

Key points
The endogenous molecular clock in skeletal muscle is necessary for maintenance of phenotype and function.
Loss of Bmal1 solely from adult skeletal muscle (iMSBmal1 −/−) results in reductions in specific tension, increased oxidative fibre type and increased muscle fibrosis with no change in feeding or activity.
Disruption of the molecular clock in adult skeletal muscle is sufficient to induce changes in skeletal muscle similar to those seen in the Bmal1 knockout mouse (Bmal1 −/−), a model of advanced ageing.
iMSBmal1−/− mice develop increased bone calcification and decreased joint collagen, which in combination with the functional changes in skeletal muscle results in altered gait.
This study uncovers a fundamental role for the skeletal muscle clock in musculoskeletal homeostasis with potential implications for ageing.
Abstract
Disruption of circadian rhythms in humans and rodents has implicated a fundamental role for circadian rhythms in ageing and the development of many chronic diseases including diabetes, cardiovascular disease, depression and cancer. The molecular clock mechanism underlies circadian rhythms and is defined by a transcription–translation feedback loop with Bmal1 encoding a core molecular clock transcription factor. Germline Bmal1 knockout (Bmal1 KO) mice have a shortened lifespan, show features of advanced ageing and exhibit significant weakness with decreased maximum specific tension at the whole muscle and single fibre levels. We tested the role of the molecular clock in adult skeletal muscle by generating mice that allow for the inducible skeletal muscle‐specific deletion of Bmal1 (iMSBmal1). Here we show that disruption of the molecular clock, specifically in adult skeletal muscle, is associated with a muscle phenotype including reductions in specific tension, increased oxidative fibre type, and increased muscle fibrosis similar to that seen in the Bmal1 KO mouse. Remarkably, the phenotype observed in the iMSBmal1−/− mice was not limited to changes in muscle. Similar to the germline Bmal1 KO mice, we observed significant bone and cartilage changes throughout the body suggesting a role for the skeletal muscle molecular clock in both the skeletal muscle niche and the systemic milieu. This emerging area of circadian rhythms and the molecular clock in skeletal muscle holds the potential to provide significant insight into intrinsic mechanisms of the maintenance of muscle quality and function as well as identifying a novel crosstalk between skeletal muscle, cartilage and bone.
Key points
The endogenous molecular clock in skeletal muscle is necessary for maintenance of phenotype and function.
Loss of Bmal1 solely from adult skeletal muscle (iMSBmal1 −/−) results in reductions in specific tension, increased oxidative fibre type and increased muscle fibrosis with no change in feeding or activity.
Disruption of the molecular clock in adult skeletal muscle is sufficient to induce changes in skeletal muscle similar to those seen in the Bmal1 knockout mouse (Bmal1 −/−), a model of advanced ageing.
iMSBmal1−/− mice develop increased bone calcification and decreased joint collagen, which in combination with the functional changes in skeletal muscle results in altered gait.
This study uncovers a fundamental role for the skeletal muscle clock in musculoskeletal homeostasis with potential implications for ageing.
Abbreviations
- EDL
extensor digitorum longus
- GTN
gastrocnemius
- H&E
haematoxylin and eosin
- KO
knockout
- MTP
metatarsal‐phalangeal
- SDH
succinate dehydrogenase
- TA
tibialis anterior
- WGA
wheat germ agglutinin
- WT
wild‐type
Introduction
Maintenance of circadian rhythms is emerging as an important new factor in human health with disruptions linked to ageing as well as to the development of many chronic diseases, including diabetes, cardiovascular disease, depression and cancer (Rajaratnam & Arendt, 2001). The molecular mechanism responsible for generating circadian rhythms is a conserved gene regulatory network composed of transcriptional–translational feedback loops referred to as the molecular clock (King & Takahashi, 2000; Schibler, 2005). Bmal1, a member of the PAS‐bHLH family of transcription factors, encodes a core component of the molecular clock. It is now clear that the molecular clock mechanism exists in all cells in the body and can function in a cell autonomous manner (Yoo et al. 2004; Schibler, 2005; Schibler & Naef, 2005; Yamazaki & Takahashi, 2005).
Studies have established that Bmal1 KO mice have a shortened lifespan, exhibit features of advanced ageing and develop cardiomyopathy and metabolic disease (Bunger et al. 2000, 2005; Kondratov et al. 2006; Antoch et al. 2008; Lefta et al. 2012). Our previous work demonstrated that skeletal muscle from Bmal1 KO mice exhibited significant weakness with decreased maximum specific tension at the whole muscle and single fibre levels at 12–14 weeks of age. We also demonstrated structural pathology with altered myofilament architecture and abnormal mitochondrial volume and function (Andrews et al. 2010).
The circadian transcriptome in skeletal muscle was first reported by Miller and colleagues in 2007 (Miller et al. 2007). Our lab followed this work and identified ∼215 mRNAs as circadian in expression in the gastrocnemius (GTN) of wild‐type (WT) C57BL/6 mice, including known core molecular clock components Bmal1, Per2 and Cry1. We also identified several skeletal muscle enriched genes as circadian in expression, including Myod1, Ucp3 and Myh1, which suggested the potential for links between the molecular clock and muscle homeostasis (McCarthy et al. 2007). Studies that employed more frequent tissue sampling (every 2 h for 48 h) have provided greater statistical power and temporal resolution of circadian mRNA expression. This work revealed that the numbers of mRNAs exhibiting a circadian pattern in skeletal muscle is greater than 800 (Pizarro et al. 2013; Hodge et al. 2015). In addition to these gene expression findings, studies have characterized the muscle clock in development, disease and ageing (Miyazaki et al. 2011; Chatterjee et al. 2013; Piovezan et al. 2015). The most recent research utilizing skeletal muscle‐specific models of molecular clock disruption have focused on metabolic effects (Dyar et al. 2014; Hodge et al. 2015).
The goal of this study was to test whether the endogenous clock in skeletal muscle is necessary for the maintenance of adult skeletal muscle phenotype and function. We generated an inducible line of mice in which Bmal1 is deleted only in adult skeletal muscle following tamoxifen treatment (iMSBmal1 −/−). Remarkably, at the time of collection, histological examination and functional measures revealed a similar phenotype to that observed in the germline Bmal1 KO mice. The phenotype observed in the iMSBmal1−/− mice was not limited to changes in muscle quality. Similar to the Bmal1 KO mice, we observed significant bone and cartilage changes throughout the body. This emerging area of circadian rhythms and the molecular clock in skeletal muscle holds the potential to provide significant new insights into mechanisms of age associated muscle weakness and the potential for novel therapeutic strategies for treatment.
Methods
Ethical approval
All animal procedures were conducted in accordance with institutional guidelines for the care and use of laboratory animals as approved by the AAALAC accredited, University of Kentucky Institutional Animal Care and Use Committees.
General experimental approaches
No data points, samples or mice were excluded from the study. Animals were randomized as stated below for vehicle or tamoxifen treatment.
Mouse model
The floxed Bmal1 mouse was purchased from Jackson laboratories and has no reported breeding, physical or behavioural abnormalities (Storch et al. 2007). The skeletal muscle‐specific Cre‐recombinase mouse (HSA‐Cre) was generated in‐house (McCarthy et al. 2012). The floxed Bmal1 mouse has loxP sites flanking exon 8 and is indistinguishable from wild‐type littermates. Breeding with the skeletal muscle‐specific Cre‐recombinase mouse generates offspring with a selective deletion of the bHLH domain of Bmal1 in skeletal muscle upon tamoxifen administration. Skeletal muscle‐specific conditional Bmal1−/ − mice were generated as follows. The Bmal1 flox/flox female was crossed with the skeletal muscle‐specific Cre‐recombinase male. This yielded an F1 generation of skeletal muscle‐specific Cre +/−; Bmal1 +/flox mice. Breeding the F1 generation males to the Bmal1 flox/flox females resulted in the skeletal muscle‐specific Cre+/ −; Bmal1 flox/flox mice needed for this study. F2 genotypes were determined by PCR using genomic DNA isolated from tail snips. Mice needed to be homozygous for the floxed Bmal1 locus; however, sufficient recombination has been shown to occur with one copy of HSA‐Cre (McCarthy et al. 2012). Activation of Cre‐recombination was done by intraperitoneal injections of tamoxifen (2 mg day−1) for five consecutive days when the mice reached between 12 and 16 weeks. This age is chosen to eliminate any effects that the lack of Bmal1 might have on skeletal muscle development and postnatal maturation. Controls were vehicle (15% ethanol in sunflower seed oil) treated Cre+/−; Bmal1 flox/flox mice. Tissue‐specific recombination data for this mouse model have been demonstrated previously (Hodge et al. 2015).
C57BL/6J (Jackson Laboratory, Bar Harbor, ME, USA) and Bmal1−/− mice (Bmal1 KO) (Jackson Laboratory), 20–22 weeks of age, were used in the current study (Andrews et al. 2010). It is important to note that the wild‐type mice used in this analysis are littermates of the Bmal1 KO mice because the Bmal1 KO mice are sterile so all breeding was done with the Bmal1+/− mice. All experimental mice were housed in temperature‐ and humidity‐controlled custom light boxes maintained on a 12 h light–12 h dark cycle with food and water ad libitum. The cardiac‐specific Bmal1 KO mouse (iCSΔBmal1) has been characterized previously (Schroder et al. 2013).
Circadian collections
Forty‐eight iMSBmal1 mice were housed in cages in light boxes and entrained to a 12 h light:dark cycle for 14 days. Mice had ad libitum access to food and water. Following the 2 week entrainment period, 24 mice, 12–16 weeks of age, were injected with vehicle and 24 mice with tamoxifen for five consecutive days, generating 24 iMSBmal1+/+ and 24 iMSBmal1−/− mice, respectively. The light schedule was kept the same during injections and for the subsequent 5 weeks. After the last normal dark cycle at 5 weeks, lights were not turned on so the mice were housed in constant darkness. Tissue collections were started after 30 h of darkness and mice were killed by cervical dislocation under dim red light. Once the mice were dead, we cut neural connections from the eyes to the retinohypothalamic tract to make sure no light would be received once lights were turned on for dissection of tissues. Gastrocnemius muscles, heart and liver were collected every 4 h for 28 h (6 time points) and flash frozen in liquid N2 for RNA and protein analysis.
RNA isolation and real‐time PCR
Total RNA was prepared from frozen tissue samples using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's directions. RNA samples were treated with TURBO DNase (Ambion, Foster City, CA, USA) to remove genomic DNA contamination. Isolated RNA was quantified by spectrophotometry (λ = 260 nm). First‐strand cDNA synthesis from total RNA was performed with a mixture of oligo(dT) primer and random hexamers using SuperScript III First‐Strand Synthesis SuperMix (Invitrogen). All isolated RNA and cDNA samples were stored at −80°C until further analysis. Real‐time quantitative PCR using TaqMan (Applied Biosystems, Carlsbad, CA, USA) assays was used to examine the gene expression of core‐clock and clock‐controlled genes. RPL26 was used as the internal calibration control. The ∆∆CT method was used for the quantification of real‐time PCR data in the circadian collections. Gene expression in each sample was shown as the relative value compared to the mean vehicle value in that tissue (GTN). Microarray sample preparation and analysis has been described previously (Hodge et al. 2015).
Immunohistochemistry
Vehicle and tamoxifen treated iMSBmal1 mice were killed by cervical dislocation and tibialis anterior (TA) muscles were collected at 58 weeks post‐treatment. Wild‐type (WT) and Bmal1 KO mice were killed and TA were collected at 22 weeks (4 WT, 5 KO) of age. Muscles were carefully dissected, pinned to a cork board at resting length, covered with a thin layer of O.C.T. Compound (Tissue‐Tek 4583, Sakura Finetek USA, Inc., Torrance, CA, USA) and snap frozen in liquid nitrogen‐cooled isopentane. The frozen TA was stored at −20°C before sectioning or stored long term at −80°C. Muscle samples were sectioned 7 μm thick and air dried. They were stained for fibre type with wheat germ agglutinin (WGA), haematoxylin and eosin (H&E) and Pax7 as previously described (McCarthy et al. 2011). Images were captured with a Zeiss upright microscope (Axio Imager.M1, Zeiss, Dublin, CA, USA). Cross sectional area and centrally nucleated fibres were determined using the H&E stained sections as previously described (McCarthy et al. 2011).
Muscle function
Immediately following cervical dislocation, the extensor digitorum longus (EDL) muscle was dissected free and removed for measures of maximum isometric tetanic force from wild‐type (n = 5), Bmal1−/− (n = 6) and iMSBmal1+/+ (n = 5), iMSBmal1−/− (n = 5) and iCSΔBmal1−/− (n = 3). Braided silk suture (4‐0) tethers were tied to the EDL at the proximal and distal tendons before the EDL was excised from the leg. The distal end was attached to a platinum wire hook inside a temperature‐controlled muscle bath (Radnoti, LLC, Monrovia, CA, USA) containing oxygenated Krebs–Ringer solution (137 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm NaH2PO4, 24 mm NaHCO3, and 2 mm CaCl2) perfused with 5% CO2–95% O2 at a pH between 7.25 and 7.35 at room temperature. The other end was attached to a lever arm (ASI 300C‐LR; Aurora Scientific, Aurora, Ontario, CA, USA). The muscle was positioned between platinum electrodes and subjected to electrical field stimulation (supramaximal voltage, 0.5 ms pulse duration) using a model 701C stimulator (Aurora). Force output was measured and recorded using ASI 610A Dynamic Muscle Control software. Optimal length (L o) was determined by adjusting the EDL position to maximize twitch force at 1 Hz and length was measured with digital calipers. Bath temperature was then increased to 37°C and the muscle was permitted to warm up for 30 min. Curare (d‐tubocurarine, 25 μm) was added after 25 min. The force–frequency relationship was determined using contractions at 2 min intervals with stimulus frequencies of 1, 15, 30, 50, 80, 120, 150, 250 and 300 Hz and a tetanic pulse duration of 500 ms. Maximal tetanic contractions were stimulated at 300 Hz between lower‐frequency contractions to monitor the change in maximal force over time, an index of stability. After each experiment, the muscle was removed from the apparatus, gently blotted dry, and weighed. Physiological cross‐sectional area was estimated according to Brooks & Faulkner (1988) and used to calculate specific force.
Myosin heavy chain gels
Mice were killed by cervical dislocation and plantaris muscles from Bmal1 KO (22 weeks old) and age matched wild‐type, or iMSBmal1−/− and iMSBmal1+/+ (58 weeks post‐treatment) were homogenized using 60 volumes of extraction buffer (50 mm Tris‐HCl, pH 8.8, 10% glycerol containing 2% SDS) (Biesiadecki et al. 2004). The diaphragm was used as control sample for visualizing different myosin heavy chain (MHC) isoforms. Samples were heated at 80°C for 5 min and spun at high speed for 5 min to remove debris. Protein concentration was measured, and samples were diluted with dilution buffer (0.38 m Tris‐HCl, pH 6.8, 2% SDS, 25 mm DTT, 0.02% bromophenol, 15% glycerol) to 0.1 μg μl− 1. A quantity of 0.75 μg protein per sample was resolved by SDS‐PAGE gel prepared as described previously (Talmadge & Roy, 1993). The gels were run at 4°C with an initial voltage of 70 V. After 2 h, voltage was increased to 100 V. Total time for running the gel was 32 h. MHC isoforms were developed using a silver stain kit (Bio‐Rad Life Science Technologies, Hercules, CA, USA).
Gait analysis
Mice were anaesthetized in an induction box using 5% isoflurane, 1 l min−1 oxygen), then maintained at 1.5% isoflurane and 500 l min−1 oxygen via a nose cone. Hair on the hindlimbs was removed using Nair (Church & Dwight Co., Inc.). This was performed at least 24 h prior to gait analysis. Mice were placed in the first lane of a motorized walkway (EXER 3/6 treadmill, Columbus Instruments, Columbus, OH, USA) and sagittal plane movements were recorded for 2 min at 250 frames s−1 (HotSHot Camera, NAC Image Technology, Simi Valley, CA, USA) to ensure accurate delineation of stance and swing phases during each gait cycle. Following the recording session, mice were returned to their cages (see Videos S1–S4 in the online Supporting information). These mice were killed approximately 10 weeks later by cervical dislocation and were included in the cohort of mice used for the bone staining protocol.
Digital image data were manually selected and extracted from the video data for 7–10 consecutive gait cycles at the moment the paw made contact and lost contact with the treadmill to calculate stance and swing phase durations during gait. Additional images were extracted during the swing and stance phases to measure maximal active dorsiflexion of the ankle and maximal passive extension of the metatarsal‐phalangeal (MTP) joints of the forefoot, respectively. Digital images were transferred to a digital microscope and imaging system (VHX‐600; Keyence Corporation, Itasca, IL, USA) and line‐segments were created for the femur, tibiofibula, foot and phalanx segments.
Bone staining
Hindlimbs for Alizarin Red/Alcian Blue staining were collected from 58 weeks post‐treatment iMSBmal1+/+ and iMSBmal1−/− mice, and muscle and connective tissue were removed. Bones were fixed in 95% ethanol for 1–2 days. They were then rocked at 37°C in Alcian Blue/Alizarin Red stain for 3–4 days. Following staining they were rinsed one time in ddH2O and then transferred to 1% KOH. They were rocked at room temperature for a minimum of 3 h. The 1% KOH was replaced and rocking continued overnight at room temperature. Depending on the intensity of the stain and the amount of tissue remaining on the bone additional KOH washes were performed as needed. They were then rocked for 24 h each in the following glycerol/KOH washes: 20% glycerol/1% KOH (24 h at room temperature on rocker); 50% glycerol/1% KOH (24 h at room temperature on rocker); 80% glycerol/1% KOH (24 h at room temperature on rocker). Bones were stored and photographed in 80% glycerol/1% KOH solution at room temperature.
Statistical analysis
Results are reported as means ± SEM. For comparison of one parameter between two strains of mice, statistical analysis was performed using 2‐tailed, unpaired Student's t test. A two‐way ANOVA was used to determine a significant interaction between factors (strain and time). If a significant interaction was detected, a Bonferroni post hoc comparison was performed. P < 0.05 was considered significant. Statistical analysis was performed with Graphpad Prism software (GraphPad Software, Inc.). Circadian analysis was performed using JTK_cycle (Hughes et al. 2010).
Results
Characterization of the mouse model
Our early studies of muscle in the Bmal1 KO mouse provided evidence to support links between disruption of circadian rhythms, the molecular clock and muscle function. However, these mice are disrupted in all cell types so they do not allow us to discern the relative contribution of the skeletal muscle clock to muscle function and weakness. To address the function of the molecular clock in skeletal muscle we generated an inducible muscle‐specific Bmal1 knockout mouse for the targeted disruption of the molecular clock mechanism only in adult skeletal muscle (Hodge et al. 2015). We carried out a circadian time course collection with vehicle and tamoxifen‐treated mice. GTN from iMSBmal1+/+ and iMSBmal1−/− mice were collected every 4 h over a 28 h period, RNA was extracted, and microarray assessment and real‐time PCR were performed to evaluate the function of the molecular clock. The time course collection allowed us to identify the mRNAs exhibiting a circadian pattern and extract quantitative data about the amplitude (difference between peak and mean value for the waveform) of the oscillation using JTK_cycle (Hughes et al. 2010). As depicted in the heat map in Fig. 1, we identified 629 non‐redundant mRNAs that exhibit a circadian pattern of expression in the GTN of vehicle treated mice. In contrast, loss of Bmal1 from skeletal muscle resulted in a significant disruption in almost every gene (Fig. 1) (Hodge et al. 2015). We performed quantitative real time PCR (qPCR) on core molecular clock genes including Per2, Cry1, Reverb, Rora, Clock and genes known to be regulated downstream of the molecular clock (i.e. clock controlled genes) including Dbp and MyoD1 to quantify the efficacy of molecular clock disruption. As expected we observed a robust, high amplitude oscillation for Bmal1 expression in the GTN of iMSBmal1 +/+ mice, which was lost in the iMSBmal1−/− mice. The low level expression and damped amplitude of Bmal1 mRNA in the iMSBmal1−/− mice is consistent with loss of expression in the muscle but continued expression in other non‐muscle cell types in the tissue. Consistent with disruption of Bmal1, we found that the amplitudes for most of the core molecular clock genes and clock controlled genes were significantly reduced (∼70–90%) in the iMSBmal1−/− GTN. Mean amplitudes with calculated high and low amplitude values as error bars (JTK_cycle), as well as the average fold change in expression over the entire time course for these genes, are graphed in Fig. 2 A, demonstrating significant reductions in amplitude and overall expression for the core molecular clock genes, Bmal1, Rora and Reverbα. Expression levels of Dbp, a well‐characterized clock controlled gene (CCG), and Myod1, a muscle‐specific CCG (Andrews et al. 2010; Zhang et al. 2012), were significantly decreased in the GTN of the iMSBmal1−/− mice. Clock, a component of the core molecular clock mechanism, was upregulated in response to loss of Bmal1, similar to what had previously been shown in the skeletal muscle (Dyar et al. 2014). Further analysis of the array data revealed that several transcription factors well known to regulate muscle‐specific and fibre‐type gene expression were disrupted. These transcription factors included Pgc1α, Sox6, Mef2a and Six1 (Fig. 2 B).
Figure 1. Core molecular clock gene expression is disrupted with loss of Bmal1 in skeletal muscle .
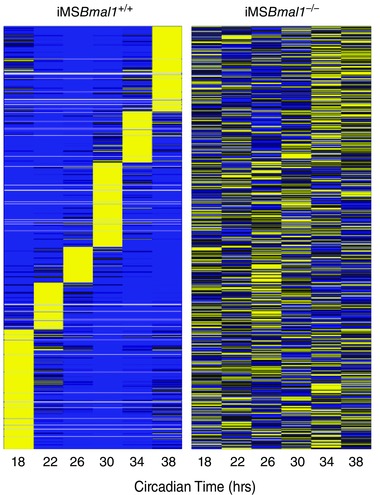
A heat map from median normalized microarray data of mRNAs expressed in a circadian manner from pooled samples (n = 4) of the iMSBmal1 +/+ and iMSBmal1 −/− GTN collected from the circadian time course is shown. Bright yellow represents mRNA expression that is increased and blue represents mRNA expression that is decreased compared to median levels. Note the significant disruption of circadian expression patterns in the GTN of the iMSBmal1 −/− mice.
Figure 2. Real time PCR confirms disruption in the expression of several core molecular clock and clock controlled genes .
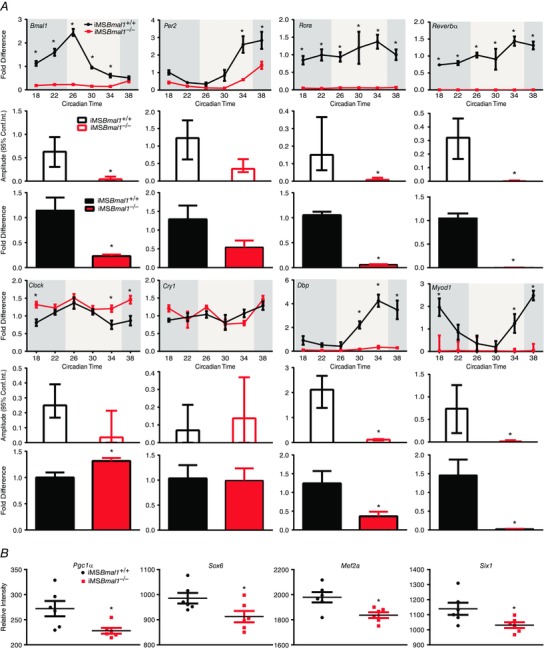
A, The circadian expression profiles of Bmal1, Per2, Rora, Reverbα, Clock, Cry1, Dbp, and Myod1 mRNA expression every 4h over 24h. The histogram below each timecourse graph provides summary data of the amplitude results obtained with JTK_cycle for each mRNA. The histograms for average fold change provide summary data over the 24h for level of expression as mean ± SEM with significant differences (P < 0.05) designated by an asterisk (∗). n = 4/time point; ∗ P < 0.05. B, The relative intensity from microarray expression data generated from pooled samples (n = 4) at each time point (6 time points) of the iMSBmal1 +/+ and iMSBmal −/− GTN collected from the circadian time course. Array data for Pgc1α, Sox6, Mef2a, and Six1 in the GTN of iMSBmal1 +/+ and iMSBmal1 −/− mice are shown; ∗ P < 0.05.
Muscle function in the iMSBmal1 and Bmal1 KO mice
The in vivo decline in human muscle strength with age and other chronic diseases can be regulated at many levels including changes in size, motor unit recruitment, innervation and excitation–contraction coupling (Brooks & Faulkner, 1994; D'Antona et al. 2003; Delbono, 2003; Morse et al. 2005; Mitchell et al. 2012). Our previous work has demonstrated that muscle force per unit cross section (specific force) measured in vitro is significantly reduced in the EDL muscles/muscle fibres of the 12‐ to 14‐week‐old Bmal1 KO mouse compared to wild‐type controls (Andrews et al. 2010). In this study we used grip strength measures as a way to monitor changes in neuromuscular function in our mice. We assessed muscle function in vivo using grip strength analysis at 55–60 weeks post‐treatment and measured a reduction in forelimb grip force in the iMSBmal1 −/− compared to iMSBmal1 +/+ mice (Fig. 3 A). Ex vivo measures of specific force in the EDL muscle demonstrated that loss of grip strength was associated with significant muscle weakness. In fact, we observed reductions in specific force in EDL muscle at both 20 and 58 weeks post‐treatment (Fig. 3 B and C). We also found that Bmal1 KO mice have significantly reduced forelimb grip strength compared to their littermate controls at 25–40 weeks of age (Fig. 3 D). Consistent with our previous studies of muscle function in young Bmal1 KO mice we found that specific force continued to be reduced in the EDL muscle Bmal1 KO mice at 20–22 weeks of age. However, unlike what we reported for muscle function in the 12‐ to 14‐week‐old Bmal1 KO mice, we observed a significant leftward shift of the force frequency curve suggesting a shift toward slower fibre types in the Bmal1 KO mice in consonance with an ageing muscle phenotype (Tikunov et al. 2001; Chan & Head, 2010) (Fig. 3 E). In contrast to the iMSBmal1 −/−, skeletal muscle from aged mice in which Bmal1 is knocked out only in cardiomyocytes (iCS∆Bmal1 −/−) (Schroder et al. 2013) revealed no changes in EDL specific force (Fig. 3 F). The results from the iCS∆Bmal1 −/− mouse demonstrate that the functional effects we report are not due to the 5 day tamoxifen treatment and are specific to loss of Bmal1 in skeletal muscle and not another striated muscle tissue.
Figure 3. Maintenance of muscle function is disrupted in the iMSBmal1−/− and Bmal1 KO mice .
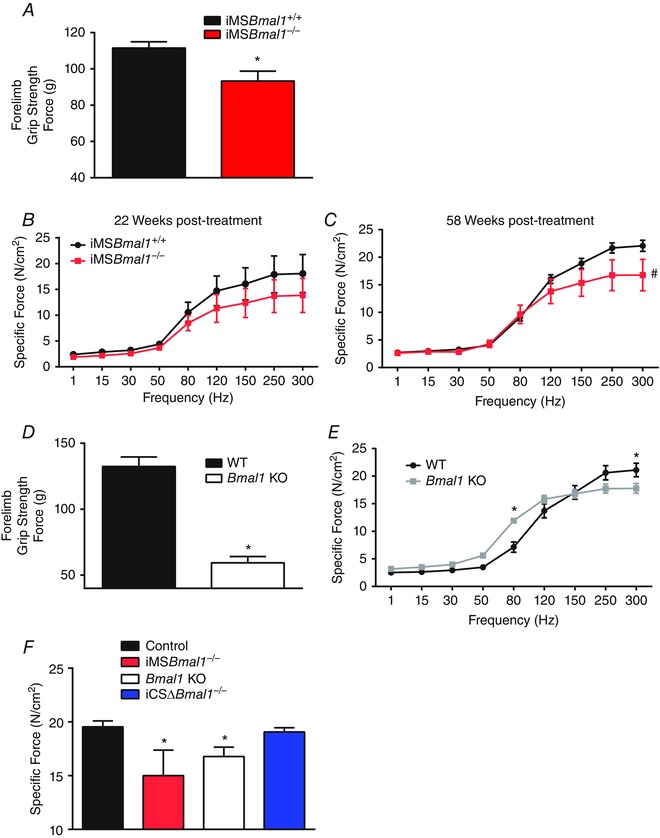
A, grip strength measured in the iMSBmal1+/+ (n = 8) and iMSBmal1−/− (n = 10) at 55–60 weeks post‐treatment was reduced in the iMSBmal1−/−. B, specific tension measured in the EDL of the iMSBmal1+/+ and iMSBmal1−/− mice at 20 weeks post‐treatment was also decreased in the iMSBmal1−/− mice (n = 3). C, specific tension measured in the EDL of the iMSBmal1+/+ and iMSBmal1−/− mice at 58 weeks post‐treatment was also decreased in the iMSBmal1−/− mice (n = 5). Three additional iMSBmal1−/− muscles ripped mid‐belly during force measures indicating changes in biological material properties in these muscles. D, grip strength measured in the WT (n = 9) and Bmal1 KO (n = 5) littermates at 25–40 weeks of age was reduced in the Bmal1 KO mice. E, specific tension measured in the EDL of WT (n = 5) and Bmal1 KO (n = 6) mice at 20–22 weeks of age was also decreased in the Bmal1 KO mice. F, peak specific force at 300 Hz measured in the EDL was significantly reduced in both the iMSBmal1−/− (58 weeks post‐treatment; n = 5) and Bmal1 KO (20–22 weeks post‐treatment; n = 6) mice but was unaffected in the cardiac‐specific Bmal1 KO (iCSΔBmal1−/−; 65 weeks post‐treatment; n = 3). Summary values are presented as means ± SEM with significant differences (P < 0.05) designated by an asterisk (*).
A well‐known feature of ageing in skeletal muscle is a shift in fibre type characterized by a decrease in the type IIB fibres with increases in the more oxidative type IIA fibres (Ciciliot et al. 2013; Miller et al. 2014; Purves‐Smith et al. 2014; Miljkovic et al. 2015). Figure 4 A shows representative images of tibialis anterior (TA) muscle immunoreacted with myosin heavy chain isoform‐specific antibodies revealing a lower percentage of type IIB fibres and a trend toward a higher percentage of type IIA. Further assessment of the oxidative potential of muscle fibres utilized immunohistochemistry to detect succinate dehydrogenase (SDH) and demonstrated a trend toward an increase in SDH‐positive fibres (Fig. 4 B). Myosin heavy chain gels of plantaris muscle quantitatively confirm increased expression of type IIA myosin heavy chain (Fig. 4 C). This shift toward a more oxidative fibre type is consistent with that observed in the Bmal1 KO. Significant decreases in the number of type IIX fibres with concomitant increases in type IIA fibres were observed in the TA from the Bmal1 KO muscle (Fig. 4 D). Figure 4 E shows representative images of SDH staining in the TA muscle from wild‐type and Bmal1 KO mice at 22 weeks of age. The percentage of SDH‐positive fibres was significantly increased in the Bmal1 KO muscle. As with the iMSBmal1−/− mice, myosin heavy chain gels confirmed the fibre type shift (Fig. 4 F). These changes in fibre type occurred without a change in cage activity (Hodge et al. 2015) and with no change in fibre cross sectional area (1265 ± 23.08 mm2 iMSBmal1 +/+ (n = 6); 1224 ± 62.58 mm2 iMSBmal1−/− (n = 5)) or the number of centrally nucleated fibres in the TA of iMSBmal1−/− mice as shown in representative H&E images (Fig. 5 A). However, in concordance with the previously demonstrated advanced ageing phenotype, H&E stained tibialis anterior (TA) muscle from wild‐type and Bmal1 KO mice revealed an increased number of centrally nucleated fibres and a decreased average fibre area in Bmal1 KO compared to wild‐type muscle at 22 weeks of age. The summary data for the image analysis (n = 5) are provided in Fig. 5 B.
Figure 4. A shift toward an oxidative fibre type is observed in the skeletal muscles of both the iMSBmal1−/− and Bmal1 KO mice .
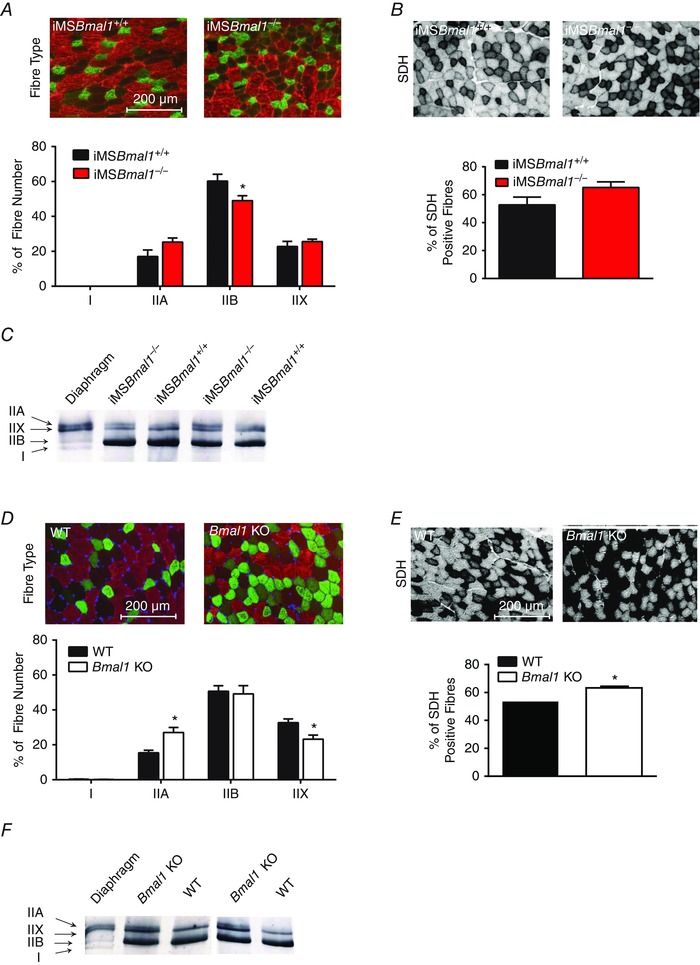
A, representative images of fibre type staining (58 weeks post‐treatment) using fibre type‐specific myosin heavy chain antibodies to distinguish type I (pink), type IIA (green), type IIB (red) and type IIX (black) fibres in the TA muscle of the iMSBmal1+/+ and iMSBmal1−/− mice is shown. A shift toward a more oxidative fibre type is seen in the summary of the fibre type data at 58 weeks post‐treatment in the iMSBmal1−/− (n = 9) when compared to iMSBmal1+/+ (n = 4) mice. B, representative images of SDH staining (58 weeks post‐treatment) in the TA of the iMSBmal1+/+ and iMSBmal1−/− mice is shown. A trend toward more oxidative fibres in the iMSBmal1−/− mice with SDH staining is seen in the summary data (iMSBmal1+/+, 4; iMSBmal1−/−, 9). C, myosin heavy chain gels revealed increased type IIA myosin in the plantaris of (58 weeks post‐treatment) iMSBmal1−/− mice (n = 3). D, representative image of fibre type staining using fibre type‐specific myosin heavy chain antibodies to distinguish type I (pink), type IIA (green), type IIB (red), and type IIX (black) fibres (22 weeks) and summary data in the TA muscle of the WT and Bmal1 KO mice are shown. A shift toward a more oxidative fibre type is observed in the Bmal1 KO muscle (n = 4). E, representative images of SDH staining in the TA of the WT and Bmal1 KO mice is shown. An increase in SDH staining is seen in the summary data for WT and Bmal1 KO mice (n = 4). F, myosin heavy chain gels revealed increased type IIA myosin in the plantaris of the Bmal1 KO mice (22 weeks) (n = 3). Summary values are presented as means ± SEM with significant differences (P < 0.05) designated by an asterisk (*).
Figure 5. The iMSBmal1−/− and Bmal1 KO mice exhibit differential effects on fibre area and centrally nucleated fibres .
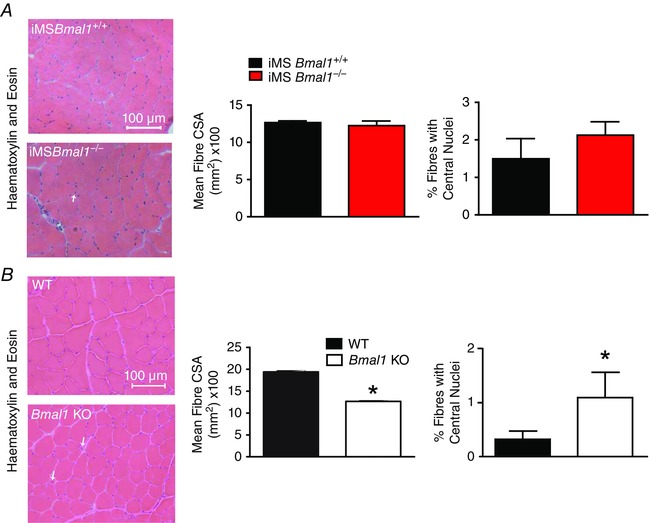
A, representative images of H&E stained TA muscle from iMSBmal1+/+ (n = 4) and iMSBmal1−/− (n = 5) mice at 58 weeks post‐treatment is shown. No differences in fibre size or the number of centrally nucleated fibres were observed as can be seen in the summary data. B, representative images of H&E stained TA muscle from WT and Bmal1 KO (n = 4) at 20–22 weeks of age. Fibre size was decreased and the number of centrally nucleated fibres was increased as can be seen in the summary data. Summary values are presented as means ± SEM with significant differences (P < 0.05) designated by an asterisk (*).
Fibrosis and skeletal phenotype in iMSBmal1 −/− and Bmal1 KO mice
One common feature in models of muscle weakness, including dystrophies, myopathies and ageing, is that increased fibrosis is often observed in muscle concomitant with the reduction in specific force as extracellular matrix components accumulate. Consistent with these observations we found an increased interstitial staining with WGA in the iMSBmal1−/− mice (Fig. 6 A). We detected more fibrosis in the muscle of iMSBmal1−/− mice when compared to the muscle of Bmal1−/− mouse (Fig. 6 B). We next asked whether the weakness and increased fibrosis seen in the iMSBmal1 −/− muscle was associated with diminished satellite cell number. In contrast to what is seen with ageing, Fig. 6 C shows no significant changes in Pax7‐positive nuclei per muscle fibre in the TA of the iMSBmal1−/− mice. This is in contrast to the reduced Pax7 staining in the Bmal1 knockout TA that shows a significant reduction in satellite cell number (Fig. 6 D).
Figure 6. The iMSBmal1−/− and Bmal1 KO mice exhibit differential effects on satellite cell number .
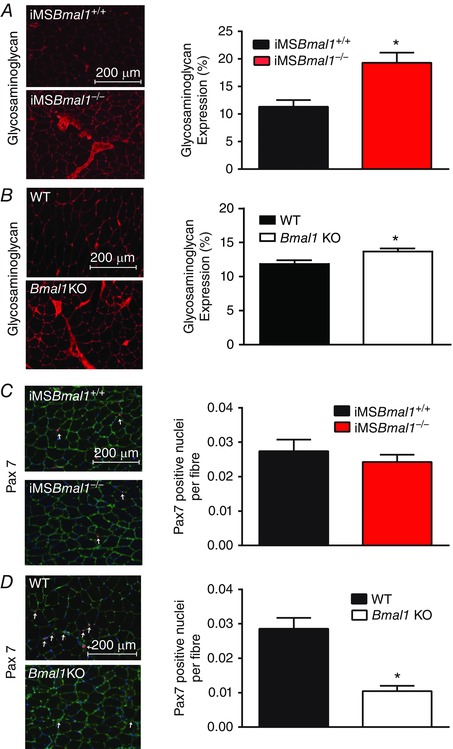
A, fibrosis was increased in the iMSBmal1−/− (n = 5) compared to iMSBmal1+/+ (n = 4) muscle as seen by increased interstitial staining using WGA. B, fibrosis was increased in the Bmal1 KO (n = 5) TA muscle compared to WT (n = 4) as seen by increased interstitial staining using WGA. C, representative Pax7 staining (red) of the TA muscle from 58 week post‐treatment iMSBmal1+/+ and iMSBmal1−/− mice is shown. Sections were counterstained with DAPI (blue) and laminin (green), and Pax7+/DAPI+ nuclei were quantified (white arrows). Pax7+/DAPI+ nuclei per muscle fibre were not different in the iMSBmal1−/− mice (n = 6) compared to iMSBmal1+/+ (n = 3). D, representative PAX7 staining (red) of the tibialis anterior muscle from 22‐week‐old WT and Bmal1 KO mice is shown. Sections were counterstained with DAPI (blue) and laminin (green), and PAX7+/DAPI+ nuclei were quantified (white arrows). PAX7+/DAPI+ nuclei per muscle fibre were reduced in the Bmal1 KO mice (n = 8) compared to WT (n = 7). Summary values are presented as mean ± SEM with significant differences (P < 0.05) between iMSBmal1+/+ and iMSBmal1−/− mice designated by an asterisk (*).
To evaluate potential factors that could contribute to the observed increases in fibrosis, we examined our microarray data from the GTN of the iMSBmal1+/+ and iMSBmal−/− mice for changes in extracellular matrix components. The microarray data are measured at 5 weeks post‐treatment and at that time we observe significant increases in the mRNA expression for collagens 4 (Col4) and 6 (Col6), and a trend toward an increase for collagen 1 (Col1) (P = 0.057). Extracellular matrix components, including the proteoglycan perlecan (Hspg2), as well as agrin (Agrn) and dystroglycan (Dag1), were also significantly increased at this time point suggesting that signalling involved in the development of fibrosis begins shortly after loss of Bmal1 in the skeletal muscle (Fig. 7 A). In addition, we used the microarray data to examine markers of fibroblasts in skeletal muscle. This analysis revealed increases in the mRNA expression of several fibroblast specific markers including Tcf‐4, scleraxis (Scx), Fgf1 and the Fgf1 receptor (Fgf1r) (Fig. 7 B). Lastly we found changes in the mRNA expression of components of the Wnt signalling pathway (e.g. Frzb, Lrp1), as well as components of the transforming growth factor β (Tgfβ) superfamily (e.g. Tgfβ1, Mstn) in the microarray data from the iMSBmal1−/− mice. Future studies are needed to determine the mechanism(s) through which loss of Bmal1 only in muscle fibres contributes to significant changes impacting extracellular matrix (ECM) formation and remodelling (Fig. 7 C).
Figure 7. mRNA expression of genes involved in fibrosis and Wnt and Tgfβ signalling are differentially regulated in the GTN of the iMSBmal1−/− mice 5 weeks post‐treatment .
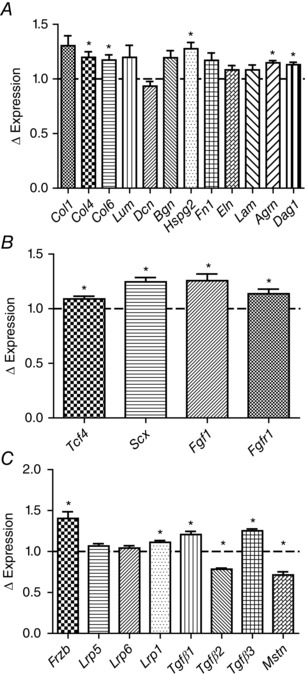
A, microarray data from the GTN of the iMSBmal1−/− mice revealed differential expression of extracellular matrix genes. B, microarray data from the GTN of the iMSBmal1−/− mice revealed differential expression of fibroblast associated genes. C, microarray data from the GTN of the iMSBmal1−/− mice revealed differential expression of the genes for secreted proteins which have been demonstrated to impact the skeletal system.
One of the more striking phenotypes was the overall outward appearance of the iMSBmal1−/− mice. As can be seen in Fig. 8 A, iMSBmal1−/− displayed loss of hair, increased prevalence of dermatitis, kyphosis, as well as tail and ear morphological abnormalities. Because of the close proximity of skeletal muscle with bone and the known arthropathy reported in the Bmal1−/− mice, we asked whether there was any bone phenotype in the iMSBmal1−/− mice. Figure 8 B provides a representative image of the lower hindlimb and foot from age‐matched iMSBmal1+/+ and iMSBmal1−/− mice stained with Alizarin Red for bone and Alcian Blue for cartilage. To our surprise, muscle weakness in this mouse model was associated with increased Alizarin Red staining in the Achilles’ tendon (see arrow in Fig. 8 B), reduced cartilage staining in the foot/ankle and the appearance of flattened tarsals and misshapen tibia and fibula. We also found that changes in the skeletal system were not limited to the limbs as we observed increased calcification and diminished cartilage staining throughout the rib cage and thoracic spine (Fig. 8 C). The arthropathy observed in the iMSBmal1−/− is similar to that observed in the germline Bmal1 KO mice indicating that loss of Bmal1 only in skeletal muscle is sufficient to induce this severe skeletal system pathology (Fig. 8 D and E).
Figure 8. Disruption of the molecular clock specifically in adult skeletal muscle is sufficient for disruption of systemic skeletal homeostasis and gait, similar to that observed in the Bmal1 KO .
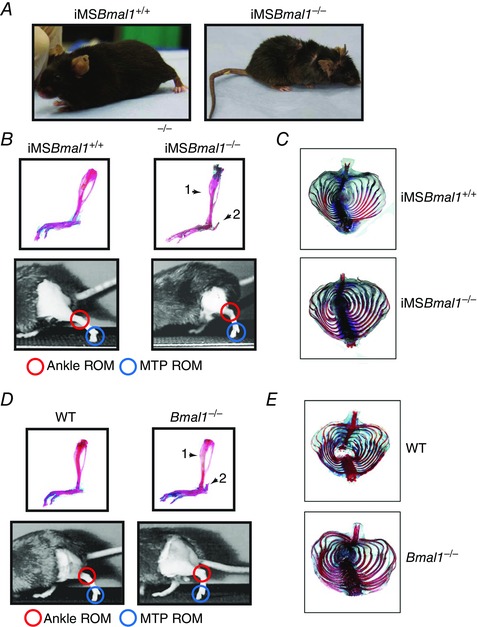
A, representative images of iMSBmal1 +/+ and iMSBmal1 −/− (58 weeks post‐treatment). Notice the smaller body mass as well as effects on coat condition and tail/ear cartilage. B, arthropathy similar to that observed in the germline Bmal1 KO is present in the iMSBmal1 −/− mice. Representative images of iMSBmal1 +/+ and iMSBmal1 −/− (58 weeks post‐treatment) lower hindlimb and foot stained with Alcian Blue (cartilage) and Alizarin Red which stains calcium (bone). Arrows mark differences in bone showing a thickening of the distal tibia (arrow 1), and calcification of the calcaneal tendon (arrow 2). Representative frames taken from high speed video of iMSBmal1 +/+ and iMSBmal1 −/− (58 weeks post‐treatment) at toe‐off during treadmill walking showing the marked reduction of range of motion in the ankle and MTP joints in the skeletal muscle‐specific knockout mice. The gait of the knockout mice was noticeably disrupted with more of a hopping behaviour at a speed at which the iMSBmal1 +/+ mice walked comfortably. In addition, gait analysis revealed decreased range of motion (ROM) in the iMSBmal1 −/− mice. C, representative images of iMSBmal1 +/+ and iMSBmal1 −/− (58 weeks post‐treatment) ribcage stained with Alcian Blue and Alizarin Red demonstrated increased calcification of the ribcage similar to that observed in the Bmal1 KO mouse. D, representative images of WT and Bmal1 KO (20–22 weeks) lower hindlimb and foot stained with Alcian Blue and Alizarin Red. Arrows mark differences in bone showing a thickening of the distal tibia (arrow 1), and calcification of the calcaneal tendon (arrow 2). Representative frames taken from high speed video of WT and Bmal1 KO (20–22 weeks) at toe‐off during treadmill walking showed the marked reduction of range of motion in the ankle and MTP joints in the global knockout mouse. E, representative images of WT and Bmal1 KO (20–22 weeks) ribcage stained with Alcian Blue and Alizarin Red demonstrated increased calcification of the ribcage.
Changes in muscle function and muscle and bone phenotype suggested potential impacts on gait. We performed gait analysis using high speed videography with mice walking on a flat treadmill. In our studies mice were forced to walk at a given pace on a treadmill but the pace selected was not faster than the slowest mouse could maintain during the recording. Biomechanical analysis of the iMSBmal1−/− mice revealed significant reductions in both ankle and MTP range of motion (Table 1) at toe‐off, common in joint pathology (see Videos S1 and S2). They also had significant reductions in swing time and stride length. Similar disruptions in gait were present in the Bmal1 KO mice (Table 2, Videos S3 and S4).
Table 1.
Gait analysis revealed decreased range of motion in the iMSBmal1−/− mice
| iMSBmal1 +/+ | iMSBmal1 −/− | |
|---|---|---|
| Ankle ROM (deg) | 76.23 ± 4.27 | 25.88 ± 7.56* |
| MTP ROM (deg) | 75.68 ± 12.28 | 33.85 ± 13.17* |
| Stride length (mm) | 19.63 ± 1.46 | 11.08 ± 2.18* |
| Stance time (ms) | 282.20 ± 42.11 | 236.10 ± 82.9 |
| Swing time (ms) | 141.32 ± 12.34 | 89.28 ± 14.09* |
| Stance/swing | 2.05 ± 0.45 | 2.68 ± 1.03 |
ROM, range of motion; MTP, metatarsal‐phalangeal. *P < 0.05 compared to strain matched control.
Table 2.
Gait analysis revealed decreased range of motion in the Bmal1 KO mice
| WT | Bmal1 KO | |
|---|---|---|
| Ankle ROM (deg) | 61.20 ± 13.06 | 36.41 ± 1.03* |
| MTP ROM (deg) | 75.46 ± 12.43 | 37.92 ± 6.38* |
| Stride length (mm) | 15.56 ± 1.86 | 14.83 ± 1.35 |
| Stance time (ms) | 220.11 ± 15.32 | 234.09 ± 4.76 |
| Swing time (ms) | 117.44 ± 10.10 | 109.81 ± 5.76 |
| Stance/swing | 1.89 ± 0.17 | 2.17 ± 0.20 |
ROM, range of motion; MTP, metatarsal‐phalangeal. *P < 0.05 compared to strain matched control.
Discussion
The recognition that the molecular clock mechanism exists in virtually all cells, including skeletal muscle, combined with the knowledge that peripheral clocks can be modulated independently of the central clock has raised significant questions about the role of the molecular clock in peripheral tissues. Genetic mouse models have provided a means to disrupt the molecular clock in a tissue‐/cell‐specific manner in order to mechanistically address the contribution of the clock to tissue/cell function with the potential to examine larger effects on systems health (McDearmon et al. 2006; Hughes et al. 2012; Paschos et al. 2012; Su et al. 2012; Schroder et al. 2013). Novel data presented in this paper provide evidence demonstrating the importance of the intrinsic skeletal muscle molecular clock in regulating the maintenance of muscle function and phenotype. Our findings are also exciting as they uncover a fundamental role for skeletal muscle molecular clock in skeletal homeostasis. We show that loss of Bmal1 in adult skeletal muscle leads to a decrease in specific tension that is evident by 20 weeks along with a shift toward a more oxidative fibre type and increases in muscle fibrosis. These properties of skeletal muscle are changing in the absence of changes in muscle mass and were not associated with behavioural changes in feeding or cage activity (Hodge et al. 2015). Surprisingly, the phenotype observed in the iMSBmal1−/− mice was not limited to changes in skeletal muscle. Similar to the germline Bmal1 KO mice, we observed significant bone and tendon/cartilage changes throughout the body. Thus, our findings implicate the endogenous skeletal muscle molecular clock as a modulator of musculoskeletal health.
The observed changes in muscle phenotype and function occurred in the absence of changes in physical activity or feeding (Hodge et al. 2015). Thus, they were directly downstream of disruption of the molecular clock machinery in adult muscle. One potential mechanism for changes in fibre type and phenotype could be through altered expression of transcription factors linked to muscle gene expression. It is well established that molecular clock factors bind to many sites across the genome (Atwood et al. 2011; Koike et al. 2012; Korenčič et al. 2014; Yoshitane et al. 2014). In addition, circadian transcriptome studies have suggested that the clock factors target the expression of many transcription factors (McCarthy et al. 2007; Andrews et al. 2010; Khapre et al. 2010; Mazzoccoli et al. 2012; Zhang et al. 2012). Analysis of our microarray time series (Hodge et al. 2015), as well as real time PCR studies, demonstrated that loss of Bmal1, in addition to disruption of the core clock mechanism, resulted in significant decreases in expression of a number of transcription factors important in regulating skeletal muscle phenotype and function, including MyoD1, Sox6, Mef2a, Six1, as well as the transcriptional co‐activator Pgc1α (Yablonka‐Reuveni & Rivera, 1997; Dunn et al. 2000; Andrews et al. 2005; Quiat et al. 2011; Richard et al. 2011; Zhang et al. 2012; Khodabukus & Baar, 2015). We propose that daily expression of Bmal1 targets these transcription factors for maintenance of a myogenic programme of gene expression. It is important to note that not all transcription factors exhibit a circadian pattern in skeletal muscle. For example both Pgc1α and Mef2a are not expressed in a circadian manner in skeletal muscle but are significantly down‐regulated following loss of Bmal1 (Pizarro et al. 2013; Hodge et al. 2015). Future studies examining the mechanism of clock control for these muscle transcription factors will provide exciting new insight into mechanisms through which muscle enriched and fibre type‐specific protein expression is maintained in vivo. In this study we observed decreased muscle function at both 22 and 58 weeks post‐treatment and a shift toward a more oxidative fibre type at 58 weeks post‐treatment in the iMSBmal1−/− mice. Our results are in contrast to recent work using a similar inducible muscle‐specific Bmal1 KO model which did not observe significant changes in fibre type composition or loss of force production (Dyar et al. 2014). We have analysed several different generations of mice and we have performed a thorough analysis of recombination specificity with important downstream molecular clock analysis providing confidence in our findings (Hodge et al. 2015). Thus, we believe that the contrasting outcomes between our studies are probably due to two differences in experimental design. Specifically, we treated our mice with tamoxifen for Bmal1 recombination between the ages of 12 and 16 weeks of age. We chose this age because it is beyond the age of rapid skeletal muscle growth during postnatal maturation (Quinlan et al. 1997; Harrison, 2011). This maturational growth is associated with satellite cell fusion so would effectively dilute the Bmal1 − muscle fibres through addition of Bmal1+ nuclei after the tamoxifen treatment. Recombination in the Dyar paper was induced at 8 weeks. The second difference between our studies is that we completed the primary functional and histological studies at 58 weeks post‐treatment compared to the 5 months/20 weeks reported in the Dyar studies.
Satellite cells, the primary resident stem cell in skeletal muscle, are required for regeneration and repair and numbers diminish with age (Montarras et al. 2013; Rai et al. 2014). Quiescent satellite cells express the transcription factor Pax7 which is subsequently down‐regulated upon satellite cell activation (Chang & Rudnicki, 2014). In accordance with the demonstrated advanced ageing phenotype of the Bmal1 KO mice we saw a reduction in the number of satellite cells per muscle fibre in the TA; however, no significant differences were observed in the iMSBmal1−/− mice. Disparities between the iMSBmal1 −/− and Bmal1 KO mice suggest that the presence of Bmal1 in the satellite cell, or timing of Bmal1 expression in the satellite cell during development, or both may regulate satellite cell number. More importantly, in the iMSBmal1 −/− mice, the observed muscle weakness and increased fibrosis are not due to any changes in satellite cell number and therefore indicate that this phenotype originates from the loss of Bmal1 within the muscle fibre. This observation is consistent with the recent findings by Fry et al. (2015) that satellite cells did not contribute to the maintenance of muscle size or fibre type composition during ageing.
Muscle and bone are intimately linked through growth and development, mechanical load, endocrine function and genetic pleiotropy (Curtis et al. 2015). Our findings presented are the first to demonstrate a fundamental and direct muscle to bone interaction that occurs not due to development but in an adult animal. The systemic increases in bone/tendon calcification in the presence of a functionally weak skeletal muscle appears to be a finding unique to mice in which circadian rhythm/molecular clock is disrupted (Bunger et al. 2005; Maronde et al. 2010). However, those studies use mice in which molecular clock components are disrupted in all cell types. Our study clearly defines that loss of Bmal1/molecular clock function only in skeletal muscle is sufficient to induce skeletal remodelling in the presence of normal molecular clock components across non‐skeletal muscle tissue. The other unusual observation is that most models of muscle weakness are associated with decreased bone calcification (Morissette et al. 2009; Arounleut et al. 2013; Isaac et al. 2013). This suggests that the observed changes in bone (increased calcification) in our mouse model are not due to altered mechanics but are more likely the result of changes in circulating factors from the iMSBmal1−/− muscle influencing gene expression/function in bone and thus effecting remodelling.
The role of muscle as an endocrine organ is becoming more widely recognized in the field (Iizuka et al. 2014; Catoire & Kersten, 2015). Our findings suggest that loss of Bmal1 in skeletal muscle leads to significant changes in the endocrine/paracrine function of muscle with implications for cartilage, tendon and bone. The gene expression results implicate the Wnt signalling pathway, which has been established to lead to a disruption of both bone and skeletal muscle (Tzahor et al. 2003; Wagers, 2008; Scimè et al. 2010; Tompkins, 2011; von Maltzahn et al. 2012; Zhang et al. 2014). We also observed changes in the expression of several secreted proteins in the Tgfβ family, which may act in a paracrine fashion in the musculoskeletal niche and are known to impact bone. These include Tgfβ1, Tgfβ2 and Tgfβ3 (Fig. 7 C) (Kawao & Kaji, 2015). Important in bone remodelling, they are responsible for complex interactions involved in coupling bone formation and resorption. In addition, expression of myostatin (Mstn), a well‐known muscle‐specific Tgfβ family member, is significantly decreased in the GTN of the iMSBmal1−/− mice. Mice with a global knockout of Mstn show increased bone density (Elkasrawy & Hamrick, 2010). Thus, future studies using tissue‐specific targeting of both Wnt and Tgfβ signalling pathways will be critical for understanding their role in the muscle to bone remodelling seen in the iMSBmal1−/− mice.
Gait disorders generally have multiple origins and tend to be related to underlying medical conditions. However, there are several hallmarks of an ageing gait, including a reduction in gait velocity and stride length, increased stance width and time, and reduced force development at push‐off (Ostir et al. 2015; Aboutorabi et al. 2015). The observed changes in muscle function and muscle and bone phenotype in the Bmal mice have measurable impacts on gait which mimic that observed in the ageing human population, including reductions in stride length and swing time, as well as loss of range of motion in the joints of the ankle and foot. Although not measured, based on assessed parameters, as well as video observation, the chosen gait velocity for the knockouts would have been significantly slower, consistent with human ageing.
Our findings provide new insight into the fundamental role of the endogenous molecular clock in skeletal muscle homeostasis, and uncover the broader function of the skeletal muscle molecular clock in musculoskeletal health. These findings provide a foundation for the inclusion of skeletal muscle assessment and development of targeted therapeutic strategies to ‘reset’ the muscle clock in ageing and disease in order to promote musculoskeletal health.
Additional information
Competing interests
None declared.
Author contributions
This work was performed at the University of Kentucky. K.A.E. conceived the study and E.A.S. helped design the study. T.S. generated the mice needed for the study and performed grip strength analysis. B.D.H., R.S. and J.M. performed histology sample preparation and analysis. Q.Y. and A.C. assisted with characterization of the animal models (genotyping and qPCR) X.Z. performed myosin heavy chain analysis and assisted with molecular biology experiments. E.M.W.‐H., J.D.S., Y.W. and L.A.R. performed in vitro muscle force measurements. J.H.E. stained and imaged bones assisted by B.A.H. T.A.B. built the gait analysis system and performed gait experimentation and analysis. J.H.E. and L.A.R assisted with gait experimentation. E.A.S. and K.A.E. wrote the manuscript with contributions from C.A.P. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH grants RC1ES018636 (National Institute of Arthritis and Musculoskeletal and Skin Diseases) and R01AR066082 (National Institute of Environmental Health Sciences) to K.A.E.
Supporting information
Video S1. Related to Fig. 8. High speed video recorded in the iMSBmal1 +/+ mice used for the assessment of gait.
Video S2. Related to Fig. 8. High speed video recorded in the iMSBmal1 −/− mice used for the assessment of gait.
Video S3. Related to Fig. 8. High speed video recorded in the WT mice used for the assessment of gait.
Video S4. Related to Fig. 8. High speed video recorded in the Bmal1 KO mice used for the assessment of gait.
References
- Aboutorabi A, Arazpour M, Bahramizadeh M, Hutchins SW & Fadayevatan R (2015). The effect of aging on gait parameters in able‐bodied older subjects: a literature review. Aging Clin Exp Res 107, DOI: 10.1007/s40520-015-0420-6. [DOI] [PubMed] [Google Scholar]
- Andrews JL, Zhang X, McCarthy JJ, McDearmon EL, Hornberger TA, Russell B, Campbell KS, Arbogast S, Reid MB, Walker JR, Hogenesch JB, Takahashi JS & Esser KA (2010). CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci USA 107, 19090–19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elseworth J, Yang L, Beal MF, Roth RH, Matthews RT & Horvath TL (2005). Uncoupling protein‐2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci 25, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, Lee C & Nikitin AY (2008). Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle 7, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arounleut P, Bialek P, Liang L‐F, Upadhyay S, Fulzele S, Johnson M, Elsalanty M, Isales CM & Hamrick MW (2013). A myostatin inhibitor (propeptide‐Fc) increases muscle mass and muscle fiber size in aged mice but does not increase bone density or bone strength. Exp Gerontol 48, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood A, DeConde R, Wang SS, Mockler TC, Sabir JS, Ideker T & Kay SA (2011). Cell‐autonomous circadian clock of hepatocytes drives rhythms in transcription and polyamine synthesis. Proc Natl Acad Sci USA 108, 18560–18565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesiadecki BJ, Schneider KL, Yu Z‐B, Chong SM & Jin J‐P (2004). An R111C polymorphism in wild turkey cardiac troponin I accompanying the dilated cardiomyopathy‐related abnormal splicing variant of cardiac troponin T with potentially compensatory effects. J Biol Chem 279, 13825–13832. [DOI] [PubMed] [Google Scholar]
- Brooks SV & Faulkner JA (1988). Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SV & Faulkner JA (1994). Skeletal muscle weakness in old age: underlying mechanisms. Med Sci Sports Exerc 26, 432–439. [PubMed] [Google Scholar]
- Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ & Bradfield CA (2005). Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal‐1 locus. Genesis 41, 122–132. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS & Bradfield CA (2000). Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catoire M & Kersten S (2015). The search for exercise factors in humans. FASEB J 29, 1615–1628. [DOI] [PubMed] [Google Scholar]
- Chan S & Head SI (2010). Age‐ and gender‐related changes in contractile properties of non‐atrophied EDL muscle. PLoS One 5, e12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang NC & Rudnicki MA (2014). Satellite cells: the architects of skeletal muscle. Curr Top Dev Biol 107, 161–181. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Nam D, Guo B, Kim JM, Winnier GE, Lee J, Berdeaux R, Yechoor VK & Ma K (2013). Brain and muscle Arnt‐like 1 is a key regulator of myogenesis. J Cell Sci 126, 2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciliot S, Rossi AC, Dyar KA, Blaauw B & Schiaffino S (2013). Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 45, 2191–2199. [DOI] [PubMed] [Google Scholar]
- Curtis E, Litwic A, Cooper C & Dennison E (2015). Determinants of muscle and bone aging. J Cell Physiol 230, 2618–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B & Bottinelli R (2003). The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol 552, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbono O (2003). Neural control of aging skeletal muscle. Aging Cell 2, 21–29. [DOI] [PubMed] [Google Scholar]
- Dunn SE, Chin ER & Michel RN (2000). Matching of calcineurin activity to upstream effectors is critical for skeletal muscle fiber growth. J Cell Biol 151, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyar KA, Ciciliot S, Wright LE, Biensø RS, Tagliazucchi GM, Patel VR, Forcato M, Paz MIP, Gudiksen A, Solagna F, Albiero M, Moretti I, Eckel‐Mahan KL, Baldi P, Sassone‐Corsi P, Rizzuto R, Bicciato S, Pilegaard H, Blaauw B & Schiaffino S (2014). Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab 3, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkasrawy MN & Hamrick MW (2010). Myostatin (GDF‐8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact 10, 56–63. [PMC free article] [PubMed] [Google Scholar]
- Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, Yang L, Mendias CL, Dupont‐Versteegden EE, McCarthy JJ & Peterson CA (2015). Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med 21, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE (2011). Life Span as a Biomarker. The Jackson Laboratory. Available at: https://www.jax.org/research‐and‐faculty/research‐labs/the‐harrison‐lab/gerontology/life‐span‐as‐a‐biomarker. [Google Scholar]
- Hodge B, Wen Y, Riley L, Zhang X, England J, Harfmann B, Schroder E & Esser KA (2015). The endogenous molecular clock orchestrates the temporal separation of substrate metabolism in skeletal muscle. Skelet Muscle 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB & Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome‐scale data sets. J Biol Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hong HK, Chong JL, Indacochea AA, Lee SS, Han M, Takahashi JS & Hogenesch JB (2012). Brain‐specific rescue of Clock reveals system‐driven transcriptional rhythms in peripheral tissue. PLoS Genet 8, e1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Machida T & Hirafuji M (2014). Skeletal muscle is an endocrine organ. J Pharmacol Sci 125, 125–131. [DOI] [PubMed] [Google Scholar]
- Isaac C, Wright A, Usas A, Li H, Tang Y, Mu X, Greco N, Dong Q, Vo N, Kang J, Wang B & Huard J (2013). Dystrophin and utrophin ‘double knockout’ dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J Orthop Res 31, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawao N & Kaji H (2015). Interactions between muscle tissues and bone metabolism. J Cell Biochem 116, 687–695. [DOI] [PubMed] [Google Scholar]
- Khapre RV, Samsa WE & Kondratov RV (2010). Circadian regulation of cell cycle: Molecular connections between aging and the circadian clock. Ann Med 42, 404–415. [DOI] [PubMed] [Google Scholar]
- Khodabukus A & Baar K (2015). Contractile and metabolic properties of engineered skeletal muscle derived from slow and fast phenotype mouse muscle. J Cell Physiol 230, 1750–1757. [DOI] [PubMed] [Google Scholar]
- King DP & Takahashi JS (2000). Molecular genetics of circadian rhythms in mammals. Annu Rev Neurosci 23, 713–742. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo S‐H, Huang H‐C, Kumar V, Lee C, Kim T‐K & Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV & Antoch MP (2006). Early aging and age‐related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev 20, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenčič A, Košir R, Bordyugov G, Lehmann R, Rozman D & Herzel H (2014). Timing of circadian genes in mammalian tissues. Sci Rep 4, 5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefta M, Campbell KS, Feng HZ, Jin JP & Esser KA (2012). Development of dilated cardiomyopathy in Bmal1‐deficient mice. Am J Physiol Heart Circ Physiol 303, H475–H485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Andrews JL, McDearmon EL, Campbell KS, Barber BK, Miller BH, Walker JR, Hogenesch JB, Takahashi JS & Esser KA (2007). Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol Genomics 31, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Mula J, Miyazaki M, Erfani R, Garrison K, Farooqui AB, Srikuea R, Lawson BA, Grimes B, Keller C, Van Zant G, Campbell KS, Esser KA, Dupont‐Versteegden EE & Peterson CA (2011). Effective fiber hypertrophy in satellite cell‐depleted skeletal muscle. Development 138, 3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Srikuea R, Kirby TJ, Peterson CA & Esser KA (2012). Inducible Cre transgenic mouse strain for skeletal muscle‐specific gene targeting. Skelet Muscle 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, Wilsbacher LD, Song EJ, Hong H‐K, Bradfield CA & Takahashi JS (2006). Dissecting the functions of the mammalian clock protein BMAL1 by tissue‐specific rescue in mice. Science 314, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maronde E, Schilling AF, Seitz S, Schinke T, Schmutz I, van der Horst G, Amling M & Albrecht U (2010). The clock genes Period 2 and Cryptochrome 2 differentially balance bone formation. PLoS One 5, e11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoccoli G, Pazienza V & Vinciguerra M (2012). Clock genes and clock‐controlled genes in the regulation of metabolic rhythms. Chronobiol Int 29, 227–251. [DOI] [PubMed] [Google Scholar]
- Miljkovic N, Lim J‐Y, Miljkovic I & Frontera WR (2015). Aging of skeletal muscle fibers. Ann Rehabil Med 39, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB & Takahashi JS (2007). Circadian and CLOCK‐controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA 104, 3342–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Callahan DM & Toth MJ (2014). Skeletal muscle myofilament adaptations to aging, disease, and disuse and their effects on whole muscle performance in older adult humans. Front Physiol 5, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell WK, Williams J, Atherton P, Larvin M, Lund J & Narici M (2012). Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front Physiol 3, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Schroder E, Edelmann SE, Hughes ME, Kornacker K, Balke CW & Esser KA (2011). Age‐associated disruption of molecular clock expression in skeletal muscle of the spontaneously hypertensive rat. PLoS One 6, e27168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras D, L'Honoré A & Buckingham M (2013). Lying low but ready for action: the quiescent muscle satellite cell. FEBS J 280, 4036–4050. [DOI] [PubMed] [Google Scholar]
- Morissette MR, Stricker JC, Rosenberg MA, Buranasombati C, Levitan EB, Mittleman MA & Rosenzweig A (2009). Effects of myostatin deletion in aging mice. Aging Cell 8, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse CI, Thom JM, Birch KM & Narici MV (2005). Changes in triceps surae muscle architecture with sarcopenia. Acta Physiol Scand 183, 291–298. [DOI] [PubMed] [Google Scholar]
- Ostir GV, Berges IM, Ottenbacher KJ, Fisher SR, Barr E, Hebel JR & Guralnik JM (2015). Gait speed and dismobility in older adults. Arch Phys Med Rehabil 96, 1641–1645. [DOI] [PubMed] [Google Scholar]
- Paschos GK, Ibrahim S, Song W‐L, Kunieda T, Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, Griffin JL, Wang F, Lawson JA & FitzGerald GA (2012). Obesity in mice with adipocyte‐specific deletion of clock component Arntl . Nat Med 18, 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovezan RD, Abucham J, dos Santos RVT, Mello MT, Tufik S & Poyares D. (2015). The impact of sleep on age‐related sarcopenia: Possible connections and clinical implications. Ageing Res Rev 23, 210–220. [DOI] [PubMed] [Google Scholar]
- Pizarro A, Hayer K, Lahens NF & Hogenesch JB (2013). CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res 41, D1009–D1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves‐Smith FM, Sgarioto N & Hepple RT (2014). Fiber typing in aging muscle. Exerc Sport Sci Rev 42, 45–52. [DOI] [PubMed] [Google Scholar]
- Quiat D, Voelker KA, Pei J, Grishin NV, Grange RW, Bassel‐Duby R & Olson EN (2011). Concerted regulation of myofiber‐specific gene expression and muscle performance by the transcriptional repressor Sox6. Proc Natl Acad Sci USA 108, 10196–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan JG, Cambier D, Lyden S, Dalvi A, Upputuri RK, Gartside P, Michaels SE & Denman D (1997). Regeneration‐blocked mdx muscle: in vivo model for testing treatments. Muscle Nerve 20, 1016–1023. [DOI] [PubMed] [Google Scholar]
- Rai M, Nongthomba U & Grounds MD (2014). Skeletal muscle degeneration and regeneration in mice and flies. Curr Top Dev Biol 108, 247–281. [DOI] [PubMed] [Google Scholar]
- Rajaratnam SMW & Arendt J (2001). Health in a 24‐h society. Lancet 358, 999–1005. [DOI] [PubMed] [Google Scholar]
- Richard A‐F, Demignon J, Sakakibara I, Pujol J, Favier M, Strochlic L, Le Grand F, Sgarioto N, Guernec A, Schmitt A, Cagnard N, Huang R, Legay C, Guillet‐Deniau I & Maire P (2011). Genesis of muscle fiber‐type diversity during mouse embryogenesis relies on Six1 and Six4 gene expression. Dev Biol 359, 303–320. [DOI] [PubMed] [Google Scholar]
- Schibler U (2005). The daily rhythms of genes, cells and organs. EMBO Rep 6, S9–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibler U & Naef F (2005). Cellular oscillators: rhythmic gene expression and metabolism. Curr Opin Cell Biol 17, 223–229. [DOI] [PubMed] [Google Scholar]
- Schroder EA, Lefta M, Zhang X, Bartos DC, Feng H‐Z, Zhao Y, Patwardhan A, Jin J‐P, Esser KA & Delisle BP (2013). The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am J Physiol Cell Physiol 304, C954–C965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimè A, Desrosiers J, Trensz F, Palidwor GA, Caron AZ, Andrade‐Navarro MA & Grenier G (2010). Transcriptional profiling of skeletal muscle reveals factors that are necessary to maintain satellite cell integrity during ageing. Mech Ageing Dev 131, 9–20. [DOI] [PubMed] [Google Scholar]
- Storch KF, Paz C, Signorovitch J, Raviola E, Pawlyk B, Li T & Weitz CJ (2007). Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Xie Z, Guo Z, Duncan MJ, Lutshumba J & Gong MC (2012). Altered clock gene expression and vascular smooth muscle diurnal contractile variations in type 2 diabetic db/db mice. Am J Physiol Heart Circ Physiol 302, H621–H633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge RJ & Roy RR (1993). Electrophoretic separation of rat skeletal muscle myosin heavy‐chain isoforms. J Appl Physiol (1985) 75, 2337–2340. [DOI] [PubMed] [Google Scholar]
- Tikunov BA, Sweeney HL & Rome LC (2001). Quantitative electrophoretic analysis of myosin heavy chains in single muscle fibers. J Appl Physiol (1985) 90, 1927–1935. [DOI] [PubMed] [Google Scholar]
- Tompkins KA (2011). Wnt proteins in mineralized tissue development and homeostasis. Connect Tissue Res 52, 448–458. [DOI] [PubMed] [Google Scholar]
- Tzahor E, Kempf H, Mootoosamy RC, Poon AC, Abzhanov A, Tabin CJ, Dietrich S & Lassar AB (2003). Antagonists of Wnt and BMP signaling promote the formation of vertebrate head muscle. Genes Dev 17, 3087–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn J, Chang NC, Bentzinger CF & Rudnicki MA (2012). Wnt signaling in myogenesis. Trends Cell Biol 22, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers AJ (2008). Wnt not, waste not. Cell Stem Cell 2, 6–7. [DOI] [PubMed] [Google Scholar]
- Yablonka‐Reuveni Z & Rivera AJ (1997). Influence of PDGF‐BB on proliferation and transition through the MyoD‐myogenin‐MEF2A expression program during myogenesis in mouse C2 myoblasts. Growth Factors 15, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S & Takahashi JS (2005). Real‐time luminescence reporting of circadian gene expression in mammals. Methods Enzymol 393, 288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M & Takahashi JS (2004). PERIOD2::LUCIFERASE real‐time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 101, 5339–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitane H, Ozaki H, Terajima H, Du N‐H, Suzuki Y, Fujimori T, Kosaka N, Shimba S, Sugano S, Takagi T, Iwasaki W & Fukada Y (2014). CLOCK‐controlled polyphonic regulation of circadian rhythms through canonical and noncanonical E‐boxes. Mol Cell Biol 34, 1776–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Choi HJ, Estrada K, Leo PJ, Li J, Pei Y‐F, Zhang Y, Lin Y, Shen H, Liu Y‐Z, Liu Y, Zhao Y, Zhang J‐G, Tian Q, Wang Y‐p, Han Y, Ran S, Hai R, Zhu X‐Z, Wu S, Yan H, Liu X, Yang T‐L, Guo Y, Zhang F, Guo Y‐f, Chen Y, Chen X, Tan L, Zhang L, Deng F‐Y, Deng H, Rivadeneira F, Duncan EL, Lee JY, Han BG, Cho NH, Nicholson GC, McCloskey E, Eastell R, Prince RL, Eisman JA, Jones G, Reid IR, Sambrook PN, Dennison EM, Danoy P, Yerges‐Armstrong LM, Streeten EA, Hu T, Xiang S, Papasian CJ, Brown MA, Shin CS, Uitterlinden AG & Deng H‐W (2014). Multistage genome‐wide association meta‐analyses identified two new loci for bone mineral density. Hum Mol Genet 23, 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Patel SP, McCarthy JJ, Rabchevsky AG, Goldhamer DJ & Esser KA (2012). A non‐canonical E‐box within the MyoD core enhancer is necessary for circadian expression in skeletal muscle. Nucleic Acids Res 40, 3419–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Related to Fig. 8. High speed video recorded in the iMSBmal1 +/+ mice used for the assessment of gait.
Video S2. Related to Fig. 8. High speed video recorded in the iMSBmal1 −/− mice used for the assessment of gait.
Video S3. Related to Fig. 8. High speed video recorded in the WT mice used for the assessment of gait.
Video S4. Related to Fig. 8. High speed video recorded in the Bmal1 KO mice used for the assessment of gait.