
amiloride sensitive short‐circuit current
- JNa
epithelial sodium absorption via the ENaC
- MAPK
mitogen‐activated protein kinase
- MR
mineralocorticoid receptor
- MRE
mineralocorticoid response element
- PI3K
phosphatidylinositol 3‐kinase
- SGK1
serum and glucocorticoid‐induced kinase 1
- TER
transepithelial resistance
- UC
ulcerative colitis
- WT
wildtype
Introduction
The extent of water absorption by healthy human colon is directly related to the absorption of solutes, especially sodium ions (Na+). Different Na+ transport systems exist along the length of the large intestine. In the proximal colon, Na+ is transported through electroneutral NaCl absorption, achieved by the sodium/hydrogen exchanger 3 (NHE3) acting in conjunction with chloride/bicarbonate antiporters (DRA or AE1). In distal colon, Na+ is absorbed predominantly via the epithelial sodium channel (ENaC).
At a molecular level, ENaC is usually assumed to be a heterotrimer of one α‐subunit, one β‐subunit and one γ‐subunit (Jasti et al. 2007; Firsov et al. 1998). In contrast to other steroid‐responsive organs, such as lung and distal nephron, in the distal colon only the β‐ and γ‐subunits are regulated, while the α‐subunit is constitutively expressed (Renard et al. 1995; Epple et al. 2000). In addition, ENaC‐dependent Na+ transport is regulated by increased recruitment as well as prolonged insertion of active channels into the apical enterocyte membrane (Greig et al. 2004).
It has been shown that the apical ENaC β‐ and γ‐subunits are down‐regulated in ulcerative colitis (UC), and therefore contribute to the pathogenesis of diarrhoea in this disease (Amasheh et al. 2004; Sullivan et al. 2009). Furthermore, Amasheh and colleagues showed that ENaC dysregulation could be attributed to elevated proinflammatory cytokines, such as TNFα and interleukin‐1β. In addition, IL‐13 has been identified as a key inflammatory cytokine in the Th2 response in UC (Fuss et al. 2004). IL‐13 leads to epithelial barrier disturbance, caused by increased epithelial apoptosis and also by an increased expression of the channel‐forming tight junction protein claudin‐2 (Heller et al. 2005). It has also been proposed that IL‐13 is linked to the pathogenesis of Th2‐cytokine mediated inflammation, since IL‐13 neutralization prevented the inflammatory response in the oxazolone‐colitis model (Heller et al. 2002). IL‐13 acts through a heterodimeric cell‐surface receptor composed of the IL‐4Rα chain and the IL‐13Rα1 chain. Its binding activates Janus‐kinases (JAK) and leads to the phosphorylation and activation of signal transducers and activators of transcription (e.g. STAT3 and STAT6), which then dimerize and translocate to the nucleus, where they interact with other transcription factors to regulate gene transcription (Takeda et al. 1996; Hershey, 2003). Recent findings suggest that IL‐13 binds to another receptor, the IL‐13Rα2 chain (thought to act only as a decoy‐receptor), which results in the activation of signalling cascades, such as AP1 or MAPK (Fichtner‐Feigl et al. 2006; Mandal & Levine, 2010), both of which have been shown to regulate ENaC (Shen & Cotton, 2003; Zeissig et al. 2008). Previous observations suggest an important role for the STAT6 pathway activated by IL‐13 in patients with UC, such as IL‐13‐induced epithelial apoptosis and claudin‐2 expression in vitro (Rosen et al. 2011; Rosen et al. 2013).
Previous studies on the role of IL‐13 in UC have only considered barrier destruction, and the influence of this cytokine on intestinal transport processes (for example, electrogenic Na+ absorption), remains unclear. In allergic airway diseases, IL‐13 acts as a potent modulator of ENaC activity in the lung through a STAT6‐dependent mechanism (Anagnostopoulou et al. 2010). However, many differences exist between ENaC regulation in the lung and in the distal colon, and between the pathogenesis of allergic airway diseases and UC. Therefore, the aim of the current study was to evaluate whether ENaC function in the colon is regulated by IL‐13, and the signalling pathways involved. In HT‐29/B6‐GR/MR cell monolayers (a novel, highly differentiated colon cell model) stably transfected with the glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) (Bergann et al. 2011), we demonstrated that intestinal sodium absorption via ENaC was significantly affected by IL‐13, a change which reflected defective up‐regulation of ENaC β‐ and γ‐subunits. Using inhibitors to differentiate between MAPK and JAK/STAT6 pathways, and STAT6‐deficient (STAT6−/−) mice, we found that IL‐13‐dependent dysregulation of ENaC relied on both MAPK (p38) and STAT6 signalling.
Methods
Ethical approval and animals
Ethical approval for animal experiments was obtained from the local ethics commitee of the LAGeSo (Landesamt für Gesundheit und Soziales, Berlin, Germany; approval number: G 0208/12). BALB/c wild‐type (Forschungseinrichtungen für experimentelle Medizin, Charité – Universitätsmedizin Berlin, Germany) and STAT6−/− mice on BALB/c background (The Jackson Laboratory, Bar Harbor, ME, USA) were maintained according to institutional standards. For cytokine stimulation experiments, 12‐ to 20‐week‐old female mice were administered an intraperitoneal (i.p.) injection of 3 μg recombinant murine IL‐13 (BioLegend GmbH) dissolved in 300 μl phosphate buffered saline (PBS) or an equal volume of vehicle alone. Seventy‐two hours later, further injections were administered once daily for three consecutive days. Twenty‐four hours after the last challenge, mice were killed by cervical dislocation. Distal colon was removed for direct measurements in Ussing chamber experiments.
Cell culture and reagents
HT‐29/B6‐GR/MR cells, a subclone of the human colorectal cancer cell line HT‐29 (Bergann et al. 2011), were cultured at 37°C in humidified 95% air/5% CO2 in 25 cm2 culture flasks in RPMI1640 medium supplemented with 10% fetal calf serum (FCS, Sigma‐Aldrich Chemie GmbH, Munich, Germany), 1% penicillin–streptomycin (Sigma‐Aldrich), 500 IU ml−1 G418 (Biochrom GmbH, Berlin, Germany) and 200 μg ml−1 hygromycin B (Life Technologies GmbH, Darmstadt, Germany). For most of the experiments cells were seeded on Millicell PCF filters (3 μm; Millipore, Schwalbach, Germany) and grown for 7 days. Experiments were performed when cell monolayers had reached transepithelial resistance (TER) values of 1500–2000 Ω cm2. Prior to stimulating the cells with DBA (a combination of dexamethasone (D, 50 nm; Sigma‐Aldrich), sodium butyrate (B, 2 mm; Merck‐Schuchardt, Hohenbrunn, Germany) and aldosterone (A, 3 nm; Sigma‐Aldrich)), they were incubated in medium containing 10% hormone‐free FCS (Sigma‐Aldrich) for 24 h. Human recombinant IL‐13 (BioLegend GmbH, Fell, Germany) was added to the culture medium at 100 ng ml−1 2 h after stimulating with DBA. In some experiments, cells were pre‐treated with inhibitors for 2 h prior to exposure to IL‐13. Specific MAPK inhibitors U0126 (which inhibits the p42/44 extracellular signal‐regulated kinase, upstream kinase MEK 1/2) and SP600125 (which inibits JNK MAPK) (both from Cell Signaling Technology, Frankfurt am Main, Germany), and SB202190 (which inhibits p38 MAPK) (Calbiochem, Darmstadt, Germany), were used at the same concentration (10 μm). Baricitinib (500 nm; Selleckchem, Houston, TX, USA) was used to inhibit JAK 1 and 2 (JAK1/2). STAT3 inhibitor V (Stattic, 10 μm, Santa Cruz, Heidelberg, Germany) and AS1517499 (10 μm; Axon Medchem, Groningen, Netherlands) were used to inhibit STAT3 and STAT6, respectively, and Ly294002 (10 μm; Sigma‐Aldrich) to inhibit the phosphatidylinositol 3‐kinase (PI3K) pathway. The IL‐13 type‐I receptor (IL‐4Rα/IL‐13Rα1) was blocked with an anti‐IL‐4R antibody (100 ng ml−1; R&D Systems, Wiesbaden, Germany).
Electrophysiological measurements
Human epithelial cell culture
After 24 to 96 h of stimulation, confluent monolayers of HT‐29/B6‐GR/MR cells grown on filters were mounted into Ussing‐type chambers (epithelial area 0.6 cm2). The bathing solution consisted of (mm): Na+ 140.0; Cl− 123.8; K+ 5.4; Ca2+ 1.2; Mg2+ 1.2; HPO4 2− 2.4; H2PO4 − 0.6 and HCO3 − 21.0. The solution was gassed with a 95% O2/5% CO2 mixture by bubble lift. Temperature was kept at 37°C, pH 7.4. Transepithelial resistance (TER, Ω cm2) and short‐circuit current (I SC; μA cm−2) were recorded using voltage clamp devices (CVC6; Fiebig, Berlin, Germany). After an equilibration period of 20 min, the ENaC‐dependent Na+ transport was measured as a drop in I SC after adding the ENaC blocker amiloride (100 μm; Sigma‐Aldrich) to the apical side of the cell monolayer. The concentration of amiloride yielding 50% ENaC inhibition (IC50) is ∼100 nm (Kellenberger & Schild, 2002). To ensure complete inhibition of ENaC in the colonic epithelium, which is covered by a mucus layer, an amiloride concentration of 100 μm was employed. This concentration is 10‐fold higher than concentrations usually used to completely block the ENaC in epithelial cell models, or in the kidney (10 μm), but still specific for Na+ transport via the ENaC, as NHE3, the other transport system for Na+ in the apical cell membrane of colonocytes, is only affected by amiloride concentrations at 1 mm.
Mouse colon
For determination of the electrogenic Na+ absorption in the colon of BALB/c and STAT6−/− mice, epithelia were mounted into miniaturized Ussing chambers with the exposed area of 0.049 cm2. The bathing solution had the same ion composition described above. Additionally, 10.0 mm d(+)‐glucose, 10.0 mm d(+)‐mannose, 2.5 mm glutamine and the antibiotics piperacillin (50 mg l−1) and imipenem (4 mg l−1) were added. After a 30 min equilibration period with heat‐inactivated hormone‐free FCS (10%), which prevented adhesion of cytokines to glass surfaces of the Ussing set‐up, IL‐13 (100 ng ml−1) was added to the serosal side of the epithelia. After an additional 30 min, the epithelia were stimulated by adding DBA to both sides of the epithelium. After 8 h, amiloride (100 μm) was added to the mucosal compartment to quantify ENaC‐dependent electrogenic sodium absorption (J Na).
RNA extraction and real‐time RT‐PCR
In accordance with the manufacturer's protocol, total RNA was isolated by using PeqGOLD RNApure (PEQLAB, Biotechnologie GmbH, Germany). Two micrograms of extracted RNA were then reverse‐transcribed into cDNA by using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Mannheim, Germany), following the manufacturer's instructions. mRNA expression of the β‐ and γ‐ENaC subunits and SGK‐1 under different conditions was evaluated by real‐time PCR, using a 7500 Fast Real‐time PCR System and TaqMan Gene Expression assays No. HS00165722_ml, HS00168918_ml and HS00178612_ml (Applied Biosystems). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (unlabelled primer, VIC/TAMARA) served as a reference gene. Relative mRNA expression for each transcript of interest was quantified using the comparative C t (ΔΔC t) method (Livak & Schmittgen, 2001).
Transfection and gene reporter assay
HT‐29/B6‐GR/MR cells were seeded with a density of 5 × 104 cells/12‐well under hormone‐free conditions. After 24 h, transient transfections were performed with Lipofectamine plus (Invitrogen, Karlsruhe, Germany). The reporter gene construct pMMTV‐Luc, containing tandem hormone response element (HRE) sequences followed by a luciferase (luc) gene, served for the detection of HRE‐driven luciferase expression. The pghENaC‐B construct (−459 to +35) was a gift of Dr Christie P. Thomas (University of Iowa, Iowa City, IA, USA), and was used to study the promoter activity of γ‐ENaC. SGK1‐driven luciferase expression was detected using a phSGK1‐3kb‐up‐Luc construct containing the SGK1 promoter sequence followed by firefly luciferase gene (a gift of Dr Tim Zierra, Bayer Schering Pharma AG, Berlin). As co‐reporter, a pGL4.70 construct (hRluc; Promega, Mannheim, Germany) containing a Renilla luciferase gene was applied. The expression level of Renilla luciferase remained unaffected under our experimental conditions, and served to normalize the transfection efficiency.
A total of 500 ng reporter plasmid and 50 ng of co‐reporter plasmid per well were used for transfection. Four hours after transfection, cells were stimulated with DBA, and 2 h later with rhIL‐13. Measurement of both firefly and Renilla luciferase activity was performed with the Dual‐Luciferase Reporter Assay System (Promega) and a centro LB 960 microplate‐luminometer (Berthold, Bad Wildbach, Germany) 72 h after transfection.
Western blot analysis
Two hours after stimulation of filter‐grown HT‐29/B6‐GR/MR cells with DBA in completely hormone‐free medium, rhIL‐13 (100 ng ml−1) was added to the basal side of the cells. They were then incubated for 96 h. The medium was then replaced by medium lacking FCS and IL‐13. Cells were incubated again for 3 h prior to a new incubation with IL‐13. Depending on the phosphorylated protein, after 5, 15, 30 or 60 min of IL‐13 incubation whole cell protein was prepared. In brief, cells were washed with ice‐cold PBS and scraped off with lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm Triton X‐100, 1 mm EDTA, 1 mm PMSF, 2.5 mm sodium pyrophosphate, 1 mm β‐glycerophosphate, 1 mm sodium orthovanadate, 1 mm EGTA, complete protease inhibitor cocktail (Roche, Mannheim, Germany)) and pushed through a 1 ml hypodermic syringe with a 0.45 × 10 mm needle. After an incubation step of 10 min on ice, the lysates were centrifuged (15,000 g, 15 min, 4°C) and the supernatants used as whole cell extracts. Proteins were resolved by SDS‐Page on a 12% gel under reducing conditions and electrotransferred onto PVDF‐ membranes. After blocking for 1 h in blocking solution (5% BSA/PBS‐T), membranes were incubated with primary antibodies overnight at 4°C. Antibodies for detection of anti‐phospho‐p42/44 MAPK (anti‐pERK; Thr202/Tyr204), anti‐phospho‐SAP/JNK (anti‐pJNK; Thr183/Tyr185), anti‐phospho‐p38 (anti‐p‐p38; Thr334), anti‐p42/44 MAPK (anti‐ERK; Thr202/Tyr204), anti‐SAPK/JNK (anti‐JNK), anti‐p38 (Thr334), and phospho‐specific and total STAT6 and STAT3 were purchased from Cell Signaling Technology. Anti‐β‐actin was purchased from Sigma‐Aldrich. Following an incubation period for 1 h at room temperature with the peroxidase (POD)‐conjugated secondary antibody (Lumi‐LightPLUS Western blotting kit; Roche), chemiluminescence signals were detected using an Fusion FX7 imaging system (Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany), and were analysed with the AIDA program package (Raytest, Berlin, Germany).
Statistical analysis
Data are presented as arithmetic means ± standard error of the mean (SEM) and were evaluated using student's t‐test for independent samples. For comparison of more than two groups, analysis of variance was performed (1‐way ANOVA) for non‐parametric data, or was adjusted with the Bonferroni correction for parametric values. P‐values < 0.05 were considered to be statistically significant.
Results
IL‐13 impairs ENaC‐dependent active electrogenic Na+ transport (J Na) in colonic HT‐29/B6‐GR/MR cells and native mouse colon
To explore the IL‐13 effect on J Na, HT‐29/B6‐GR/MR cells were treated with DBA (D: dexamethasone, 50 nm; B: sodium butyrate, 2 mm; A: aldosterone, 3 nm) in order to stimulate J Na. After 96 h with and without IL‐13, Ussing measurements were performed. IL‐13 (100 ng ml−1) reduced the DBA‐induced J Na to 49 ± 9% (Fig. 1 A). Interestingly, this pronounced effect of IL‐13 was observed only after 96 h (Fig. 1 B), whereas the transepithelial resistance (TER) was already reduced after 48 h (1527 ± 116 Ω cm2 for DBA versus 1094 ± 132 Ω cm2 for DBA+IL‐13; P < 0.05). Furthermore, the IL‐13 influence on J Na was dose dependent, with significant effects starting at 50 ng ml−1 (Fig. 1 C). Since malignant transformation may alter electrogenic Na+ transport in colonic cell lines, we also studied the effect of IL‐13 in native large intestine from BALB/c mice. Wild‐type (WT) mice were i.p. pre‐treated with either IL‐13 or vehicle for 1 week. Ex vivo distal colon specimens were then stimulated with DBA in the Ussing chamber (Fig. 1 D and E). Similar to the effects in HT‐29/B6‐GR/MR cells, J Na was also decreased by IL‐13 in distal mouse colon (1.8 ± 0.4 μmol h−1 cm−2 for IL‐13 versus 4.0 ± 1.0 μmol h−1 cm−2 for vehicle; P < 0.01).
Figure 1. IL‐13 impairs ENaC‐dependent Na+ absorption in HT‐29/B6‐GR/MR cells as well as in native intestinal tissue .
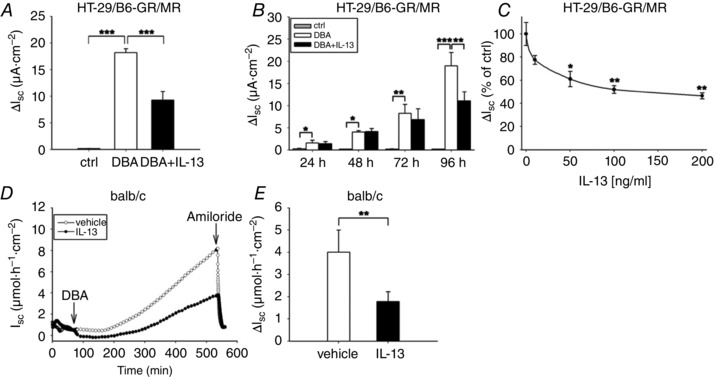
A–C, cells were non‐treated (ctrl) or triple‐incubated (DBA, dexamethasone (50 nm), butyrate (2 mm), aldosterone (3 nm)) and treated with or without IL‐13. A, ENaC‐dependent Na+ absorption after 96 h of incubation (J Na) was measured as drop in I SC after addition of amiloride (100 μm). B, time‐dependent progress of J Na. C, IL‐13 dose dependency of J Na. Data in A–C represent means ± SEM; n = 4–10, *P < 0.05, **P < 0.01, ***P < 0.001. D, time course of a representative experiment of DBA‐induced active electrogenic Na+ transport in the distal colon of WT mice that were pretreated with PBS (ctrl) or IL‐13 as described in Methods. Tissues were stimulated with a triple combination of DBA for 8 h and then ENaC‐dependent Na+ absorption (J Na) was determined as drop in amiloride‐sensitive I SC. E, J Na of WT mice treated as described in D given as means ± SEM of 8 mice per group; *P < 0.05, **P < 0.01, ***P < 0.001.
IL‐13 effects on ENaC activity are apoptosis independent but due to reduced transcription of β‐ and γ‐ENaC subunits and SGK1
In order to exclude the possibility that IL‐13‐suppression of J Na simply resulted from cell damage due to induction of epithelial apoptosis, caspase‐3 cleavage was quantified. However, IL‐13 did not activate apoptosis in HT‐29/B6‐GR/MR cells, at least at the dose of 100 ng ml−1 and an incubation time of 96 h (Fig. 2 A). To analyse whether the observed effects of IL‐13 could be attributed to changes in ENaC mRNA expression, the expression level of ENaC subunit mRNAs was examined by real time‐PCR. The increase in β‐ and γ‐ENaC mRNAs was smaller in HT‐29/B6‐GR/MR cells incubated with DBA+IL‐13 (321 ± 70‐fold of control for β‐ENaC, and 601 ± 161‐fold of control for γ‐ENaC) than in cells incubated solely with DBA (610 ± 98‐fold for β‐ENaC and 1116 ± 155‐fold for γ‐ENaC) (Fig. 2 B). By contrast, the DBA‐induced increase in α‐ENaC subunit expression was marginal and not affected by the additional incubation of IL‐13 (2.9 ± 0.4‐fold of control for DBA vs. 3.3 ± 0.6‐fold for DBA with IL‐13, P = 0.5).
Figure 2. ENaC‐mediated Na+ absorption is inhibited by IL‐13 which was apoptosis independent but due to decreased ENaC subunit and SGK1 transcription .
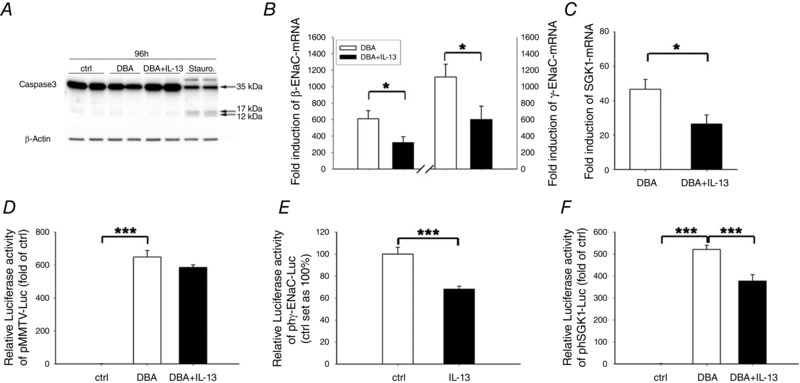
A, apoptosis control. HT‐29/B6‐GR/MR cells were incubated without (ctrl) or with DBA and with or without IL‐13 (100 ng ml−1), and caspase‐3 activation/cleavage was studied by Western blot analyses. Staurosporine served as positive control and human β‐actin as loading control. n = 4. B and C, cells were incubated with or without DBA and with or without IL‐13, and mRNA expression of β‐ and γ‐ENaC subunits (B) and SGK1 (C) was measured after an incubation period of 72–96 h. D–F, for gene reporter assay, cells were transfected with either pMMTV‐Luc (D), pgh‐ENaC‐B (E), or phSGK1‐3kb‐up‐Luc (F) and incubated for 72 h with or without DBA in the presence or absence of IL‐13 (100 ng ml−1). Data given are means ± SEM of x‐fold induction over controls; n = 4–5, *P < 0.05, ***P < 0.001.
An important intracellular regulator of J Na is the serum and glucocorticoid‐regulated kinase‐1 (SGK1), which triggers the apical localization of ENaC and is regulated by glucocorticoids (GC) and mineralocorticoids (MC) (Chen et al. 1999; Itani et al. 2002). Determination of the SGK1 expression level demonstrated the DBA‐induced mRNA expression to be attenuated by IL‐13 (47 ± 6‐fold of control for DBA versus 26 ± 5‐fold of control for DBA+IL‐13; P < 0.05; Fig. 2 C). To further investigate whether the effect of IL‐13 on β‐ and γ‐ENaC subunits or SGK1 gene expression was due to altered transactivation by the GR or MR, HT‐29/B6‐GR/MR cells were transiently transfected with the plasmid pMMTV‐Luc, containing consensus HRE sequences in the promoter region of a luciferase gene. However, even after an incubation period of 72 h, IL‐13 did not modify the DBA‐dependent upregulation of HRE‐driven luciferase expression (Fig. 2 D). To study the effect of IL‐13 on γ‐ENaC promoter, HT‐29/B6‐GR/MR cells were transiently transfected with the plasmid pghENaC‐B (phγENaC‐Luc), linking an appropriate γ‐ENaC promoter fragment to a luciferase reporter gene. Spontaneous transcription activity of γ‐ENaC was significantly reduced in the presence of IL‐13 (68 ± 3%; P < 0.001 compared to control; Fig. 2 E), indicating a regulatory effect of IL‐13 on γ‐ENaC transactivation. To investigate the possibility of associated changes in SGK1 promoter activity, HT‐29/B6‐GR/MR cells were transfected with the phSGK1‐3kb‐up‐Luc plasmid (phSGK1‐Luc) and incubated with DBA with and without IL‐13. Cells exposed to IL‐13 and DBA showed decreased luciferase expression levels (377 ± 29‐fold for DBA+IL‐13 versus 521 ± 19‐fold for DBA; P < 0.001). This indicates that IL‐13 led to decreases in both γ‐ENaC subunit and SGK1 mRNA expression, which could be attributed to IL‐13‐mediated changes of promoter activity.
IL‐13 influences all MAPKs but only p38 signalling is involved in J Na regulation
MAPK pathways can be activated by IL‐13 (David et al. 2001; Mandal & Levine, 2010) and MAPKs are involved in transcriptional, translational and post‐translational ENaC regulation (Rogatsky et al. 1998; Shen & Cotton, 2003; Yang et al. 2006; Zeissig et al. 2008). To elucidate whether IL‐13 affected MAPK signalling in our HT‐29/B6‐GR/MR cell model, MAPK phosphorylation/activation was examined with phospho‐specific antibodies. All three MAPKs were affected by IL‐13 (Fig. 3 A and B). ERK1/2 and JNK phosphorylation was augmented by IL‐13 (ERK1/2: 62 ± 2% of control for DBA versus 98 ± 2% of control for DBA+IL‐13; JNK: 96 ± 5% of control for DBA versus 229 ± 14% of control for DBA+IL‐13, Fig. 3 B). By contrast, p38 phosphorylation, which was strongly increased after stimulation with DBA, was inhibited by IL‐13 (598 ± 78% of control for DBA versus 262 ± 44% of control for DBA+IL‐13; P < 0.01). Having analysed MAPK phosphorylation, we tested whether IL‐13 modulation of MAPKs was also reflected in J Na by performing Ussing experiments with specific MAPK inhibitors. Neither the ERK1/2 inhibitor (U0126) nor the JNK inhibitor (SP600125) prevented the IL‐13‐mediated suppression in J Na, and both inhibitors also had no significant effect on J Na in the absence of IL‐13 (Fig. 3 C). However, pre‐incubation with the p38 inhibitor SB202190 markedly decreased the DBA‐induced J Na (Fig. 3 D). Under these conditions, the IL‐13 effect on ENaC activity was completely abrogated (20 ± 1% of DBA for DBA with inhibitor versus 22 ± 4% of DBA for DBA with inhibitor plus IL‐13. It should be emphasized that despite the substantial decrease in DBA‐stimulated J Na produced by p38 inhibition, J Na remained significantly increased compared with control cells.
Figure 3. ENaC‐mediated Na+ absorption is suppressed by IL‐13 via inhibition of p38 activity .
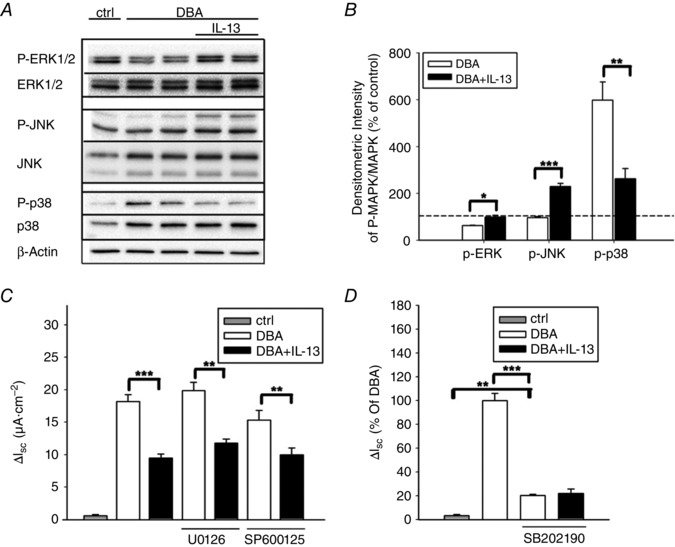
A, representative Western blot analysis of both phosphorylated and total ERK1/2, JNK, or p38 proteins after IL‐13 exposition. HT‐29/B6‐GR/MR cells were treated with or without DBA and with or without IL‐13 for 96 h, then rested in media lacking FCS and IL‐13 for 3 h and stimulated again with IL‐13 for 5 min (ERK), 15 min (JNK) or 60 min (p38). Then, total cellular protein was extracted and analysed. B, densitometric measurements were obtained from 3–4 blot membranes representing independent experiments. Data given as means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001. C, determination of ENaC‐dependent Na+ absorption in the presence or absence of ERK1/2 (U0126) or JNK (SP600125) inhibitors. D, determination of ENaC‐dependent Na+ transport with or without the p38 inhibitor SB202190. Data are given as means ± SEM; n = 6–7, **P < 0.01, ***P < 0.001.
The JAK/STAT(6) pathway mediates the IL‐13‐mediated inhibition of J Na
To elucidate the mechanisms underlying the IL‐13‐mediated decrease in J Na, we investigated the STAT6‐pathway, which is one of the major effector pathways of IL‐13 (Hebenstreit et al. 2006). In addition, there are a few reports which describe an IL‐13‐dependent phosphorylation of STAT3 (Umeshita‐Suyama et al. 2000). Thus, we performed Western blot studies with phospho‐specific antibodies for both STAT pathways in HT‐29/B6‐GR/MR cells. STAT6 and STAT3 were both phosphorylated/activated by IL‐13, whereas this phosphorylation was not induced by DBA (Fig. 4 A).
Figure 4. The JAK1/2/STAT signalling pathway is stimulated after IL‐13 treatment in HT‐29/B6‐GR/MR cells and is responsible for the drop in ENaC‐dependent Na+ absorption .
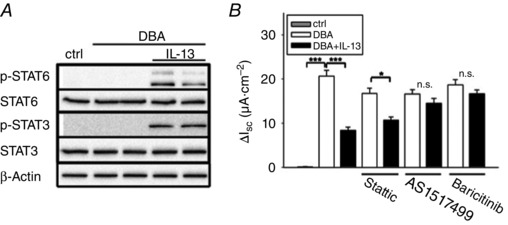
A, representative Western blot analyses of both phosphorylated and total STAT6 and STAT3. Cells were stimulated with DBA and with or without IL‐13 for 96 h, then rested FCS and IL‐13 free for 3 h and treated again with IL‐13 for 30 min. Then, total cellular protein was extracted, fractionated by SDS‐Page and immuno‐probed for phospho‐specific and total STAT6 and STAT3 expression. B, measurement of ENaC‐dependent Na+ absorption with or without IL‐13 in presence of DBA and inhibitors of the JAK1/2/STAT signalling. Results are means ± SEM; n = 5–7, *P < 0.05, ***P < 0.001.
Next we determined if the IL‐13‐dependent STAT activation was associated with changes in J Na. As shown in Fig. 4 B, Ussing experiments revealed that blocking STAT3 signalling by the STAT3 inhibitor V (Stattic) had no influence on the impaired J Na after IL‐13 exposure. By contrast, the STAT6‐specific inhibitor AS1517499 (Chiba et al. 2009) led to complete disinhibition of the IL‐13‐mediated reduction in J Na (16.6 ± 0.9 without IL‐13 versus 14.5 ± 1.1 μA cm−2 with IL‐13; P = 0.322). The JAK1/2 inhibitor baricitinib also abolished the IL‐13‐mediated decrease in J Na (18.7 ± 1.2 without IL‐13 versus 16.7 ± 0.8 μA cm−2 with IL‐13; P = 0.056).
IL‐13 signals through the IL‐13‐ Rα1/IL‐4Rα receptor in HT‐29/B6‐GR/MR
Because the IL‐13‐induced activation of the JAK1/2–STAT6 pathway is widely attributed to the heterodimeric type‐I receptor (IL‐13Rα1/IL‐4Rα), we performed inhibitor experiments with an antibody against the IL‐4Rα to confirm this in our cell line. This inhibitory antibody blocked the IL‐13 effect on J Na (DBA+IL‐13 in the presence of the IL‐4Rα‐antibody: 86 ± 9% of DBA with antibody; DBA+IL‐13 in the absence of the IL‐4Rα‐antibody: 45 ± 12% of solely DBA; n = 6 each; P < 0.05).
Since PI3K signalling has also been described by IL‐13, we analysed the phosphorylation status of Akt (p‐Akt Thr308), a downstream molecule of PI3K. In Western blots, this pathway was also activated by IL‐13 in our cell model. However, when examining J Na in the presence of the PI3K inhibitor Ly294002, no effect on IL‐13‐induced changes by PI3K inhibition was detected (18.8 ± 1.9 for DBA versus 13.9 ± 2.2 μA cm−2 for DBA+IL‐13; P < 0.05).
STAT6 mediates the IL‐13‐induced inhibition of the p38 MAPK
In an earlier study, Jang and co‐workers reported a relationship between p38 and STAT6 activation in a P388D1 macrophage cell line, as well as in BALB/c macrophages (Jang et al. 2009). To monitor the influence of STAT6 on the different MAP kinases, we analysed their phosphorylation status in cells stimulated with DBA with and without IL‐13 in the presence or absence of baricitinib. Whereas baricitinib did not inhibit IL‐13‐induced ERK1/2 and JNK phosphorylation, the IL‐13‐mediated decrease in p38 phosphorylation was significantly attenuated by baricitinib (170 ± 13% of control for DBA versus 85 ± 3% of control for DBA+IL‐13; P < 0.01; compared to 139 ± 9% of control for DBA+IL‐13 with baricitinib; P < 0.01; Fig. 5 A and B). The inhibition of JAK1/2 by baricitinib was accompanied by inhibition of STAT6 phosphorylation (Fig. 5 A).
Figure 5. STAT6 mediates IL‐13‐dependent inhibition of p38 phosphorylation .
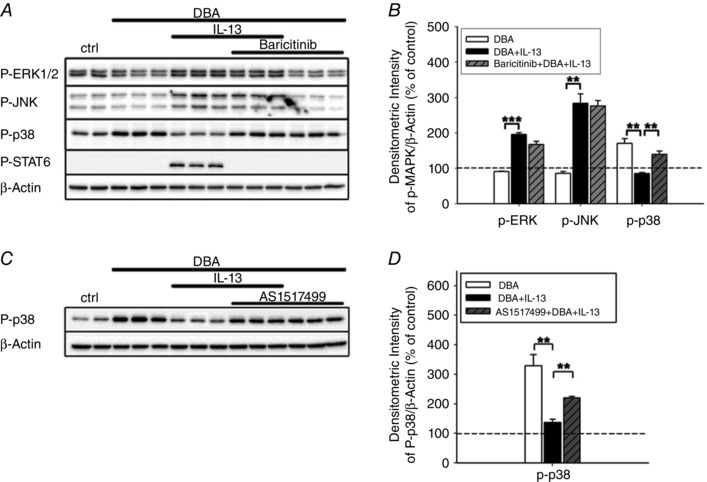
HT‐29/B6‐GR/MR cells were stimulated with or without DBA and with or without IL‐13 in the presence or absence of baricitinib or AS1517499 for 96 h, then rested FCS and IL‐13 free for 3 h and treated again with IL‐13 for 30 min. Then, total cellular protein was extracted, fractionated by SDS‐Page, and immuno‐probed for phospho‐specific ERK1/2, JNK, p38, or STAT6 expression. Human β‐actin served as loading control. A and B, representative Western blot analyses of the indicated proteins in the absence or presence of baricitinib (A) and the corresponding densitometric measurements (B). C and D, representative Western blot analyses of phospho‐specific p38 MAPK in the absence or presence of AS1517499 (C) and the corresponding densitometric measurements (D). Results in B and D are means ± SEM; n = 3, **P < 0.01, ***P < 0.001.
To exclude a STAT6‐independent effect on p38 phosphorylation mediated via JAK1/2 inhibition, we monitored p38 phosphorylation under the influence of the STAT6‐specific inhibitor AS1517499. Similar to JAK1/2 inhibition by baricitinib, AS1517499 led to disinhibition of p38 phosphorylation (329 ± 37% of control for DBA versus 137 ± 11% of control for DBA+IL‐13; P < 0.01; compared to 220 ± 5% of control for DBA+IL‐13 with AS1517499; P < 0.01; Fig. 5 C and D).
To identify the order of STAT6 and p38 in the IL‐13‐induced signalling, we monitored STAT6 phosphorylation in response to IL‐13 either with or without the p38 inhibitor SB202190. When present, STAT6 phosphorylation was still detectable at levels comparable to those without SB202190 (DBA+IL‐13+SB202190: 88 ± 12% of DBA+IL‐13 without SB202190; n = 4 each; P = 0.48). Thus, the IL‐13‐dependent inhibition of p38 phosphorylation was mediated via STAT6 activation.
IL‐13 impairs J Na in mouse distal colon via STAT6
For confirmation of the role of STAT6, we performed experiments in STAT6−/− BALB/c mice. Incubation with either vehicle or IL‐13 led to a similar increase in J Na in the knockout mouse (Fig. 6 A). Direct comparison of the response to IL‐13 in WT and STAT6−/− mice showed that the IL‐13‐mediated repression of J Na was prevented in the STAT6 knockout mouse (WT: with IL‐13 45 ± 11% of control; P < 0.01; STAT6−/−: with IL‐13 86 ± 12% of control; P = 0.45; Fig. 6 B). These findings corroborate the results obtained in HT‐29/B6‐GR/MR cells, confirming that STAT6 signalling is essential for the IL‐13 effect on J Na.
Figure 6. In mouse distal colon IL‐13 affects the DBA‐induced ENaC‐dependent Na+ absorption in a STAT6‐dependent manner .
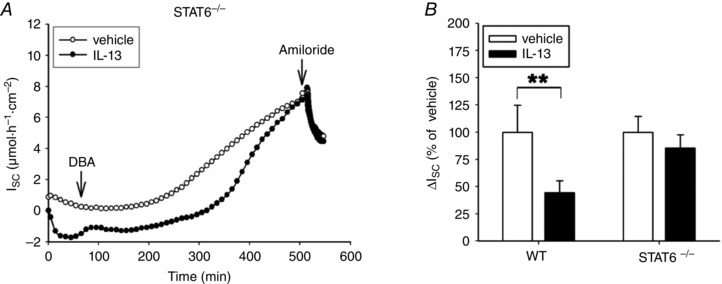
A, time course of a representative experiment on electrogenic Na+ transport (I SC) in the distal colon of STAT6−/− mice, which were pre‐treated with PBS (ctrl) or IL‐13 as described in Methods. The tissue was stimulated with DBA for 8 h and then ENaC‐dependent Na+ absorption was determined as drop in I SC. B, comparison of ENaC‐dependent Na+ absorption in WT and STAT6−/− mice treated as described in A. Data given as means ± SEM of 5–8 mice per group, **P < 0.01.
Discussion
IL‐13 is known to affect the intestinal epithelium, particularly by impairing epithelial barrier function (Heller et al. 2005; Prasad et al. 2005). It is produced by CD1‐reactive NKT cells (Fuss, 2004), and is considered to be a key effector cytokine in Th2‐driven colitis. Here, we show for the first time that IL‐13 affects active electrogenic Na+ transport (the most important Na+ transport system in the human distal colon) as well as intestinal epithelial barrier function. Exposure to IL‐13 reduced DBA‐stimulated ENaC‐dependent Na+ absorption (J Na) in our HT‐29/B6‐GR/MR cell model, and this was supported by ex vivo investigations in BALB/c mice. This effect of IL‐13 was mediated by activation of STAT6, a second important finding of our present study, again supported by additional inhibitor and knockout experiments.
Under physiological conditions, intestinal electrogenic Na+ transport occurs at only modest levels, although it plays a more important role during electrolyte and water loss in diarrhoeal states (Schultheis et al. 1998; Tatum et al. 2010). Under those conditions, ENaC is abundantly expressed and located in the apical membrane of enterocytes as the result of steroid action. Although different cellular processes, such as reduced ubiquitination, contribute to this up‐regulation of ENaC activity, we and others have previously shown that regulation occurs to a significant extent via transcriptional induction of β‐ and γ‐ENaC subunits, while the α‐ENaC subunit is constitutively expressed (Renard et al. 1995; Epple et al. 2000).
To simulate conditions found in native intestine during induction of diarrhoea, and to characterize the maximum transport capacity of this transport system, we stimulated ENaC in our experimental models with a triple‐combination of dexamethasone, butyrate and aldosterone (DBA). Butyrate has been shown to be involved in enterocyte differentiation and to act as a histone deacetylation inhibitor in the transcriptional up‐regulation of ENaC (Cuff & Shirazi‐Beechey, 2004). However, a detailed analysis revealed butyrate alone to be insufficient to activate ENaC‐dependent Na+ absorption in the absence of steroids (Zeissig et al. 2008; Bergann et al. 2009 a). Although corticosteroids (for example, dexamethasone) alone can stimulate Na+ absorption, their effect on electrogenic Na+ absorption is synergistically enhanced by butyrate. In addition, earlier in vivo perfusion studies on adrenectomized rats demonstrated that corticosteroids represent a conditio sine qua non cofactor for maximum induction of ENaC activity by aldosterone (Fromm et al. 1990); indeed, basal levels of circulating glucocorticoid are always present in vivo (Fromm et al. 1990). Thus, the mineralocorticosteroid aldosterone, which is the most important regulator of electrogenic Na+ absorption in the colon (especially during electrolyte and water losses associated with diarrhoea), requires co‐stimulatory inputs from glucocorticoids and butyrate for maximum ENaC activation.
When glucocorticoids, butyrate and aldosterone were present at physiological concentrations, we found a substantial increase in J Na in our HT‐29/B6‐GR/MR cell model that was severely impaired by IL‐13, both changes reflecting regulation of β‐ and γ‐ENaC subunit mRNA. Only ENaC molecules comprising all three subunits inserted into the apical colonocyte membrane allow Na+ transport in an effective manner (Canessa et al. 1994; Renard et al. 1995). Our data obtained in the colon are consistent with findings of Anagnostopoulou and co‐workers, who analysed ion transport processes in airway epithelium of mice after instillation of IL‐13 (Anagnostopoulou et al. 2010), also a key effector molecule in the pathogenesis of allergic airway inflammation (Wills‐Karp et al. 1998). Constitutively active in the lung, ENaC contributes to the generation of an osmotic Na+ gradient that clears fluid from the alveolar space (Matthay et al. 2002), and administration of IL‐13 also led to impaired Na+ transport. Resembling the results of our present study, this observation was also associated with a down‐regulation of ENaC subunit mRNAs.
However, an increase in ENaC‐mediated Na+ absorption does not depend only on transcriptional up‐regulation of ENaC subunits; important also are a number of regulatory proteins which contribute to rapid ENaC turnover in the apical cell membrane. In this regard, our finding of IL‐13‐mediated down‐regulation of SGK1 mRNA indicated an effect of IL‐13 on ENaC activity at the post‐translational level. The stimulatory impact of SGK1 on ENaC is mediated via an inhibitory interaction with Nedd4‐2 (neural precursor cell‐expressed, developmentally down‐regulated protein 4‐2). As shown by others, Nedd4‐2 facilitates the ubiquitination and subsequently the removal of ENaC from the apical cell membrane, leading to degradation of ENaC complexes (Debonneville et al. 2001).
Nevertheless, the relative contribution of IL‐13‐induced alterations of transcriptional and post‐translational events to the measured decrease in ENaC‐mediated Na+ absorption has been unclear. On the one hand, as mentioned above, it is possible that decreased β‐ and γ‐ENaC expression levels are responsible for ENaC dysfunction. On the other hand, it is also possible that down‐regulation of SGK1 is the rate limiting factor. To distinguish between these possibilities, additional approaches would have been needed with pharmacological inhibitors of SGK1 or siRNAs at various levels of ENaC activation.
IL‐13 actions via the heterodimeric type‐I (IL‐4Rα/IL‐13Rα1) receptor, as identified here in our present study using neutralizing antibodies, have been shown for different target genes and in different tissues to involve phosphorylation of JAK1/2 and subsequently phosphorylation and activation of the transcription factors STAT3 and STAT6 (Umeshita‐Suyama et al. 2000). Additionally, STAT‐independent activation of the PI3K signalling cascade has been described (Nelms et al. 1999), but in our study did not appear to have a role for the inhibition of J Na, as indicated by the absence of an effect of Ly294002. In the subsequent analysis, a role for STAT6, but not for STAT3, could be detected for ENaC activation in the colon.
In addition to these classical pathways, IL‐13 is also known to signal via MAP kinases (David et al. 2001; Moynihan et al. 2008). Furthermore, MAP kinases have previously been shown to influence ENaC‐dependent Na+ absorption in intestinal cells (Zeissig et al. 2008; Bergann et al. 2009 b; Kuntzsch et al. 2012). We found all three MAPKs were modified by IL‐13, but inhibitor experiments indicated that only p38 MAPK was involved in the regulation of ENaC. Our finding that inhibition of JAK1/2 as well as STAT6 disinhibited this IL‐13 effect, suggests that JAK/STAT6‐mediated p38 MAPK suppression is involved, as shown in Fig. 7. Other investigators have also proposed a link between p38 MAPK and the JAK1/2–STAT6 pathway, as cytokine‐mediated activation of p38 MAPK targeted STAT6 signals in a P388D1 macrophage cell line and in murine peritoneal macrophages (Jang et al. 2009; Jimenez‐Garcia et al. 2015). Our present data lead us to conclude that in the intestine, STAT6 causes p38 MAPK inhibition, since STAT6 phosphorylation was still visible during p38 inhibition.
Figure 7. Proposed model for IL‐13 action on ENaC‐dependent Na+ absorption .
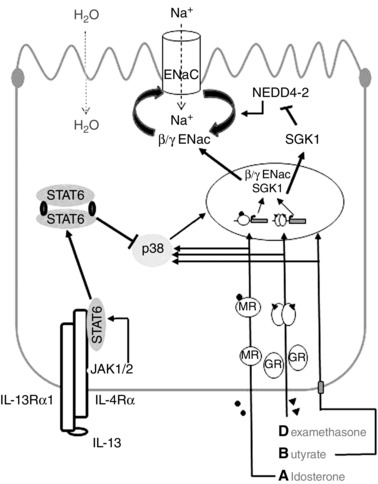
The stimulation with DBA (dexamethasone, butyrate and aldosterone) leads to the up‐regulation of β‐ and γ‐ENaC subunit as well as SGK1 expression. On the one hand this is achieved via activation of the p38 MAPK and on the other hand by transactivation of target genes via activated mineralocorticoid receptor (MR) and glucocorticoid receptor (GR). IL‐13 binds to the IL‐13Rα1/IL‐4Rα type‐I receptor leading to the activation of JAK1/2. Subsequently, STAT6 is phosphorylated and this in turn leads to the inhibition of the DBA‐stimulated activation of p38 MAPK. These signalling events promote the decrease in DBA‐stimulated ENaC activity due to reduction of β‐ and γ‐ENaC and/or SGK1 expression. Technical terms used in this figure are also listed in the abbreviations.
How might STAT6, or the inhibition of p38, contribute to impaired ENaC activity in intestinal cells? Activation of the GR and MR by glucocorticoids or mineralocorticoids, respectively, and the subsequent transcription of target genes, is the most important physiological regulatory process for ENaC induction in the intestine (Epple et al. 2000). As also postulated for other STAT proteins (Stocklin et al. 1996), Biola et al. reported that in murine T‐lymphocytes, STAT6 and GR interact to mutually antagonize transactivation (Biola et al. 2000). However, our data from reporter gene assays using a representative HRE‐driven pMMTV‐Luc plasmid argue against the hypothesis of STAT6 affecting GR/MR binding to their respective HREs in intestinal cells. Furthermore, in silico analyses (ALLGEN‐PROMO, transfac database) failed to reveal STAT6 binding elements within the γ‐ENaC promoter fragment (−459 to +35), despite an inhibitory effect of IL‐13. By contrast, putative binding sites for transcription factors were predicted, which are known targets of p38 MAPK signalling, e.g. Elk1 or CHOP.
Although further studies are required to analyse the precise mechanism by which p38 targets ENaC activity, our data clearly show a significant role of this MAPK in both ENaC activation by DBA and in IL‐13‐mediated ENaC inhibition. Indeed, there are several reports of p38 MAPK activation leading to upregulation of β‐ as well as γ‐ENaC subunit expression (Itani et al. 2003; Niisato et al. 2007). Moreover, numerous studies have suggested involvement of p38 MAPK in SGK1 activation (Waldegger et al. 2000). For example, Bell and co‐workers reported p38 MAPK‐dependent stimulation of SGK promoter activity in NMuMg mouse mammary epithelial cells (Bell et al. 2000).
In summary, our data show that IL‐13 downregulates ENaC‐dependent Na+ absorption in the intestine by coordinated modulation of JAK1/2–STAT6–p38 signalling. This demonstrates that IL‐13 in the intestine acts as a potent modulator of transcellular ion transport, in addition to its well‐recognized ability to regulate paracellular transport.
Additional information
Competing interests
The authors declare that they have no conflict of interest associated with this manuscript.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. P.D., J.D.S. and A.F. developed the conception and design of the experiments. P.D. and A.F. performed the experiments and analysed and interpreted the data. P.D. wrote the manuscript. T.B., R.B., C.B., S.K., M.F. and J.D.S. critically reviewed the manuscript. The experiments were performed in the laboratory of the Department of Gastroenterology, Infectious Diseases and Rheumatology; Charité – University Medicine Berlin, Campus Benjamin Franklin, Berlin, Germany.
Funding
This study was supported by Deutsche Forschungsgemeinschaft (DFG) grants Schu 559/10‐2 and the Sonnenfeld‐Stiftung Berlin.
Author's present address
Theresa Bergann: Department of Biochemistry, Charité Berlin, Berlin, Germany.
Translational perspective
In addition to its well‐recognized ability to regulate epithelial barrier function in the intestine, interleukin‐13 (IL‐13) acts as a potent modulator of transcellular ion transport. It does this by downregulating sodium transport mediated by epithelial sodium channels (ENaC) in the distal large intestine, through a coordinated modulation of JAK1/2, STAT6 and p38–MAP kinase signalling. In healthy colon, basal ENaC activity is low, but is readily increased during significant fluid and electrolyte losses. Thus, during diarrhoeal disorders in which disturbances of the intestinal epithelial barrier results in consecutive leak fluxes of solutes and water, ENaC is stimulated by increased circulating levels of aldosterone to conserve sodium and water. However, this can be hampered by IL‐13, for example in ulcerative colitis, a Th2‐driven inflammatory bowel disease. A fuller understanding of the mechanisms involved in IL‐13‐dependent transport and barrier defects may therefore provide new therapeutic strategies for diarrhoeal diseases in which IL‐13 plays a key role. It is also feasible that novel multimodal therapeutic approaches may be required in intestinal diseases where pathophysiological changes in epithelial function reflect the simultaneous effects of several proinflammatory cytokines.
Acknowledgements
We thank Prof. Geoffrey Sandle (Leeds, UK) for his great help in preparing the manuscript. We acknowledge the excellent technical support of Detlef Sorgenfrei (deceased February 2015).
Please note that Figures 1, 3, 4, 5 and 6 that appear in this final copy‐edited version have been updated since the paper was published online as an Accepted Article. The new figures have been supplied by the authors and have been approved by the Editor. The conclusions of the study remain the same.
References
- Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, Fromm M & Schulzke JD (2004). Cytokine‐dependent transcriptional down‐regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology 126, 1711–1720. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulou P, Dai L, Schatterny J, Hirtz S, Duerr J & Mall MA (2010). Allergic airway inflammation induces a pro‐secretory epithelial ion transport phenotype in mice. Eur Respir J 36, 1436–1447. [DOI] [PubMed] [Google Scholar]
- Bell LM, Leong ML, Kim B, Wang E, Park J, Hemmings BA & Firestone GL (2000). Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum‐ and glucocorticoid‐inducible protein kinase (Sgk) by a p38 MAPK‐dependent pathway. J Biol Chem 275, 25262–25272. [DOI] [PubMed] [Google Scholar]
- Bergann T, Fromm A, Borden SA, Fromm M & Schulzke JD (2011). Glucocorticoid receptor is indispensable for physiological responses to aldosterone in epithelial Na+ channel induction via the mineralocorticoid receptor in a human colonic cell line. Eur J Cell Biol 90, 432–439. [DOI] [PubMed] [Google Scholar]
- Bergann T, Ploger S, Fromm A, Zeissig S, Borden SA, Fromm M & Schulzke JD (2009. a). A colonic mineralocorticoid receptor cell model expressing epithelial Na+ channels. Biochem Biophys Res Commun 382, 280–285. [DOI] [PubMed] [Google Scholar]
- Bergann T, Zeissig S, Fromm A, Richter JF, Fromm M & Schulzke JD (2009. b). Glucocorticoids and tumor necrosis factor‐α synergize to induce absorption by the epithelial sodium channel in the colon. Gastroenterology 136, 933–942. [DOI] [PubMed] [Google Scholar]
- Biola A, Andreau K, David M, Sturm M, Haake M, Bertoglio J & Pallardy M (2000). The glucocorticoid receptor and STAT6 physically and functionally interact in T‐lymphocytes. FEBS Lett 487, 229–233. [DOI] [PubMed] [Google Scholar]
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD & Rossier BC (1994). Amiloride‐sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467. [DOI] [PubMed] [Google Scholar]
- Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F & Pearce D (1999). Epithelial sodium channel regulated by aldosterone‐induced protein sgk. Proc Natl Acad Sci USA 96, 2514–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Todoroki M, Nishida Y, Tanabe M & Misawa M (2009). A novel STAT6 inhibitor AS1517499 ameliorates antigen‐induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol 41, 516–524. [DOI] [PubMed] [Google Scholar]
- Cuff MA & Shirazi‐Beechey SP (2004). The importance of butyrate transport to the regulation of gene expression in the colonic epithelium. Biochem Soc Trans 32, 1100–1102. [DOI] [PubMed] [Google Scholar]
- David M, Ford D, Bertoglio J, Maizel AL & Pierre J (2001). Induction of the IL‐13 receptor α2‐chain by IL‐4 and IL‐13 in human keratinocytes: involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 20, 6660–6668. [DOI] [PubMed] [Google Scholar]
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J & Staub O (2001). Phosphorylation of Nedd4‐2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20, 7052–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epple HJ, Amasheh S, Mankertz J, Goltz M, Schulzke JD & Fromm M (2000). Early aldosterone effect in distal colon by transcriptional regulation of ENaC subunits. Am J Physiol Gastrointest Liver Physiol 278, G718–724. [DOI] [PubMed] [Google Scholar]
- Fichtner‐Feigl S, Strober W, Kawakami K, Puri RK & Kitani A (2006). IL‐13 signaling through the IL‐13α2 receptor is involved in induction of TGF‐β1 production and fibrosis. Nat Med 12, 99–106. [DOI] [PubMed] [Google Scholar]
- Firsov D, Gautschi I, Merillat AM, Rossier BC & Schild L (1998). The heterotetrameric architecture of the epithelial sodium channel (ENaC). EMBO J 17, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm M, Schulzke JD & Hegel U (1990). Aldosterone low‐dose, short‐term action in adrenalectomized glucocorticoid‐substituted rats: Na, K, Cl, HCO3, osmolyte, and water transport in proximal and rectal colon. Pflugers Arch 416, 573–579. [DOI] [PubMed] [Google Scholar]
- Fuss IJ (2004). Treatment of ulcerative colitis with infliximab: are we there yet? J Pediatr Gastroenterol Nutr 38, 247–249. [DOI] [PubMed] [Google Scholar]
- Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner‐Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P & Strober W (2004). Nonclassical CD1d‐restricted NK T cells that produce IL‐13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest 113, 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig ER, Boot‐Handford RP, Mani V & Sandle GI (2004). Decreased expression of apical Na+ channels and basolateral Na+,K+‐ATPase in ulcerative colitis. J Pathol 204, 84–92. [DOI] [PubMed] [Google Scholar]
- Hebenstreit D, Wirnsberger G, Horejs‐Hoeck J & Duschl A (2006). Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 17, 173–188. [DOI] [PubMed] [Google Scholar]
- Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, Zeitz M, Fuss I, Strober W & Schulzke JD (2005). Interleukin‐13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129, 550–564. [DOI] [PubMed] [Google Scholar]
- Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS & Strober W (2002). Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL‐13‐producing NK‐T cells. Immunity 17, 629–638. [DOI] [PubMed] [Google Scholar]
- Hershey GK (2003). IL‐13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol 111, 677–690; quiz 691. [DOI] [PubMed] [Google Scholar]
- Itani OA, Auerbach SD, Husted RF, Volk KA, Ageloff S, Knepper MA, Stokes JB & Thomas CP (2002). Glucocorticoid‐stimulated lung epithelial Na+ transport is associated with regulated ENaC and sgk1 expression. Am J Physiol Lung Cell Mol Physiol 282, L631–641. [DOI] [PubMed] [Google Scholar]
- Itani OA, Cornish KL, Liu KZ & Thomas CP (2003). Cycloheximide increases glucocorticoid‐stimulated α‐ENaC mRNA in collecting duct cells by p38 MAPK‐dependent pathway. Am J Physiol Renal Physiol 284, F778–787. [DOI] [PubMed] [Google Scholar]
- Jang YS, Kim HA, Park SR, Lee MR, Park JB & Kim PH (2009). IL‐4 stimulates mouse macrophages to express APRIL through p38MAPK and two different downstream molecules, CREB and Stat6. Cytokine 47, 43–47. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB & Gouaux E (2007). Structure of acid‐sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323. [DOI] [PubMed] [Google Scholar]
- Jimenez‐Garcia L, Herranz S, Luque A & Hortelano S (2015). Critical role of p38 MAPK in IL‐4‐induced alternative activation of peritoneal macrophages. Eur J Immunol 45, 273–286. [DOI] [PubMed] [Google Scholar]
- Kellenberger S & Schild L (2002). Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82, 735–67. [DOI] [PubMed] [Google Scholar]
- Kuntzsch D, Bergann T, Dames P, Fromm A, Fromm M, Davis RA, Melzig MF & Schulzke JD (2012). The plant‐derived glucocorticoid receptor agonist endiandrin A acts as co‐stimulator of colonic epithelial sodium channels (ENaC) via SGK‐1 and MAPKs. PloS One 7, e49426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔ C T method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Mandal D & Levine AD (2010). Elevated IL‐13Rα2 in intestinal epithelial cells from ulcerative colitis or colorectal cancer initiates MAPK pathway. Inflamm Bowel Dis 16, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matthay MA, Folkesson HG & Clerici C (2002). Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 82, 569–600. [DOI] [PubMed] [Google Scholar]
- Moynihan B, Tolloczko B, Michoud MC, Tamaoka M, Ferraro P & Martin JG (2008). MAP kinases mediate interleukin‐13 effects on calcium signaling in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 295, L171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms K, Keegan AD, Zamorano J, Ryan JJ & Paul WE (1999). The IL‐4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 17, 701–738. [DOI] [PubMed] [Google Scholar]
- Niisato N, Taruno A & Marunaka Y (2007). Involvement of p38 MAPK in hypotonic stress‐induced stimulation of β‐ and γ‐ENaC expression in renal epithelium. Biochem Biophys Res Commun 358, 819–824. [DOI] [PubMed] [Google Scholar]
- Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT & Collins JE (2005). Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest 85, 1139–1162. [DOI] [PubMed] [Google Scholar]
- Renard S, Voilley N, Bassilana F, Lazdunski M & Barbry P (1995). Localization and regulation by steroids of the alpha, beta and gamma subunits of the amiloride‐sensitive Na+ channel in colon, lung and kidney. Pflugers Arch 430, 299–307. [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Waase CL & Garabedian MJ (1998). Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase‐3 (GSK‐3). Species‐specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK‐3 phosphorylation. J Biol Chem 273, 14315–14321. [DOI] [PubMed] [Google Scholar]
- Rosen MJ, Chaturvedi R, Washington MK, Kuhnhein LA, Moore PD, Coggeshall SS, McDonough EM, Weitkamp JH, Singh AB, Coburn LA, Williams CS, Yan F, Van Kaer L, Peebles RS Jr & Wilson KT (2013). STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin‐2 and Th2‐inducing cytokines. J Immunol 190, 1849–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MJ, Frey MR, Washington MK, Chaturvedi R, Kuhnhein LA, Matta P, Revetta FL, Wilson KT & Polk DB (2011). STAT6 activation in ulcerative colitis: a new target for prevention of IL‐13‐induced colon epithelial cell dysfunction. Inflamm Bowel Dis 17, 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN & Shull GE (1998). Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 19, 282–285. [DOI] [PubMed] [Google Scholar]
- Shen JP & Cotton CU (2003). Epidermal growth factor inhibits amiloride‐sensitive sodium absorption in renal collecting duct cells. Am J Physiol Renal Physiol 284, F57–64. [DOI] [PubMed] [Google Scholar]
- Stocklin E, Wissler M, Gouilleux F & Groner B (1996). Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383, 726–728. [DOI] [PubMed] [Google Scholar]
- Sullivan S, Alex P, Dassopoulos T, Zachos NC, Iacobuzio‐Donahue C, Donowitz M, Brant SR, Cuffari C, Harris ML, Datta LW, Conklin L, Chen Y & Li X (2009). Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD‐associated diarrhea. Inflamm Bowel Dis 15, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kamanaka M, Tanaka T, Kishimoto T & Akira S (1996). Impaired IL‐13‐mediated functions of macrophages in STAT6‐deficient mice. J Immunol 157, 3220–3222. [PubMed] [Google Scholar]
- Tatum R, Zhang Y, Salleng K, Lu Z, Lin JJ, Lu Q, Jeansonne BG, Ding L & Chen YH (2010). Renal salt wasting and chronic dehydration in claudin‐7‐deficient mice. Am J Physiol Renal Physiol 298, F24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeshita‐Suyama R, Sugimoto R, Akaiwa M, Arima K, Yu B, Wada M, Kuwano M, Nakajima K, Hamasaki N & Izuhara K (2000). Characterization of IL‐4 and IL‐13 signals dependent on the human IL‐13 receptor α chain 1: redundancy of requirement of tyrosine residue for STAT3 activation. Int Immunol 12, 1499–1509. [DOI] [PubMed] [Google Scholar]
- Waldegger S, Gabrysch S, Barth P, Fillon S & Lang F (2000). h‐sgk serine‐threonine protein kinase as transcriptional target of p38/MAP kinase pathway in HepG2 human hepatoma cells. Cell Physiol Biochem 10, 203–208. [DOI] [PubMed] [Google Scholar]
- Wills‐Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL & Donaldson DD (1998). Interleukin‐13: central mediator of allergic asthma. Science 282, 2258–2261. [DOI] [PubMed] [Google Scholar]
- Yang LM, Rinke R & Korbmacher C (2006). Stimulation of the epithelial sodium channel (ENaC) by cAMP involves putative ERK phosphorylation sites in the C termini of the channel's β‐ and γ‐subunit. J Biol Chem 281, 9859–9868. [DOI] [PubMed] [Google Scholar]
- Zeissig S, Bergann T, Fromm A, Bojarski C, Heller F, Guenther U, Zeitz M, Fromm M & Schulzke JD (2008). Altered ENaC expression leads to impaired sodium absorption in the noninflamed intestine in Crohn's disease. Gastroenterology 134, 1436–1447. [DOI] [PubMed] [Google Scholar]