

A systems genetics approach identifies gene regulatory networks associated with fatty acid composition in Brassica rapa seed.
Abstract
Fatty acids in seeds affect seed germination and seedling vigor, and fatty acid composition determines the quality of seed oil. In this study, quantitative trait locus (QTL) mapping of fatty acid and transcript abundance was integrated with gene network analysis to unravel the genetic regulation of seed fatty acid composition in a Brassica rapa doubled haploid population from a cross between a yellow sarson oil type and a black-seeded pak choi. The distribution of major QTLs for fatty acids showed a relationship with the fatty acid types: linkage group A03 for monounsaturated fatty acids, A04 for saturated fatty acids, and A05 for polyunsaturated fatty acids. Using a genetical genomics approach, expression quantitative trait locus (eQTL) hotspots were found at major fatty acid QTLs on linkage groups A03, A04, A05, and A09. An eQTL-guided gene coexpression network of lipid metabolism-related genes showed major hubs at the genes BrPLA2-ALPHA, BrWD-40, a number of seed storage protein genes, and the transcription factor BrMD-2, suggesting essential roles for these genes in lipid metabolism. Three subnetworks were extracted for the economically important and most abundant fatty acids erucic, oleic, linoleic, and linolenic acids. Network analysis, combined with comparison of the genome positions of cis- or trans-eQTLs with fatty acid QTLs, allowed the identification of candidate genes for genetic regulation of these fatty acids. The generated insights in the genetic architecture of fatty acid composition and the underlying complex gene regulatory networks in B. rapa seeds are discussed.
The species Brassica rapa displays enormous morphological variation, as illustrated by the diversity of crops, including leafy vegetables, turnips, and oil types (Zhao et al., 2005). B. rapa ssp. oleifera (oil-type rape) consists of the annual oil crops yellow sarson and brown sarson with high seed oil content (greater than 42%; Lühs et al., 1999; Kumar et al., 2011). In the past, both sarsons were preferred as oil crops over Brassica napus in Asia, Canada, and other parts of the world, as they mature earlier and have a higher level of shattering resistance and spring frost tolerance (Kadkol et al., 1986; Karim et al., 2014). However, B. rapa rape seed was gradually replaced by B. napus, mainly because of the latter’s higher oilseed yield and the availability of double-low genotypes, which are low in glucosinolate content as well as erucic acid content (Rahman et al., 2001; Karim et al., 2014). Still, B. rapa has been used to widen the genetic variation for the improvement of B. napus (Qian et al., 2006; Karim et al., 2014).
Lipids are a group of naturally occurring molecules that include fats, glycerolipids, fatty acids (FAs), glycerophospholipids, sphingolipids, waxes, and others. De novo synthesized FAs are modified by desaturation and elongation reactions and form triacylglycerols, which are the major storage form of seed oil in plants (Guschina and Harwood, 2007). For both nutritional and industrial purposes, the composition of FAs determines the economic value of seed oil (Yan et al., 2011; Sanyal and Randal Linder, 2012). For food or feed, oil that is high in the level of human health-beneficial oleic acid (C18:1) is preferred. This, in turn, can be easily desaturated to linoleic and linolenic acids or elongated to erucic acid. Oil with high erucic acid (C22:1) has a health risk but can be used for industrial purposes, while high linolenic acid (C18:3) negatively affects oil storability (Yan et al., 2011). To breed for optimal FA composition and high oil yield, understanding the genetic regulation of FA composition and the FA regulatory network is crucial.
Quantitative trait locus (QTL) analysis and gene functional studies have been performed to unravel the genetics of FA composition in Arabidopsis (Arabidopsis thaliana; Sanyal and Randal Linder, 2012), soybean (Glycine max; Wang et al., 2014b), jatropha (Jatropha curcas; Liu et al., 2011), and B. napus (Peng et al., 2010; Yan et al., 2011). In Arabidopsis, almost all genes and transcription factors involved in lipid metabolism and storage oils have been identified (Beisson et al., 2003; Le et al., 2010; Peng and Weselake, 2011). The B. rapa genome (A genome), like the Brassica oleracea genome (C genome), is syntenic to Arabidopsis but underwent a genome triplication (Wang et al., 2011; Liu et al., 2014). B. napus is an amphidiploid resulting from natural hybridization between B. rapa (A genome) and B. oleracea (C genome). These genome triplications, resulting in many genes with paralogs, add another level of complexity to the genetic regulation of FA composition in Brassica spp. In a recent publication, the allopolyploid B. napus oilseed genome sequence was described, with special attention to the cross talk between the An and Cn genomes. Evidence was presented for the expansion of oil biosynthesis genes, which exceeds that of other known oilseed plants, with a total of 1,097 and 1,132 genes annotated on the An and Cn subgenomes, respectively (Chalhoub et al., 2014). In another recent study, the differential regulation of the genes and transcription factors involved in lipid biosynthesis in seeds and in leaves of oilseed B. napus was described, including details of FA-specific pathways (Chen et al., 2015). Across a number of studies, a large number of QTLs for FAs and oil content have been reported, suggesting a complex genetic architecture (Yan et al., 2011; Sanyal and Randal Linder, 2012). Genetic studies have identified the FA desaturase genes BnaFAD2 and BnaFAD3 as the major genes for the regulation of C18:1 (oleic acid) and C18:3 (linolenic acid) content in B. napus (Peng et al., 2010; Yang et al., 2012; Lee et al., 2013). BnaFAE1 is a candidate gene for erucic acid and total oil content in B. napus seed (Peng et al., 2010), while BrFAD3 is a candidate gene for the synthesis of linolenic acid in seed triacylglycerols in B. rapa ssp. oleifera (Tanhuanpää and Schulman, 2002). To our knowledge, this is the first genome-wide genetic study for the FA composition and transcriptional regulation of developing seeds of B. rapa.
Many genes involved in different metabolic processes are regulated in a coordinated fashion during seed development in Arabidopsis (Ruuska et al., 2002), B. napus (Yu et al., 2010), and B. rapa (Basnet et al., 2013). The combined study of phenotypic QTLs and expression quantitative trait loci (eQTLs) provides a basis to investigate the molecular mechanism and to understand the regulatory networks of genes involved in pathways of specific phenotypic traits in different organisms (Civelek and Lusis, 2014). Genome-wide mapping of gene transcripts in a segregating population was first proposed by Jansen and Nap (2001) and was named genetical genomics. Using genetical genomics, candidate genes were identified in B. rapa for flowering time and leaf development (Xiao et al., 2013, 2014), phytonutrient content (Pino Del Carpio et al., 2014), and phosphorus use efficiency (Hammond et al., 2011).
The aim of this study was to identify QTLs for FA content and composition in B. rapa seeds using a doubled haploid (DH) population from a cross between an oil-type yellow sarson and a vegetable pak choi. In order to understand the lipid gene regulatory network in B. rapa, we followed a genetical genomics approach combining QTLs for FAs in mature seeds with eQTLs for genes related to the lipid metabolism in developing seeds: FA biosynthesis and elongation, triacylglycerol biosynthesis, glycerol synthesis, and lipid degradation. Based on gene expression variation, a gene coexpression network was constructed for genes involved in lipid metabolism and relative content of FAs. Because of the economic importance of erucic acid, oleic acid, linoleic acid, and linolenic acid, individual subnetworks of those FAs were also derived. Finally, eQTL results were integrated with known FA pathways to unravel the regulation of genes involved in the composition of FAs in B. rapa seeds. This resulted in the identification of a number of QTL hotspots and key regulatory genes that are of importance for breeding purposes.
RESULTS
Variation of FAs in Seed
The most abundant FAs in the seed lots were three monounsaturated fatty acids (MUFAs), erucic acid (C22:1), oleic acid (C18:1), and eicosenoic acid (C20:1), and two polyunsaturated fatty acids (PUFAs), linoleic acid (C18:2) and linolenic acid (C18:3). Together, these accounted for 71.6% to 74.2% (MUFAs) and 16.9% to 19.2% (PUFAs) of the total oil concentration (Fig. 1; Supplemental Table S1). Erucic acid (C22:1) was the most predominant FA in both years’ seed lots and had a higher level in yellow sarson (YS143; 55.8%) than in pak choi (47%). Oleic acid (C18:1) was the second most abundant FA, but it was higher in pak choi (17.3%) than in yellow sarson (13.7%). Linoleic and linolenic acids had comparable levels in both parents: 8.2% to 11% in pak choi and 6.8% to 10.8% in yellow sarson. The content of the FAs was very similar in the two different years. Total oil concentration was higher in yellow sarson (44.2%) than in pak choi (29.3%). For most of the FAs and total oil, a number of DH lines had higher or lower content than the two parents, indicating transgressive segregation in this population.
Figure 1.

Box plots showing the distribution of FAs (A) and total oil content (B) of ripe seeds of the B. rapa DH population from the 2009 and 2011 seed lots. FAs were measured in mass percentage of the total oil content and the total oil content was measured in mass percentage on the basis of whole seed dry matter (zero moisture).
Correlations between Years and among FAs
For most FAs, there were high positive Pearson correlation coefficients (r = 0.5–0.9) between 2009 and 2011 (Fig. 2). A notable exception was mead acid (C20:3), with a much lower positive correlation (r = 0.20) between years. However, its abundance was near the detection limit for both years. A combined analysis from both seed lots shows high positive correlations among saturated fatty acids (SFAs), PUFAs, and MUFAs, with the exception of the two MUFAs nervonic acid (C24:1) and the predominant FA erucic acid (C22:1). Nervonic acid and erucic acid were both negatively correlated with SFAs and MUFAs and positively correlated with several PUFAs, but they were not significantly correlated with each other (Fig. 2).
Figure 2.

Heat map of Pearson correlation coefficients of FAs and total oil content from the 2009 and 2011 seed lots. The name of each FA, its molecular structure, and the year are indicated. #, SFAs; *, MUFAs; ** and ***, PUFAs with two and three double bonds, respectively.
QTL Mapping of FAs
We performed QTL analyses for 17 different FAs from the 2009 and 2011 seed lots. We observed more QTLs for FAs (hereafter called faQTLs) from the 2009 seed lot when plants flowered without synchronization than from the 2011 seed lot, harvested from plants that flowered synchronously. In the 2009 seed lot, 56 faQTLs were detected, and 24 of these faQTLs (43%) had at least 10% explained variance (mean, 11%; maximum, 36%). For the 2011 seed lot, only 32 faQTLs were detected, but for 2011, a much higher percentage, 24 faQTLs (75% of the faQTLs) had an explained variance of at least 10% (mean, 15.8%; maximum, 46%; Supplemental Tables S3 and S4).
Major faQTLs (log of the odds [LOD] scores greater than 10, explained variances greater than 15%) were observed on linkage groups A03, A04, and A05 (Fig. 3). faQTLs were observed across all 10 linkage groups; faQTLs for 11 FAs colocated with a major flowering QTL at the genomic region (16.7 centimorgan [cM]) of the BrFLC2 gene-targeted marker on A02 in the 2009 seed lot (Fig. 3; Supplemental Table S3). For the 2009 and 2011 seed lots, faQTLs were mainly mapped on A03, A04, A05, and A07 (Fig. 3). In both seed lots, at major faQTLs detected for SFAs (myristic acid and behenic acid) on A04 and for PUFAs (linoleic acid, eicosadienoic acid, and docosadienoic acid) on A05, the yellow sarson allele was associated with a higher concentration (Fig. 3). For the erucic acid QTL on A03 and A05, the pak choi allele was associated with higher concentrations, while on A02, A07, and A09, the yellow sarson allele was associated with higher concentrations (Fig. 3).
Figure 3.

Heat map of QTL profiles for FAs in the 2009 and 2011 seed lots. The darker the color intensity, the higher the LOD score. Yellow indicates a QTL effect where the yellow sarson allele is associated with higher abundance, while blue indicates an effect where the pak choi allele is associated with higher abundance. The red stars indicate the positions of QTL peak markers. The black tick marks indicate marker positions, and the vertical dashed lines separate the linkage groups. The horizontal dotted black lines separate traits, and red lines separate SFAs, MUFAs, and PUFAs. Dotted boxes indicate QTLs present in both years’ seed lots.
eQTL Mapping of Transcript Abundance of Lipid-Related Genes
A total of 1,568 probes (of 921 BraIDs) for genes related to lipid metabolism, lipid signaling, lipid storage proteins, and lipid transfer proteins were used for eQTL analyses. Those lipid-related genes were selected according to MapMan annotation. Out of the 1,568 probes, 760 probes (representing 537 BraIDs) had at least one eQTL at −10log of the P value (LOP) > 3 and 270 probes (194 BraIDs) had at least one eQTL at LOP > 5. In total, 1,118 eQTLs were detected for 760 probes (537 BraIDs), including 304 cis-eQTLs (27%) and 814 trans-eQTLs (73%; Table I). A total of 146 probes (115 BraIDs) had both cis- and trans-eQTLs. The majority of probes (745 probes) had one to three eQTLs, while only 12 probes had four eQTLs and three had a maximum of five eQTLs per probe. Five linkage groups, A03, A04, A05, A07, and A09, had significant eQTL hotspots (more than 42 eQTLs; Fig. 4, left).
Table I. Numbers and percentages of cis- and trans-eQTLs detected in each linkage group before and after correction for seed color.
| Linkage Group | Before Correction |
After Correction |
||||||
|---|---|---|---|---|---|---|---|---|
| Cis-eQTLs | Trans-eQTLs | Total | Percentage eQTL | Cis-eQTLs | Trans-eQTLs | Total | Percentage eQTL | |
| A01 | 36 | 48 | 84 | 7.5 | 31 | 46 | 77 | 8.1 |
| A02 | 19 | 43 | 62 | 5.6 | 20 | 48 | 68 | 7.2 |
| A03 | 64 | 89 | 153 | 13.7 | 55 | 98 | 153 | 16.2 |
| A04 | 17 | 103 | 120 | 10.7 | 19 | 96 | 115 | 12.2 |
| A05 | 43 | 154 | 197 | 17.6 | 39 | 133 | 172 | 18.2 |
| A06 | 16 | 23 | 39 | 3.5 | 15 | 25 | 40 | 4.2 |
| A07 | 14 | 76 | 90 | 8.1 | 12 | 56 | 68 | 7.2 |
| A08 | 26 | 54 | 80 | 7.2 | 27 | 61 | 88 | 9.3 |
| A09 | 53 | 199 | 252 | 22.6 | 36 | 89 | 125 | 13.2 |
| A10 | 16 | 24 | 40 | 3.6 | 19 | 21 | 40 | 4.2 |
| Total | 304 (27.2%) | 813 (72.8%) | 1,117 | 273 (28.9%) | 673 (71.1%) | 946 | ||
Figure 4.

Genome-wide distribution of eQTLs in the developing seeds (28 d after pollination) of a B. rapa DH population: before (left) and after (right) correction for seed color. A, Frequency of eQTLs at each genetic marker along the 10 linkage groups, separated by dashed lines. The y axes represent the number of eQTLs. The red line indicates the threshold for declaring a significant eQTL hotspot. B, Scatterplots of cis-/trans-eQTLs of probes related to lipid metabolism before (left) and after (right) correction. The y axes represent the order of probes according to physical positions in the genome, and the x axes indicate the order of genetic markers in the genetic map. eQTLs on the diagonal represent cis-eQTLs, and off-diagonal eQTLs are trans-eQTLs. Significant eQTLs associated with higher transcript abundance from the yellow sarson allele or from the pak choi allele are shown in red and blue color gradients, respectively. The significance of eQTLs was determined at LOP 3 [−log10(P value)]. Vertical dashed lines separate the linkage groups in the genetic map, and horizontal dashed lines separate the linkage groups in the physical map.
Most of the cis-eQTLs had a higher significance (maximum LOP 29 and mean LOP 7) than the trans-eQTLs (maximum LOP 18 and mean LOP 4). The largest trans-eQTL hotspots were on A05 (19% of total trans-eQTL) and A09 (25% of trans-eQTL; Fig. 4B, left). The trans-eQTL hotspot at A09 colocates with a major QTL for seed coat color, which explains 33% of the color variation (data not shown).
QTL Mapping after Correcting for Seed Color Differences
A possible confounding effect of seed color on transcript abundance was corrected for using a simple linear regression model, as described in “Materials and Methods.” After correction for seed color, 946 eQTLs were observed for 662 probes (488 BraIDs) across the genome, 273 probes (194 BraIDs) had cis-eQTLs (29%), and 513 probes (397 BraIDs) had 673 trans-eQTLs (71%; Table I). Most of the probes (641 probes) had one to three eQTLs, while seven probes had four eQTLs and three probes had a maximum of five eQTLs per probe.
eQTL hotspots were now detected on A03 (153 eQTLs, 16% of total eQTLs), A04 (115 eQTLs, 12%), A05 (172 eQTLs, 18%), A07 (68 eQTLs, 7%), and A09 (125 eQTLs, 13%; Table I; Fig. 4, right). As in the analysis before correction, cis-eQTLs had higher significance than trans-eQTLs.
Comparison of eQTLs before and after Correcting for Seed Color
A large number of probes (630 probes for 461 BraIDs) with significant eQTLs were in common between both analyses. After correction, an additional 32 probes (28 BraIDs), belonging to eight different pathways, such as lipid degradation, FA synthesis and elongation, seed storage proteins, and FA desaturation, had eQTLs. eQTLs of the 130 probes (110 BraIDs) that were lost after correction could be either false positives in the analysis before correction or false negatives after correction due to overcorrection for seed color, because their expression variation correlated with seed color variation (e.g. due to linkage or a pleiotropic effect). χ2 tests were performed for each linkage group to test the significance of the differences in number of eQTLs before and after correction. There were no significant changes in the number of cis-eQTLs (P > 0.05), but the number of trans-eQTLs was significantly changed on A03 (from 89 to 98 trans-eQTLs; P = 0.03) and A09 (from 199 to 89 trans-eQTLs; P < 0.0001; Table I; Fig. 4).
eQTLs from the Reverse Transcription-Quantitative PCR Gene Expression Studies
The expression values of 23 genes obtained by reverse transcription-quantitative PCR (RT-qPCR) were also subjected to correction for seed color and then eQTL analysis to validate microarray eQTL results. For 16 out of 23 genes, at least one of the eQTLs was detected at the same position in both microarray and RT-qPCR experiments (Supplemental Fig. S1). In the case of BrCER8, BrLACS2, BrCRU3 (Bra011036), and Br006444, eQTL profiles did not correspond between RT-qPCR and microarrays. The expression of FLOWERING LOCUS C (BrFLC2; a major regulator of flowering time in B. rapa) and the TRANSPARENT TESTA8 gene (BrTT8; a major regulator of seed color in B. rapa), which map under the faQTL hotspot on A02 in 2009 and the eQTL hotspot on A09, respectively, was also measured with RT-qPCR. For BrFLC2, a cis-eQTL on A02 was confirmed in both RT-qPCR and microarray experiments, but an additional trans-eQTL was detected on A05 only in the RT-qPCR experiment. For BrTT8, a cis-eQTL was detected on A09 under the trans-eQTL hotspot only for RT-qPCR (Supplemental Fig. S1). For almost all genes, eQTLs detected for expression profiles measured by RT-qPCR were stronger (higher explained variance) than in the microarray.
Colocalization of Metabolite QTLs and eQTLs
Major faQTLs detected in both seed lots were compared with eQTL hotspots observed after correction for seed color (Figs. 3 and 4, right). On A03, faQTLs of the MUFAs oleic acid, eicosenoic acid, and erucic acid from both seed lots colocalized with an eQTL hotspot. On A04, major faQTLs of the SFAs behenic acid and myristic acid and a minor faQTL for palmitic acid, also from both seed lots, colocalized with an eQTL hotspot. Major faQTLs for the PUFAs linoleic acid and eicosadienoic acid and minor faQTLs for docosadienoic acid for both seed lots were detected at the same region on A05, where a gene-targeted marker for the BrFAD2 gene mapped with its cis-eQTL and where also an eQTL hotspot was located (Fig. 4, right; Supplemental Tables S3 and S4). If we also consider minor faQTLs, the association of A03, A04, and A05 with MUFAs, SFAs, and PUFAs, respectively, is not perfect.
The eQTL hotspots on A03, A04, and A05 are interesting for further investigation toward candidate genes for lipid metabolism and Brassica spp. oil crop improvement. At the major eQTL hotspot on A09 (89 trans-eQTLs), where the cis-regulated BrTT8 gene for seed color is located, we did not detect major faQTLs.
We looked for colocation of eQTLs of the genes BrFAE1, TAG1 (also called DGAT1), BrFAD2, and other BrFAD genes with faQTLs, as these genes were reported as candidate genes for the synthesis of linoleic acid, linolenic acid, erucic acid, oleic acid, or total oil content in Arabidopsis and B. napus (Peng et al., 2010; Yang et al., 2012; Lee et al., 2013). BrFAE1 has two paralogs on A01 and A03, which only have trans-eQTLs on A02, A03, A05, and A07, and BrTAG1 on A07 has a trans-eQTL on A05 (Supplemental Fig. S1). Trans-eQTLs of BrFAE1 colocated mainly with faQTLs of MUFAs on A03, the PUFA linolenic acid on A05, and SFAs and MUFAs on A07, while a trans-eQTL of BrTAG1 was colocated with an eQTL hotspot on A05, mainly with faQTLs of PUFAs (Fig. 3). The BrFAD series genes (BrFAD2, BraFAD5, and BrFAD7) from the FA desaturation pathway that are colocated within a physical range of 19.5 to 21.6 Mb on A05 all had a cis-eQTL on A05 at the map position (89.1 cM) of a BrFAD2 gene-targeted marker, colocating with faQTLs for the SFA lignoceric acid, the MUFAs palmitoleic, oleic, eicosenoic, and erucic acids, and the PUFAs linoleic, eicosadienoic, and docosadienoic acids (Fig. 3).
Coexpression Network of Lipid-Related Genes and FAs
Pearson correlation coefficients were calculated based on transcript abundance (after correction for seed color) of 662 probes (488 BraIDs), with at least one eQTL, and 17 FAs (2011 seed lot) to construct a coexpression network (Supplemental Fig. S2). This figure shows that many genes from the lipid metabolic pathways are involved in the regulation of FAs. We here not only show the interactions between genes but also whether they are cis- or trans-regulated. The coexpression network was constructed using two approaches: in the first one, correlations among all probes and FAs were considered (Supplemental Fig. S2), while in the second approach, we considered only the probes associated with FAs and using weighted gene coexpression network analysis (WGCNA; we call this an FA-centered network). In the first approach, a higher degree of connection among genes and FAs indicates a major hub gene that could potentially be a major regulator of lipid metabolism. In the second, the degree of connection of genes indicates the numbers of significant associations with only FAs, where a gene with a high degree of connection could be potentially a major gene involved in the regulation of those FAs.
In both analyses, a very similar set of genes with a high degree of connection was observed. The gene BrPLA2-ALPHA (PHOSPHOLIPASE A2; BraID Bra038125) with a cis-eQTL on A05 had the highest degree of connection in both network analyses (Fig. 5; Table II), suggesting that this is an essential gene for lipid metabolism. Since BrPLA2-ALPHA is one of the major hub genes, a BrPLA2-ALPHA-centered subnetwork (Fig. 5) was extracted from the whole gene-FA coexpression network (Supplemental Fig. S2). Figure 5 shows that genes from the lipid metabolic pathways and seed storage interact with this major hub gene. From the top 25 genes, based on their degree of connection from both analyses, 16 were selected in both (Table II). Among those 16 genes, three genes had a cis-eQTL on A05 and two genes had a cis-eQTL on A09, while 11 genes had a trans-eQTL on A09, four genes had a trans-eQTL on A03, and only four genes from single analyses had a trans-eQTL on A04 (Table II).
Figure 5.

Major hub gene BrPLA2-ALPHA-centered gene coexpression network using probes with at least one eQTL (after correction). Only genes that are connected with BrPLA2-ALPHA in the whole gene-metabolite coexpression network (Supplemental Fig. S2) are shown. Nodes represent genes or FAs. Genes from different pathways are shown in different node colors, while metabolites are shown in triangle-shaped nodes in purple. Edges represent high absolute correlations (r > 0.5). Edges connecting the major hub gene and genes from the same pathway (lipid degradation) are in blue, edges between the major hub gene and genes from the other pathways are in red, and edges connecting genes and FAs are in green. Edges connecting genes (other than with the major hub gene) of different pathways have been left out to improve the visibility of the network. The shapes of gene nodes indicate cis-eQTLs (chevrons), trans-eQTLs (squares), and cis-/trans-eQTLs (diamonds). Node names are coded by concatenating the gene name, cis-/trans-regulation, and the linkage group. For example, node FAD2_C-5 indicates gene FAD2, C for cis-eQTL, and 5 for linkage group A05, where the cis-eQTL was detected. In the case of a cis-/trans-eQTL, CER10_CT-9_1 indicates gene CER10 and CT-9_1 indicates a cis-eQTL on A09 and trans-eQTLs on A01. Solid lines indicate positive correlations, and dotted lines indicate negative correlations. All the gene names are prefixed with Br because of B. rapa gene nomenclature. Multiple occurrence of the same gene name represents genes with multiple paralogs or probes.
Table II. List of the top 25 genes based on degree of connection in each of two network approaches, NetworkAnalyzer (Cytoscape) and WGCNA (FA centered).
Sixteen genes are in common between the two lists, resulting in a total of 35 genes over the two approaches. √ indicates a significant association of a gene with FAs in WGCNA. The dashes indicate that there is no significant association of a gene with the specific FA in WGCNA. For the MapMan pathway, 1 = exotics, 2 = FA synthesis and FA elongation, 3 = glycerol metabolism, 4 = lipid degradation, 5 = lipid signaling, 6 = lipid transfer proteins, and 7 = phospholipid synthesis.
| bhhhhhhhh | Gene Symbol | BraID | Network Analysis Method | Degree of Connection |
eQTL |
SFAs |
MUFAs |
PUFAs |
MapMan Pathway | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NetworkAnalyzer | WGCNA | Cis- | Trans- | Myristic | Stearic | Palmitic | Arachidic | Behenic | Palmitoleic | Oleic | Eicosenoic | Erucic | Docosadienoic | Linoleic | Eicosadienoic | |||||
| 1 | PLA2-ALPHA | Bra038125 | Both | 91 | 5 | A05 | – | √ | – | – | – | – | √ | – | – | – | √ | √ | √ | 4 |
| 2 | Alcohol oxidase | Bra013391 | Both | 90 | 2 | – | A09 | √ | – | – | – | – | √ | – | – | – | – | – | – | 4 |
| 3 | Choline monooxygenase | Bra024118 | Both | 88 | 3 | – | A02 | √ | √ | – | – | – | √ | – | – | – | – | – | – | 7 |
| 4 | Lysophospholipases | Bra003814 | Both | 87 | 4 | – | A09 | – | √ | √ | – | – | √ | – | – | √ | – | – | – | 4 |
| 5 | Choline kinase | Bra028032 | Both | 87 | 4 | A09 | A03, A08 | √ | – | √ | – | – | √ | – | – | √ | – | – | – | 7 |
| 6 | Hydrolase | Bra016558 | Both | 87 | 2 | – | A03, A09 | √ | – | – | – | – | √ | – | – | – | – | – | – | 4 |
| 7 | Alcohol oxidase | Bra012548 | Both | 87 | 2 | A03 | A09 | – | – | √ | – | – | √ | – | – | – | – | – | – | 4 |
| 8 | LPAT3 | Bra030448 | Both | 84 | 6 | A05 | A09 | √ | √ | √ | – | – | √ | – | √ | √ | – | – | – | 7 |
| 9 | CER10 | Bra007154 | Both | 81 | 3 | A09 | A01 | – | – | √ | – | – | √ | – | – | √ | – | – | – | 1 |
| 10 | IBR10 | Bra039860 | Both | 79 | 2 | A08 | A09 | √ | – | – | – | – | √ | – | – | – | – | – | – | 4 |
| 11 | NPC4 | Bra021355 | Both | 78 | 4 | – | A03, A09 | √ | – | √ | – | – | √ | – | – | √ | – | – | – | 4 |
| 12 | Stearoyl-ACP desaturase | Bra021427 | Both | 78 | 2 | – | A09 | √ | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 13 | Lipases | Bra035263 | Both | 77 | 3 | – | A03, A07, A09 | – | – | √ | – | – | √ | – | – | √ | – | – | – | 4 |
| 14 | AT3BETAHSD | Bra015621 | Both | 74 | 2 | A10 | A05 | – | – | √ | – | – | √ | – | – | – | – | – | – | 1 |
| 15 | GPDHC1 | Bra029669 | Both | 73 | 2 | A05 | A07, A09 | √ | – | – | – | – | √ | – | – | – | – | – | – | 3 |
| 16 | SDP6 | Bra035180 | Both | 69 | 3 | A07 | A05, A08, A09 | √ | – | √ | – | – | √ | – | – | – | – | – | – | 3 |
| 17 | MOD1 | Bra013159 | NetworkAnalyzer | 86 | 1 | A03 | A04, A05, A09 | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 18 | Triacylglycerol lipase | Bra007686 | NetworkAnalyzer | 86 | 1 | A09 | – | – | – | – | – | – | √ | – | – | – | – | – | – | 4 |
| 19 | CAC3 | Bra000037 | NetworkAnalyzer | 85 | 1 | A03 | – | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 20 | ACP dehydratase | Bra038539 | NetworkAnalyzer | 84 | 1 | A09 | A02 | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 21 | Stearoyl-ACP desaturase | Bra008631 | NetworkAnalyzer | 77 | 1 | – | A09 | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 22 | ACP1 | Bra039471 | NetworkAnalyzer | 77 | 1 | – | A09 | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 23 | ACBP4 | Bra039439 | NetworkAnalyzer | 75 | 1 | A05 | A01, A09 | – | – | – | – | – | √ | – | – | – | – | – | – | 2 |
| 24 | Lipases | Bra002174 | NetworkAnalyzer | 73 | 1 | – | A04 | – | – | – | – | – | √ | – | – | – | – | – | – | 4 |
| 25 | LTP5 | Bra038908 | NetworkAnalyzer | 71 | 1 | A01 | A06, A09 | – | – | – | – | – | √ | – | – | – | – | – | – | 6 |
| 26 | MD-2 | Bra001421 | WGCNA | 69 | 3 | A03 | A05, A09 | – | – | – | – | – | – | √ | – | √ | – | √ | – | 8 |
| 27 | WD-40 | Bra001726 | WGCNA | 62 | 5 | A03 | A05 | – | √ | – | √ | – | – | √ | √ | √ | – | – | – | 4 |
| 28 | LTP4 | Bra020323 | WGCNA | 52 | 3 | – | A06, A08, A09 | – | √ | – | – | – | – | – | √ | √ | – | – | – | 6 |
| 29 | ACBP3 | Bra019240 | WGCNA | 51 | 3 | A03 | A05, A07 | – | √ | – | – | – | – | – | √ | √ | – | – | – | 2 |
| 30 | LTP5 | Bra012847 | WGCNA | 31 | 3 | A03 | – | – | √ | – | – | – | – | – | √ | √ | – | – | – | 6 |
| 31 | Protein kinase | Bra034040 | WGCNA | 13 | 3 | – | A04, A10 | – | – | √ | – | √ | – | – | √ | – | – | – | – | 4 |
| 32 | mtACP2 | Bra035355 | WGCNA | – | 3 | – | A04 | – | – | √ | – | – | – | – | √ | √ | – | – | – | 2 |
| 33 | phosphoethanolamine NMT2 | Bra018740 | WGCNA | – | 5 | No eQTL | No eQTL | – | √ | √ | – | – | √ | – | √ | √ | – | – | – | 7 |
| 34 | Unknown | Bra001486 | WGCNA | – | 3 | No eQTL | No eQTL | – | – | √ | – | – | – | – | √ | √ | – | – | – | 5 |
| 35 | ATVPS34 | Bra027152 | WGCNA | – | 3 | No eQTL | No eQTL | – | √ | – | – | – | – | – | √ | √ | – | – | – | 5 |
Subnetworks were extracted for economically important FAs, erucic acid, oleic acid, linoleic acid, and linolenic acid, which were also the most predominant FAs in this population, as shown in Figure 1. All the FAs and gene nodes that were directly linked with each of these four metabolites were included in the subnetworks.
Oleic Acid (C18:1)
The MUFA oleic acid had faQTLs on A03, A05, and A10 in both years’ seed lots with explained variance ranging from 8% to 18%, plus additional QTLs on A01 and A02 in 2009 (Fig. 3; Supplemental Tables S3 and S4). In the oleic acid-centered subnetwork, approximately 19 genes were connected to oleic acid, with cis- and/or trans-acting regulation (Fig. 6A). The eQTLs of these genes colocalized with several oleic acid faQTLs. Overall, seven genes had a cis-eQTL and three genes had a trans-eQTL on A03 (Fig. 6A). This included two lipid degradation genes, two FA synthesis genes (among a total of three genes), an FA elongation gene, and three out of seven seed storage protein genes (Fig. 6A). Four genes had a cis-eQTL and four had a trans-eQTL on A05. These included two lipid degradation genes, BrPLA2-ALPHA and BrWD-40, one FA desaturation gene, BrFAD7, and the glycerol metabolism gene BrGPDHC1. Only one gene, BrGPAT6, had a trans-eQTL on A10 and also on A01, A03, and A09 (Fig. 6A). For some of the genes, a cis- or trans-eQTL was detected also on A01, A04, A07, A09, and A10 (Fig. 6A).
Figure 6.



FA-centered gene-metabolite coexpression subnetwork. A, Oleic acid. B, Erucic acid. C, Linoleic and linolenic acids. All the nodes that have a connection with an FA (oleic acid, erucic acid, and linoleic and linolenic acids) were extracted from the main network shown in Supplemental Figure S2. Multiple occurrence of the same gene name represents genes with multiple paralogs or probes.
Erucic Acid (C22:1)
The main FA, the MUFA erucic acid, had faQTLs on A03 and A09 for the 2011 seed lot with explained variance of 16% and 14%, respectively (Fig. 3; Supplemental Table S4). For the 2009 seed lot, putative faQTLs (LOD scores between 2 and 3) were detected on A03 and A09 as well, with additional faQTLs on A01, A02, A05, and A07 (Fig. 3; Supplemental Table S3). In the erucic acid-centered subnetwork, 16 out of the more than 50 genes had a cis- or trans-eQTL on A03 (Fig. 6B); in general, genes related to seed storage proteins, FA synthesis, and elongation and lipid degradation had cis-eQTLs, while genes related to phospholipid synthesis, lipid binding, and transcription factor had trans-eQTLs (Fig. 6B). On A09, six phospholipid synthesis and seed storage protein genes and BrCER10 (a very-long-chain FA elongation gene) had a cis-eQTL. Twenty-four genes, of which the majority are involved in lipid degradation, had a trans-eQTL on A09 (Fig. 6B).
Linoleic Acid (C18:2) and Linolenic Acid (C18:3)
faQTLs were mapped for the PUFAs linoleic and linolenic acids. A major faQTL of explained variance 36% to 45% was detected for linoleic acid in both experiments on A05, where we also mapped a gene-targeted marker for BrFAD2 and its cis-eQTL (Supplemental Tables S3 and S4). This faQTL overlapped with the eQTL hotspot on A05 (Figs. 3 and 4, right). In the linoleic and linolenic acids-centered subnetwork, more than 25 genes were associated with linoleic acid (Fig. 6C). Among these linoleic acid-associated genes, nine genes had a cis-eQTL and five genes had a trans-eQTL on A05. In general, the genes related to FA synthesis and elongation, phospholipids, lipid degradation (including BrPLA2-ALPHA), and FA desaturation (BrFAD7) had cis-eQTLs, while seed storage genes and a transcription factor (BrMD-2) had trans-eQTLs on A05 (Fig. 6C). Genes such as BrPLA2-ALPHA had a high degree of connection (more than 69 edges) based on their coexpression (r ≥ 0.5) with other genes and FAs (Table II).
In the case of linolenic acid, only three seed protein-encoding genes were connected: two with a cis-eQTL on A03 and one with trans-eQTLs on A04 and A09 (Fig. 6C), while its minor faQTLs were detected on A05, A08, and A10, so none of them colocalized (Fig. 3).
Genetic Coregulation of Erucic Acid, Oleic Acid, and Linoleic and Linolenic Acids
On A05, a major faQTL for linoleic acid and minor faQTLs for erucic and oleic acids were detected, together with major QTLs for the two PUFAs eicosadienoic acid and docosadienoic acid, colocating with an eQTL hotspot (Figs. 3 and 4, right). In the network analyses for each of these FAs, genes having either cis- or trans-acting eQTLs on A05 had a high degree of connection and also high correlations with these FAs (Table II; Fig. 5; Supplemental Fig. S2). These results suggest that these genes are likely major hub genes and have an essential role in the metabolic pathway of these FAs.
Two lipid degradation genes, BrPLA2-ALPHA and BrWD-40, one seed storage protein gene (Bra019067), and the transcription factor BrMD-2 were present in all three subnetworks of erucic, oleic, and linoleic and linolenic acids (Fig. 6). BrPLA2-ALPHA had a cis-eQTL on A05 and had the highest degree of connection with 91 nodes in the whole network in Cytoscape, while WGCNA (FA-centered network) also identified this gene as being associated with five FAs: the SFA myristic acid, the MUFA palmitoleic acid, and the PUFAs linoleic acid, eicosadienoic acid, and docosadienoic acid, each with an faQTL on A05.
On A03, QTLs for the three FAs erucic acid, oleic acid, and linoleic acid, plus additional QTLs for another four to five FAs, were mapped (Fig. 3). The seed storage protein-encoding gene (Bra019067), BrWD-40, and the transcription factor BrMD-2 had a cis-eQTL on A03; in addition, BrWD-40 and BrMD-2 genes had trans-eQTLs on A05 and A07 or A09 (Fig. 6), where additional faQTLs for one or more of these four FAs mapped. These genes had a high degree of connection (Table II). In WGCNA, the phospholipid synthesis gene BRASSICA RAPA LYSOPHOSPHATIDYL ACYLTRANSFERASE3 (BrLPAT3; Bra030448) had the highest degree of connection, and this gene had a cis-eQTL on A05, overlapping with the cis-eQTL of the BrPLA2-ALPHA gene and major faQTLs of the PUFAs linoleic acid (C18:2), eicosadienoic acid (C20:2), and docosadienoic acid (C22:2) and minor QTLs of the MUFAs palmitoleic acid (C16:1), oleic acid (C18:1), eicosenoic acid (C20:1), and erucic acid (C22:1; Table II; Fig. 3).
DISCUSSION
Seed FA composition and content per se are important for seed oil crops but also as a source of energy for the emerging seedling (Wang et al., 2010; Zhang et al., 2012). In this article, we describe the FA composition in seeds of a DH progeny from a cross between an oil-type and a vegetable-type B. rapa. We combined the QTLs for FA composition in seeds with eQTL analysis followed by gene coexpression network analysis with the aim to identify major regulatory genes.
Systems genetics has been widely used as an approach to integrate data at the metabolic and gene expression levels in segregating populations. Interestingly, major faQTLs for MUFAs, SFAs, and PUFAs mapped on different linkage groups on A03, A04, and A05, respectively (Fig. 3), which may suggest some level of genome organization according to the FA types (MUFAs, SFAs, and PUFAs). However, the biosynthetic processes of these FAs share part of their biochemical pathways; therefore, there could be common regulatory genes or genetic interactions in the regulation of these different FAs. This could be indicated by the fact that, in some cases, minor faQTLs of one type colocate with a major faQTL of a different FA type. In this study, we observed that SFAs in general were positively correlated with MUFAs, apart from erucic and nervonic acids, but PUFAs had a low correlation with SFAs and MUFAs (Fig. 2).
In genome-wide genetic studies, the presence of hidden confounding factors, such as unobserved covariates or unknown subtle environmental perturbations, can lead to spurious marker-trait associations or mask real genetic association signals. These confounding factors can be introduced or are inherent to the data at different steps while conducting experiments (Fusi et al., 2012). In this study, we observed the effects of two such confounding factors. The first was flowering time and, related to that, the timing of seed set and seed maturation on faQTLs (metabolite level) at A02 (colocalizing gene-targeted marker for BrFLC2 and a major flowering QTL; Figs. 1 and 3). Flowering time variation is very obvious in this DH population, and the BrFLC2 gene at A02 (16.7 cM) is the major regulator of flowering time, with a nonfunctional allele in the yellow sarson parent (Wu et al., 2012; Xiao et al., 2013). In the 2009 seed lot, when flowering was asynchronous, many faQTLs colocated with the BrFLC2 gene, which can point to pleiotropy or linkage of QTLs for flowering time and FAs. However, the synchronization of flowering time in the 2011 experiment removed this confounding effect on faQTL detection. The synchronization of flowering time of all DH lines resulted in similar environmental conditions during seed development, which is important to study the genetic variation of seed metabolites and seed quality-related traits. Other studies also reported the possibility of such confounding or pleiotropic effects of major genes on many developmental traits, for example at the ERECTA gene in Arabidopsis (Stinchcombe et al., 2009) and the EARLINESS locus in potato (Solanum tuberosum; Hurtado-Lopez, 2012). Despite the differences during seed development in 2009 and 2011 (asynchronous versus synchronous), very high correlations between the two seed lots (2009 and 2011) for each FA across the DH genotypes were observed (Fig. 2). Major faQTLs were also always detected in both years, while minor faQTLs varied between years.
The second confounding factor was the effect of seed color on eQTLs (transcript level) at A09 (Fig. 4, left). In addition to variation in morphotypes, and, as a result, in many other morphological and biochemical traits (Zhao et al., 2005; Pino Del Carpio et al., 2011; Xiao et al., 2013, 2014), the yellow sarson and pak choi parents have contrasting seed coat color (Basnet et al., 2013). A strong seed color QTL with 33% explained variance was mapped on A09 (data not shown); the causal gene, the basic helix-loop-helix transcription factor BrTT8, was cloned, and its role in seed color was functionally validated in B. rapa (Li et al., 2012) and in other species (Padmaja et al., 2014). At this BrTT8 position, a large trans-eQTL hotspot was mapped, even after correction for seed color (Fig. 4, left). In contrast with the eQTL hotspot, only minor faQTLs for erucic acid and eicosadienoic acid and a few additional minor faQTLs colocalized with this seed color QTL (Fig. 3). A possible explanation for the confounding effect of seed color only at the transcript level but not at the metabolite level might be the fact that genetic regulation of the metabolic process is not always completely hierarchical in translating gene expression variation to metabolite regulation. ter Kuile and Westerhoff (2001) reported a lack of hierarchical genetic control over metabolic flux in their study on the regulation of the glycolytic pathway. Additionally, the absence of a strong one-transcript, one-metabolite relationship is quite common in the regulation of biological processes due to the complexity involved in the extrapolation of gene expression variation to changes in metabolite content, such as posttranscriptional modification and epigenetic regulation. Another explanation could be that FA metabolites and transcripts were measured in different developmental stages: in mature ripe seeds and in developing seeds (28 d after pollination), respectively, which could lead to different levels of interactions at different stages of seed development.
In this genetical genomics study, we were able to subset only those genes that have eQTLs, detect eQTL hotspots, and identify cis- and trans-acting eQTLs (Fig. 4). Following this genetical genomics approach, an eQTL-guided gene coexpression network was constructed that allowed us to identify (possible) candidate genes and their regulatory interactions for lipid metabolism.
Genes with a high degree of connection in a network could possibly be major regulators of a pathway. Those genes could be essential genes in the sense that variation in these genes could change the pathway. In contrast, genes with a lower degree of connection could play a role in modifying FA content or composition. From the two types of network analyses carried out in this study (using Cytoscape and WGCNA), the top 25 genes with a high degree of connection were selected, which are likely to be key drivers in lipid metabolism; 16 genes were in common between these two lists, illustrating that these approaches were quite effective in selecting the most essential genes. These 16 genes belong to pathways such as lipid degradation, FA synthesis and elongation, phospholipid synthesis, glycerol metabolism, and lipid transfer proteins (Table II). Interestingly, those top-ranking genes were from different pathways, indicating an extensive coordination among biosynthetic pathways in lipid metabolism in B. rapa seed. Those top 16 genes had cis- and trans-eQTLs mainly colocalized with major faQTLs on A03, A05, and A09 (Table II), suggesting that those regions harbor the possible key regulator genes. Among the top selected genes, the lipid degradation pathway gene BrPLA2-ALPHA (Bra038125) had the highest degree of connection and a cis-eQTL on A05, colocating with major PUFA faQTLs for linoleic acid (C18:2) and eicosadienoic acid (C20:2) and minor faQTLs for other FAs (Table II). Ryu et al. (2005) functionally characterized the BrPLA2-ALPHA gene and concluded that it has an acyl preference for linoleic acid over palmitic acid in phospholipid hydrolysis in Arabidopsis. They also reported a role of this gene in the release of free FAs and lysophospholipids from membrane phospholipids. However, we have not found any other study reporting this gene as a potential regulator of seed FA composition in Arabidopsis, B. napus, or other oil crops. However, many studies did report that cis-eQTLs for master regulator genes mapped under trans-eQTL hotspots (Civelek and Lusis, 2014; Wang et al., 2014a), similar to what we found for BrPLA2-ALPHA on A05.
The explanation that such key regulator genes, like BrPLA2-ALPHA in this study, are generally not reported as potential regulators of, in our case, seed FA composition is that these genes are often highly conserved in regulatory networks during evolution (Khurana et al., 2013) and are less likely to be genetically perturbed in mapping populations (Mäkinen et al., 2014). In our study, we still found a cis-eQTL for the highly connected gene BrPLA2-ALPHA, which might be due to the different selection history of oil and vegetable types.
Additional genes from the top 25 were genes in the FA synthesis and FA elongation pathways, such as BrCAC3, BrMOD1, ACP dehydratase, and two paralogs of ACP desaturase, BrACP1 and BrACBP4 (Table II), whose functional roles are described for converting acetyl-CoA to malonyl-CoA chain at the beginning of the pathway in the Kyoto Encyclopedia of Genes and Genome pathway database.
The MUFA erucic acid was the most abundant FA (47%–55.8% of total dry weight) in both parents (yellow sarson and pak choi) and their DH progeny (Fig. 1; Supplemental Table S1). Lühs et al. (1999) also reported high erucic acid content (54.8%) in yellow sarson seeds. Breeding for low erucic acid can cause a decrease in the total oil content if low erucic acid is not compensated for by an increase of other FAs. Even though the MUFA oleic acid is a substrate for both erucic acid and for the PUFA linoleic acid, oleic acid was strongly negatively correlated only with linoleic acid but not with erucic acid (Fig. 2). Linoleic acid shares the genomic region of its major faQTL on A05 (36%–46% explained variance; yellow sarson effect) with a QTL for its precursor oleic acid (9%–13% explained variation) and the chain-elongated erucic acid (9% explained variation) with opposite allelic effects (Fig. 3; Supplemental Tables S3 and S4), suggesting that these FAs share regulatory elements. Erucic acid, oleic acid, linoleic acid, and linolenic acid are the most predominant FAs (Fig. 1) and are economically important for oil quality. Therefore, we further looked into eQTL-guided coexpression networks particularly associated with those four FAs (called subnetworks), with the aim to unravel the underlying gene regulatory networks. The six genes BrPLA2-ALPHA, BrWD-40, three seed storage genes (Bra024983, Bra019067 and Bra024983), and the transcription factor BrMD-2 were in common among all three subnetworks (Fig. 6), indicating essential roles of these genes in the biosynthesis of these FAs. BrWD-40 has a protein domain that regulates chromatin dynamics and the transcription of the evolutionarily well-conserved gene ADIPOSE (ADP). The loss-of-function allele of ADP promotes triacylglycerol storage in Drosophila spp. flies (Häder et al., 2003). MD-2 also has a conserved domain present across plants, animals, and bacteria and is involved in lipopolysaccharide binding (Inohara and Nuñez, 2002). Most of the genes are shared by at least two of the three subnetworks (Fig. 6) and might have pleiotropic effects on lipid metabolism. There were also many genes present only in the erucic acid subnetwork (Fig. 6B). Erucic acid has a larger gene metabolite coexpression network (more than 50 genes) than oleic acid (approximately 20 genes) and linoleic and linolenic acids (more than 25 genes) and also has many minor faQTLs (Fig. 6), implying a polygenic inheritance and more complex gene network (Fig. 6B). For an eQTL-based gene network, the genetic composition of the mapping population could pose a limiting factor in selecting genes for network construction; therefore, QTL mapping for FAs and gene expression in multiple populations from genetically diverse parents could confirm our network components.
Well-studied genes such as FAE1, TAG1, and FAD2 in Arabidopsis, B. napus, and other oilseed crops were reported as candidate genes for the synthesis of linoleic acid, linolenic acid, erucic acid, oleic acid, or total oil content (Peng et al., 2010; Yang et al., 2012; Lee et al., 2013). Cis-eQTL of BrFAD2 and other BrFAD genes (BrFAD5 and BrFAD7) and trans-eQTL of BrFAE1 and BrTAG1 genes colocalized with faQTLs for oleic acid (C18:2), linoleic acid (C18:2), erucic acid (C22:1), and other FAs (Fig. 3; Supplemental Fig. S1). It is likely that those genes are regulated by some of the key regulators present on A05; those genes were not highlighted in the network analyses due to their low degrees of connection, but they could play a role in modifying FA content or composition. The expression profile of BrFAD7 was correlated with linoleic acid and oleic acid content, while BrFAD5 was correlated with erucic acid content (Supplemental Fig. S2). BrFAD2 had only a weak correlation (r < 0.5) with any of the FAs, but its expression was correlated with that of BrFAD5 and BrFAD7 (r > 0.5). Several studies in B. napus and Arabidopsis reported that BrFAD2 regulates the conversion of oleic acid to linoleic acid in the endoplasmic reticulum, while BrFAD5 desaturates C18:0 acyl carrier protein to oleic acid and BrFAD7 desaturates linoleic acid to linolenic acid in plastids (Zhang et al., 2012). Therefore, there could be interactive roles among BrFAD genes.
In conclusion, we were able to identify major regulatory genes involved in the genetic regulation of lipid metabolism and those genes belonging to the different lipid metabolic pathways: lipid degradation, FA synthesis and elongation, phospholipid synthesis, glycerol metabolism, transfer protein, signaling, and very-long-chain elongation (Table II). Those results suggest the need for a global study of lipid metabolism rather than a strict focus on the FA biosynthesis pathway per se. This study gives a starting point for understanding the genetic regulation of lipid metabolism by the identification of a number of key regulatory genes, identified as major hub genes, and candidate genes for faQTLs. Finally, the data generated in this study will be valuable in Brassica spp. breeding, as they offers tools to breed for optimal oil composition.
MATERIALS AND METHODS
Plant Materials and Growth Conditions
A Brassica rapa DH population of 163 DH lines developed from a cross of a yellow sarson (YS143; accession no. FIL500) as a female parent and a pak choi (PC175; cv Nai Bai Cai; accession no. VO2B0226) as a male parent was used in this study (Basnet et al., 2013; Xiao et al., 2013). YS143 is a self-compatible annual oil crop with yellow seed color, while PC175 is a self-incompatible leafy vegetable with brown/black seed color. These two parents differ in seed size, seed color, oil and FA content, and many morphological and developmental traits (Zhao et al., 2005; Pino Del Carpio et al., 2011; Basnet et al., 2013).
The experiments were carried out in two years, 2009 and 2011, using two different experimental designs, without and with synchronization of time of flowering of the DH lines, respectively. In both years, seeds of DH lines and parents were sown in pressed soil cubes of a standard soil mixture of 85% peat and 15% clay (Lentse Potgrond no. 4; Lentse Potgrond) for germination in a greenhouse at the Unifarm facility of Wageningen University. Two plants per DH plus parents were transplanted to plastic pots (diameter, 17 cm) filled with the same standard soil mixture in the greenhouse, and later, only one plant was kept for harvesting developing seeds. In 2009, seeds were sown on March 27 and DH lines flowered over a period from the first week of May to the second week of June. In 2011, the population was evaluated again, but this time, the lines were sown staggered at different dates from the second week of January to the last week of February to synchronize the flowering of the lines. The aim of synchronizing flowering time is to avoid different environmental conditions during seed development. All lines flowered during the first two weeks of April. The ripe seeds were harvested per plant, dried, stored in a certified manner (ISO-certified method 9001:2008) at 13°C temperature and 30% relative humidity, and later used for FA measurements. Transcript abundance measurements were done in the developing seeds of the DH lines from 2011.
FA Measurements
About 0.2 to 2 g of seeds was used to determine oil content using near-infrared reflectance spectroscopy (Foss NIRS system; Tillmann, 1997) and calibrated with oil seed extracted with hexane following the standard protocol described by Raney et al. (1987). The oil content was calculated as a mass percentage of whole seed dry matter (zero moisture). Seed oil was analyzed for FA composition using gas chromatography following the preparation of FA methyl esters by base-catalyzed methanolysis (Thies, 1971). The individual FAs were reported as mass percentages of total FAs.
These relative contents of 18 FAs were studied in mature, dry seeds from 2009 and 2011 seed lots. In 2009, FAs were measured in 135 DH lines; due to a lack of availability of seeds, in 2011, only of a subset of 92 DH lines and two to three biological replicates of each of the two parents were studied. The 18 FAs consisted of seven SFAs, five MUFAs, and six PUFAs (Supplemental Table S1). Unidentified FAs were categorized as other FAs. Lauric acid (C12:0) was excluded from the data analysis because its concentration was below the detection limit in almost all DH lines, including the parents.
RNA Isolation for Gene Expression Studies
In earlier studies, we observed that at 28 d after pollination, a large subset of genes related to lipid metabolism was differentially expressed between the two parents as well as between two selected DH lines (Basnet et al., 2013). Therefore, siliques from a subset of 118 DH lines from the 2011 experiment, selected on the basis of genotypic contrast, were harvested at 28 d after pollination and kept in liquid nitrogen (−196°C); seeds were taken from the siliques under dry ice, and around 150 to 200 seeds were ground in liquid nitrogen (−196°C) using RNase-free mortar and pestle. Seeds and RNA samples were stored at −80°C. Since Brassica spp. seeds have high concentrations of oils, organic acids, and proteins, 5% (w/v) polyvinylpyrrolidone-40 (Sigma) was added to RLC lysis buffer (Qiagen) and kept overnight at 65°C to dissolve properly. After adding RLC lysis buffer in each tube, the powdered seed materials were incubated for 30 min at 65°C in a water bath. The total RNA was extracted with the RNeasy Plant Mini Kit (Qiagen) following the manufacturer’s instructions and purified using the RNA Clean-up protocol for RNeasy columns (RNeasy Mini Kit; Qiagen) with on-column digestion with the DNase Kit (Qiagen) to remove residual genomic DNA. The quantity of RNA samples was measured by using an ND-100 UV-visible light spectrophotometer (NanoDrop Technologies), and quality was assessed by A260/A280 and A260/A230 ratios (NanoDrop Technologies) and by 1% agarose gels.
Distant Pair Design and Microarray Hybridization
A distant pair design was used to find an optimal combination of pairs of DH lines with maximum genetic contrast for the microarray analysis (Fu and Jansen, 2006). Missing marker data were imputed based on the genetic positions of flanking markers using the R/qtl package (Broman et al., 2003). Using the marker information, the pairs of DH lines to hybridize on an array were designed using the R package designGG (Li et al., 2009). The copy RNA samples were labeled with Cy3 (green) and Cy5 (red) dyes using the QuickAmp Labeling Kit (Agilent Technologies) and hybridized on our 8- × 60-K custom-made B. rapa array in a two-color Agilent platform as described by Basnet et al. (2013). Arrays were then washed and scanned on an Agilent scanner, according to the manufacturer’s instructions. Data files were generated using the Agilent Feature Extraction Software (version 10.10.1.1). In total, 59 arrays for 118 DH lines and four arrays for two parents and their biological replicates were used. The raw data were normalized without background correction, using loess for within-array normalization and quantile normalization between arrays using the limma package in R (Smyth, 2005; R Core Team, 2012).
Gene Expression Measurements by RT-qPCR
The transcript abundance of 21 genes, including genes for FA synthesis and FA elongation, FA desaturation, lipid degradation, seed storage proteins, and lipid degradation, was measured using RT-qPCR to validate microarray transcript abundance using complementary DNA from 28-d developing seeds collected in 2011. Genes in each pathway were selected based on the literature, and primers for each gene and for the reference gene are listed in Supplemental Table S2. The genes BrFLC2 and BrTT8 were also included because this population segregates for flowering time and seed color, which both have confounding effects on QTL mapping for FAs and transcript abundance. RT-qPCR was performed with paralog-specific primers for the genes that have paralogs. The detailed procedure was as described by Xiao et al. (2013); we used the β-actin gene as a reference gene to estimate the normalized gene expression (∆∆CT) of each gene and each sample.
QTL Mapping of FAs (faQTL) and Transcript Abundance (eQTL)
In this study, an integrated genetic map was constructed (Xiao et al., 2013) and used for QTL mapping of FAs and transcript abundance in developing seeds. This integrated map comprised 435 molecular markers: amplified fragment length polymorphism, myb-targeted, microsatellite (single sequence repeat), and gene-targeted markers from the flowering time, FA, and glucosinolate pathways. QTL interval mapping was performed for 17 FAs from the two years’ seed lots with the R/qtl package (Broman et al., 2003), followed by multiple QTL mapping with marker cofactors. Initially, cofactors were selected from the peak markers of significant QTLs in interval mapping. After that, a backward elimination was performed to select the final set of cofactors. LOD thresholds for the significance of QTLs were determined at the 95th percentile of 10,000 permutations of each of the FAs. For QTL analysis, FA abundance was transformed using either a reciprocal or a log transformation, depending on the observed distribution of FA abundance values. In the 2009 seed lot, a log transformation was done for stearic acid while a reciprocal transformation was used for arachidic acid, behenic acid, lignoceric acid, and palmitoleic acid. For the 2011 seed lot, a reciprocal transformation was done for palmitic acid, stearic acid, arachidic acid, and behenic acid and a log transformation was done for lignoceric acid. The same procedure as described above for mapping QTLs for FAs was followed to map eQTLs for the transcript abundance of genes measured using RT-qPCR.
eQTL analysis was performed using single marker regression analysis as proposed by Fu and Jansen (2006). This method relates the log ratios of each probe (transcript abundance contrast of a pair of genotypes) to DNA markers on the linkage map. The following regression model was used to regress the log ratios of each probe against each marker as:
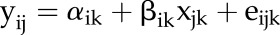 |
where yij is the log ratio of transcript abundance of pair j for gene i and xjk denotes the marker allele contrast for the pair j at marker k, with the following marker values: 1 for yellow sarson/pak choi, −1 for pak choi/yellow sarson, and 0 for yellow sarson/yellow sarson and pak choi/pak choi. The regression coefficient βik represents the allele substitution effect at marker k for probe i. The intercept αik is also estimated in the regression approach, but it should be close to zero unless there is a dye bias; eijk is the residual error.
In total, 61,551 probes (representing 40,904 B. rapa gene models, called BraID) for the two-color Agilent microarray platform were designed using gene models predicted based on the reference genome sequence of B. rapa ‘Chiifu’ (a vegetable-type inbred line), published by Wang et al. (2011). A slightly modified version of the array designed for a microarray study by Basnet et al. (2013) was used in this study. All probes were annotated into 35 functional categories or BINS, as defined by MapMan software. However, only 1,568 probes related to lipid metabolism, lipid signaling, lipid storage proteins, and lipid transfer proteins were extracted and subjected to eQTL analysis in this study. Significant eQTLs were declared using a genome-wide threshold of α = 0.001 [or −log10(0.001) = 3]. The −log10(P value) are here denoted as LOP values (to increase readability); the interpretation of the LOP score is different from a LOD score as used in the faQTL analysis, since the LOD is obtained by likelihood estimation whereas the LOP is from least-squares estimation using per-marker regression. The estimated regression coefficients of markers (β) represent the estimated additive effects of hypothetical QTLs at the marker position; the sign of β gives the direction of the effect of a parental allele (with a positive value indicating a higher mean for the yellow sarson allele and a negative value indicating a higher mean for the pak choi allele).
Correction for the Effect of Seed Color on eQTL Mapping
The two parents were contrasting in seed color, YS143 being yellow seeded and PC175 being black seeded. Yellow-colored seeds are associated with a high content of oil and protein and low fiber in the meal of Brassica spp. oil seeds (Chen and Heneen, 1992; Rahman et al., 2001). Since a large number of eQTLs were mapped on A09 in the vicinity of a major QTL for seed color, we were also interested in the eQTL results after correcting for a possibly confounding effect of seed color in addition to the results without such a correction. The correction for seed color was done by regressing transcript abundance (per probe) on the image pixel size of color intensity (a quantitative score of seed color) from image analysis. A higher intensity indicates yellow seed coat color, and a lower intensity indicates a black seed coat color. The residuals of transcript abundance of each probe were then used for eQTL mapping, instead of the original values.
Cis- and Trans-eQTLs
In this study, an eQTL was defined as cis-acting if the eQTL was observed in the same linkage group of the physical position of the probe. An eQTL that was mapped to another linkage group was defined as a trans-eQTL. This broad definition is likely to result in an overestimation of the number of cis-eQTLs, since also distant eQTLs on the same chromosome are now considered to be cis-regulated. On the other hand, it could result in a possible underestimation of the total number of eQTLs and trans-eQTLs if more eQTLs were on the same chromosome.
Significance of eQTL Hotspots
The significance of the presence of eQTL hotspots at each genetic marker was tested using the hotspots package in R software (Darrouzet-Nardi, 2012). First, the number of eQTLs (LOP > 3) was counted at each genetic marker. The hotspots package then uses this eQTL number as its input variable. This package calculates a hotspot cutoff for the eQTL number distribution across the genome based on deviance from the normal distribution of a variable, the number of eQTLs in this case. Statistical as well as computational details of this package are as described by Darrouzet-Nardi (2012).
Construction of Coexpression Networks of Genes and FAs
All the probes that were annotated as being involved in lipid metabolic processes in the MapMan annotation (Usadel et al., 2005; Basnet et al., 2013) were selected for eQTL mapping, and among those selected probes, only the probes with an eQTL (LOP > 3) were used to calculate Pearson correlation coefficients in R software (R Core Team, 2012) and for the construction of a correlation network using Cytoscape (Shannon et al., 2003). For correlation-based coexpression analysis, gene expression measured by RT-qPCR as well as the relative abundance of 17 different FAs were included. FAs from the 2011 experiment were used for the construction of this gene-FA coexpression network because the RNA transcripts were also measured from the 2011 seed lot. For network visualization, correlation coefficients were considered to be significant at an absolute value of 0.3 at P < 0.05. In the network, nodes represent probes or metabolites and edges represent significant correlation coefficients. Since all the genes are from the lipid metabolism, most were significantly correlated to each other, which makes it difficult to visualize the network due to the large number of edges. To increase the visibility of the network, only absolute correlation coefficients greater than 0.5 were shown. A NetworkAnalyzer plugin available in Cytoscape was used to calculate relevant network parameters, such as the degree of connection (Assenov et al., 2008). The degree of connection measures the number of incoming and outgoing edges of a node. A higher degree of connection indicates a node with a higher number of edges, which identifies a gene as a major hub of the network; these genes may represent major regulating genes of the pathway.
WGCNA to Correlate Gene Expression to FA Abundance
A weighted correlation-based network was used to complement the correlation coexpression network. Unlike the correlation coexpression network, in the WGCNA approach, not only the probes with an eQTL were used but all the genes related to lipid metabolism. WGCNA constructs a network based on correlation coefficients between genes (expression values after correction) and further classifies gene modules (groups of highly correlated genes). Finally, this method calculates the significance of the association of gene modules and each FA. A detailed description of the WGCNA approach was given by Horvath and Dong (2008), and the analysis was performed in R software using the WGCNA package (Langfelder and Horvath, 2008). In this WGCNA approach, the network was constructed based on significant associations of genes with FAs. The number of times that a probe was related to different FAs was calculated and compared with the degree of connection as calculated from the correlation coexpression network.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Heat map showing the eQTL LOD scores for gene expression of the probes measured by RT-qPCR.
Supplemental Figure S2. Gene-metabolite coexpression network using probes with at least one eQTL (after correction for seed color).
Supplemental Table S1. Summary statistics of FAs measured in ripe seeds of the B. rapa DH population, including the parents yellow sarson and pak choi, in the 2009 and 2011 seed lots.
Supplemental Table S2. List of genes with their primer sequences used in RT-qPCR.
Supplemental Table S3. Summary of QTLs detected for the relative abundance of FAs from the 2009 seed lot.
Supplemental Table S4. Summary of QTLs detected for the relative abundance of FAs from the 2011 seed lot.
Supplementary Material
Acknowledgments
We thank the members of the Unifarm facility of Wageningen University and Research Centre for taking care of the plants and all the necessary support as well as students and colleagues for their assistance during pollination.
Glossary
- FA
fatty acid
- QTL
quantitative trait locus
- eQTL
expression quantitative trait locus
- DH
doubled haploid
- MUFA
monounsaturated fatty acid
- PUFA
polyunsaturated fatty acid
- SFA
saturated fatty acid
- faQTL
quantitative trait locus for fatty acids
- LOD
log of the odds
- cM
centimorgan
- LOP
−10log of the P value
- RT-qPCR
reverse transcription-quantitative PCR
- WGCNA
weighted gene coexpression network analysis
Footnotes
This work was supported by the Center for BioSystems Genomics, which is a part of the Netherlands Genomics Initiative.
References
- Assenov Y, Ramírez F, Schelhorn SE, Lengauer T, Albrecht M (2008) Computing topological parameters of biological networks. Bioinformatics 24: 282–284 [DOI] [PubMed] [Google Scholar]
- Basnet RK, Moreno-Pachon N, Lin K, Bucher J, Visser RGF, Maliepaard C, Bonnema G (2013) Genome-wide analysis of coordinated transcript abundance during seed development in different Brassica rapa morphotypes. BMC Genomics 14: 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisson F, Koo AJK, Ruuska S, Schwender J, Pollard M, Thelen JJ, Paddock T, Salas JJ, Savage L, Milcamps A, et al. (2003) Arabidopsis genes involved in acyl lipid metabolism: a 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a Web-based database. Plant Physiol 132: 681–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890 [DOI] [PubMed] [Google Scholar]
- Chalhoub B, Denoeud F, Liu S, Parkin IAP, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, et al. (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345: 950–953 [DOI] [PubMed] [Google Scholar]
- Chen B, Heneen W (1992) Inheritance of seed colour in Brassica campestris L. and breeding for yellow-seeded B. napus L. Euphytica 59: 157–163 [Google Scholar]
- Chen J, Tan RK, Guo XJ, Fu ZL, Wang Z, Zhang ZY, Tan XL (2015) Transcriptome analysis comparison of lipid biosynthesis in the leaves and developing seeds of Brassica napus. PLoS ONE 10: e0126250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek M, Lusis AJ (2014) Systems genetics approaches to understand complex traits. Nat Rev Genet 15: 34–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrouzet-Nardi A (2012) hotspots: an R package version 1.0.2. http://CRAN.R-project.org/package=hotspots
- Fu J, Jansen RC (2006) Optimal design and analysis of genetic studies on gene expression. Genetics 172: 1993–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi N, Stegle O, Lawrence ND (2012) Joint modelling of confounding factors and prominent genetic regulators provides increased accuracy in genetical genomics studies. PLoS Comput Biol 8: e1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschina IA, Harwood JL (2007) Complex lipid biosynthesis and its manipulation in plants. In Ranalli P, ed, Improvement of Crop Plants for Industrial End Uses. Springer, Dordrecht, The Netherlands, pp 253–279 [Google Scholar]
- Häder T, Müller S, Aguilera M, Eulenberg KG, Steuernagel A, Ciossek T, Kühnlein RP, Lemaire L, Fritsch R, Dohrmann C, et al. (2003) Control of triglyceride storage by a WD40/TPR-domain protein. EMBO Rep 4: 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JP, Mayes S, Bowen HC, Graham NS, Hayden RM, Love CG, Spracklen WP, Wang J, Welham SJ, White PJ, et al. (2011) Regulatory hotspots are associated with plant gene expression under varying soil phosphorus supply in Brassica rapa. Plant Physiol 156: 1230–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Dong J (2008) Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol 4: e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Lopez P (2012) Investigating genotype by environment and QTL by environment interactions for developmental traits in potato. PhD thesis. Wageningen University, Wageningen, The Netherlands [Google Scholar]
- Inohara N, Nuñez G (2002) ML: a conserved domain involved in innate immunity and lipid metabolism. Trends Biochem Sci 27: 219–221 [DOI] [PubMed] [Google Scholar]
- Jansen RC, Nap JP (2001) Genetical genomics: the added value from segregation. Trends Genet 17: 388–391 [DOI] [PubMed] [Google Scholar]
- Kadkol G, Beilharz V, Halloran G, Macmillan R (1986) Anatomical basis of shatter-resistance in the oilseed Brassicas. Aust J Bot 34: 595–601 [Google Scholar]
- Karim MM, Siddika A, Tonu NN, Hossain DM, Meah MB, Kawanabe T, Fujimoto R, Okazaki K (2014) Production of high yield short duration Brassica napus by interspecific hybridization between B. oleracea and B. rapa. Breed Sci 63: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chen J, Gerstein M (2013) Interpretation of genomic variants using a unified biological network approach. PLoS Comput Biol 9: e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Anubha, Vishwakarma M, Lal J (2011) Morphological and molecular characterization of Brassica rapa ssp yellow sarson mutants. Journal of Oilseed Brassica 2: 1–6 [Google Scholar]
- Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, et al. (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA 107: 8063–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KR, In Sohn S, Jung JH, Kim SH, Roh KH, Kim JB, Suh MC, Kim HU (2013) Functional analysis and tissue-differential expression of four FAD2 genes in amphidiploid Brassica napus derived from Brassica rapa and Brassica oleracea. Gene 531: 253–262 [DOI] [PubMed] [Google Scholar]
- Li X, Chen L, Hong M, Zhang Y, Zu F, Wen J, Yi B, Ma C, Shen J, Tu J, et al. (2012) A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE 7: e44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Swertz MA, Vera G, Fu J, Breitling R, Jansen RC (2009) designGG: an R-package and web tool for the optimal design of genetical genomics experiments. BMC Bioinformatics 10: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Wang CM, Li L, Sun F, Liu P, Yue GH (2011) Mapping QTLs for oil traits and eQTLs for oleosin genes in jatropha. BMC Plant Biol 11: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IAP, Zhao M, Ma J, Yu J, Huang S, et al. (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun 5: 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lühs WW, Voss A, Seyis F, Friedt W (1999) Molecular genetics of erucic acid content in the genus Brassica In Wratten N, Salisbury P, eds, New Horizons for an Old Crop: Proceedings of the 10th International Rapseed Congress, Canberra, Australia. Regional Institute Limited, Gosford, New South Wales, Australia [Google Scholar]
- Mäkinen VP, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segrè AV, Ghosh S, Vivar J, et al. (2014) Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet 10: e1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmaja LK, Agarwal P, Gupta V, Mukhopadhyay A, Sodhi YS, Pental D, Pradhan AK (2014) Natural mutations in two homoeologous TT8 genes control yellow seed coat trait in allotetraploid Brassica juncea (AABB). Theor Appl Genet 127: 339–347 [DOI] [PubMed] [Google Scholar]
- Peng FY, Weselake RJ (2011) Gene coexpression clusters and putative regulatory elements underlying seed storage reserve accumulation in Arabidopsis. BMC Genomics 12: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Hu Y, Wei R, Zhang Y, Guan C, Ruan Y, Liu C (2010) Simultaneous silencing of FAD2 and FAE1 genes affects both oleic acid and erucic acid contents in Brassica napus seeds. Plant Cell Rep 29: 317–325 [DOI] [PubMed] [Google Scholar]
- Pino Del Carpio D, Basnet RK, Arends D, Lin K, De Vos RCH, Muth D, Kodde J, Boutilier K, Bucher J, Wang X, et al. (2014) Regulatory network of secondary metabolism in Brassica rapa: insight into the glucosinolate pathway. PLoS ONE 9: e107123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino Del Carpio D, Basnet RK, De Vos RCH, Maliepaard C, Visser R, Bonnema G (2011) The patterns of population differentiation in a Brassica rapa core collection. Theor Appl Genet 122: 1105–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Meng J, Li M, Frauen M, Sass O, Noack J, Jung C (2006) Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus L.), with emphasis on the evolution of Chinese rapeseed. Theor Appl Genet 113: 49–54 [DOI] [PubMed] [Google Scholar]
- R Core Team (2012) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna [Google Scholar]
- Rahman MH, Joersbo M, Poulsen MH (2001) Development of yellow-seeded Brassica napus of double low quality. Plant Breed 120: 473–478 [Google Scholar]
- Raney JP, Love HK, Rakow GFW, Downey RK (1987) An apparatus for rapid preparation of oil and oil-free meal from Brassica seed. Lipid 89: 235–237 [Google Scholar]
- Ruuska SA, Girke T, Benning C, Ohlrogge JB (2002) Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 14: 1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SB, Lee HY, Doelling JH, Palta JP (2005) Characterization of a cDNA encoding Arabidopsis secretory phospholipase A2-α, an enzyme that generates bioactive lysophospholipids and free fatty acids. Biochim Biophys Acta 1736: 144–151 [DOI] [PubMed] [Google Scholar]
- Sanyal A, Randal Linder C (2012) Quantitative trait loci involved in regulating seed oil composition in Arabidopsis thaliana and their evolutionary implications. Theor Appl Genet 124: 723–738 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. (2005) Limma: linear models for microarray data. In R Gentleman, V Carey, W Huber, R Irizarry, S Dudoit, eds, Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer, New York, pp 397–420 [Google Scholar]
- Stinchcombe JR, Weinig C, Heath KD, Brock MT, Schmitt J (2009) Polymorphic genes of major effect: consequences for variation, selection and evolution in Arabidopsis thaliana. Genetics 182: 911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanhuanpää P, Schulman A (2002) Mapping of genes affecting linolenic acid content in Brassica rapa ssp. oleifera. Mol Breed 10: 51–62 [Google Scholar]
- ter Kuile BH, Westerhoff HV (2001) Transcriptome meets metabolome: hierarchical and metabolic regulation of the glycolytic pathway. FEBS Lett 500: 169–171 [DOI] [PubMed] [Google Scholar]
- Thies W. (1971) Schnelle und einfache Analysen der Fettsäurezusammensetzung in einzelnen Raps-Kotyledonen. 1. Gaschromatographische und papierchromatographische Methoden. Z Pflanzenzuchtg 65: 181–202 [Google Scholar]
- Tillmann P. (1997) Recent experiences with NIRS analysis in rapeseed. GCIRC Bull 13: 84–87 [Google Scholar]
- Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D, et al. (2005) Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol 138: 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yu H, Weng X, Xie W, Xu C, Li X, Xiao J, Zhang Q (2014a) An expression quantitative trait loci-guided co-expression analysis for constructing regulatory network using a rice recombinant inbred line population. J Exp Bot 65: 1069–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Liu M, Li D, Wu J, Li X, Yang Y (2010) Overexpression of FAD2 promotes seed germination and hypocotyl elongation in Brassica napus. Plant Cell Tissue Organ Cult 102: 205–211 [Google Scholar]
- Wang X, Jiang GL, Green M, Scott RA, Hyten DL, Cregan PB (2014b) Quantitative trait locus analysis of unsaturated fatty acids in a recombinant inbred population of soybean. Mol Breed 33: 281–296 [Google Scholar]
- Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, et al. (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43: 1035–1039 [DOI] [PubMed] [Google Scholar]
- Wu J, Wei K, Cheng F, Li S, Wang Q, Zhao J, Bonnema G, Wang X (2012) A naturally occurring InDel variation in BraA.FLC.b (BrFLC2) associated with flowering time variation in Brassica rapa. BMC Plant Biol 12: 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Wang H, Basnet RK, Zhao J, Lin K, Hou X, Bonnema G (2014) Genetic dissection of leaf development in Brassica rapa using a genetical genomics approach. Plant Physiol 164: 1309–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Zhao JJ, Hou XL, Basnet RK, Carpio DPD, Zhang NW, Bucher J, Lin K, Cheng F, Wang XW, et al. (2013) The Brassica rapa FLC homologue FLC2 is a key regulator of flowering time, identified through transcriptional co-expression networks. J Exp Bot 64: 4503–4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Li J, Wang R, Jin M, Chen L, Qian W, Wang X, Liu L (2011) Mapping of QTLs controlling content of fatty acid composition in rapeseed (Brassica napus). Genes Genomics 33: 365–371 [Google Scholar]
- Yang Q, Fan C, Guo Z, Qin J, Wu J, Li Q, Fu T, Zhou Y (2012) Identification of FAD2 and FAD3 genes in Brassica napus genome and development of allele-specific markers for high oleic and low linolenic acid contents. Theor Appl Genet 125: 715–729 [DOI] [PubMed] [Google Scholar]
- Yu B, Gruber M, Khachatourians GG, Hegedus DD, Hannoufa A (2010) Gene expression profiling of developing Brassica napus seed in relation to changes in major storage compounds. Plant Sci 178: 381–389 [Google Scholar]
- Zhang J, Liu H, Sun J, Li B, Zhu Q, Chen S, Zhang H (2012) Arabidopsis fatty acid desaturase FAD2 is required for salt tolerance during seed germination and early seedling growth. PLoS ONE 7: e30355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Wang X, Deng B, Lou P, Wu J, Sun R, Xu Z, Vromans J, Koornneef M, Bonnema G (2005) Genetic relationships within Brassica rapa as inferred from AFLP fingerprints. Theor Appl Genet 110: 1301–1314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.