

Abstract
T cell suppression in sepsis is a well-known phenomenon; however, the underlying mechanisms are not fully understood. Previous studies have shown that T cell stimulation up-regulates mitochondrial adenosine triphosphate (ATP) production to fuel purinergic signaling mechanisms necessary for adequate T cell responses. Here we show that basal mitochondrial ATP production, ATP release, and stimulation of P2X1 receptors represent a standby purinergic signaling mechanism that is necessary for antigen recognition. Inhibition of this process impairs T cell vigilance and the ability of T cells to trigger T cell activation, up-regulate mitochondrial ATP production, and stimulate P2X4 and P2X7 receptors that elicit interleukin 2 production and T cell proliferation. T cells of patients with sepsis lack this standby purinergic signaling system owing to defects in mitochondrial function, ATP release, and calcium signaling. These defects impair antigen recognition and T cell function and are correlated with sepsis severity. Pharmacological targeting of these defects may improve T cell function and reduce the risk of sepsis.
Keywords: mitochondrial dysfunction, purinergic signaling, sepsis, T cell suppression
Sepsis is a leading cause of death worldwide, with mortality rates ranging from 20% to 40% [1, 2]. Despite intensive research for several decades, our current understanding of the underlying mechanisms is limited and no effective treatments are available. This problem is underscored by the disappointing results of numerous clinical trials [1, 3]. Most conventional treatment strategies for sepsis in these trials have focused on blocking the cytokine-mediated hyperinflammatory response that causes systemic inflammation and multiorgan dysfunction. However, this initial hyperinflammatory response is offset by a protracted immunosuppressive phase with lethal consequences because it disables the cellular immune system that helps patients cope with primary and hospital-acquired infections that can progress to sepsis [4, 5].
This immunosuppressive phase is characterized by increased plasma levels of anti-inflammatory mediators such as interleukin 10 and by T cell suppression that impairs cytokine production and the proliferation of T cells in response to antigen stimulation [6–9]. The mechanisms involved in T cell suppression are unclear [10, 11]. It has only recently been recognized that T cell activation is accompanied by the release of cellular adenosine triphosphate (ATP) and autocrine feedback mechanisms involving purinergic receptors located on the cell surface of T cells [12–15]. Autocrine stimulation of P2X1, P2X4, and P2X7 receptors contributes to the influx of extracellular calcium ions (Ca2+), which is required for the initiation of effector functions such as interleukin 2 (IL-2) production and T cell proliferation [14, 15].
More recently, it was shown that mitochondria play a central role in these autocrine purinergic signaling mechanisms [12]. T cell stimulation rapidly increases mitochondrial activity and ATP formation and induces the translocation of mitochondria to the immunological synapse (IS) that T cells form with antigen-presenting cells. The ATP generated by mitochondria is released into the IS, where it drives autocrine purinergic signaling events that contribute to Ca2+ influx. In the current study, we investigated whether defects in these purinergic signaling processes are responsible for T cell suppression in sepsis.
We found that not only stimulated but also resting T cells require purinergic signaling mechanisms. In T cells from healthy subjects, basal mitochondrial activity and ATP release constitute a resting or basal purinergic signaling loop that maintains T cell vigilance and allows T cells to recognize and effectively respond to antigen stimulation. In patients with sepsis, this basal purinergic signaling mechanism is defective, resulting in impaired T cell vigilance, T cell suppression, and an inability of T cells to mount appropriate functional responses to infections.
MATERIALS AND METHODS
Reagents
Fluo-4-AM, tetramethylrhodamine ethyl ester (TMRE), dihydrorhodamine123 (DHR123) and MitoTracker Green-FM were purchased from Molecular Probes (Life Technologies). Mouse anti–human CD4–allophycocyanin (APC) antibodies were obtained from Biolegend, and goat anti–mouse immunoglobulin G (IgG) Fc antibodies from Pierce (Thermo Scientific). Polystyrene particles (3.0–3.9 µm; Spherotech) or Dynabeads (Invitrogen; Life Technologies) coated with goat anti–mouse IgG antibodies were labeled with mouse anti–human CD3 and anti–human CD28 antibodies (BD Biosciences). Suramin, NF023, NF157, 5-BDBD, A438079, KN62, oxidized ATP (o-ATP), and NF340 were from Tocris Bioscience. All other reagents were from Sigma-Aldrich, unless otherwise stated.
Patients
Eligible patients included adults (>18 years) presenting to the emergency department of the Beth Israel Deaconess Medical Center (BIDMC) with a diagnosis of sepsis or septic shock based on published criteria [16]. Heparinized blood samples (4 mL) were obtained within 12 hours of sepsis diagnosis and before initiation of medical therapy with antibiotics or steroids as needed. Initial Sequential Organ Failure Assessment (SOFA) scores were calculated at admission [17]. Nonseptic patients presenting to the emergency department for benign complaints with noninfective causes and healthy volunteers served as control groups. Demographic patient characteristics are given in Supplementary Tables 1 and 2. All studies involving human subjects were approved by the Institutional Review Board of BIDMC, and written informed consent was obtained before blood samples were collected.
Cells
CD4+ T cells were purified from heparinized venous blood, as described elsewhere [18]. Leukocyte-rich plasma was obtained immediately after blood sampling by spontaneous sedimentation of red blood cells over Ficoll-PaqueTM PLUS (GE Healthcare) for 30 minutes at 37°C. The human T cell line Jurkat (clone E6-1; American Type Culture Collection) was cultured as described elsewhere [12]. In some experiments, cells were treated with inhibitors of mitochondrial function or purinergic signaling. These treatments did not change viability, as determined with trypan blue dye exclusion (viability, >95%).
Imaging of Cytosolic and Mitochondrial Ca2+
Jurkat cells expressing the Ca2+ biosensors G-GECO1.1 and mito-CAR-GECO1 (Addgene plasmids 46 022 and 32 445; Addgene) [19] were generated by electroporation (Neon transfection system; Life Technologies), cultured for 4–6 hours, and placed into fibronectin-coated 8-well glass-bottom dishes (Lab-Tek). Fluorescence live-cell imaging was performed with an inverted Leica DMI6000B microscope (Leica Microsystems) equipped with a temperature-controlled stage incubator (Harvard Apparatus) and a Leica DFC365 FX camera. Changes in cytosolic and mitochondrial Ca2+ after stimulation with anti-CD3 antibodies (0.5 µg/mL) were monitored in cells treated or not treated with suramin (100 µmol/L) for 20 minutes. Fluorescence images were captured through a ×63 oil objective with a nominal aperture of 1.4 using fluorescein isothiocyanate (FITC) and tetrarhodamine isothiocyanate (TRITC) filter sets (Leica Microsystems) and Leica LAS microscope imaging software. Images were analyzed with ImageJ software (National Institutes of Health).
Live-cell Imaging of Cell/Bead Interactions
The capability of Jurkat cells to form immune synapses with anti-CD3/CD28 antibody–coated beads after treatment with carbonyl cyanide m-chlorophenylhydrazone (CCCP; 10 µmol/L) for 20 minutes was monitored by bright field imaging with the microscope system mentioned above.
Assessment of Mitochondrial Membrane Potential, Reactive Oxygen Species Formation, and Mitochondrial Mass
Mitochondrial membrane potential (ΔΨm) was assessed with TMRE, mitochondrial reactive oxygen species (ROS) formation with DHR123 and mitochondrial content with MitoTracker Green-FM. Cell preparations were diluted 1:10 in Hank's buffered salt solution; incubated with TMRE (100 nmol/L), DHR123 (10 µmol/L), or MitoTracker Green-FM (1 µmol/L) and anti–CD4-APC monoclonal antibodies (1:1000) for 15 minutes at 37°C; and immediately analyzed using a BD FACSCalibur flow cytometer (BD Biosciences). CD4+ T cells were identified by characteristic forward and side scattering and CD4 staining. In some experiments, cells were stimulated for 10 minutes with anti-CD3/CD28 antibody coated polystyrene beads or anti-CD3/CD28 antibodies cross-linked by goat anti–mouse IgG. Jurkat cells were treated with inihibitors as described above, stained with TMRE (100 nM), and ΔΨm was imaged using the microscope system described above.
Intracellular Ca2+, ATP Release, and Messenger RNA Expression
Cytosolic Ca2+ levels were determined in purified CD4+ T cells or enriched leukocytes using the Ca2+ indicator Fluo-4-AM, as described elsewhere [12]. In experiments with enriched leukocytes, CD4+ T cells were identified by forward or side scattering and CD4-APC staining. ATP measurements in cell culture supernatants and imaging of ATP release using the cell-surface–targeting fluorescent ATP probe 2-2Zn(II) [20] were done as described elsewhere [12]. The expression of IL-2, P2X1, P2X4, and P2X7 messenger RNA (mRNA) was determined using quantitative polymerase chain reaction, as described elsewhere [14, 18].
Statistical Analyses
Unless otherwise stated, data are expressed as mean values (with standard deviations) from ≥3 independent experiments. Statistical analyses were performed using unpaired Student t tests for 2 groups or 1-way analysis of variance and post hoc Holm-Sidak test for multiple comparisons. Pearson analysis was used to test for correlation between parameters. Differences were considered statistically significant at P < .05.
RESULTS
Purinergic Regulation of Ca2+ Signaling and Mitochondrial Activity in Stimulated T Cells
Autocrine purinergic signaling is an essential mechanism in T cell activation [13, 15, 21]. It was recently demonstrated that mitochondria accumulate at the IS of stimulated Jurkat T cells, where they generate large amounts of ATP that drives autocrine purinergic signaling processes [12]. ATP released at the IS stimulates P2X1 and P2X4 receptors that promote Ca2+ influx, which is essential for T cell activation [14, 22]. In the current study, we investigated whether these purinergic signaling processes also regulate mitochondrial ATP production. Stimulation of primary human CD4+ T cells by T cell receptor (TCR)/CD28 cross-linking induced a significant increase in cytosolic Ca2+ levels that was virtually completely blocked by pretreatment with the general P2 receptor inhibitor suramin (Figure 1A). These findings highlight the critical role of autocrine purinergic signaling in the regulation of Ca2+ signaling during T cell stimulation. To study whether purinergic signaling is also involved in mitochondrial activation, we cotransfected Jurkat cells with the Ca2+ biosensors G-GECO1.1 and mito-CAR-GECO1 that allow monitoring of real-time changes in cytosolic and mitochondrial Ca2+ levels after T cell stimulation (Supplementary Video 1). Using this approach, we found that suramin blocked the cytosolic Ca2+ response as well as mitochondrial Ca2+ uptake (Figure 1B).
Figure 1.
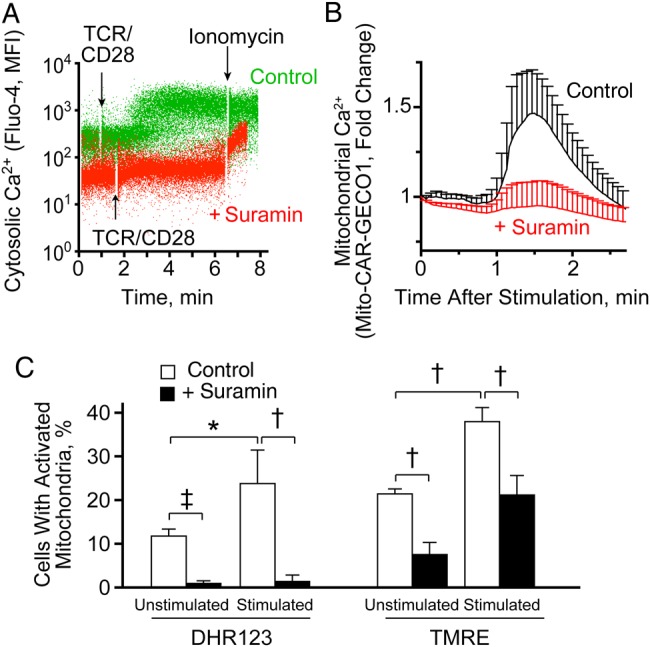
Positive feedback through autocrine purinergic signaling stimulates calcium (Ca2+) signaling and mitochondrial activity during T cell activation. A, Purified CD4+ T cells (1 × 106/mL) were loaded with Fluo-4 and incubated for 10 minutes with or without (control) suramin (100 µmol/L). Then cells were stimulated by T cell receptor (TCR)/CD28 cross-linking, and Ca2+ signaling was analyzed with flow cytometry. Ionomycin was used to assess the maximum response. Representative dot plots of separate experiments (n = 3) with similar results are shown. B, Jurkat T cells transiently expressing the mitochondrial Ca2+ biosensor mito-CAR-GECO1 were treated or not treated (control) with suramin (100 µmol/L) for 20 minutes. Changes in mitochondrial Ca2+ after TCR stimulation were recorded using time-lapse fluorescence video microscopy and analyzed using National Institutes of Health ImageJ software. Results are shown as means (with standard deviations [SDs]) from different cells (n = 10–15) and are representative of separate experiments (n = 4) with similar results (see also Supplementary Video 1). C, Whole blood leukocytes were treated or not treated (control) with suramin (200 µmol/L) for 10 minutes and stimulated by TCR/CD28 cross-linking for another 10 minutes. Then cells were stained with DHR123 or tetramethylrhodamine ethyl ester (TMRE), and mitochondrial activation (DHR123) and changes in mitochondrial membrane potential were analyzed with flow cytometry. CD4+ T cells were identified using allophycocyanin-CD4 labeling; the percentages of cells with increased TMRE and DHR123 fluorescence are shown as means and SDs from separate experiments with cells from different donors (n = 3). *P < .05; †P < .01; ‡P < .001 (all Student t test). Abbreviation: MFI, mean fluorescence intensity.
Ca2+ uptake by mitochondria is known to promote mitochondrial ATP formation through oxidative phosphorylation [23]. Recently, it was demonstrated that autocrine purinergic signaling of stimulated T cells requires mitochondrial ATP production and that this process is paralleled by increases in mitochondrial membrane potential (ΔΨm) and formation of mitochondrial ROS [12]. Using CD4+ T cells, we found that suramin pretreatment blocked the increase in ΔΨm and ROS formation in response to T cell stimulation (Figure 1C). However, to our surprise, we noticed that suramin also significantly reduced baseline ΔΨm and ROS production in unstimulated CD4+ T cells (Figure 1C), which suggests that purinergic feedback mechanisms maintain basal mitochondrial activity in resting T cells.
Maintenance of T Cell Vigilance by Basal Purinergic Signaling
Using a novel membrane-anchoring fluorescent ATP sensor, 2-2Zn(II) [20], it was shown that T cells release ATP at the IS [12]. In the current study, we found that resting T cells also release ATP from local hot spots associated with filopodia that seem to probe the extracellular environment for antigen-presenting cells (Figure 2A; Supplementary Video 2). On recognition of beads coated with anti-CD3/CD28 antibodies, ATP release intensified and concentrated at the IS (Figure 2A; Supplementary Video 3). Treating cells for 20 minutes with the mitochondrial inhibitor CCCP or with suramin impaired the interaction of cells with beads (Supplementary Video 4 and Supplementary Figure 1A and 1B). Suramin significantly reduced mitochondrial activity and the number of T cells binding to beads and responding with mitochondrial ATP production (Figure 2B and 2C; Supplementary Figure 1B and 1C). These findings indicate that T cells require purinergic signaling and mitochondrial ATP production to recognize antigens, form immune synapses, and to up-regulate mitochondrial activity. In support of this conclusion, we found that suramin or CCCP treatment abolished mitochondrial Ca2+ uptake and translocation of activated mitochondria to the IS (Supplementary Video 5). Taken together, these findings demonstrate that resting T cells have a basal purinergic signaling mechanism that maintains T cells at a level of vigilance that allows them to probe their extracellular environment and up-regulate mitochondrial activity and IS formation in response to antigen recognition.
Figure 2.

Autocrine purinergic signaling maintains basal functions of resting T cells. A, Jurkat T cells were stained with the adenosine triphosphate (ATP) probe 2-2Zn(II) and basal ATP release or ATP release in response to T cell receptor/CD28 stimulation with anti-CD3/CD28–coated beads was recorded using fluorescence microscopy. Apyrase (10 U/mL) was added as a control to remove extracellular ATP (eATP) (×63 oil objective; nominal aperture, 1.4; scale bar, 5 µm) (see also Supplementary Videos 2 and 3). B, C, Jurkat cells were treated or not treated (control) with 100 µmol/L suramin for 10 minutes, stimulated with anti-CD3/CD28 antibody–coated beads, and stained with DHR123; then mitochondrial activity (reactive oxygen species production) and the percentage of cells interacting with beads were analyzed with flow cytometry. Cells attached to beads were identified by their increased side scatter (SSC) signal (B; see also Supplementary Figure 1B). The area under the curve in SSC-positive cells was used to quantify changes in the percentage of cells bound to beads. Cells with active mitochondria were identified by DHR123 staining, as shown in Supplementary Figure 1C. Means and standard deviations from different experiments (n = 6) are shown in C. *P < .05; †P < .001 (Student t test).
Control of Basal Mitochondrial Activity by Basal Purinergic Signaling
It was previously demonstrated that mitochondria and pannexin-1 channels (panx1) contribute to the ATP release at the IS [12, 14]. Therefore, we tested whether mitochondria and panx1 are also involved in the purinergic signaling mechanism of resting T cells. Inhibition of panx1 or mitochondria with carbenoxolone (CBX) or CCCP decreased ATP release in resting Jurkat cells in a dose-dependent manner (Figure 3A). Inhibition of mitochondria and ATP release also reduced cytosolic Ca2+ levels in CD4+ T cells (Figure 3B). Likewise removal of extracellular ATP by apyrase or blocking of P2 receptor signaling with suramin decreased basal Ca2+ levels to a similar extent (Figure 3B). Because up-regulation of mitochondrial activity is part of the purinergic feedback cycle in stimulated T cells, as described above, we studied next how suramin affects ΔΨm in resting cells. Suramin markedly reduced ΔΨm in unstimulated Jurkat cells about as much as the uncoupling agent CCCP (Figure 3C and 3D; Supplementary Video 6). Together with the findings above, these results demonstrate that basal ATP release, basal purinergic signaling, and basal mitochondrial activity maintain Ca2+ homeostasis in resting T cells.
Figure 3.

Purinergic signaling maintains calcium homeostasis and basal mitochondrial activity in resting T cells. A, Jurkat T cells were treated for 10 minutes with different concentrations of carbonyl cyanide m-chlorophenylhydrazone (CCCP) or carbenoxolone (CBX), and extracellular adenosine triphosphate (eATP) concentrations in the supernatant were determined with a luciferin/luciferase assay; values represent means and standard deviations (SDs) (n = 2–5). †P < .001 (vs control). B, Primary CD4+ T cells were treated or not treated (control) with apyrase (20 U/mL), CBX (50 µmol/L), suramin (200 µmol/L), or CCCP (50 µmol/L) for 10 minutes. Then cells were loaded with Fluo-4, and cytosolic calcium ion (Ca2+) levels were analyzed with flow cytometry. CD4+ T cells were identified using anti-CD4 staining. Data represent means and SDs from 3–6 experiments. *P < .05 (vs control; 1-way analysis of variance. C, D, Jurkat cells were treated or not treated (control) with suramin (100 µmol/L) or CCCP (10 µmol/L) for 20 minutes, and changes in mitochondrial membrane potential (ΔΨm) were assessed with tetramethylrhodamine ethyl ester (TMRE) and live-cell fluorescence imaging. C shows representative images, and D shows increases in TMRE fluorescence (gray values), as means and SDs of different cells (n = 13–18); data are representative of 3 separate experiments (×100 oil objective; nominal aperture, 1.3; scale bar, 5 µm) (see also Supplementary Video 6). Abbreviation: MFI, mean fluorescence intensity.
Maintenance of T Cell Vigilance by P2X1 but Not P2X4 or P2X7 Receptors
To further define the basal autocrine purinergic signaling mechanisms that maintain T cell vigilance, we studied how different P2 receptor antagonists affect ΔΨm and mitochondrial ATP production in resting CD4+ T cells. Similar to the results with Jurkat cells shown above, we found that the nonspecific P2 receptor antagonist suramin reduced both ΔΨm and mitochondrial ROS production in a dose-dependent manner (Figure 4A). Similar effects were observed with the P2X1 receptor antagonists NF023 and NF157 (Figure 4B and 4C) but not with the P2Y11 antagonist NF340. Previous work had shown that P2X1 along with P2X4 and P2X7 receptors contribute to cytosolic Ca2+ signaling in stimulated T cells [14, 15]. Our current studies indicate that neither P2X4 nor P2X7 receptors contribute to the basal purinergic signaling that maintains mitochondrial function in resting T cells. Neither inhibition of P2X4 receptors with 5-BDBD nor inhibition of P2X7 receptors with KN62 or A438079 reduced the mitochondrial activity in resting CD4+ T cells (Figure 4C).
Figure 4.

P2X1 receptors contribute to basal mitochondrial activity of resting T cells, but neither P2X4 nor P2X7 receptors do. A, B, Primary CD4+ T cells were treated for 20 minutes with the indicated concentrations of the general P2 receptor inhibitor suramin (A) or the specific P2X1 receptor inhibitor NF023 (B). Cells were then stained with tetramethylrhodamine ethyl ester (TMRE) or DHR123, and mitochondrial membrane potential (ΔΨm) or mitochondrial activity (reactive oxygen species [ROS] production) was analyzed by means of fluorescence-activated cell sorting, as described for Figure 1C. C, Primary CD4+ T cells were treated for 20 minutes with the general P2 receptor inhibitor suramin (200 µmol/L), the specific P2X1 receptor inhibitors NF023 (20 µmol/L) or NF157 (20 µmol/L), the P2X4 receptor inhibitor 5-BDBD (10 µmol/L), the P2X7 inhibitors KN62 (50 nmol/L) or A438079 (10 µmol/L), or the P2Y11 receptor inhibitor NF340 (20 µmol/L). Then cells were stained with TMRE or DHR123, and ΔΨm and ROS production were analyzed as described above. Results are expressed as means and standard deviations of 3–5 separate experiments with cells from different donors. Results in B and C are shown as the percentage of cells with active mitochondria relative to untreated controls and cells treated with suramin (200 µmol/L). Statistical analyses were performed with 1-way analysis of variance. *P < .05 (vs control). Abbreviation: MFI, mean fluorescence intensity.
Basal Purinergic Signaling and Functional T cell Responses
The findings described above demonstrate that basal autocrine purinergic signaling via P2X1 receptors maintains mitochondrial function and the immune vigilance of unstimulated T cells. In support of this notion, we found that inhibition of mitochondrial ATP production with CCCP, removal of released ATP with apyrase, or inhibition of P2X1 receptors with suramin or NF023, but not of P2X4 or P2X7 receptors with 5-BDBD or A438079, blocked ΔΨm in resting CD4+ T cells (Figure 5A). The inhibition of purinergic signaling also decreased basal cytosolic Ca2+ levels and impaired Ca2+ signaling in response to TCR/CD28 stimulation (Figure 5B). Although the P2X7 receptor antagonists A438079 did not significantly affect basal cytosolic Ca2+ levels, it blocked Ca2+ signaling after TCR stimulation. Inhibition of purinergic signaling with either P2X1 (NF023) or P2X7 (A438079 or o-ATP) receptor inhibitors reduced IL-2 mRNA synthesis of stimulated T cells (Figure 5B and 5C). Taken together with previous reports [14, 15], these results indicate that there are 2 distinct autocrine purinergic signaling systems: one facilitating T cell vigilance through P2X1 receptors and a second one regulating functional T cell responses via P2X1, P2X4, and P2X7 receptors.
Figure 5.

P2X1 receptors together with P2X7 receptors contribute to the functional responses of activated T cells. A, CD4+ T cells were treated or not treated (control) for 20 minutes with carbonyl cyanide m-chlorophenylhydrazone (CCCP; 50 µmol/L), apyrase (10 U/mL), suramin (200 µmol/L), NF023 (10 µmol/L), 5-BDBD (10 µmol/L), or A438079 (10 µmol/L), and mitochondrial membrane potential (ΔΨm) was assessed with tetramethylrhodamine ethyl ester (TMRE) and flow cytometry; values are presented as means and standard deviations (SDs) (n = 3–5). *P < .05 (vs control; 1-way analysis of variance [ANOVA]). B, CD4+ T cells were treated or not treated (control) with apyrase (20 U/mL), suramin (200 µmol/L), or A438079 (10 µmol/L) for 10 minutes and loaded with Fluo-4; cytosolic calcium ion (Ca2+) levels were assessed before and 3 minutes after cell stimulation via T cell receptor (TCR)/CD28 cross-linking using flow cytometry. Values represent means and SDs of 3–6 experiments. *P < .05 (vs noninhibitor control; 1-way ANOVA). C, CD4+ T cells were treated or not treated (control) for 10 minutes with the indicated concentrations of apyrase, suramin, the P2X1 receptor inhibitor NF023, or the P2X7 receptor inhibitor oxidized ATP (o-ATP). Then cells were stimulated via TCR/CD28 using anti-CD3/CD28 antibody–coated beads for 1 hour, and interleukin 2 (IL-2) messenger RNA (mRNA) expression was determined by means of quantitative polymerase chain reaction. Values represent means and SDs of 3 separate experiments with cells from different donors. *P < .05 (vs control).
Defective Basal Purinergic Signaling, T Cell Vigilance, and T Cell Function in Sepsis
T cell suppression in sepsis is a well-known phenomenon [24], but its underlying mechanisms are unclear. We wondered whether sepsis impairs the basal purinergic signaling events that maintain T cell vigilance. To test this possibility, we assessed basal ΔΨm and mitochondrial ROS production in CD4+ T cells from patients admitted to the emergency department at BIDMC. We found that both mitochondrial parameters were markedly lower in cells from patients with sepsis than in healthy controls or patients without sepsis, whereas there were no significant differences in total mitochondrial content among these 3 cohorts (Figure 6A–C; Supplementary Figure 2A). Furthermore, alterations in P2X receptor expression levels did not seem to contribute to T cell suppression, because mRNA expression was either unchanged (P2X4, P2X7) or up-regulated (P2X1) in T cells of patients with sepsis compared with control cells (Supplementary Figure 2B). The decreased mitochondrial activity levels in T cells of septic patients were paralleled by significantly lower basal and stimulated Ca2+ levels (Figure 7A). These findings suggest that impaired basal mitochondrial function and purinergic signaling deplete the mechanism that maintains T cell vigilance and the ability of T cells to mount appropriate responses to antigen stimulation.
Figure 6.

Mitochondrial function is decreased in lymphocytes of septic patients. A–C, CD4+ T cells of healthy subjects (n = 8–16) or patients admitted to the emergency department with sepsis (n = 9–14) or without sepsis (n = 7–10) were stained with tetramethylrhodamine ethyl ester (TMRE) to assess mitochondrial membrane potential (ΔΨm) or with DHR123 to assess reactive oxygen species (ROS) formation as a measure of mitochondrial activity. Cells were analyzed with flow cytometry, and CD4+ T cells were identified by forward/side scattering and CD4 staining. A, B, Representative histograms of CD4+ T cells stained with TMRE (A) or DHR123 (B). C, TMRE mean fluorescence intensity (MFI) values or the percentage of DHR123-positive cells (as defined in B) among CD4+ T cells derived from healthy subjects and patients with or without sepsis. *P < .05 (1-way analysis of variance).
Figure 7.

Impaired calcium ion (Ca2+) homeostasis in CD4+ T cells is correlated with sepsis severity. A, CD4+ T cells of healthy subjects (n = 13–14) or of patients admitted to the emergency department with (n = 10–12) or without (n = 6–7) sepsis were stained with Fluo-4, and intracellular Ca2+ levels were assessed with flow cytometry before and 3 minutes after cell stimulation by T cell receptor (TCR)/CD28 cross-linking or addition of phytohemagglutinin (PHA; 5 µg/mL). CD4+ T cells were identified by forward/side scattering and CD4 staining. *P < .05 (1-way analysis of variance). B, C, Correlation between stimulated (B) or basal (C) cytosolic Ca2+ levels and the Sequential Organ Failure Assessment (SOFA) score, a measure of sepsis severity, determined on day 1 of sepsis diagnosis, was assessed with Pearson correlation analysis. Abbreviation: MFI, mean fluorescence intensity.
Septic patients were on average older than the subjects in either of the control groups (Supplementary Tables 1 and 2). The age range in the control groups was 23–74 years (median, 35 and 36.5 years in healthy controls and nonseptic patients, respectively), compared with 22–98 years (median, 66 years) for patients with sepsis. Nevertheless, we found no correlations between age and the parameters of mitochondrial function or Ca2+ signaling tested (Supplementary Figure 3A–C). Similarly, gender did not seem to be associated with any of our results (Supplementary Figure 3D–E).
Mitochondrial ROS formation correlated significantly with cytosolic Ca2+ levels, which further suggests a causal relationship between depleted basal purinergic signaling and defective T cell vigilance and function in septic patients (Supplementary Figure 2C). In addition, we found strong correlations between the SOFA score, a clinical parameter that closely predicts mortality and outcome in septic patients [25], and the basal and stimulated cytosolic Ca2+ levels (Figure 7B and 7C) and mitochondrial ROS production by CD4+ T cells (Supplementary Figure 2D). Taken together, our results demonstrate that depleted mitochondrial function and impaired Ca2+ homeostasis of CD4+ T cells are tightly associated with clinical signs of sepsis, underscoring that impaired T cell vigilance is causally linked to the T cell suppression that renders septic patients unable to cope with their infections.
DISCUSSION
Purinergic signaling is a central regulator of T cell function [14, 15]. Recently, it was shown that stimulation of T cells results in a rapid burst of mitochondrial ATP production, the localized release of ATP at the IS and Ca2+ influx through P2X receptors that promote T cell activation [12].
Surprisingly, the current study revealed that unstimulated T cells possess a designated basal purinergic signaling system that maintains basic functions to support antigen recognition. We identified 3 distinct stages of T cell activity depending on the levels of cytosolic Ca2+ signaling, mitochondrial ATP production, and cellular ATP release (Supplementary Figure 4). Resting T cells are in standby mode and expend little energy but are constantly surveying their environment for signs of infection. In this stage of vigilance, mitochondria provide ATP that stimulates P2X1 receptors in order to maintain basal cytosolic Ca2+ homeostasis. When stimulated, vigilant T cells quickly recognize and effectively respond to danger signals in their surroundings. They increase mitochondrial ATP production and cellular ATP release to stimulate P2X1, P2X4, and P2X7 receptors, resulting in sustained cytosolic Ca2+ levels that are necessary for prolonged T cell activation and appropriate functional responses, such as IL-2 production and cell proliferation [14, 15].
This activated state empowers T cells of healthy subjects to effectively cope with infections. However, we found that autocrine purinergic signaling is defective in T cells of patients with sepsis. Resting cells from such patients lack the autocrine feedback loops that maintain basal cytosolic Ca2+ levels and mitochondrial function, resulting in a state of cellular paralysis that impairs vigilance and precludes proper functional responses to T cell stimulation. This suppression of purinergic signaling is not due to P2X receptor down-regulation. P2X receptor mRNA levels were unchanged or elevated in patients with sepsis.
The incidence of severe sepsis is higher in elderly individuals [26]. Subjects in both of our control groups were significantly younger than patients in the sepsis group. This limitation of our study should be addressed in future larger studies with age-matched control groups. However, we did not find that any of the parameters tested were significantly correlated with age. Instead, we found that defects in mitochondrial function and basal purinergic signaling strongly correlated with sepsis severity. However, additional studies with larger patient cohorts will be necessary to determine how these changes impact survival. Furthermore, although samples were obtained immediately after the diagnosis of sepsis and before the initiation of further medical treatment, we did not investigate how medications administered before sepsis was diagnosed might have influenced our results.
We conclude that defective mitochondrial function and basal purinergic signaling in sepsis result in T cell suppression [5, 24, 27]. Our findings are in agreement with other reports of depletion of cellular oxygen consumption and mitochondrial function in sepsis [28–33]. Previous studies have shown that sepsis dramatically depletes cytosolic Ca2+ levels of resting and stimulated T cells [34, 35]. Our finding that mitochondrial function and purinergic signaling are closely linked to cellular Ca2+ homeostasis provides an explanation for those surprising findings.
Previous studies have also shown that the activation of IL-2 production and proliferation of T cells requires P2X1, P2X4, and P2X7 receptors [13–15]. In the current study, we found that, of these receptors, only P2X1 contributes to T cell vigilance. P2X1 receptors display strong affinity for ATP (100–700 nmol/L [36]), but considerably higher ATP concentrations are needed to stimulate P2X4 (micromolar [37]) and P2X7 receptors (>1 mmol/L [38, 39]). Release of low amounts of ATP from resting T cells would therefore preferentially stimulate P2X1 receptors, whereas ATP concentrations high enough for P2X4 and P2X7 receptor stimulation require TCR/CD28 stimulation and up-regulated mitochondrial ATP production. Further studies applying genetic approaches such as receptor silencing will be needed to confirm our results and to dissect the specific roles of P2X receptor subtypes in the regulation of unstimulated and stimulated T cells.
Despite intensive research and numerous clinical trials during the last 2 decades, there has been little improvement in the treatment of sepsis. Therapeutic approaches targeting triggers of the hyperinflammatory phase have been explored but none of these approaches showed clinical efficacy in patients [3]. This has led to a reassessment of the pathology of sepsis [10, 24]. Our current study defines a novel mechanism of T cell suppression, whereby mitochondrial dysfunction depletes purinergic signaling, disrupts Ca2+ homeostasis, and impairs the vigilance of T cells and their ability to mount functional responses to T cell stimulation. This new concept points to new strategies designed to improve T cell function and immune defense in septic patients. These strategies may include approaches to avoid mitochondrial dysfunction, to bolster ATP production by mitochondria, and to promote T cell function by enhancing purinergic feedback mechanisms in order to restore T cell vigilance and prevent immunosuppression in sepsis.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Drs Yasutaka Kurishita and Itaru Hamachi for kindly providing the fluorescent ATP probe 2-2Zn(II).
Financial support. This work was supported in part by the National Institutes of Health (grants GM-51477, GM-60475, AI-080582, and T32GM103702 to W. G. J.) and the German Research Foundation (grant LE-3209/1-1 to C. L.).
Potential conflicts of interest. All authors: No potential conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA 2011; 306:2614–5. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. New Engl J Med 2003; 348:1546–54. [DOI] [PubMed] [Google Scholar]
- 3.Wenzel RP, Edmond MB. Septic shock—evaluating another failed treatment. New Engl J Med 2012; 366:2122–4. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. New Engl J Med 2003; 348:138–50. [DOI] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013; 13:862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carson WFT, Cavassani KA, Ito T et al. Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur J Immunol 2010; 40:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Tinsley KW, Swanson PE et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 2001; 166:6952–63. [DOI] [PubMed] [Google Scholar]
- 8.van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, Frolich M. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 1998; 351:950–3. [DOI] [PubMed] [Google Scholar]
- 9.Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492–9. [DOI] [PubMed] [Google Scholar]
- 10.Boomer JS, To K, Chang KC et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011; 306:2594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013; 13:260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledderose C, Bao Y, Lidicky M et al. Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling. J Biol Chem 2014; 289:25936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenk U, Westendorf AM, Radaelli E et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal 2008; 1:ra6. [DOI] [PubMed] [Google Scholar]
- 14.Woehrle T, Yip L, Elkhal A et al. Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 2010; 116:3475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yip L, Woehrle T, Corriden R et al. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 2009; 23:1685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy MM, Fink MP, Marshall JC et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2003; 31:1250–6. [DOI] [PubMed] [Google Scholar]
- 17.Vincent JL, Moreno R, Takala J et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996; 22:707–10. [DOI] [PubMed] [Google Scholar]
- 18.Yip L, Cheung CW, Corriden R, Chen Y, Insel PA, Junger WG. Hypertonic stress regulates T-cell function by the opposing actions of extracellular adenosine triphosphate and adenosine. Shock 2007; 27:242–50. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, Liu L, Matsuda T et al. Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem Neurosci 2013; 4:963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurishita Y, Kohira T, Ojida A, Hamachi I. Organelle-localizable fluorescent chemosensors for site-specific multicolor imaging of nucleoside polyphosphate dynamics in living cells. J Am Chem Soc 2012; 134:18779–89. [DOI] [PubMed] [Google Scholar]
- 21.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol 2001; 19:497–521. [DOI] [PubMed] [Google Scholar]
- 23.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 1990; 70:391–425. [DOI] [PubMed] [Google Scholar]
- 24.Ward PA. Immunosuppression in sepsis. JAMA 2011; 306:2618–9. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira FL, Bota DP, Bross A, Melot C, Vincent JL. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 2001; 286:1754–8. [DOI] [PubMed] [Google Scholar]
- 26.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–10. [DOI] [PubMed] [Google Scholar]
- 27.Walker MA, Volpi S, Sims KB, Walter JE, Traggiai E. Powering the immune system: mitochondria in immune function and deficiency. J Immunol Res 2014; 2014:164309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belikova I, Lukaszewicz AC, Faivre V, Damoisel C, Singer M, Payen D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit Care Med 2007; 35:2702–8. [DOI] [PubMed] [Google Scholar]
- 29.Brealey D, Brand M, Hargreaves I et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–23. [DOI] [PubMed] [Google Scholar]
- 30.Fredriksson K, Hammarqvist F, Strigard K et al. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am J Physiol Endocrinol Metab 2006; 291:E1044–50. [DOI] [PubMed] [Google Scholar]
- 31.Japiassu AM, Santiago AP, d'Avila JC et al. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5′-triphosphate synthase activity. Crit Care Med 2011; 39:1056–63. [DOI] [PubMed] [Google Scholar]
- 32.Reichenbach J, Schubert R, Horvath R et al. Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr Res 2006; 60:321–6. [DOI] [PubMed] [Google Scholar]
- 33.Walker MA, Slate N, Alejos A et al. Predisposition to infection and SIRS in mitochondrial disorders: 8 years’ experience in an academic center. J Allergy Clin Immunol Pract 2014; 2:465-8.e1. [DOI] [PubMed] [Google Scholar]
- 34.Choudhry MA, Ahmad S, Thompson KD, Sayeed MM. T-lymphocyte Ca2+ signalling and proliferative responses during sepsis. Shock 1994; 1:466–71. [DOI] [PubMed] [Google Scholar]
- 35.Hoyt DB, Junger WG, Loomis WH, Liu FC. Effects of trauma on immune cell function: impairment of intracellular calcium signaling. Shock 1994; 2:23–8. [DOI] [PubMed] [Google Scholar]
- 36.Evans RJ, Lewis C, Buell G, Valera S, North RA, Surprenant A. Pharmacological characterization of heterologously expressed ATP-gated cation channels (P2x purinoceptors). Mol Pharmacol 1995; 48:178–83. [PubMed] [Google Scholar]
- 37.Jones CA, Chessell IP, Simon J et al. Functional characterization of the P2X(4) receptor orthologues. Br J Pharmacol 2000; 129:388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 2011; 63:641–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan Z, Khadra A, Li S, Tomic M, Sherman A, Stojilkovic SS. Experimental characterization and mathematical modeling of P2X7 receptor channel gating. J Neurosci 2010; 30:14213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.