

Assembly into stable trimers provides strong evidence for 18 protein subunits to assemble in a cellulose synthesis complex that synthesizes an 18-chain cellulose microfibril.
Abstract
A cellulose synthesis complex with a “rosette” shape is responsible for synthesis of cellulose chains and their assembly into microfibrils within the cell walls of land plants and their charophyte algal progenitors. The number of cellulose synthase proteins in this large multisubunit transmembrane protein complex and the number of cellulose chains in a microfibril have been debated for many years. This work reports a low resolution structure of the catalytic domain of CESA1 from Arabidopsis (Arabidopsis thaliana; AtCESA1CatD) determined by small-angle scattering techniques and provides the first experimental evidence for the self-assembly of CESA into a stable trimer in solution. The catalytic domain was overexpressed in Escherichia coli, and using a two-step procedure, it was possible to isolate monomeric and trimeric forms of AtCESA1CatD. The conformation of monomeric and trimeric AtCESA1CatD proteins were studied using small-angle neutron scattering and small-angle x-ray scattering. A series of AtCESA1CatD trimer computational models were compared with the small-angle x-ray scattering trimer profile to explore the possible arrangement of the monomers in the trimers. Several candidate trimers were identified with monomers oriented such that the newly synthesized cellulose chains project toward the cell membrane. In these models, the class-specific region is found at the periphery of the complex, and the plant-conserved region forms the base of the trimer. This study strongly supports the “hexamer of trimers” model for the rosette cellulose synthesis complex that synthesizes an 18-chain cellulose microfibril as its fundamental product.
Cellulose, the most abundant biopolymer on Earth, is composed of linear chains of β-1,4 linked d-Glc monomers with repeating structural units of the disaccharide cellobiose. Numerous cellulose polymers cocrystallize to form microfibrils, which provide mechanical strength and rigidity to plants. Its natural abundance makes it an attractive target for many industrial applications, including paper and pulping, construction, and textile manufacture. More recently, cellulose has been used for production of biofuels, such as ethanol (Ragauskas et al., 2006; Langan et al., 2014), and in the form of nanocellulose as a component in advanced composite materials (Reddy et al., 2013; Habibi, 2014). Cellulose microfibrils are synthesized by a large membrane-bound protein complex. In the land plants and charophycean algae, the cellulose synthesis complex (CSC) has a “rosette” shape (Mueller et al., 1976; Mueller and Brown, 1980b; Kimura et al., 1999), and the entire CSC has reported diameters between 24 to 30 nm (Lerouxel et al., 2006). This structural information was revealed by freeze-fracture transmission electron microscopy, showing six lobes in a hexagonal arrangement at the point where the transmembrane helices of multiple cellulose synthase proteins (CESAs) cross the plasma membrane. Recently KORRIGAN, a protein with cellulase activity, has also been implicated as an integral component of the CSC (Vain et al., 2014).
Vascular plants produce several different CESA isoforms. For example, Arabidopsis (Arabidopsis thaliana) has 10 different isoforms with 64% to 98% sequence identity (Holland et al., 2000; Richmond, 2000; McFarlane et al., 2014). The different CESA isoforms play specific roles in cellulose synthesis during plant development. In Arabidopsis, CESA1, CESA3, and CESA6 are required for primary cell wall synthesis, while CESA4, CESA7, and CESA8 are required for secondary cell wall synthesis (Gardiner et al., 2003; Taylor et al., 2003; Persson et al., 2007). CESA2, CESA5, and CESA9 play roles in tissue-specific processes and are partially redundant with CESA6, whereas CESA10 is closely related to AtCESA1 but evidently has a minor role in plant development (Somerville, 2006). The absolute number of CESA proteins present in a CSC remains a subject of much speculation, largely because the stoichiometry of the cellulose microfibril remains unresolved (Cosgrove, 2014). The traditional representation of the microfibril has 36 cellulose chains, and based on this, one would expect that each lobe of the rosette CSC contains six CESA proteins responsible for the synthesis of six glucan chains for a total of 36 CESA proteins per rosette CSC (Herth, 1983; Perrin, 2001; Doblin et al., 2002). However, recent studies using different analytical techniques combined with computation report 18 to 24 cellulose chains per microfibril (Fernandes et al., 2011; Thomas et al., 2013; Oehme et al., 2015). A study of cellulose from mung bean (Vigna radiata) primary cell walls, using x-ray diffraction, solid-state NMR, and computational analysis, supports an 18-chain model for a cellulose microfibril (Newman et al., 2013). This implies that the CSC is composed of fewer than 36 CESA proteins or that not all of the proteins in a CSC are simultaneously active. Further, it has been recently reported that the stoichiometry of CESAs 1, 3, and 6 and CESAs 4, 7, and 8 in the primary and secondary cell walls, respectively, is 1:1:1 (Gonneau et al., 2014; Hill et al., 2014). Together, these reports suggest a rosette CSC composed of 18 CESA proteins with three CESAs per lobe as the most likely composition of a rosette CSC to account for an 18-chain cellulose microfibril (Newman et al., 2013; Gonneau et al., 2014; Hill et al., 2014). In addition, it should also be noted that 24 CESA proteins in a rosette CSC with four proteins per lobe is incompatible with a 1:1:1 CESA stoichiometry.
Numerous efforts to isolate active CESA proteins directly from plants or by recombinant expression have not been successful, preventing a detailed structural analysis of CESA proteins or the mechanism of plant cellulose synthesis. In contrast, the recently reported crystal structure of cellulose synthase from Rhodoobacter sphaeroides (Morgan et al., 2013) clearly showed that only a single cellulose synthase polypeptide is required for glucan polymerization and also identified the conserved sequence motifs responsible for catalysis. In addition, based on the presence of an 18-residue glucan chain in the protein tunnel, a mechanism for cellulose synthesis and translocation across a cytoplasmic membrane was proposed that also addressed how the alternate d-Glc molecules are inverted during polymer synthesis (Morgan et al., 2013; Omadjela et al., 2013). However, this structure cannot provide insight into the formation of microfibrils from the cellulose chains synthesized by single polypeptides of CESA.
The CESA proteins of land plants and their charophycean algal relatives are multidomain single polypeptide chains of approximately 1000 amino acids. They are predicted to have eight transmembrane helices and to have their N- and C-terminal regions facing the cytoplasm (Pear et al., 1996). Although they share sequence similarity with the bacterial counterpart, they also have unique structural features not found in the bacterial enzymes. The N-terminal domain contains a Zn-binding site that may play a role in oligomerization of CESA proteins (Kurek et al., 2002). The putative cytosolic domain, which is flanked by a two-helix N-terminal transmembrane domain and a six-helix C-terminal transmembrane domain (McFarlane et al., 2014; Slabaugh et al., 2014), has D, D, D, QxxRW motifs that are conserved substrate binding and catalytic residues in the glycosyltransferase-2 superfamily (Nagahashi et al., 1995; Pear et al., 1996; Saxena and Brown, 1997; Yoshida et al., 2000). This domain also has a plant-conserved region (P-CR) and a class-specific region (CSR) that are only found in CESAs that form rosette CSCs. Although the roles of these regions are unknown, they are proposed to be involved in regulatory functions, such as interactions with other proteins and oligomerization to form the rosette shape. In the Arabidopsis CESAs, the sequence identity within the P-CR regions is greater than 80%, while in CSR regions, it is only about 40%. A recent computational model of the cytosolic domain of cotton (Gossypium hirsutum) CESA1 provides the first detailed structural model of the catalytic domain of CESA (Sethaphong et al., 2013). This model structure aligns well with the crystal structure of the bacterial cellulose synthase, indicating that a common mechanism exists for cellulose synthesis in bacteria and plants and that CESAs within rosette CSCs contain a single active synthetic site. In addition, this model made it possible to test possible configurations for the assembly of CESA monomers into a functional rosette CSC (Newman et al., 2013; Sethaphong et al., 2013).
Our understanding of the mechanism of cellulose biosynthesis in plants at the molecular level is hampered by the lack of an atomic level CESA model. To gain deeper insight into the structure and role of the catalytic domain of CESA in rosette formation, we carried out a structural characterization of the cytosolic domain of Arabidopsis CESA1, a protein that is essential for cellulose synthesis in the primary cell wall (Arioli et al., 1998). The recombinant protein was purified from Escherichia coli in a two-step process that allowed us to obtain low-resolution structural information about the monomeric and trimeric forms of the recombinant protein using small-angle scattering (SAS) techniques. This study provides the first experimental evidence to support the self-assembly of CESAs into a stable trimer complex, revealing the possible role of the catalytic domain in the formation of the rosette CSC. Comparison of the size of the catalytic domain trimer with dimensions of rosette CSCs obtained from TEM studies strongly supports the “hexamer of trimers” model for rosette CSCs. Computational analysis of the scattering data suggested configurations for how the monomers, including the plant-specific P-CR and CSR domains, may be arranged in the trimeric lobes of the rosette CSC. Knowledge of how CESA proteins assemble in the CSC will enable approaches for rational genetic manipulation of plant cell wall synthesis, which offers enormous opportunities to improve feedstocks for the production of sustainable fuels and chemicals.
RESULTS
Protein Expression and Purification
The recombinant AtCESA1CatD, encoding the catalytic domain of CESA1, was overexpressed in E. coli BL21-RIL as described in “Materials and Methods.” Although AtCESA1CatD is predicted to be a soluble cytosolic domain of ATCESA1, based on analysis of the amino acid sequence (Delmer, 1999), it was, in fact, expressed in the insoluble fraction of the bacterial cells. Attempts to solubilize AtCESA1CatD under denaturing conditions using urea or lithium dodecyl sulfate were unsuccessful, resulting in protein aggregation during the refolding process. However, the protein was solubilized under non-denaturing conditions using a low concentration of sodium lauroyl sarcosine according to a previously reported protocol (García-Fruitós et al., 2005; Jevsevar et al., 2005; Peternel et al., 2008). The purified protein was >95% pure after size exclusion chromatography as judged by SDS-PAGE analysis (Fig. 1). Mass spectrometric analysis confirmed the protein’s identity with a confidence limit of >90% and determined the mass of the protein to be 59.7 kD, which is in agreement with the mass calculated based on the amino acid sequence (Gasteiger et al., 2005).
Figure 1.

Physicochemical analysis of recombinant AtCESA1CatD. A, SDS-PAGE analysis: lane 1, protein marker; lane 2, purified AtCESA1CatD. B, CD spectrum of the purified AtCESA1CatD.
Secondary Structure and Thermal Melting
The circular dichroism (CD) spectrum of AtCESA1CatD is characteristic of a folded protein (Fig. 1C). Table I presents the secondary structural elements in AtCESA1CatD calculated from the CD spectrum (Compton and Johnson, 1986; Whitmore and Wallace, 2008), the predicted secondary structure of catalytic domain from Oryza sativa CESA8 (OsCESA8CatD; Cole et al., 2008), and the computational model of GhCESA1CatD (de Beer et al., 2014) The predicted secondary structures of the catalytic domains of AtCESA1CatD and GhCESA1CatD are included for comparison. The secondary structure of AtCESA1CatD calculated from its CD spectrum and predicted secondary structure of other CESACatD domains is comparable for α-helix and random coil content. The β-Sheet content calculated from the CD spectrum is higher than the value obtained from JPred3, which may reflect known inaccuracies in secondary structure prediction and CD analysis programs (Montgomerie et al., 2006). Overall, these data provide confidence that the protein is in a folded conformation. CD thermal melting studies showed that the melting temperature (Tm) of the protein increased from 69.5°C in the absence of ligands to 72.4°C and 74.5°C when the protein was incubated with UDP-Glc or UDP, respectively (Fig. 2). The increase in the Tm of the protein in presence of ligands provides further evidence that the ATCESACatD domain is in a native conformation. UDP appears to interact more strongly than the UDP-Glc, similar to a recent report where UDP has been shown to block the binding of UDP-Glc (Olek et al., 2014). Addition of cellobiose did not alter the Tm of the protein, revealing the specificity of interaction only with UDP and UDP-Glc, which are known ligands.
Table I. Comparison of the secondary structural elements of CESA catalytic domains.
| Organism | α-Helix | β-Sheet | Turn/Random |
|---|---|---|---|
| % | |||
| AtCESA1a | 28 | 22 | 50 |
| AtCESA1b | 34.6 | 10.1 | 55.3 |
| GhCESA1c | 29.6 | 4 | 66.4 |
| GhCESA1b | 36.6 | 9.2 | 44.2 |
| OsCESA8b | 40.7 | 9.5 | 49.8 |
Determined from the CD spectrum.
Calculated from the amino acid sequence as described in “Materials and Methods.”
Calculated from the computational model as described in “Materials and Methods.”
Figure 2.

CD thermal melting studies of AtCESA1CatD. AtCESA1CatD only (white circles) and in presence of UDP (black circles) and UDP-Glc (black squares) monitored at 222 nm. Solid lines are fit to a two-state model for thermal denaturation (Cohen and Pielak, 1994).
Solution Structure of AtCESA1CatD
Small-angle neutron scattering (SANS) was used to study the solution structure of the recombinant AtCESA1CatD domain. The SANS intensity profile, obtained in the Q-range from 0.01 Å−1 < Q < 0.2 Å−1 is shown in Figure 3A. The plateau in the low-Q region of the scattering profile is indicative of a monodisperse scattering particle without evidence of higher-order protein aggregates. The radius of gyration (Rg) determined by the Guinier approximation (Guinier and Fournet, 1955) is 18.7 ± 0.5 Å (Supplemental Fig. S3). The pair-distance distribution function, P(r), calculated from the scattering profile using GNOM (Svergun, 1992) exhibits a single maximum at 23 Å and has a maximum dimension (Dmax) of 60 Å (Fig. 3A). The Rg, calculated as the second moment of P(r) is 19.5 ± 0.2 Å, similar to the value obtained from the Guinier analysis. The molecular mass of the protein calculated from the scattering profile is 58.7 ± 1.9 kD and is almost identical to the mass of AtCESA1CatD determined by mass spectrometry (59.7 kD), indicating that the protein is a monomer. Kratky analysis of the scattering data (Supplemental Fig. S4) shows a single peak that decays at high Q and is characteristic of a compact scattering object, consistent with the protein being in a folded state (Glatter and Kratky, 1982).
Figure 3.

SANS analysis of AtCESA1CatD monomers. SANS profile of AtCESA1CatD (black circles) overlaid with simulated scattering curve for GhCESA1CatD (white circles). Inset shows P(r) plot derived from experimental scattering data of AtCESA1CatD.
The experimental scattering profile of AtCESA1CatD was compared with a theoretical scattering curve of GhCESA1CatD (Sethaphong et al., 2013) calculated using CRYSON (Svergun et al., 1998). The theoretical curve overlays the experimental data well in the low Q-region but deviates in the mid-Q region (Fig. 3). This indicates that the sizes of the computational GhCESA1CatD model and AtCESA1CatD are similar but the experimental structure is more compact than the model structure, perhaps due to small differences in the local packing of the polypeptide chains. Such differences are also reflected in the somewhat larger Rg of GhCESA1CatD (22.1 ± 0.1 Å, obtained using CRYSON) compared to AtCESA1CatD (19.5 ± 0.2 Å), even though their molecular masses are almost identical, at 56.5 kDa and 59.7 kD, respectively.
An ab initio model of the AtCESA1CatD was calculated using GASBOR (Fig. 4) and was superimposed with the computational model of GhCESA1CatD using SUPCOMB (Kozin and Svergun, 2001). As expected, the models overlay each other reasonably well, providing experimental validation of the accuracy of the GhCESA1CatD model (Sethaphong et al., 2013). However, several stretches of amino acids that include the CSR and P-CR regions from GhCESA1CatD protrude from the low-resolution model.
Figure 4.

Ab initio shape reconstruction of AtCESA1CatD monomer. Ab initio model of the AtCESA1CatD overlaid with GhCESA1CatD (cartoon) in two different orientations. The P-CR and CSR regions in GhCESA1CatD are colored in violet and blue, respectively.
Oligomerization of AtCESA1CatD
A systematic study of the effect of buffer reagents on the oligomeric state of AtCESA1CatD was carried out using dynamic light scattering. It was discovered that removal of lauroyl sarcosine from the buffer resulted in an increase in the hydrodynamic radius of the particles from approximately 5 nm to approximately 8 nm, indicating a change in its molecular mass (Supplemental Fig. S2). In contrast, the oligomeric state of AtCESA1CatD is insensitive to the presence of dithiothreitol (DTT) up to a concentration of 20 mm. Small-angle x-ray scattering (SAXS) analysis of the samples with increased hydrodynamic radius shows that the shape of the scattering curve is consistent with a monodisperse scattering particle (Fig. 5). The Rg calculated by Guinier analysis was 42.9 ± 0.2 Å (Supplemental Fig. S5) and the Rg and Dmax calculated from the P(r) analysis were 43.8 ± 0.1 Å and 175 Å, respectively. These values are clearly larger than the corresponding values for the monomer, indicating the presence of a larger scattering particle with a maximum dimension of approximately 11.3 nm calculated from the Rg value above. The molecular mass of the scattering particle, calculated directly from the scattering curve (Rambo and Tainer, 2013), was determined to be 185 ± 1.1 kD, providing compelling evidence for the presence of a trimer of AtCESA1CatD, since the monomer mass is approximately 60 kD. In addition, a Kratky plot (Glatter and Kratky, 1982) showed that the protein is in a compact conformation (Supplemental Fig. S6). A low-resolution ab initio model of AtCESA1CatD trimer was calculated using GASBOR with the assumption of P3 symmetry (Fig. 8).
Figure 5.

Analysis of SAXS data of the AtCESA1CatD trimer. SAXS profile for AtCESA1CatD trimer. Inset shows P(r) curve derived from scattering data of AtCESA1CatD trimer.
Figure 8.

Ab initio models of AtCESA1CatD trimer superposed with ROSETTA models. The ab initio models calculated from the SAXS data using GASBOR are represented by light gray (averaged) and dark gray (filtered) surface models with the superposed ROSETTA models shown in orange. A and B show the top and side views of AtCESA1CatD-m1 and AtCESA1CatD-m12 ROSETTA models superposed in the SAXS ab initio models, respectively.
Computational Modeling of AtCESA1CatD Trimers
To gain insight into possible arrangements of the AtCESA1CatD monomers, a series of AtCESA1CatD trimers was generated computationally and compared with the SAXS data. For this, a homology model of the AtCESA1CatD monomer (see “Materials and Methods”) was used as input for generating 1000 symmetric trimer configurations using the ROSETTA SymDock algorithm (André et al., 2007). Theoretical SAXS curves were calculated using CRYSOL for each of the 1000 protein trimer configurations for comparison with the experimental SAXS data. The χ value was computed to quantify the fit of the theoretical SAXS curves to the experimental SAXS data. Comparison of the χ values with the ROSETTA energy score (E) in ROSETTA Energy Units (REU) of each trimeric model shows that, even for the trimer models with a low E value, there is large variation in the quality of the fit to the experimental SAXS data (Fig. 6A). Furthermore, structurally very different models can have similar χ scores. This is exemplified in Figure 6B that compares the root-mean-square deviation (RMSD) of each trimer model to the model with the lowest χ value (AtCESA1CatD-m1; χ = 4.39). Based on this observation, it is clear that one cannot rule out any particular arrangement of monomers based on the fit to the SAXS data alone.
Figure 6.

Analysis of ROSETTA-generated AtCESA1CatD trimer models. A, Plot of χ score obtained from fit of AtCESA1CatD trimer theoretical SAXS curves to experimental SAXS data versus the energy score of ROSETTA models. B, A plot of χ versus RMSD of the ROSETTA models computed using AtCESA1CatD-m1 as a reference model. The models below the red line (χ < 9.10) were included in the analysis.
Since it is not possible to rule out a particular trimer model using solely the χ value, an additional constraint was needed to identify the most likely configurations among the best fitting, low E trimer models. Of the initial 1000 models generated by ROSETTA SymDock, 30 models were selected for detailed analysis based on the χ score (χ ≤ 9.1) obtained from the fits of the theoretical SAXS curves to the experimental SAXS data. In all cases, the theoretical scattering curves are similar in the low Q-region and fit the experimental data well, indicating that all trimer models capture the overall size of the scattering particle. However, theoretical curves deviate in the mid and high Q region (Q > ∼0.08 Å−1), suggesting that differences in the arrangement of the monomers in the trimer are captured in that Q-range. We can anticipate where the β-1,4-glucan chains will emerge from the monomers, given the structural similarity between the invariant DD, DCD, and QVLRW motifs that constitute the catalytic core in the GhCESA1CatD computational model (Sethaphong et al., 2013) and the crystal structure of R. sphaeroides bacterial cellulose synthase (BcsA) that also contains an emerging glucan chain (Morgan et al., 2013). We thus evaluated the candidate trimer models according to where the glucan chains would be predicted to emerge from them.
The fit to the experimental data for the best-fit trimer model (AtCESA1CatD-m1) is shown in Figure 7. Superimposition of this model with the GASBOR ab initio model indicates that the overall size and shape of the computational and experimental structure are similar (Fig. 8). In this trimer model, the CSR regions are at the interfaces of the monomers, and the P-CR regions project outward into the cytoplasm (Fig. 9, A and B). Each monomer-monomer interface has approximately 60 amino acids and a total of 964 Å2 of buried surface area, which is a reasonable value based on previously reported studies analyzing interfaces in protein complexes (Tsuchiya et al., 2008; Krissinel and Henrick, 2007). Interestingly, a similar arrangement of the CSR regions was reported for the OsCESA8CatD dimer (Olek et al., 2014). However, structural alignment with BcsA shows that the catalytic cores are projected radially inward in AtCESA1CatD-m1, such that the emergent glucan chains are at an acute angle to the membrane (Fig. 9C). This orientation is not optimal for translocation of the cellulose chains across the plasma membrane. Analysis of most other trimer models resulted in a similar outcome, having the emergent cellulose chains from the catalytic domain projecting away from the membrane.
Figure 7.

Comparison of theoretical scattering curves of the ROSETTA models with experimental AtCESA1CatD SAXS profile. The theoretical scattering profiles for the model AtCESA1CatD-m1 and the model AtCESA1CatD-m12 are shown in orange and magenta, respectively. The AtCESA1CatD SAXS curve is shown as open circles. Inset shows the magnified fit for Q in the range of 0.06 to 0.1.
Figure 9.

The ROSETTA models of AtCESA1CatD trimers. Left and right panels represent AtCESA1CatD-m1 and AtCESA1CatD-m12, respectively. A, P-CR regions are highlighted as spheres. B, CSR regions are highlighted as spheres. C, The models are rotated 90° to provide a side view of the emergent glucan chains based on structural superposition of the bacterial cellulose synthase (PDB code, 4HG6).
However, notable exceptions are models AtCESA1CatD-m12, -m-13, -m-15, and -m-28 (see “Supplemental Data”), for which χ scores resulting from the SAXS curve fitting are between 6.33 and 9.02. The fit of the theoretical SAXS curve for one of these models, AtCESA1CatD-m12 (χ = 6.33), is shown in Figure 7, and the trimer model is shown in Figure 9. Structural alignment with BcsA shows that the catalytic residues are oriented such that the emergent glucan chains would be near to each other and directed toward the membrane (Fig. 9C). In this trimer model, the highly conserved P-CR regions form the base of the catalytic trimer pointing toward the cytosol, and the CSR regions project radially outward and do not participate in any interfaces within the trimer (Fig. 9, A and B). Each monomer-monomer interface has approximately 32 amino acid residues and a total of 750 Å2 of buried surface area. This value is lower than that obtained for AtCESA1CatD-m1, but is a reasonable value for a stable protein-protein interface (Tsuchiya et al., 2008; Krissinel and Henrick, 2007). The majority of the interfacial residues (26 residues) are from the highly conserved portions of the GT domain between the P-CR and CSR regions. Superimposition of the AtCESA1CatD-m12 with the GASBOR ab initio model indicates that both structures overlay well, as was observed for the AtCESA1CatD-m1structure (Fig. 8).
DISCUSSION
A significant achievement of this study is the expression and purification of a well-behaved, soluble portion of a CESA protein representative of the isoforms found within rosette CSCs. We were able to obtain monodisperse preparations of the AtCESA1CatD in a two-step process from inclusion bodies after overexpression in E. coli. Using a mild detergent at low concentration, it was possible to solubilize AtCESA1CatD in a folded conformation with the implication that the protein was already folded in the inclusion bodies and the detergent acted to disrupt hydrophobic interactions, resulting in a soluble monomeric protein preparation. Upon removal of detergent, the protein underwent oligomerization to form stable trimers. Apparently, breaking the purification into these two stages prevented nonspecific aggregation, which plagued our early attempts and those of many others. CD and SAS methods clearly showed that both monomer and trimer forms could be studied as monodisperse, well-folded proteins that bind to UDP and UDP-Glc. Although it bound substrate, we did not detect any catalysis of cellulose synthesis, suggesting other CESA components missing from this cytosolic fragment are required for function.
Solution Structure of AtCESA1CatD
The monodisperse nature of the monomer and higher-ordered oligomer form of AtCESA1CatD that we were able to purify enabled us to obtain SAS data of both forms. Because it is possible to directly derive a Mr for the scattering particles from SAS data, we were able to determine that removing detergent from the monomer yielded trimers. Since the scattering data showed no indications of oligomer forms beyond a trimer, we can conclude that this domain of ATCESA1 has determinants that contribute to lobe formation, but likely makes no direct contribution to the assembly of lobes into CSCs. Genetic and biochemical studies have shown that in Arabidopsis CESA1, -3, and -6 and CESA4, -7, and -8 isoforms are required for primary and secondary wall cellulose synthesis, respectively (Gardiner et al., 2003; Taylor et al., 2003; Persson et al., 2007). Other CESAs that play roles in tissue-specific processes are partially redundant with CESA6, as described in the introduction (Gardiner et al., 2003; Taylor et al., 2003; Somerville, 2006; Persson et al., 2007). Furthermore, the stoichiometry of the CESA isoforms responsible for primary and secondary cell wall synthesis in Arabidopsis has recently been reported to be 1:1:1 (Gonneau et al., 2014; Hill et al., 2014). Consequently, we cannot rule out the possibility that interaction of the cytosolic domain of ATCESA1 with other isoforms of CESA or other factors in the cytoplasm contributes to further assembly of lobes into rosettes.
Low-resolution structural models for the monomer and trimeric forms of the AtCESA1CatD were able to be calculated directly from the SAS data. When superimposed with the GhCESA1CatD, the monomer model provided experimental validation of the overall accuracy of the GhCESA1CatD model (Sethaphong et al., 2013). However, several stretches of amino acids that include the CSR and P-CR regions from GhCESA1CatD protrude from the low-resolution model. Further studies will be required to determine if the differences reflect structural differences between the cotton and Arabidopsis CESAs or are an artifact of either limitations of de novo modeling of the CSR and P-CR domains of the GhCESA1CatD or flexibility in the AtCESA1CatD structure that is not captured in the low-resolution SAS models. The level of sequence identity in the CSR regions is 47%, low enough for such structural variation to exist.
A previous study of recombinant OsCESA8CatD reported that it exists as a mixture of monomer and homodimer species (Olek et al., 2014). As noted by the authors, the scattering curves of those samples are indicative of mixed oligomers, whereas the current study of AtCESA1CatD indicates monodisperse monomers or trimers, depending whether detergent was present or absent. Also, the trimer form of the AtCESA1CatD was not sensitive to reducing agents, and the computational models did not reveal Cys residues in the interface regions that can form disulfide linkages. In contrast, the OsCESA8CatD dimers were reduced to monomers in the presence of DTT (Olek et al., 2014). It is not clear why the two proteins behaved differently, although it may reflect the two-stage purification process in which detergent prevented hydrophobic surfaces in the monomer from interacting until a slow removal of the detergent allowed for further, specific trimer assembly without nonspecific aggregation. Alternatively, it could reflect differences in CESA proteins from these two plant species.
AtCESA1CatD Trimer Models
To gain insight into arrangement of AtCESA1CatD monomers in the trimer, we constructed a series of computational AtCESA1CatD trimers from a homology model of the AtCESA1CatD monomer. By examining the fit between 1000 symmetric trimers and the scattering data, it became clear that a very small subset of low energy models could be identified, but that it was not possible to rule out a particular arrangement of monomers based on the fit to the SAXS data alone. The close agreement at low Q and discrepancies in the mid-Q and high-Q region revealed that these models largely differed in how the monomers were juxtaposed in the trimer geometry. We propose that it is possible to further select from among these low energy models by considering a priori structural information. Using the alignment of our computational models with the atomic structure of R. sphaeroides cellulose synthase, inclusive of a glucan chain passing from catalytic core through its transmembrane domain, we are able to suggest that four of the candidate models for ATCESACatD trimers were most likely to be correct. Of these, we chose ATCESACatD-m12 as the most reasonable model for cytosolic domain trimer based on its fit to the experimental data. In this trimer, the majority of the interfacial residues (26 residues) are from the highly conserved regions within the GT domain. The A549V mutation reported for the temperature-sensitive mutant Arabidopsis rsw1 is associated with disruption of the rosette CSC formation (Arioli et al., 1998). A549 is located far from the monomer interfaces and trimer surface, suggesting that it does not directly disrupt the formation of trimers in individual lobes of the rosette CSC.
Implications for the Assembly of Rosette CSC
The previously reported size of a rosette CSC obtained where the transmembrane domain across the plasma membrane is 24 to 30 nm, with individual lobes measuring 8 nm in diameter (Mueller and Brown, 1980a; Rudolph and Schnepf, 1988; Rudolph et al., 1989; Kimura et al., 1999). In addition, Bowling and Brown (2008) reported that a 45 to 50 nm hexagonal structure represented the catalytic part of the CSC of the cytosolic side of the plasma membrane, and they predicted a maximum of four CESA proteins per lobe of the CSC. Assuming the arrangement of the lobes of the CSC is the same on both sides of the membrane, we can estimate a diameter of 15 nm for a lobe at the cytosolic side of the CSC (Supplemental Fig. S7). In this work, the size of ATCESACatD trimer calculated from the computational trimer models presented above is approximately 14 nm, which is in reasonable agreement with the estimated sized of a lobe at the cytosolic side of the CSC, given the absence of the N-terminal domain and also potential contributions from other accessory proteins to the overall size of each lobe at the cytosolic side of the complex (Lei et al., 2012; Gu and Somerville, 2010). Increasing the number of CESA proteins to six per lobe, as proposed in the “hexamer of hexamers” model (Herth, 1983; Doblin et al., 2002; Perrin, 2001), would clearly occupy too much area to fit in an individual lobe. Although fewer CESA proteins per lobe is theoretically possible (Bowling and Brown, 2008), our data unequivocally demonstrates a stable trimer of CESAs without any evidence of higher order oligomers. Therefore, we propose a trimer of CESAs forms the individual lobe of a rosette CSC.
As described above, the P-CR and CSR regions are hypothesized to play a role in specific functions such as interactions with accessory proteins or formation of the rosette CSC (Somerville, 2006). In this study, the highly conserved P-CR residues form the base of the selected catalytic trimer and point toward the cytosol suggesting that this region may be involved in communicating with accessory proteins. In contrast, the CSR regions that are less conserved among CESAs project radially outward and do not participate in any of the interfaces within the trimer. This observation supports a role for the CSR region in facilitating trimer-trimer interactions in rosette CSC formation. This is in contrast to the OsCESA8CatD study that reports that the CSR regions form the monomer-monomer interface, raising questions about CESA isoform and species dependent differences in the mechanism of CESA oligomerization (Olek et al., 2014). There is no evidence of the assembly of trimers into higher order oligomers, which supports previous studies that report at least three different CESA proteins are required to assemble a functional CSC (Doblin et al., 2002; Taylor, 2008; Timmers et al., 2009; Hill et al., 2014). However, in an interesting departure, the current study presents the possibility that homotrimers are also a possible configuration for the stoichiometry of each lobe rather than the heterotrimers (Hill et al., 2014) or heterohexamers (Taylor, 2008; Timmers et al., 2009) that have previously been proposed (Fig. 10). A CSC composed of three different homotrimers would fulfill the requirements laid out by Hill et al. (2014) for the stoichiometry of a CSC. It should be noted the assembly of the rosette lobes into heterotrimers cannot be excluded because the amino acid residues that are found at the interfaces of AtCESA1CatD share high sequence similarity with other CESA proteins.
Figure 10.

Schematic representation of potential arrangements of CESA proteins in a cellulose synthase complex. The black, gray, and white circles represent the three different isoforms of CESAs required for an active CSC in a 1:1:1 ratio (Gonneau et al., 2014, Hill et al., 2014).
CONCLUSION
Recent studies of cellulose structure have estimated 18 to 24 glucan chains per microfibril (Fernandes et al., 2011; Newman et al., 2013), contrary to the more widely accepted value of a 36 mer of glucan chains comprising a microfibril (Herth, 1983; Delmer, 1999; Doblin et al., 2002). As a consequence, the stoichiometry of the cellulose synthase complex has been revisited, and there is speculation that the individual particles of the hexameric rosette, identified in TEM studies, could contain as few as three CESA proteins (Newman et al., 2013; Hill et al., 2014). This study provides the first experimental evidence to support the self-assembly of CESAs into a stable trimer complex. Comparison of the trimer complex obtained using SAXS with the dimensions of rosette CSCs from TEM images provides compelling evidence that each lobe of a CSC contains three CESAs rather than the long-standing model of six CESAs within each lobe of a rosette CSC. In summary, our data strongly support the “hexamer of trimers” model for rosette CSCs with the implication that an 18-chain cellulose microfibril is the fundamental product of cellulose synthesis in the land plants that mainly comprise the cellulosic raw materials used in industry.
MATERIALS AND METHODS
Cloning, Expression, and Purification
A gene encoding amino acids 341 to 845 in the glycosyl transferase domain of Arabidopsis (Arabidopsis thaliana) cellulose synthase 1 (AtCESA1CatD) was amplified using the PCR from a synthetic Arabidopsis CESA1 (AtCESA1) gene (Genscript, Inc.) optimized for expression in Escherichia coli. The amplified gene was cloned into pENTR (Novagen), followed by subcloning into pET300NT (Life Technologies). Optimal protein expression was obtained in strain BL21-RIL (Agilent Technologies) grown in Luria Bertani medium supplemented with 0.25 m sorbitol. Cultures (1.5 L) were grown at 37°C to OD600 = 0.4 before cooling to 16°C, inducing protein expression with 0.2 mm IPTG at OD600 = 0.6, and incubating for a further 10 to 12 h. Cells were harvested by centrifuging at 7000g for 15 min, followed by resuspending in 30 mL of lysis buffer [20 mm Tris, pH 8.0, 200 mm NaCl supplemented with lysozyme (0.2 mg/mL)] and incubation at 25°C for 30 min. The cell suspension was then sonicated (6 pulses × 30 s) using a 1.3 cm (diameter) horn and 400 watt sonicator (Branson Digital sonicator model-450) at 50% power. The recombinant AtCESA1CatD domain was present in the insoluble fraction, which was separated from the lysate by centrifugation at 36,000g for 30 min and washed three times with wash buffer 1 (20 mm Tris, pH 8.0, 1% sodium deoxycholic acid, 1% Nonidet-P40, 10 mm β-mercaptoethanol) and then three times with wash buffer 2 (20 mm Tris, pH 8.0, and 1% Triton X-100). The pellet was resuspended in solubilization buffer [50 mm CAPS, pH 10.5, 0.2 m Glc, 3% glycerol, 5 mm DTT, and 0.2% (w/v) sodium lauroyl sarcosine] with gentle mixing. The protein was purified to homogeneity using a Superdex 200 chromatography column pre-equilibrated with the solubilization buffer. The protein was exchanged into the same buffer, except the pH value was 8.0 for further experiments. The identity of the protein and its molecular mass were determined by mass spectrometry. AtCESA1CatD trimers were formed by dialysis against 20 mm HEPES (pH 8.0), 50 mm NaCl, 10% glycerol, and 5 mm DTT for 48 h using a 10 kD cutoff dialysis membrane (Slide-A-Lyzer, Thermo Scientific, Inc.).
CD, Secondary Structure, and Thermal Melting
CD measurements were carried out using a Jasco J-810 spectropolarimeter equipped with a Peltier temperature controller from 190 to 260 nm at a scanning speed of 20 nm/min with a bandwidth of 2 nm using a 1 mm optical path quartz cuvette and are shown as an average of five independent spectra. The protein concentrations were 1 to 2 μm in 20 mm HEPES (pH 8.0). CD thermal melting studies were carried out with 1 μm purified AtCESA1CatD in 20 mm HEPES (pH 8.0) in the absence and presence of 100 μm UDP, 100 μm UDP-Glc, and 100 μm cellobiose. The thermal denaturation profiles were recorded at 222 nm from 25°C to 80°C at a heating rate of 1°C /min. The data were analyzed using a two-state model to determine the Tm value (Cohen and Pielak, 1994).
Secondary structure analysis of the CD data was carried out using Dichroweb suite of programs (Whitmore and Wallace, 2008). The reported secondary structure content was obtained using CDSSTR program and reference set number 4 (Compton and Johnson, 1986). JPred3 was used for secondary structure prediction from amino acid sequences (Cole et al., 2008), and the amino acid sequence alignments were performed using ClustalW (Larkin et al., 2007). The secondary structure elements in the cotton (Gossypium hirsutum) CESA1 (GhCESA1CatD) computational model (Sethaphong et al., 2013) were calculated using the program PDBSum (de Beer et al., 2014).
Computational Modeling
A homology model of AtCESA1CatD was generated with the program MODELER (Sali and Blundell, 1993) using the GhCESA1CatD computational model (Sethaphong et al., 2013) as a template. The RMSD value between the two models was 0.30 Å. The Symmetric Docking tool (SymDock) in the ROSETTA modeling software (Andre et al., 2007) was used to generate multiple configurations of symmetric trimers of the AtCESA1CatD model that were ranked based on a ROSETTA-generated energy score. Simulated SAXS profiles of 1000 AtCESA1CatD trimer models with the lowest energy score were calculated using CRYSOL (Svergun et al., 1995) and were fit to the experimental AtCESA1CatD SAXS profile. Based on this analysis, 30 trimer models with χ values smaller than 9.1 were chosen for further study. The χ value measures the discrepancy between experimentally determined and theoretically predicted SAS data. The x-ray crystal structure of cellulose synthase from Rhodoobacter sphaeroides (PDB code, 4HG6; Morgan et al., 2013) was structurally superposed on each monomer in the 30 trimer models using the DALILITE pairwise alignment tool (Hasegawa and Holm, 2009). The program PISA (Krissinel and Henrick, 2007) was used to calculate the surface area in the interfaces in the trimers.
SAS Data Collection and Analysis
SANS experiments were performed using the EQ-SANS (extended Q-range small-angle neutron scattering) instrument at the Spallation Neutron Source (Zhao et al., 2010) and the BioSANS instrument at the High Flux Isotope Reactor at Oak Ridge National Laboratory (Heller et al., 2014). EQ-SANS was operated at a 30 Hz mode, which provided wavelength bands of 2.5 to 6.1 Å and 9.4 to 13.4 Å. A sample-to-detector distance of 4 m was used to obtain an effective momentum transfer vector, Q, of 0.005 to 0.45 Å−1 (Q = 4π sin (θ) / λ for scattering angle θ and wavelength, λ). BioSANS was operated using 6.0 Å neutrons at two sample-to-detector distances (1.1 m and 6.8 m), thus obtaining a Q-range of 0.007 to 0.45 Å−1. All the SANS measurements of AtCESA1CatD were performed in 1 mm path cylindrical quartz cuvettes at 20°C using 5 mg/mL protein in 50 mm CAPS (pH 8.0), 200 mm Glc, 3% glycerol, 0.2% lauroyl sarcosine, in the absence and presence of 5 mm DTT. The same buffers without protein were used for background subtraction. Data reduction was carried out using MantidPlot (Wignall and Bates, 1987). Data analysis was performed with the ATSAS suite of programs (Petoukhov et al., 2012) using data with a Q-range of 0.007 to 0.18 Å−1.
SAXS experiments were done at the beamline X9 at the National Synchrotron Light Source at Brookhaven National Laboratory. The instrument utilizes x-rays of wavelength 0.98 Å and is equipped with a Photonic Science WAXS detector and a MarCCD SAXS detector that provide a combined Q-range from 0.005 Å−1 to 2.0 Å−1. SAXS data were acquired for 10 mg/mL AtCESA1CatD in 20 mm HEPES (pH 8.0), 50 mm NaCl, 10% glycerol, and 5 mm DTT and the appropriate background measurements. For some measurements, the DTT concentration was increased to 20 mm to investigate its effect on the oligomerization of AtCESA1CatD. Software pyXS, developed at beamline X9, was used for data processing. The protein scattering data were corrected for the buffer background and were radially averaged and scaled to obtain a relative scattering intensity, I(Q). Data analysis was performed with the ATSAS suite of programs (Petoukhov et al., 2012) using data with a Q-range of 0.005 Å−1 to 0.3 Å−1.
Rg was calculated by using the Guinier approximation (Guinier and Fournet, 1955),
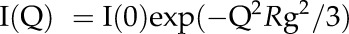 |
where I(0) is the forward scattering at momentum transfer Q = 0 Å−1, which is a shape-independent function of the total scattering power of the sample. By performing a linear fit to a plot of ln[I(Q)] versus Q2, I(0) and Rg can be determined from the intercept and slope, respectively. The program PRIMUS was used to carry Guinier analysis (Konarev et al., 2003). The maximum dimension of the scattering particle was obtained using 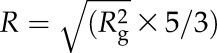 , where R is the radius. The molecular mass of AtCESA1CatD was calculated from the scattering profiles, as described previously (Rambo and Tainer, 2013). The pair-distance distribution function, P(r), was calculated using the indirect Fourier transform method implemented in the program GNOM (Svergun, 1992). The output from GNOM was used as input for GASBOR (reciprocal space version; Svergun et al., 2001), a program for ab initio protein shape determination. The output of GASBOR is a chain-like ensemble of dummy residues that satisfies the scattering profile of the scattering object. Since multiple structures can fit the scattering curve, 20 runs of GASBOR were performed for the SANS and SAXS data and the resulting set of models were used to calculate the averaged model using DAMAVER program (Volkov and Svergun, 2003). The DAMFILT program was used to generate the most probable model using a contact threshold cut off value of 9 (Volkov and Svergun, 2003). P3 symmetry and an oblate shape were applied in GASBOR to generate the molecular structure of AtCESA1CatD from the SAXS data. The SUPCOMB program (Kozin and Svergun, 2001) was used to superimpose the computational models on the ab initio molecular structures generated using GASBOR. All figures of cartoon and surface models were generated in PYMOL (Schrodinger, 2010).
, where R is the radius. The molecular mass of AtCESA1CatD was calculated from the scattering profiles, as described previously (Rambo and Tainer, 2013). The pair-distance distribution function, P(r), was calculated using the indirect Fourier transform method implemented in the program GNOM (Svergun, 1992). The output from GNOM was used as input for GASBOR (reciprocal space version; Svergun et al., 2001), a program for ab initio protein shape determination. The output of GASBOR is a chain-like ensemble of dummy residues that satisfies the scattering profile of the scattering object. Since multiple structures can fit the scattering curve, 20 runs of GASBOR were performed for the SANS and SAXS data and the resulting set of models were used to calculate the averaged model using DAMAVER program (Volkov and Svergun, 2003). The DAMFILT program was used to generate the most probable model using a contact threshold cut off value of 9 (Volkov and Svergun, 2003). P3 symmetry and an oblate shape were applied in GASBOR to generate the molecular structure of AtCESA1CatD from the SAXS data. The SUPCOMB program (Kozin and Svergun, 2001) was used to superimpose the computational models on the ab initio molecular structures generated using GASBOR. All figures of cartoon and surface models were generated in PYMOL (Schrodinger, 2010).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Multiple sequence alignment of CESA catalytic domains from different organisms.
Supplemental Figure S2. Dynamic light scattering analysis of AtCESA1CatD.
Supplemental Figure S3. Guinier analysis of AtCESA1CatD monomer.
Supplemental Figure S4. Kratky plot derived from scattering data of AtCESA1CatD trimer.
Supplemental Figure S5. Guinier analysis of AtCESA1CatD trimer.
Supplemental Figure S6. Kratky plot derived from scattering data of AtCESA1CatD trimer.
Supplemental Figure S7. Schematic of hexagonal arrangement of lobes of a rosette CSC overlaid with a TEM image of the transmembrane domain of the rosette CSC.
Supplementary Material
Acknowledgments
Molecular biology and structural characterization carried out by H.O., V.G.V., Q.Z., W.T.H., L.P., U.K., J.C.S., P.L., and L.C. was supported by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory (ORNL), managed by UT-Batelle, LLC, for the U.S. Department of Energy under Contract Number DE–AC05–00OR22725. Small-angle scattering and computational analysis performed by H.O., V.G.V., L.P., B.T.N., and C.H.H. were supported by the Center for Lignocellulose Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. J.M. and D.K.P. acknowledge the support of a High Performance Computing Grant from Oak Ridge Associated Universities. Bio-SANS is operated by the Center for Structural Molecular Biology at ORNL, supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research Project ERKP291. EQ-SANS at the Spallation Neutron Source and the High Flux Isotope Reactor are sponsored by the Scientific User Facilities Division, Basic Energy Sciences, U.S. Department of Energy.
The authors acknowledge Dr. Sai Venkatesh Pingali for assistance with the operation of Bio-SANS and data reduction and Dr. Paul Abraham and the BioEnergy Science Center proteomics facilities for validation of purified proteins. The BioEnergy Science Center is supported by the Office of Biological and Environmental Research in the U.S. Department of Energy. Mass spectrometry analysis was carried out by the U.S. Department of Energy Office of Biological and Environmental Research supported Bioenergy Research Center proteomics pipeline. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract Number DE–AC02–98CH10886.
Glossary
- CSC
cellulose synthesis complex
- P-CR
plant-conserved region
- CSR
class-specific region
- SAS
small-angle scattering
- CD
circular dichroism
- Tm
temperature
- SANS
small-angle neutron scattering
- SAXS
small-angle x-ray scattering
- RMSD
root-mean-squared deviation
- BcsA
bacterial cellulose synthase
- EQ-SANS
extended Q-range small-angle neutron scattering
Footnotes
This work has been authored by UT-Battelle, LLC, under Contract Number DE–AC05–00OR22725 with the U.S. Department of Energy.
References
- André I, Bradley P, Wang C, Baker D (2007) Prediction of the structure of symmetrical protein assemblies. Proc Natl Acad Sci USA 104: 17656–17661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arioli T, Peng L, Betzner AS, Burn J, Wittke W, Herth W, Camilleri C, Höfte H, Plazinski J, Birch R, et al. (1998) Molecular analysis of cellulose biosynthesis in Arabidopsis. Science 279: 717–720 [DOI] [PubMed] [Google Scholar]
- Bowling AJ, Brown RM Jr (2008) The cytoplasmic domain of the cellulose-synthesizing complex in vascular plants. Protoplasma 233: 115–127 [DOI] [PubMed] [Google Scholar]
- Cohen DS, Pielak GJ (1994) Stability of yeast iso-1-ferricytochrome c as a function of pH and temperature. Protein Sci 3: 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36: W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton LA, Johnson WC Jr (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal Biochem 155: 155–167 [DOI] [PubMed] [Google Scholar]
- Cosgrove DJ. (2014) Re-constructing our models of cellulose and primary cell wall assembly. Curr Opin Plant Biol 22: 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer TAP, Berka K, Thornton JM, Laskowski RA (2014) PDBsum additions. Nucleic Acids Res 42: D292–D296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmer DP. (1999) Cellulose biosynthesis: exciting times for a difficult field of study. Annu Rev Plant Physiol Plant Mol Biol 50: 245–276 [DOI] [PubMed] [Google Scholar]
- Doblin MS, Kurek I, Jacob-Wilk D, Delmer DP (2002) Cellulose biosynthesis in plants: from genes to rosettes. Plant Cell Physiol 43: 1407–1420 [DOI] [PubMed] [Google Scholar]
- Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci USA 108: E1195–E1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Fruitós E, González-Montalbán N, Morell M, Vera A, Ferraz RM, Arís A, Ventura S, Villaverde A (2005) Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb Cell Fact 4: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner JC, Taylor NG, Turner SR (2003) Control of cellulose synthase complex localization in developing xylem. Plant Cell 15: 1740–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy Server. In JM Walker, ed, The Proteomics Protocols Handbook. Humana Press, New York, pp 571–607 [Google Scholar]
- Glatter O, Kratky O (1982) Small-Angle X-Ray Scattering. Academic, London: [DOI] [PubMed] [Google Scholar]
- Gonneau M, Desprez T, Guillot A, Vernhettes S, Höfte H (2014) Catalytic subunit stoichiometry within the cellulose synthase complex. Plant Physiol 166: 1709–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Somerville C (2010) Cellulose synthase interacting protein: a new factor in cellulose synthesis. Plant Signal Behav 5: 1571–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinier A, Fournet G (1955) Small-Angle Scattering of X-Rays. Wiley, New York [Google Scholar]
- Habibi Y. (2014) Key advances in the chemical modification of nanocelluloses. Chem Soc Rev 43: 1519–1542 [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Holm L (2009) Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol 19: 341–348 [DOI] [PubMed] [Google Scholar]
- Heller WT, Urban VS, Lynn GW, Weiss KL, O’Neill HM, Pingali SV, Qian S, Littrell KC, Melnichenko YB, Buchanan MV, et al. (2014) The Bio-SANS instrument at the High Flux Isotope Reactor of Oak Ridge National Laboratory. J Appl Cryst 47: 1238–1246 [Google Scholar]
- Herth W. (1983) Arrays of plasma-membrane “rosettes” involved in cellulose microfibril formation of Spirogyra. Planta 159: 347–356 [DOI] [PubMed] [Google Scholar]
- Hill JL Jr, Hammudi MB, Tien M (2014) The Arabidopsis cellulose synthase complex: a proposed hexamer of CESA trimers in an equimolar stoichiometry. Plant Cell 26: 4834–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland N, Holland D, Helentjaris T, Dhugga KS, Xoconostle-Cazares B, Delmer DP (2000) A comparative analysis of the plant cellulose synthase (CesA) gene family. Plant Physiol 123: 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevsevar S, Gaberc-Porekar V, Fonda I, Podobnik B, Grdadolnik J, Menart V (2005) Production of nonclassical inclusion bodies from which correctly folded protein can be extracted. Biotechnol Prog 21: 632–639 [DOI] [PubMed] [Google Scholar]
- Kimura S, Laosinchai W, Itoh T, Cui X, Linder CR, Brown RM Jr (1999) Immunogold labeling of rosette terminal cellulose-synthesizing complexes in the vascular plant vigna angularis. Plant Cell 11: 2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst 36: 1277–1282 [Google Scholar]
- Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Cryst 34: 33–41 [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Kurek I, Kawagoe Y, Jacob-Wilk D, Doblin M, Delmer D (2002) Dimerization of cotton fiber cellulose synthase catalytic subunits occurs via oxidation of the zinc-binding domains. Proc Natl Acad Sci USA 99: 11109–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan P, Petridis L, O’Neill HM, Pingali SV, Foston M, Nishiyama Y, Schulz R, Lindner B, Hanson BL, Harton S, et al. (2014) Common processes drive the thermochemical pretreatment of lignocellulosic biomass. Green Chem 16: 63–68 [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. (2007) Clustal W and clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Lei L, Li S, Gu Y (2012) Cellulose synthase interactive protein 1 (CSI1) mediates the intimate relationship between cellulose microfibrils and cortical microtubules. Plant Signal Behav 7: 714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouxel O, Cavalier DM, Liepman AH, Keegstra K (2006) Biosynthesis of plant cell wall polysaccharides - a complex process. Curr Opin Plant Biol 9: 621–630 [DOI] [PubMed] [Google Scholar]
- McFarlane HE, Döring A, Persson S (2014) The cell biology of cellulose synthesis. Annu Rev Plant Biol 65: 69–94 [DOI] [PubMed] [Google Scholar]
- Montgomerie S, Sundararaj S, Gallin WJ, Wishart DS (2006) Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinformatics 7: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JLW, Strumillo J, Zimmer J (2013) Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature 493: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, Brown RM Jr (1980a) Evidence for an intramembrane component associated with a cellulose microfibril-synthesizing complex in higher plants. J Cell Biol 84: 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, Brown RM Jr (1980b) Evidence for an intramembrane component associated with a cellulose microfibril-synthesizing complex in higher plants. J Cell Biol 84: 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, Brown RM Jr, Scott TK (1976) Cellulosic microfibrils: nascent stages of synthesis in a higher plant cell. Science 194: 949–951 [DOI] [PubMed] [Google Scholar]
- Nagahashi S, Sudoh M, Ono N, Sawada R, Yamaguchi E, Uchida Y, Mio T, Takagi M, Arisawa M, Yamada-Okabe H (1995) Characterization of chitin synthase 2 of Saccharomyces cerevisiae. Implication of two highly conserved domains as possible catalytic sites. J Biol Chem 270: 13961–13967 [DOI] [PubMed] [Google Scholar]
- Newman RH, Hill SJ, Harris PJ (2013) Wide-angle x-ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol 163: 1558–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehme DP, Downton MT, Doblin MS, Wagner J, Gidley MJ, Bacic A (2015) Unique aspects of the structure and dynamics of elementary Iβ cellulose microfibrils revealed by computational simulations. Plant Physiol 168: 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olek AT, Rayon C, Makowski L, Kim HR, Ciesielski P, Badger J, Paul LN, Ghosh S, Kihara D, Crowley M, et al. (2014) The structure of the catalytic domain of a plant cellulose synthase and its assembly into dimers. Plant Cell 26: 2996–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omadjela O, Narahari A, Strumillo J, Mélida H, Mazur O, Bulone V, Zimmer J (2013) BcsA and BcsB form the catalytically active core of bacterial cellulose synthase sufficient for in vitro cellulose synthesis. Proc Natl Acad Sci USA 110: 17856–17861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear JR, Kawagoe Y, Schreckengost WE, Delmer DP, Stalker DM (1996) Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc Natl Acad Sci USA 93: 12637–12642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RM. (2001) Cellulose: how many cellulose synthases to make a plant? Curr Biol 11: R213–R216 [DOI] [PubMed] [Google Scholar]
- Persson S, Paredez A, Carroll A, Palsdottir H, Doblin M, Poindexter P, Khitrov N, Auer M, Somerville CR (2007) Genetic evidence for three unique components in primary cell-wall cellulose synthase complexes in Arabidopsis. Proc Natl Acad Sci USA 104: 15566–15571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peternel S, Jevsevar S, Bele M, Gaberc-Porekar V, Menart V (2008) New properties of inclusion bodies with implications for biotechnology. Biotechnol Appl Biochem 49: 239–246 [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Cryst 45: 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL, et al. (2006) The path forward for biofuels and biomaterials. Science 311: 484–489 [DOI] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MM, Vivekanandhan S, Misra M, Bhatia SK, Mohanty AK (2013) Biobased plastics and bionanocomposites: current status and future opportunities. Prog Polym Sci 38: 1653–1689 [Google Scholar]
- Richmond T (2000) Higher plant cellulose synthases. Genome Biol 1: 3001.1–3001.6 [DOI] [PMC free article] [PubMed]
- Rudolph U, Gross H, Schnepf E (1989) Investigations of the Turnover of the Putative Cellulose-Synthesizing Particle Rosettes within the Plasma-Membrane of Funaria-Hygrometrica Protonema Cells. 2. Rosette Structure and the Effects of Cycloheximide, Actinomycin-D, 2.6-Dichlorobenzonitrile, Biofluor, Heat-Shock, and Plasmolysis. Protoplasma 148: 57–69 [Google Scholar]
- Rudolph U, Schnepf E (1988) Investigations of the Turnover of the Putative Cellulose-Synthesizing Particle Rosettes within the Plasma-Membrane of Funaria-Hygrometrica Protonema Cells. 1. Effects of Monensin and Cytochalasin-B. Protoplasma 143: 63–73 [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Saxena IM, Brown RM (1997) Identification of cellulose synthase(s) in higher plants: sequence analysis of processive beta-glycosyltransferases with the common motif ‘D, D, D35Q(R,Q)XRW’. Cellulose 4: 33–49 [Google Scholar]
- Schrodinger LLC (2010) The PyMOL Molecular Graphics System, Version 1.3r1. Schrodinger LLC, New York
- Sethaphong L, Haigler CH, Kubicki JD, Zimmer J, Bonetta D, DeBolt S, Yingling YG (2013) Tertiary model of a plant cellulose synthase. Proc Natl Acad Sci USA 110: 7512–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabaugh E, Davis JK, Haigler CH, Yingling YG, Zimmer J (2014) Cellulose synthases: new insights from crystallography and modeling. Trends Plant Sci 19: 99–106 [DOI] [PubMed] [Google Scholar]
- Somerville C. (2006) Cellulose synthesis in higher plants. Annu Rev Cell Dev Biol 22: 53–78 [DOI] [PubMed] [Google Scholar]
- Svergun D, Barberato C, Koch MHJ (1995) CRYSOL - a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst 28: 768–773 [Google Scholar]
- Svergun DI. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst 25: 495–503 [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80: 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI, Richard S, Koch MHJ, Sayers Z, Kuprin S, Zaccai G (1998) Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc Natl Acad Sci USA 95: 2267–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NG. (2008) Cellulose biosynthesis and deposition in higher plants. New Phytol 178: 239–252 [DOI] [PubMed] [Google Scholar]
- Taylor NG, Howells RM, Huttly AK, Vickers K, Turner SR (2003) Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc Natl Acad Sci USA 100: 1450–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LH, Forsyth VT, Sturcová A, Kennedy CJ, May RP, Altaner CM, Apperley DC, Wess TJ, Jarvis MC (2013) Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Physiol 161: 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers J, Vernhettes S, Desprez T, Vincken JP, Visser RGF, Trindade LM (2009) Interactions between membrane-bound cellulose synthases involved in the synthesis of the secondary cell wall. FEBS Lett 583: 978–982 [DOI] [PubMed] [Google Scholar]
- Tsuchiya Y, Nakamura H, Kinoshita K (2008) Discrimination between biological interfaces and crystal-packing contacts. Adv Appl Bioinform Chem 1: 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vain T, Crowell EF, Timpano H, Biot E, Desprez T, Mansoori N, Trindade LM, Pagant S, Robert S, Höfte H, et al. (2014) The cellulase KORRIGAN is part of the cellulose synthase complex. Plant Physiol 165: 1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst 36: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89: 392–400 [DOI] [PubMed] [Google Scholar]
- Wignall GD, Bates FS (1987) Absolute calibration of small-angle neutron-scattering data. J Appl Cryst 20: 28–40 [Google Scholar]
- Yoshida M, Itano N, Yamada Y, Kimata K (2000) In vitro synthesis of hyaluronan by a single protein derived from mouse HAS1 gene and characterization of amino acid residues essential for the activity. J Biol Chem 275: 497–506 [DOI] [PubMed] [Google Scholar]
- Zhao JK, Gao CY, Liu D (2010) The extended Q-range small-angle neutron scattering diffractometer at the SNS. J Appl Cryst 43: 1068–1077 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.