

Key points
Fluid and HCO3 − secretion is essential for all epithelia; aberrant secretion is associated with several diseases.
Carbonic anhydrase XII (CA12) is the key carbonic anhydrase in epithelial fluid and HCO3 − secretion and works by activating the ductal Cl−–HCO3 − exchanger AE2.
Delivery of CA12 to salivary glands increases salivation in mice and of the human mutation CA12(E143K) markedly inhibits it.
The human mutation CA12(E143K) causes disease due to aberrant CA12 glycosylation, and misfolding resulting in loss of AE2 activity.
Abstract
Aberrant epithelial fluid and HCO3 − secretion is associated with many diseases. The activity of HCO3 − transporters depends of HCO3 − availability that is determined by carbonic anhydrases (CAs). Which CAs are essential for epithelial function is unknown. CA12 stands out since the CA12(E143K) mutation causes salt wasting in sweat and dehydration in humans. Here, we report that expression of CA12 and of CA12(E143K) in mice salivary glands respectively increased and prominently inhibited ductal fluid secretion and salivation in vivo. CA12 markedly increases the activity and is the major HCO3 − supplier of ductal Cl−–HCO3 − exchanger AE2, but not of NBCe1‐B. The E143K mutation alters CA12 glycosylation at N28 and N80, resulting in retention of the basolateral CA12 in the ER. Knockdown of AE2 and of CA12 inhibited pancreatic and salivary gland ductal AE2 activity and fluid secretion. Accordingly, patients homozygous for the CA12(E143K) mutation have a dry mouth, dry tongue phenotype. These findings reveal an unsuspected prominent role of CA12 in epithelial function, explain the disease and call for caution in the use of CA12 inhibitors in cancer treatment.
Key points
Fluid and HCO3 − secretion is essential for all epithelia; aberrant secretion is associated with several diseases.
Carbonic anhydrase XII (CA12) is the key carbonic anhydrase in epithelial fluid and HCO3 − secretion and works by activating the ductal Cl−–HCO3 − exchanger AE2.
Delivery of CA12 to salivary glands increases salivation in mice and of the human mutation CA12(E143K) markedly inhibits it.
The human mutation CA12(E143K) causes disease due to aberrant CA12 glycosylation, and misfolding resulting in loss of AE2 activity.
Abbreviations
- AE2
Cl−–HCO3 − anion exchanger isoform 2
- CA7
carbonic anhydrase 7
- CA12
carbonic anhydrase 12
- Co‐IP
co‐immunoprecipitation
- ER
endoplasmic reticulum
- NBCe1‐B
Na+–HCO3 − cotransporter isoform e1‐B
Introduction
Fluid and HCO3 − secretion are vital functions of all epithelia, regulating systemic and cellular pH, cell volume, facilitating solubilization of macromolecules and guarding against tissue dehydration (Lee et al. 2012). In secretory epithelia, fluid and HCO3 − secretion occurs in two steps. The serous acini secrete NaCl‐rich fluid and the ducts or their equivalent absorb the Cl− and secrete HCO3 − and some or most of the fluid (Lee et al. 2012; Catalan et al. 2014; Lee & Foskett, 2014). The major transporters mediating acinar secretion include basolateral Cl− influx mediated by the Na+–K+ –2Cl− cotransporter NKCC1 and luminal Cl− efflux mediated by the Ca2+‐activated Cl− channel ANO1 (Lee et al. 2012; Jang & Oh, 2014; Lee & Foskett, 2014; Catalan et al. 2015). A recent study reported additional basolateral Cl− influx by the Cl−–HCO3 − exchanger AE4 in submandibular acinar cells and, surprisingly, no role for the Cl−–HCO3 − exchanger AE2 (Pena‐Munzenmayer et al. 2015). Ductal fluid and HCO3 − secretion involve basolateral HCO3 − influx by the 1Na+–2HCO3 − cotransporter NBCe1‐B and luminal HCO3 − efflux and Cl− absorption by the coordinated action of the 1Cl−–2HCO3 − exchanger slc26a6 and the Cl− channel CFTR (Lee et al. 2012; Ahuja et al. 2014). The role of AE2 in ductal secretion by pancreatic or salivary glands has not been examined before. Unlike the findings in salivary gland (Pena‐Munzenmayer et al. 2015), colonic anion secretion requires AE2 (Gawenis et al. 2010) and Cl− influx by AE2 has a major role in airway fluid and HCO3 − secretion (Huang et al. 2012; Shan et al. 2012). Hence, the contribution of AE2 appears to be tissue specific and needs to be examined further in various epithelia.
The activity of all transporters involved in fluid and HCO3 − secretion is affected by the local HCO3 − concentration, which controls both HCO3 − availability and the pH at plasma membrane surfaces. Local HCO3 − concentration is determined by the function of carbonic anhydrases (CAs) that catalyse the hydration of CO2 (McKenna & Frost, 2014). Of the mammalian CAs, several are cytoplasmic (CA2 and CA7) and several are plasma membrane anchored (CA4, CA12 and CA14) with the catalytic site at the extracellular surface, regulating HCO3 − concentration at the basolateral (CA4 and CA12) or the luminal (CA4) membrane surfaces (Frost, 2014). The plasma membrane localized CAs interact with H+ and HCO3 − transporters, including NBCe1 (Orlowski et al. 2012) and AE2 (Morgan et al. 2007), and regulate their activity (Becker et al. 2014).
Mutations in HCO3 − transporters like CFTR, AE2 and NBCe1 (Lee et al. 2012) lead to aberrant fluid and HCO3 − secretion and diseases, including cystic fibrosis (Yang et al. 2009; Quinton, 2010), pancreatitis (Lee et al. 2012; Maleth & Hegyi, 2014) and Sjögren's syndrome (Almstahl & Wikstrom, 2003; Lee et al. 2012). Similarly, mutations in several CAs are associated with diseases; mutations in CA4 cause autosomal dominant retinitis pigmentosa (Rebello et al. 2004) and changes in CA9 and CA12 are associated with several cancers (Parks et al. 2013). Moreover, recently we (Muhammad et al. 2011; Feinstein et al. 2014) and others (Feldshtein et al. 2010) have reported that the Glu143Lys mutation in CA12 (CA12(E143K)) is the cause of an autosomal recessive form of salt wasting, which leads to hyponatraemia with hyperkalaemia, high sweat Cl−, dehydration and failure to thrive. The role of CA12 in epithelial ion transport and how the E143K mutation causes the disease are not clear. Modelling suggested that the CA12(E143K) mutation affect coordination of the Zn2+ in the enzyme active site (Muhammad et al. 2011), while measuring the activity of recombinant CA12(E143K) suggested altered sensitivity to anions (Feldshtein et al. 2010). However, whether these potential effects account for the disease and the role of CA12 in epithelial function are not known.
Here, we explored the role of CA12 and CA12(E143K) in epithelial transport and the associated disease. Our findings reveal an unrecognized role of CA12 in epithelial fluid and HCO3 − secretion through regulation of ductal AE2. The CA12(E143K) mutation causes misfolding and mistargeting of CA12 and inhibition of epithelial AE2 and consequently fluid secretion. Accordingly, patients with the CA12(E143K) mutation have dry mouth phenotype.
Methods
Antibodies, plasmids and solutions
The polyclonal CA12 and polyclonal CA2 antibodies were purchased from ProteinTech Inc. (Chicago, IL, USA). Monoclonal and polyclonal HA antibodies were purchased from Cell Signalling Inc. (Beverly, MA, USA). Polyclonal GFP, monoclonal ZO‐1, and mKate antibody were from Invitrogen (Carlsbad, CA, USA). Monoclonal Flag and monoclonal β‐actin antibodies were obtained from Sigma (St Louis, MO, USA).
Human wild‐type CA7 and CA12 cDNA in pCMV6‐AC‐Myc DDK vector was purchased from Origene Technologies (Rockville, MD, USA). Human CA2 and CA4 were generously provided by Dr Joseph Casey (University of Alberta, Canada) and have been described previously (Sterling & Casey, 1999; Alvarez et al. 2003). The HA‐tagged human AE2 was generously provided by Dr Seth Alper (Harvard Medical School, Boston, MA, USA). The cDNA encoding CA12 and NBCe1‐B were excised from the original vectors and transferred to pCMV6‐AC‐mKate (Origene Technologies ) and pEGFP‐C1, respectively. To create all single or double mutation constructs of CAs, PCR splicing was performed with QuikChange lightning enzyme (Agilent Biotech., Santa Clara, CA, USA), using primers to mutate desired amino acid. All constructs were verified by sequencing of the entire open reading frames to ensure lack of unwanted mutations. The standard bath solution (solution A) contained (mm) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 Hepes, and 10 glucose, adjusted to pH 7.4 with NaOH. Na+‐free solutions were prepared by replacing Na+ with N‐methyl‐d‐glucamine (NMDG). HCO3 –‐buffered solutions were prepared by replacing 25 mm NaCl with 25 mm NaHCO3 and reducing Hepes to 2.5 mm. HCO3 –‐containing, Cl−‐free solutions were prepared by replacing NaCl with NMDG‐gluconate and NaHCO3 with choline‐HCO3. HCO3 –‐buffered solutions were gassed with 5% CO2 and 95% O2. The osmolarity of all solutions was adjusted to 310 mosmol l−1with the major salt.
Adenovirus vector (ADCA12 and ADCA12(E143K) construction and administration
The AdCA12 and AdCA12(E143K) vectors used in this study are a first generation, E1‐deleted, replication‐defective, serotype 5 recombinant adenoviral vector with a human CA12 or CA12(E143K) cDNA. Briefly, the CA12 and CA12(E143K) fragments were generated by PCR and ligated into pAC‐IRES‐RFP to generate pAC‐CA12‐IRES‐RFP and pAC‐CA12(E143K). The AdCA12 and AdCA12(E143K) vectors were produced by homologous recombination of pAC‐CA12‐IRES‐RFP and pAC‐CA12(E143K) with pJM17 in C7 cells. The titres (particles per litre) of purified vectors were determined by QPCR using primers from the E2 region (Zheng & Baum, 2005). Viral delivery and salivary secretion in mice were approved by the National Institute of Dental and Craniofacial Research Animal Care and Use Committee. Viruses were delivered to 8‐week‐old C57 mice. The mice were anaesthetized with ketamine (60 mg kg−1) and xylazine (8 mg kg−1) i.m. AdCA12 or AdCA12(E143K) were delivered to both submandibular and parotid glands at 5 × 109 particles per gland by retrograde ductal instillation (Baum et al. 2002 and Fig. 6 A). During the cannulation, 0.5 mg kg−1 of atropine was used i.m. to inhibit saliva secretion in order to increase transduction efficiency.
Figure 6. Role of CA12 in pancreatic duct NBCe1‐B and AE2 activity and fluid secretion .
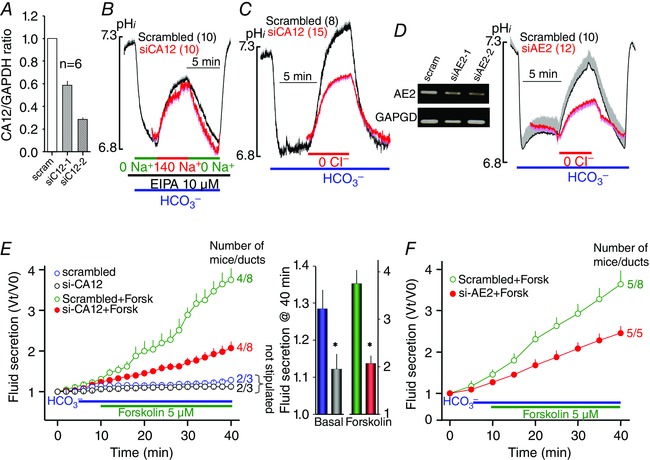
A, the efficiency of 48 h treatment with two siRNA duplexes in reducing CA12 mRNA expression in sealed pancreatic ducts. B–D, sealed pancreatic ducts were treated with scrambled siRNA (black traces) or siRNA targeting CA12 (B and C) or AE2 (D) for 48 h and were used to measure Na+–HCO3 − cotransport (B) or Cl−–HCO3 − exchange activity (C and D), as in Figs 4 and 3, respectively. The blot in D shows the effect of two siRNA duplexes in reducing AE2 mRNA expression in sealed pancreatic ducts. The blot is one of two similar experiments. The traces are the means ± SEM of the number of ducts listed in parenthesis next to the traces. E and F, sealed pancreatic ducts were treated with siRNA duplexes 2 of CA12 (E) or AE2 (F) and used to measure fluid secretion in response to stimulation with 5 μm forskolin (red green trances) or kept unstimulated (black blue). * denotes p < 0.05 with respect to scrambled siRNA. E, the 40 min secretion in the form of columns to better show inhibition of basal secretion by CA12(E143K). The results are means ± SEM of the number of experiments indicated next to the traces (second number) obtained from the indicated number of mice (first number).
Isolation and culture of parotid and pancreatic ducts
All killing procedures and experimental protocols with the mice followed NIH guidelines and were approved by the Animal Care and Use Committee of National Institutes of Health. Cultured, sealed parotid and pancreatic ducts were prepared as described previously (Yang et al. 2009; Park et al. 2013). In brief, male mice (20–25 g) were killed by cervical dislocation. The parotid glands or pancreas were removed and injected with a digestion buffer consisting of serum‐free DMEM, containing 50 U ml−1 collagenase, 400 U ml−1 hyaluronidase, 0.2 mg ml−1 soybean trypsin inhibitor (STI), and 2 mg ml−1 bovine serum albumin (BSA). The tissue was minced and incubated at 37°C for 30 min and then incubated in fresh digestion buffer for a further 45 min. After a wash with DMEM containing 0.2 mg ml−1 STI and 3% BSA, ducts were microdissected from the partially digested tissue to remove acini and connective tissue. The ducts were cultured in DMEM supplemented with 10% fetal bovine serum at 37°C on filter membranes and treated with scrambled or the desired siRNA for 48 h before use.
Treatment with siRNA duplexes and qPCR OF CA12 and AE2 genes
Total RNA was extracted from microdissected cultured sealed pancreatic ducts using RNeasy Mini Kit from Qiagen. Expression levels of CA12 or AE2 were evaluated by analysis of relative gene expression using SYBR Green based quantitative PCR (qPCR) from ACGT Inc. (Wheeling, IL, USA). Expression level of the target gene was compared to that of housekeeping gene (GAPDH) in pooled samples. The CA12 siRNA duplex sequences used were as follows: duplex 1, AGAUGUCAAAUCGUGGUUUAGAUCA; duplex 2, CCAUUAUAACUCAGACCUUUAUCCT. The AE2 siRNA duplex sequences used were as follows: duplex 1, AGAUGUCACUUACGUCAA; duplex 2, UGAAAUGUCUGGAUGCUA. The negative control duplex (10 nm) was used as scrambled RNA. Ducts were transfected within 1 h after dissection and were kept in 35‐mm dishes containing 2 ml of DMEM with 10% FBS. In total, 100 pmol siRNA was diluted in 250 μl of Opti‐MEM I, and 5 μl Lipofectamine 2000 was diluted in 250 μl of the same medium. The diluted siRNA and Lipofectamine 2000 were mixed and after 25 min were added to the dish. After 24 h, the medium was replaced with a fresh medium without the siRNA, and the ducts were cut into half fragments to release the accumulated fluid and tension. The ducts were used 48 h after the beginning of the transfection after their resealing.
Measurement of fluid secretion by the sealed ducts
Fluid secretion was measured by video microscopy as described previously (Yang et al. 2009; Park et al. 2013). The sealed ducts were transferred to a perfusion chamber and perfused with Hepes‐ and then HCO3 –‐buffered media and stimulated with 5 μm forskolin in HCO3 –‐buffered media. Images were captured at 2–5 min intervals, obtained up to 40 min, and analysed offline by calculating the lumen volume. Due to the variation in size between the microdissected ducts, a normalization procedure was used with the volume of the first image (V 0) set as 1. Secretion is represented as the ratio V t/V 0, which was calculated using the equation V t/V 0 = (A t/A 0)3/2, with A as the duct area (Szalmay et al. 2001; Fernandez‐Salazar et al. 2004).
pHi measurements
pHi was measured with BCECF by recording BCECF fluorescence at excitation wavelengths of 490 and 440 nm and calibrating the fluorescence as described previously (Yang et al. 2009; Park et al. 2013). The ducts or HEK cells were loaded with BCECF by 15 min incubation at room temperature, with 6 μm of BCECF/AM (Teflabs Inc., Austin, TX, USA) and subsequent perfusion with dye free solution for at least 10 min. After stabilization of the fluorescence ratio, Na+–HCO3 –‐ cotransport was measured by incubating the ducts and cells with 10 μm S‐(N‐ethyl‐N‐isopropyl) amiloride (EIPA) to inhibit all Na+–H+ exchangers and in Na+‐free, HCO3 –‐buffered media to acidify the cytosol. Na+–HCO3 – cotransporter activity was initiated by perfusing the ducts or HeLa cells with HCO3 –‐buffered solution containing 140 mm Na+. Na+–HCO3 – cotransport activity was estimated from the slope of changes in pHi and is given as averaged percentage of control. Cl−–HCO3 – exchange activity was measured by incubating the ducts and cells with HCO3 –‐buffered media and after stabilization of pHi Cl−–HCO3 – exchanger activity was initiated by perfusing the ducts or cells with Cl–‐free, HCO3 –‐buffered solution containing 120 mm sodium gluconate. Cl−–HCO3 – exchanger activity was estimated from the slope of change in pHi.
Confocal microscopy and immunocytochemistry
HeLa cells expressing AE2 or NBCe1‐B were grown on glass coverslips and fixed by incubation with cold methanol or with 3% paraformaldehyde and permeabilized by incubation with 0.05% Triton X‐100 at room temperature for 10 min. After fixation, non‐specific sites were blocked with 5% goat serum. Cells were stained with primary antibody overnight and then for 1 h at room temperature with fluorescent secondary antibody. Coverslips were mounted on glass slides with Fluoromount‐G (Electron Microscopy Sciences, Hatfield, PA, USA) and analysed using an Olympus confocal microscope. The images were recorded with a ×40 objective using FluoView 1000 software (FV10‐ASW). All images are maximum intensity z‐projections. MDCK cells co‐expressing GFP‐NBCe1‐B and mKate‐CA12 were grown on a Corning transwell membrane system. To evaluate the integrity of the cell monolayers, transepithelial electric resistance was measured. The images of MDCK cells were reconstituted by Velocity software (Mountain View, CA, USA).
Surface biotinylation, co‐immunoprecipitation and Western blotting
For biotinylation experiments, cells were incubated with 0.5 mg ml−1 EZ‐LINK Sulfo‐NHS‐LC‐biotin (Thermo Scientific, Waltham, MA, USA) for 30 min on ice, then incubated with 100 mm glycine for 10 min to quench the free biotin and washed with PBS. Lysates were prepared in lysis buffer (contained (mm) 20 Tris, 150 NaCl, 2 EDTA, with 1% Triton X‐100, and a protease inhibitor mixture) by passing 10–12 times through a 27‐gauge needle after sonication on ice. The lysates were centrifuged at 11,000 rpm for 20 min at 4°C, and protein concentration in the supernatants was determined. Biotinylated AE2 and NBCe1‐B were isolated with Avidin beads via incubation of lysates with the appropriate antibodies for 4 h at room temperature, the beads were washed with lysis buffer and proteins were recovered by heating with sample buffer at 37°C for 30 min.
For co‐immunoprecipitation, cell extracts were incubated with Flag and β‐actin (Sigma‐Aldrich), GFP (Invitrogen), and HA (Cell signalling) antibodies overnight at 4°C and incubated with protein G Sepharose beads (Sigma) for 4 h at 0°C. Beads were collected and washed three times with lysis buffer, and proteins were recovered by heating in sample buffer at 37°C for 30 min. The heated samples were subjected to SDS‐PAGE and subsequently transferred to methanol‐soaked PVDF membranes.
To assay for CA12 and CA12(E143K) stability, HeLa cells were grown in 6‐well plate for 24 h were transfected at 80% confluence using the TransIT‐LT1 Transfection reagent (Mirus, Madison, WI, USA). Twenty four hours after transfection 10 μg ml−1 cycloheximide was added and cells were harvested after 4, 8, and 14 h by washing three times with PBS × 1 and adding lysis buffer (50 mm TrisHCl pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP‐40, 0.1% SDS). Protein concentration was determined by the Bradford dye‐binding procedure (Bio‐Rad). Thirty micrograms of cell lysate was separated in 10% SDS‐polyacrylamide gel and transferred onto nitrocellulose membrane. The membrane was blocked by 5% non‐fat milk in TTBS and incubated with monoclonal antibodies against myc (Sigma M4439) and GAPDH (Millipore AB2302) overnight at 4°C with gentle agitation. After three washes in TTBS, the blot incubated for 1 h with HRP‐anti mouse secondary antibody (1:20000) at room temperature. Densitometry analysis was performed with ImageJ software.
Analysis of saliva Na+ and CI− content
Saliva was collected from mice stimulated with 10 mg kg−1 pilocarpine. Collected saliva samples were stored at −80°C until further analysis. Na+ and Cl− concentration in the salivary fluid were analysed as previously described (Romanenko et al. 2008).
Results and discussion
Abnormal CA12(E143K) glycosylation and targeting
CA12 is expressed in the basolateral membrane (Kyllonen et al. 2003). Figure 1 A shows similar basolateral expression in parotid gland acinar and duct cells. In addition, mKate‐tagged CA12 and GFP‐tagged NBCe1‐B co‐expressed in MDCK cells grown on filter supports are targeted to the basolateral membrane (Fig. 1 B, left image). The Glu mutated in CA12 that causes hyponatraemia is conserved in all mammalian CAs (not shown). The CA12(E143K) mutation affects protein expression and localization. In Fig. 1 B, the right image shows mistargeting of CA12(E143K) and its retention in intracellular compartments in MDCK cells. Figure 1 D shows that CA12(E143K) expression is reticular like and co‐localized with the ER marker KDEL‐CFP. Figure 2 A shows that CA12(E143K) expressed in HEK cells has a higher mobility, indicative of altered posttranslational modification, such as glycosylation. Indeed, treatment of CA12 with EndoF (PNGase) that hydrolyses all sugars resulted in a lower band, while treatment with EndoH had minimal effect. Sensitivity to EndoH provides unequivocal evidence that CA12(E143K) is retained in the ER since resistance to EndoH is acquired in the Golgi. As a control, Fig. 2 A shows that EndoF and EndoH had no effect on the mobility of the cytoplasmic CA7. CA12(E143K) migration was between the fully glycosylated and unglycosylated CA12. The level of CA12(E143K) was lower than that of CA12, suggesting partial degradation. Indeed, assaying protein stability by cycloheximide chase showed high degradation rate of CA12(E143K) (Fig. 2 B).
Figure 1. Localization of native and expressed CA12 and mutants .
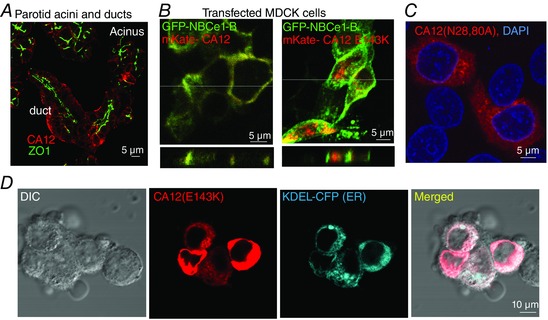
A, localization of CA12 (red) and ZO1 (green) in dispersed parotid acini and ducts. B, x–y (upper panels) and z scan (lower panels, taken at the white line in the upper panels) images of MDCK cells transfected with mKate‐CA12 (red) and GFP‐NBCe1‐B (green) (left image) or with mKate‐CA12(E143K) and GFP‐NBCe1‐B (right panel). C, HEK cells transfected with CA12(N28,80A) (red) and stained for DAPI (blue). D, co‐localization of CA12(E143K) (red) and the ER marker KDEL‐CFP (blue) in transfected in HEK cells.
Figure 2. Interaction of CA12 and CA12(E143K) with AE2 .
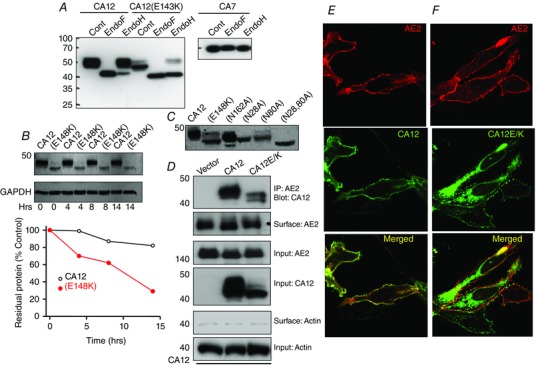
A, wild‐type CA12 and CA7 and their E/K mutants were transfected in HEK cells and extracted into lysis buffer. The extracts were treated with EndoF (PNGase) and Endo H for 20 min at room temperature and analysed by Western blots. B, the reduced stability of CA12(E143K). Cells expressing CA12 or CA12(E143K) were exposed to cycloheximide at time 0 to halt protein synthesis and extracted at the indicated times to analyse protein levels. C, the effect of mutating the putative N‐glycosylation sites N162A, N28A, N80A and N28,80A on migration of CA12. D, the co‐immunoprecipitation (Co‐IP) of CA12 and CA12(E143K) with AE2 and the effects of CA12 and CA12(E143K) on surface expression of AE2. Surface actin was used as a control. AE2 is tagged with HA and CAs with flag. Anti‐HA and anti‐Frag were used for the Co‐IP and proteins detection. E and F, example images of HEK cells transfected with the indicated plasmids showing the localization of AE2 and CA12 (E) or CA12(E143K) (F).
Potential N‐glycosylation sites in CA12 are predicted by the NetNGlyc 1.0 program (Gupta & Brunak, 2002) to be N28, N89 and N162. Figure 2 C shows that the N162A mutation slightly reduced CA12 mobility but did not prevent glycosylation. N28A and N80A each partially reduced glycosylation and the double mutant CA12(N28,80A) was not glycosylated. Glycosylation of membrane proteins often affects their targeting. Accordingly, when expressed in MDCK (Fig. 1 B) or HEK cells (Fig. 2 F) most CA12(E143K) was retained in the ER, although a small amount did make it to the plasma membrane (see also below). Similarly, CA12(N28,80A) was retained in the ER (Fig. 1 C).
CA12(E143K) inhibits the HCO3 − transporters that are located in the basolateral membrane of epithelial cells
Mistargeting of CA12, rather than only a change in activity, can explain the disease phenotype, in particular if the mutant protein affects the activity of the HCO3 − transporters found in the basolateral membrane of epithelial cells AE2 and NBCe1‐B. Therefore, we first set out to determine the effect of the CA12, CA12(E143K) and of other CAs on the interaction, surface expression and activity of AE2 and NBCe1‐B. The co‐immunoprecipitation (Co‐IP) in Fig. 2 D shows interaction between CA12, CA12(E143K) and AE2 and that CA12(E143K) did not appreciably affect surface expression of AE2 (0.94 ± 0.11, n = 3), as determined by surface biotinylation assay. Co‐expression experiments similarly show plasma membrane co‐localization of AE2 and CA12 (Fig. 2 E), and mostly ER localization of CA12(E143K) (Fig. 2 F).
CA12 and CA12(E143K) also interact with NBCe1‐B, with CA12(E143K) reducing surface expression of NBCe1‐B by 41 ± 8% (n = 4, P < 0.05) (Fig. 3 A–C) by trapping some of it intracellularly (Fig. 3 C). To further test the effect of mutating the conserved Glu (E143 in CA12), we tested the effect of mutating the same residue in the cytoplasmic CA7 (E118A). Figure 3 A shows that the level of CA7(E118A) is lower than wild‐type CA7, again suggesting reduced stability and partial degradation of the conserved glutamate mutant. Nevertheless, CA7 and CA7(E118A) interact with NBCe1‐B.
Figure 3. Interaction of CA12 and CA12(E143K) with NBCe1‐B .
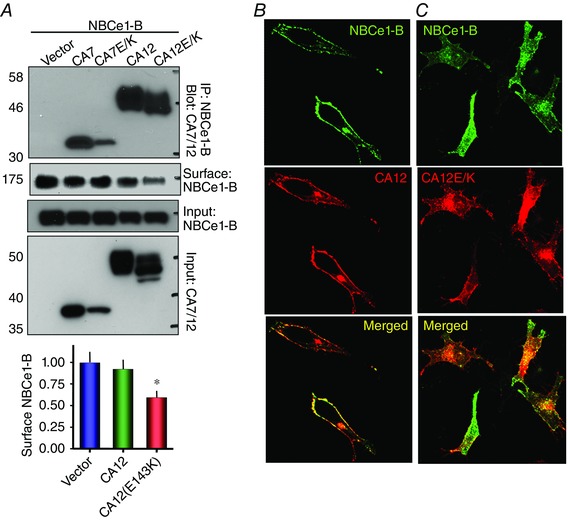
A, the Co‐IP of CA7, CA7(E118K), CA12 and CA12(E143K) with NBCe1‐B and the effects of CA7, CA7(E118K), CA12 and CA12(E143K) on surface expression of NBCe1‐B in transfected HEK cells. NBCe1‐B is tagged with GFP and anti‐GFP and anti‐Flag antibodies were used for the Co‐IP and detection of proteins. The columns show the means ± SEM of the reduction in NBCe1‐B surface expression (n = 4, P < 0.05, indicated by asterisk). B and C, example images of localization of NBCe1‐B and CA12 (B) or CA12(E143K) (C) in transfected HeLa cells.
The activities of AE2 and NBCe1 were reported to be increased by CA2 (Becker & Deitmer, 2007; Gonzalez‐Begne et al. 2007) and that of AE2 also by CA4 (Sterling et al. 2002), although an effect of CA2 on NBCe1‐A was questioned (Lu et al. 2006; Piermarini et al. 2007). Therefore, to evaluate the role of the various CAs in regulating these HCO3 − transporters we tested the effect of two plasma membrane localized (CA12 and CA4) and two cytoplasmic localized (CA2 and CA7) CAs and of their conserved Glu mutants on transport activity. The activity of the Cl−–HCO3 − exchanger AE2 was measured as the change in pHi in response to removal and re‐addition of external Cl− in HeLa cells perfused with HCO3 −‐buffered solutions and transfected with the respective constructs. The findings for AE2 are shown in Fig. 4. Expression of CA12 and CA12(E143K) had no significant effect on resting pHi of cells bathed in HCO3 −‐buffered solutions (control 7.03 ± 0.03, CA12 7.08 ± 0.04, CA12(E143K) 7.05 ± 0.03), probably because other HCO3 − transporters set normal initial pHi and CA12(E143K) only partially inhibits AE2. Notably, CA2 and CA4 only modestly increased while CA12 markedly increased the activity of AE2. Moreover, CA12(E143K) reduced basal AE2 activity by about 50%. Interestingly, CA4(E138A) and CA2(E117A) also reduced AE2 activity while CA7 and CA7(E118A) had no significant stimulatory or inhibitory effects, respectively. Overall, the findings in Fig. 4 suggest that CA12 is the major CA that generates HCO3 − near the AE2 HCO3 − transport site.
Figure 4. Effect of several CAs and their conserved Glu mutants on AE2 activity .
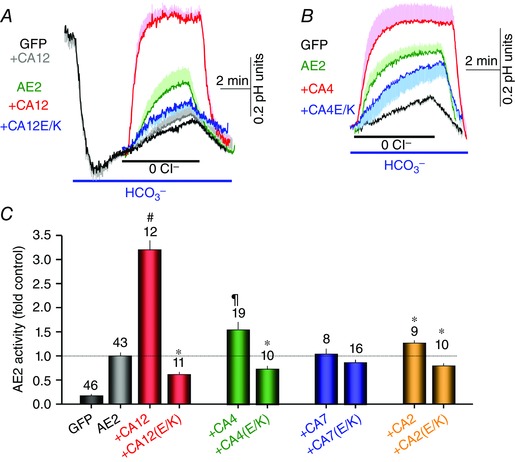
A and B, AE2 activity in HeLa cells transfected with the indicated plasmids. Cells in Hepes‐buffered solutions were exposed to HCO3 −‐buffered solutions. After stabilization of pHi, the cells were superfused with Cl−‐free and then Cl−‐containing solutions to assay for Cl−–HCO3 − exchange activity. The traces are the means ± SEM of the number of experiments indicated in the columns under each condition and the effect of CA12 and CA12(E143K) (A) or CA4 and CA4(E138K) (B) on AE2 activity. A, grey trace is control showing the effect of CA12 on native AE2 activity. The times of removal of external Cl− were aligned to facilitated comparison between experiments. C, the means ± SEM of AE2 rates obtained from the initial slop of the pHi increase on removal of external Cl− and normalized to the rate measured with AE2 alone. The numbers in the columns indicate the number of experiments obtained from at least 4 separate transfections. * denotes p < 0.05 and # denotes p < 0.01 with respect to AE2 alone and ¶ denotes p < 0.01 with respect to AE2+CA12.
The same CAs and their Glu mutants had quite different effects on NBCe1‐B activity. Figure 5 shows that CA12, CA4 and CA7 all potently increased the activity of NBCe1‐B while, as reported before for the NBCs (Piermarini et al. 2007), CA2 had no effect. As was found for AE2, the conserved Glu mutants CA12(E143K), CA4(E138A) and CA7(E118A) inhibited basal NBCe1‐B activity, while CA2(E117A) had no effect. As a control for the effect of CA12 on the activity of the transporters, Fig. 5 E and F shows that CA12 and CA12(E143K) had no effect on the Cl−–HCO3 − exchanger slc26a6 (Shcheynikov et al. 2006; Ohana et al. 2011).
Figure 5. Effect of several CAs and their conserved Glu mutants on NBCe1‐B activity .
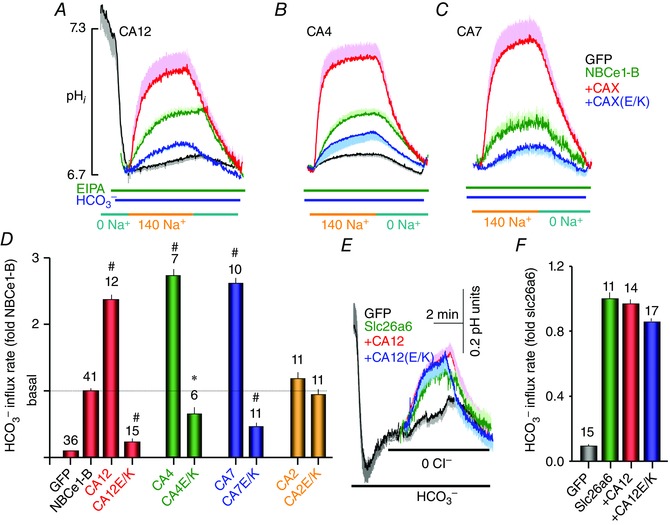
A—C, the effects of CA12 and CA12(E143K) (A), CA4 and CA4(E138K) (B) and CA7 and CA7(E118K) on NBCe1‐B activity. HeLa cells transfected with the indicated plasmids and in Hepes‐buffered solutions were exposed to Na+‐free, HCO3 −‐buffered solutions to stably acidify the cytosol. The solutions also included 10 μm of the Na+–H+ exchangers inhibitor EIPA to inhibit all Na+–H+ exchangers. The Na+‐dependent HCO3 − influx was initiated by exposing the acidified cells to a HCO3 −‐buffered solutions containing 10 μm EIPA and 140 mm Na+. The times of addition of external Na+ were aligned to facilitated comparison between experiments. The traces are the means ± SEM of number of experiments indicated in the columns under each condition. D, the means ± SEM of NBCe1‐B rates obtained from the initial slops of the pHi increase on re‐addition of Na+ in the presence of HCO3 − and of EIPA. The rates were normalized to the rate measured with NBCe1‐B alone. * denotes p < 0.05 and # denotes p < 0.01 with respect to NBCe1‐B alone. E and F, the means ± SEM traces (E) and rates (F) of slc26a6‐mediated Cl−–HCO3 − exchange are not affected by CA12 and CA12(E143K) in transfected HEK cells. The numbers in the columns in D and F indicate the number of experiments obtained from at least 4 separate transfections.
The results in Figs 4 and 5 suggest that several CAs can affect the activity of AE2 and NBCe1‐B. The physiological effects in epithelia of some of them are obvious. The two basolateral CA12 and CA4 (Schwartz et al. 1999) can supply HCO3 − to the extracellular HCO3 − site of NBCe1‐B. However, at present it is not clear how CA12 and CA4 in the case of AE2 and the cytoplasmic CA7 in the case of NBCe1‐B can increase the activity of AE2 when mediating Cl− o–HCO3 − i exchange and of NBCe1‐B when mediating Na+ o–HCO− o co‐transport. One possibility is that these CAs convert the exiting (AE2) or entering (NBCe1‐B) HCO3 − to CO2 or shuttle it away from the plasma membrane surface to reduce HCO3 − concentration at this site and facilitate HCO3 − fluxes. Generation of pH gradients at membrane surfaces and their clearance by CAs has been documented (Musa‐Aziz et al. 2014a,b). This raised the possibility that in polarized secretory cells the cytoplasmic CAs facilitate shuttling of HCO3 − across the cells from the basolateral to the luminal side.
CA12 is required for the activity of the native ductal AE2 but not NBCe1‐B
Considering the potent activation of AE2 by CA12 (Fig. 4) and the activation of NBCe1‐B by multiple CAs (Fig. 5), we examined the relative importance of CA12 in the activity of the native ductal AE2 and NBCe1‐B. For this CA12 was depleted by treating isolated pancreatic ducts in primary culture with CA12 siRNA and measuring NBCe1‐B and AE2 activity. Figure 6 A shows the efficiency of the siRNA in depletion of CA12 mRNA. Figure 6 B shows that knockdown of CA12 had no effect on native NBCe1‐B activity measured as Na+‐ and HCO3 −‐dependent recovery from an acid load while inhibiting all Na+–H+ exchangers with EIPA. By contract, knockdown of CA12 reduced Cl−–HCO3 − exchange activity by about 50%. The duct expresses AE2 and slc26a6 (Lee et al. 2012), but since slc26a6 is not regulated by CA12 (Fig. 5 E and F) the reduction in Cl−–HCO3 − exchange activity is mostly due to reduced AE2 activity. This is further supported by the results in Fig. 6 D which show that knockdown of AE2 reduced ductal Cl−–HCO3 − exchange activity similarly to knockdown of CA12.
CA12 and AE2 are required for ductal fluid section
NBCe1‐B is considered the main HCO3 − influx mechanism for secretory gland fluid and HCO3 − secretion. At the same time, all available models suggest that inhibition of Cl−–HCO3 − exchange by AE2 is essential for ductal HCO3 − secretion to prevent HCO3 − efflux across the basolateral membrane (Sohma et al. 2001; Steward et al. 2005; Steward & Ishiguro, 2009). However, this assumption was never tested experimentally. The importance of CA12‐mediated control of HCO3 − concentration in the AE2 domain (Fig. 4) and of salt wasting by patients with the CA12(E143K) mutation (Muhammad et al. 2011; Feinstein et al. 2014) raised the question of whether CA12 and AE2 activities participate in secretory gland fluid and HCO3 − secretion. To address this question we measured ductal fluid secretion using the sealed pancreatic duct system (Argent et al. 1986; Szalmay et al. 2001). Figure 6 E shows that knockdown of CA12 markedly reduced both basal and forskolin stimulated pancreatic duct fluid secretion. Opposite to what the models suggest and general assumption (Sohma et al. 2001; Steward et al. 2005; Steward & Ishiguro, 2009), Fig. 6 F shows similar inhibition of pancreatic duct fluid secretion by knockdown of AE2. Thus, it is clear that CA12 and AE2 activities are required for maximal ductal fluid secretion. However, it is necessary to indicate that inhibition of AE2 is required during the final stage of HCO3 − secretion and no information is available for the requirement of AE2 activity at the early stage of HCO3 − secretion. It is possible that the requirement for ductal AE2 activity shown here represent this early stage of ductal fluid and HCO3 − secretion.
The findings in Fig 6 E and F raise a conundrum. Activation of a basolateral HCO3 − efflux mechanism like AE2 was expected to inhibit fluid and HCO3 − secretion since the only net osmolyte secreted by the duct is HCO3 − (Lee et al. 2012), and inhibition of HCO3 − loading stops fluid secretion when measured in the presence of HCO3 − (Fernandez‐Salazar et al. 2004). Therefore, our findings indicate that AE2 has a specific role in the duct. One possible function is supplying the duct with Cl−, required for fluid secretion, if the Cl− supply cannot be fully satisfied by CFTR‐mediated recirculation of the absorbed luminal Cl−. In this case CA12 may consume the exiting HCO3 − to facilitate Cl− o–HCO3 − i exchange. However another possibility that cannot be excluded is clearance of cytoplasmic Cl− by AE2 that is absorbed by the luminal slc26a6, which mediates ductal Cl− absorption (Lee et al. 2012). Otherwise, Cl− will accumulate in the duct and stop the coupled luminal Cl− absorption and HCO3 − secretion. In this case, AE2 will function in reverse as a Cl− i–HCO3 − o exchanger to further support luminal ductal Cl− absorption and HCO3 − secretion. Ductal fluid and electrolyte secretion starts when luminal Cl− o is 100 mm, Cl− i is about 30–40 mm (Ishiguro et al. 2002; Lee et al. 2012) and HCO3 − i is about 10 mm (resting pHi of 7.03; see above). CA12 needs to increase basolateral surface HCO3 − to only about 30 mm to mediate Cl− i–HCO3 − o exchange by basolateral AE2. Additional work is needed to distinguish these possibilities. Independent of the exact role of AE2 in the duct, our findings indicate that AE2 activity fueled by CA12 is required for ductal secretion and provide further support to the conclusion that AE2 plays an essential role in epithelia anion and fluid secretion (Gawenis et al. 2010; Huang et al. 2012; Shan et al. 2012).
Relevance to disease
Previous studies reported altered sweat gland Na+ and Cl− absorption with no apparent change in saliva electrolyte composition in patients with CA12(E143K) (Muhammad et al. 2011). However, considering the wide expression of CA12 in epithelia (Kyllonen et al. 2003; Frost, 2014) and some similarity between the mechanism of sweat and salivary gland electrolyte transport (Quinton, 2007; Lee et al. 2012), we reevaluated salivary gland function in patients with the CA12(E143K) mutation. The study was approved by the Soroka Medical University Centre Institutional Review Board and conformed to the standards set by the Declaration of Helsinki, and all patients or their guardians provided written informed consent, before participating. Six patients diagnosed with the CA12(F143K) mutation, who exhibited dehydration and high sweat test, were evaluated for a dry mouth phenotype. As was reported for sweat electrolytes (Muhammad et al. 2011; Feinstein et al. 2014), the patients evaluated show considerable variability, with 3/6 patients having repeated episodes of dry mouth and dry tongues (Fig. 7).
Figure 7. Dry mouth and dry tongue of patients carrying the CA12(E143K) mutation .
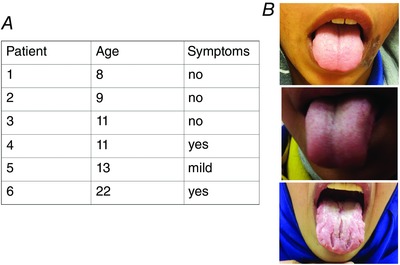
A, the age of the patients and their report of dry mouth episodes. B, the tongue of the three symptomatic patients during visit to the clinic.
Based on the clinical findings we proceeded with viral delivery of CA12 and CA12(E143K) to all major salivary glands through the ductal openings to the oral cavity. Figure 8 A shows an anaesthetized mouse with the two submandibular and the two parotid glands cannulated. Saline (30 μl for the parotid glands and 50 μl for the submandibular glands) containing AdRFP (RFP only for the control), AdCA12 or AdCA12(E143K) was then delivered. After 7 days to allow expression of the genes and recovery from the virally caused inflammation (Park et al. 2013), the parotid glands were removed and the ducts microdissected and cultured for 24 h to allow sealing. The images in Fig. 8 B show RFP expressing sealed ducts that also express wild‐type CA12 or CA12(E143K). Salivary gland ducts absorb the Na+ and Cl− secreted by acinar cells and secrete K+ and HCO3 − but are expected to secrete no or a small volume of fluid (Schneyer et al. 1972; Melvin et al. 2005; Lee et al. 2012). However, salivary gland ductal fluid secretion has never been tested directly. Figure 8 B shows that when stimulated with cAMP, sealed parotid ducts do secrete fluid at a rate about 40–50% that of the pancreatic ducts, which secretes most of the fluid in the pancreatic juice. Notably, ductal overexpression of CA12 almost doubled the rate of fluid secretion, while ductal expression of CA12(E143K) inhibited fluid secretion by about 60%.
Figure 8. CA12 is required for fluid secretion by salivary glands .
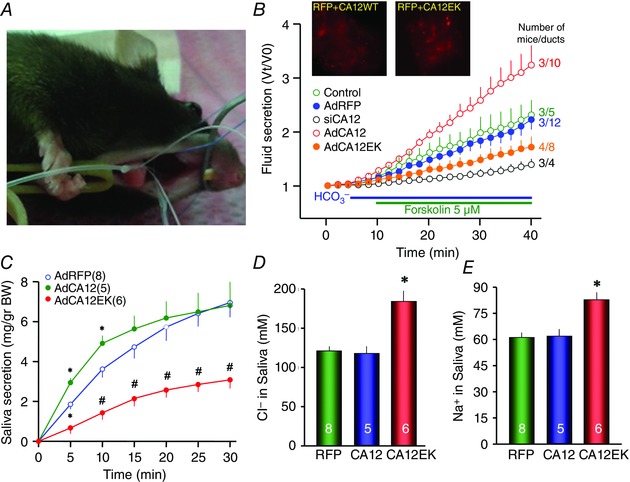
A, cannulation of all the major mouse salivary gland, the procedure used to deliver the adenoviruses. B, ducts were microdissected from the parotid glands of untreated mice (green), mice infected with AdRFP (blue, black) and in addition treated with siCA12 in culture (black), mice infected with AdCA12 (red) and mice infected with AdCA12(E143K). After 48 h in culture, the sealed ducts were used to measure fluid secretion. The results are means ± SEM of the number of ducts (second number) obtained from the number of mice indicated by the first number. C, mice infected with the indicated adenoviruses were used to measure the rate of salivary secretion in response to i.p. injection of 1 mg kg−1 pilocarpine. The collected saliva (C) was used to measure the concentrations of Cl− (D) or Na+ (E). The results are means ± SEM of the number of mice indicated in the figure. * denotes p < 0.05 and # denotes p < 0.01 with respect to mice infected with AdRFP.
To further analyse the impact of CA12 in epithelial fluid and electrolyte secretion, we measured saliva secretion and electrolyte composition of the secreted saliva. Figure 8 C shows that overexpression of CA12 increased the initial rate of pilocarpine‐stimulated salivation in mice, while expression of CA12(E143K) markedly inhibited salivation. Patients with CA12(E143K) mutation lose Na+ and Cl− in the sweat, probably due to impaired absorption (Muhammad et al. 2011). The salivary duct absorbs the Na+ and Cl− secreted by acinar cells (Melvin et al. 2005; Lee et al. 2012). Figure 8 D and E shows that while overexpression of CA12 had no effect on salivary Na+ and Cl− content, expression of CA12(E143K) increased both Na+ and Cl− concentration in the saliva, reproducing the findings with patients’ sweat.
Conclusions
The present studies reveal a key role for CA12 and its activation of AE2 in epithelial fluid and electrolyte secretion and explain the phenotype of patients carrying the CA12(E143K) mutation. The CA12(E143K) mutation is an autosomal recessive form of salt wasting, with hyponatraemia, hyperkalaemia, high sweat Cl−, dehydration and failure to thrive (Feldshtein et al. 2010; Muhammad et al. 2011; Feinstein et al. 2014). Although the E143K mutation may affect Zn2+ interaction with the CA12 active site (Muhammad et al. 2011) and the effect of anions on CA12 activity (Feldshtein et al. 2010), the present findings show that the major effect of the mutation is mistargeting of CA12(E143K) due to aberrant glycosylation and partial degradation (Figs 1 and 2). Interestingly, the Glu is conserved in the mammalian CAs and its mutation appears to have the same effect in all CAs tested, in that they are partially degraded (Fig. 3 for CA7 and not shown for CA4), raising the possibility that mutation of the conserved Glu causes misfolding. Importantly, the Glu mutants, including CA12(E143K), not only do not activate the basolateral HCO3 − transporters, they also inhibit their basal activity. Although the mechanism by which the E/K mutants inhibit the transporters is not known at present, it may be due to a dominant negative effect since they interact with the transporters, thereby preventing interaction of native CAs with the transporters.
The main effect of CA12 in regulating fluid and HCO3 − secretion appears to be due to stimulation of AE2, although we cannot exclude an additional effect on NBCe1‐B. These findings were unexpected in view of the recent report that deletion of salivary gland acinar cells AE2 had no effect on ex vivo salivary secretion, although it has prominent effect on cellular Cl−–HCO3 − exchange activity (Pena‐Munzenmayer et al. 2015). Two major differences can account for the different findings. In our studies AE2 was depleted acutely and it was depleted from the duct and, if at all, only partially from acinar cells, as adenoviruses delivered through the duct transduce the duct very well but acinar cells poorly (Voutetakis et al. 2005), while the knockout was specifically in acinar cells (Pena‐Munzenmayer et al. 2015). In addition, the AE2 knockout may result in adaptation, such as an increased role of AE4 in gland function. AE4 is expressed in the basolateral membrane of acinar cells (Ko et al. 2002; Pena‐Munzenmayer et al. 2015). The function of ductal AE2 and CA12 appears to be critical for ductal function and their impairment results in reduced salivation. This is supported by the findings of increased Na+ and Cl− concentrations in the saliva (Fig. 8), indicative of impaired ductal function associated with marked inhibition of saliva secretion.
The importance of CA12 and AE2 functions in ductal and salivary gland secretion, together with the effect of the E143K mutation on CA12(E143K) glycosylation and targeting explain well the phenotype of the disease caused by the mutation and at the same time reveal new aspects of the molecular mechanism of secretory gland fluid and electrolyte secretion. Another aspect of CA12 biology is the marked upregulation of CA12 expression in many cancers (Parks et al. 2013). Indeed, inhibitors of CA12 have been developed and are being evaluated for cancer treatment (Morris et al. 2011; Kopecka et al. 2015). Our findings call for caution in the use of these inhibitors, considering their deleterious effect on epithelial secretion, which can have significant side‐effects.
Additional information
Competing interests
All authors declare no conflict of interests.
Author contributions
J.H.H and E.M. performed and interpreted experiments, C.Z. prepared viruses and delivered to the mice and interpreted data, E.H., S.A. and N.L. performed clinical evaluations, R.P. and S.M. conceived and directed the studies and S.M. wrote the manuscript with contribution by all authors. Experiments were performed at the NIH, Beer Sheva and Inchon. All authors approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by Intramural NIH/NIDCR grant DE000735‐04, grant 3000005152 from the Ministry of Health, Israel and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2014R1A1A3049477).
J. Hee Hong and E. Muhammad contributed equally to this work.
References
- Ahuja M, Jha A, Maleth J, Park S & Muallem S (2014) cAMP and Ca2+ signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almstahl A & Wikstrom M (2003) Electrolytes in stimulated whole saliva in individuals with hyposalivation of different origins. Arch Oral Biol 48, 337–344. [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Loiselle FB, Supuran CT, Schwartz GJ & Casey JR (2003) Direct extracellular interaction between carbonic anhydrase IV and the human NBC1 sodium/bicarbonate co‐transporter. Biochemistry 42, 12321–12329. [DOI] [PubMed] [Google Scholar]
- Argent BE, Arkle S, Cullen MJ & Green R (1986) Morphological, biochemical and secretory studies on rat pancreatic ducts maintained in tissue culture. Q J Exp Physiol 71, 633–648. [DOI] [PubMed] [Google Scholar]
- Baum BJ, Wellner RB & Zheng C (2002) Gene transfer to salivary glands. Int Rev Cytol 213, 93–146. [DOI] [PubMed] [Google Scholar]
- Becker HM & Deitmer JW (2007) Carbonic anhydrase II increases the activity of the human electrogenic Na+/HCO3 − cotransporter. J Biol Chem 282, 13508–13521. [DOI] [PubMed] [Google Scholar]
- Becker HM, Klier M & Deitmer JW (2014) Carbonic anhydrases and their interplay with acid/base‐coupled membrane transporters. Subcell Biochem 75, 105–134. [DOI] [PubMed] [Google Scholar]
- Catalan MA, Kondo Y, Pena‐Munzenmayer G, Jaramillo Y, Liu F, Choi S, Crandall E, Borok Z, Flodby P, Shull GE & Melvin JE (2015) A fluid secretion pathway unmasked by acinar‐specific Tmem16A gene ablation in the adult mouse salivary gland. Proc Natl Acad Sci USA 112, 2263–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalan MA, Pena‐Munzenmayer G & Melvin JE (2014) Ca2+‐dependent K+ channels in exocrine salivary glands. Cell Calcium 55, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein Y, Yerushalmi B, Loewenthal N, Alkrinawi S, Birk OS, Parvari R & Hershkovitz E (2014) Natural history and clinical manifestations of hyponatremia and hyperchlorhidrosis due to carbonic anhydrase XII deficiency. Horm Res Paediatr 81, 336–342. [DOI] [PubMed] [Google Scholar]
- Feldshtein M, Elkrinawi S, Yerushalmi B, Marcus B, Vullo D, Romi H, Ofir R, Landau D, Sivan S, Supuran CT & Birk OS (2010) Hyperchlorhidrosis caused by homozygous mutation in CA12, encoding carbonic anhydrase XII. Am J Hum Genet 87, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Salazar MP, Pascua P, Calvo JJ, Lopez MA, Case RM, Steward MC & San Roman JI (2004) Basolateral anion transport mechanisms underlying fluid secretion by mouse, rat and guinea‐pig pancreatic ducts. J Physiol 556, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost SC (2014) Physiological functions of the alpha class of carbonic anhydrases. Subcell Biochem 75, 9–30. [DOI] [PubMed] [Google Scholar]
- Gawenis LR, Bradford EM, Alper SL, Prasad V & Shull GE (2010) AE2 Cl−/HCO3 − exchanger is required for normal cAMP‐stimulated anion secretion in murine proximal colon. Am J Physiol Gastrointest Liver Physiol 298, G493–G503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Begne M, Nakamoto T, Nguyen HV, Stewart AK, Alper SL & Melvin JE (2007) Enhanced formation of a HCO3 − transport metabolon in exocrine cells of Nhe1−/− mice. J Biol Chem 282, 35125–35132. [DOI] [PubMed] [Google Scholar]
- Gupta R & Brunak S (2002) Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput: 310–322. [PubMed] [Google Scholar]
- Huang J, Shan J, Kim D, Liao J, Evagelidis A, Alper SL & Hanrahan JW (2012) Basolateral chloride loading by the anion exchanger type 2, role in fluid secretion by the human airway epithelial cell line Calu‐3. J Physiol 590, 5299–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Naruse S, Kitagawa M, Mabuchi T, Kondo T, Hayakawa T, Case RM & Steward MC (2002) Chloride transport in microperfused interlobular ducts isolated from guinea‐pig pancreas. J Physiol 539, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y & Oh U (2014) Anoctamin 1 in secretory epithelia. Cell Calcium 55, 355–361. [DOI] [PubMed] [Google Scholar]
- Ko SB, Luo X, Hager H, Rojek A, Choi JY, Licht C, Suzuki M, Muallem S, Nielsen S & Ishibashi K (2002) AE4 is a DIDS‐sensitive Cl−/HCO3 − exchanger in the basolateral membrane of the renal CCD and the SMG duct. Am J Physiol Cell Physiol 283, C1206–C1218. [DOI] [PubMed] [Google Scholar]
- Kopecka J, Campia I, Jacobs A, Frei AP, Ghigo D, Wollscheid B & Riganti C (2015) Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 6, 6776–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyllonen MS, Parkkila S, Rajaniemi H, Waheed A, Grubb JH, Shah GN, Sly WS & Kaunisto K (2003) Localization of carbonic anhydrase XII to the basolateral membrane of H+‐secreting cells of mouse and rat kidney. J Histochem Cytochem 51, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Lee MG, Ohana E, Park HW, Yang D & Muallem S (2012) Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev 92, 39–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RJ & Foskett JK (2014) Ca2+ signaling and fluid secretion by secretory cells of the airway epithelium. Cell Calcium 55, 325–336. [DOI] [PubMed] [Google Scholar]
- Lu J, Daly CM, Parker MD, Gill HS, Piermarini PM, Pelletier MF & Boron WF (2006) Effect of human carbonic anhydrase II on the activity of the human electrogenic Na/HCO3 cotransporter NBCe1‐A in Xenopus oocytes. J Biol Chem 281, 19241–19250. [DOI] [PubMed] [Google Scholar]
- McKenna R & Frost SC (2014) Overview of the carbonic anhydrase family. Subcell Biochem 75, 3–5. [DOI] [PubMed] [Google Scholar]
- Maleth J & Hegyi P (2014) Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium 55, 337–345. [DOI] [PubMed] [Google Scholar]
- Melvin JE, Yule D, Shuttleworth T & Begenisich T (2005) Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol 67, 445–469. [DOI] [PubMed] [Google Scholar]
- Morgan PE, Pastorekova S, Stuart‐Tilley AK, Alper SL & Casey JR (2007) Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. Am J Physiol Cell Physiol 293, C738–C748. [DOI] [PubMed] [Google Scholar]
- Morris JC, Chiche J, Grellier C, Lopez M, Bornaghi LF, Maresca A, Supuran CT, Pouyssegur J & Poulsen SA (2011) Targeting hypoxic tumor cell viability with carbohydrate‐based carbonic anhydrase IX and XII inhibitors. J Med Chem 54, 6905–6918. [DOI] [PubMed] [Google Scholar]
- Muhammad E, Leventhal N, Parvari G, Hanukoglu A, Hanukoglu I, Chalifa‐Caspi V, Feinstein Y, Weinbrand J, Jacoby H, Manor E, Nagar T, Beck JC, Sheffield VC, Hershkovitz E & Parvari R (2011) Autosomal recessive hyponatremia due to isolated salt wasting in sweat associated with a mutation in the active site of Carbonic Anhydrase 12. Hum Genet 129, 397–405. [DOI] [PubMed] [Google Scholar]
- Musa‐Aziz R, Occhipinti R & Boron WF (2014a) Evidence from simultaneous intracellular‐ and surface‐pH transients that carbonic anhydrase II enhances CO2 fluxes across Xenopus oocyte plasma membranes. Am J Physiol Cell Physiol 307, C791–C813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa‐Aziz R, Occhipinti R & Boron WF (2014b) Evidence from simultaneous intracellular‐ and surface‐pH transients that carbonic anhydrase IV enhances CO2 fluxes across Xenopus oocyte plasma membranes. Am J Physiol Cell Physiol 307, C814–C840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana E, Shcheynikov N, Yang D, So I & Muallem S (2011) Determinants of coupled transport and uncoupled current by the electrogenic SLC26 transporters. J Gen Physiol 137, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski A, De Giusti VC, Morgan PE, Aiello EA & Alvarez BV (2012) Binding of carbonic anhydrase IX to extracellular loop 4 of the NBCe1 Na+/HCO3 − cotransporter enhances NBCe1‐mediated HCO3 − influx in the rat heart. Am J Physiol Cell Physiol 303, C69–C80. [DOI] [PubMed] [Google Scholar]
- Park S, Shcheynikov N, Hong JH, Zheng C, Suh SH, Kawaai K, Ando H, Mizutani A, Abe T, Kiyonari H, Seki G, Yule D, Mikoshiba K & Muallem S (2013) Irbit mediates synergy between Ca2+ and cAMP signaling pathways during epithelial transport in mice. Gastroenterology 145, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks SK, Chiche J & Pouyssegur J (2013) Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer 13, 611–623. [DOI] [PubMed] [Google Scholar]
- Pena‐Munzenmayer G, Catalan MA, Kondo Y, Jaramillo Y, Liu F, Shull GE & Melvin JE (2015) Ae4 (Slc4a9) anion exchanger drives Cl− uptake‐dependent fluid secretion by mouse submandibular gland acinar cells. J Biol Chem 290, 10677–10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piermarini PM, Kim EY & Boron WF (2007) Evidence against a direct interaction between intracellular carbonic anhydrase II and pure C‐terminal domains of SLC4 bicarbonate transporters. J Biol Chem 282, 1409–1421. [DOI] [PubMed] [Google Scholar]
- Quinton PM (2007) Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda) 22, 212–225. [DOI] [PubMed] [Google Scholar]
- Quinton PM (2010) Role of epithelial HCO3 − transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol 299, C1222–C1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebello G, Ramesar R, Vorster A, Roberts L, Ehrenreich L, Oppon E, Gama D, Bardien S, Greenberg J, Bonapace G, Waheed A, Shah GN & Sly WS (2004) Apoptosis‐inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA 101, 6617–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Nakamoto T, Catalan MA, Gonzalez‐Begne M, Schwartz GJ, Jaramillo Y, Sepulveda FV, Figueroa CD & Melvin JE (2008) Clcn2 encodes the hyperpolarization‐activated chloride channel in the ducts of mouse salivary glands. Am J Physiol Gastrointest Liver Physiol 295, G1058–G1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneyer LH, Young JA & Schneyer CA (1972) Salivary secretion of electrolytes. Physiol Rev 52, 720–777. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Olson J, Kittelberger AM, Matsumoto T, Waheed A & Sly WS (1999) Postnatal development of carbonic anhydrase IV expression in rabbit kidney. Am J Physiol Renal Physiol 276, F510–F520. [DOI] [PubMed] [Google Scholar]
- Shan J, Liao J, Huang J, Robert R, Palmer ML, Fahrenkrug SC, O'Grady SM & Hanrahan JW (2012) Bicarbonate‐dependent chloride transport drives fluid secretion by the human airway epithelial cell line Calu‐3. J Physiol 590, 5273–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheynikov N, Wang Y, Park M, Ko SB, Dorwart M, Naruse S, Thomas PJ & Muallem S (2006) Coupling modes and stoichiometry of Cl−/HCO3 − exchange by slc26a3 and slc26a6. J Gen Physiol 127, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohma Y, Gray MA, Imai Y & Argent BE (2001) 150 mM HCO3 −– how does the pancreas do it? Clues from computer modelling of the duct cell. JOP 2, 198–202. [PubMed] [Google Scholar]
- Sterling D, Alvarez BV & Casey JR (2002) The extracellular component of a transport metabolon. Extracellular loop 4 of the human AE1 Cl−/HCO3 − exchanger binds carbonic anhydrase IV. J Biol Chem 277, 25239–25246. [DOI] [PubMed] [Google Scholar]
- Sterling D & Casey JR (1999) Transport activity of AE3 chloride/bicarbonate anion‐exchange proteins and their regulation by intracellular pH. Biochem J 344, 221–229. [PMC free article] [PubMed] [Google Scholar]
- Steward MC & Ishiguro H (2009) Molecular and cellular regulation of pancreatic duct cell function. Curr Opin Gastroenterol 25, 447–453. [DOI] [PubMed] [Google Scholar]
- Steward MC, Ishiguro H & Case RM (2005) Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol 67, 377–409. [DOI] [PubMed] [Google Scholar]
- Szalmay G, Varga G, Kajiyama F, Yang XS, Lang TF, Case RM & Steward MC (2001) Bicarbonate and fluid secretion evoked by cholecystokinin, bombesin and acetylcholine in isolated guinea‐pig pancreatic ducts. J Physiol 535, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutetakis A, Bossis I, Kok MR, Zhang W, Wang J, Cotrim AP, Zheng C, Chiorini JA, Nieman LK & Baum BJ (2005) Salivary glands as a potential gene transfer target for gene therapeutics of some monogenetic endocrine disorders. J Endocrinol 185, 363–372. [DOI] [PubMed] [Google Scholar]
- Yang D, Shcheynikov N, Zeng W, Ohana E, So I, Ando H, Mizutani A, Mikoshiba K & Muallem S (2009) IRBIT coordinates epithelial fluid and HCO3 − secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. J Clin Invest 119, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C & Baum BJ (2005) Evaluation of viral and mammalian promoters for use in gene delivery to salivary glands. Mol Ther 12, 528–536. [DOI] [PubMed] [Google Scholar]