

Abstract
Although G protein-coupled receptors (GPCRs) are targeted by more clinically used drugs than any other type of protein, their ligand development is particularly challenging. Humans have four neuropeptide Y receptors: hY1R and hY5R are orexigenic, while hY2R and hY4R are anorexigenic, and represent important anti-obesity drug targets. We show for the first time that PEGylation and lipidation, chemical modifications that prolong the plasma half-lives of peptides, confer additional benefits. Both modifications enhance pancreatic polypeptide preference for hY2R/hY4R over hY1R/hY5R. Lipidation biases the ligand towards arrestin recruitment and internalization, whereas PEGylation confers the opposite bias. These effects were independent of the cell system and modified residue. We thus provide novel insights into the mode of action of peptide modifications and open innovative venues for generating peptide agonists with extended therapeutic potential.
Keywords: biased ligands, lipidation, PEGylation, peptides, receptors
The neuropeptide Y (NPY) hormone family signals through cell-surface receptors belonging to class A GPCRs.[1] With four Y receptors expressed in humans (hY1R, hY2R, hY4R, hY5R) and the three endogenous agonists NPY, peptide YY, and pancreatic polypeptide (PP),[2] this system is involved in many physiological processes, such as food intake.[3] Both hY1R and hY5R mediate orexigenic effects, whereas activation of hY2R and hY4R produce anorexigenic signals. Clearly, selective stimulation or blocking of the differentially acting Y receptors are favorable approaches for anti-obesity therapeutics.[4] Such a potential anti-obesity drug is Obinepitide (7TM Pharma), which is the human PP-derived ligand [Q34]hPP with a dual specific agonism for hY4R and hY2R.[5] Although Obinepitide was active in vivo, it was hampered by a poor pharmacokinetic profile because of its rapid clearance and degradation.[6]
Here, we investigated the impact of two commonly used chemical modifications—methoxy polyethylene glycol (PEGylation) and fatty acids (lipidation)[7]—on Obinepitide stability and, for the first time, on receptor signaling and endocytosis. The peptide core sequence [Q34]hPP was modified with palmitic acid (Pam) and diverse PEGs (5 kDa (PEG5) and 22 kDa (PEG22)) at the Nɛ groups of lysine-22, along with N-terminal labeling with fluorescent 6-carboxy-tetramethylrhodamine (TAMRA) (Figure 1). The high purity of all the peptides and maintenance of the α-helical character were confirmed, while in vitro studies using human blood plasma verified the increased metabolic stability of the PEGylated and palmitoylated analogues (see Tables S1 and S2 as well as Figure S1 in the Supporting Information).
Figure 1.
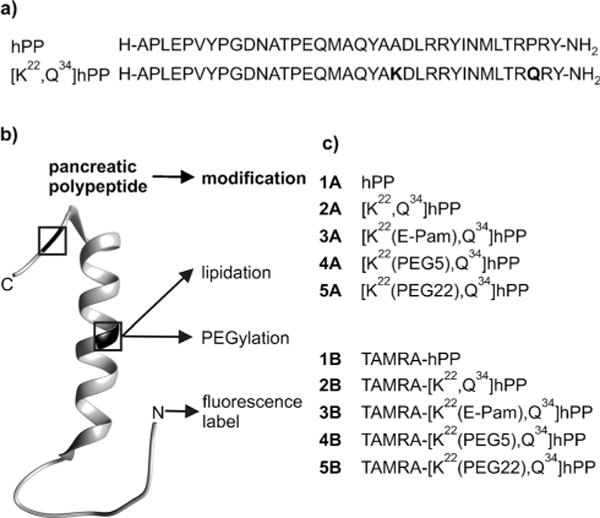
a) Sequences and b) ribbon model (PDB ID: 1LJV) with highlighted positions of modifications. c) hPP analogues modified by palmitoylation, PEGylation, and TAMRA.
The lead peptide and all the analogues were potent agonists at the hY4R (see Figure S2 in the Supporting Information), as indicated by the pEC50 values being comparable to those of hPP, and are also relatively well-tolerated by the hY2R, similar to its native ligand NPY (Table 1). Importantly, the same analogues showed low activities at hY1R. Nanomolar potencies are mostly observed at hY5R. However, as hY5R is predominantly expressed in the brain, it is not necessarily accessible to peripherally administered peptide drugs. Radioligand displacement experiments showed that the palmitoylated ligand maintained nanomolar affinity for hY4R (Figure 2; IC50 values: 1A: 0.7, 2A: 0.5, 3A: 1.2 nM). However, the addition of a PEG moiety with an increased size significantly reduced the hY4R affinity (IC50: 4A: 36.8, 5A: 94.2 nm). As reported earlier, restricted binding[6,8] of PEGylated peptides might result from steric hindrance of agonist–receptor interactions.
Table 1.
Signal transduction determined by the inositol phosphate accumulation assay, for all peptide–receptor combinations.
| compound | EC50/nM (pEC50 ± S.E.M.) | |||
|---|---|---|---|---|
| hY1R | hY2R | hY4R | hY5R | |
| pNPY | 3.3 (8.5±0.1) |
0.3 (9.6±0.1) |
44.7 (7.4±0.3) |
4.7 (8.3±0.1) |
| 1A | 76.0 (7.1±0.1) |
341.4 (6.5±0.1) |
1.3 (8.9±0.1) |
11.4 (7.9±0.1) |
| 2A | 51.6 (7.3±0.1) |
0.4 (9.4±0.2) |
1.0 (9.0±0.2) |
6.5 (8.2±0.2) |
| 3A | 83.5 (7.1±0.0) |
2.8 (8.6±0.2) |
2.1 (8.7±0.1) |
6.7 (8.2±0.3) |
| 4A | > 1000 (5.2±0.1) |
11.7 (7.9±0.1) |
2.9 (8.5±0.1) |
195.3 (6.7±0.2) |
| 5A | > 1000 (4.7±0.1) |
32.4 (7.5±0.1) |
13.8 (7.9±0.1) |
> 1000 (5.9±0.1) |
Figure 2.
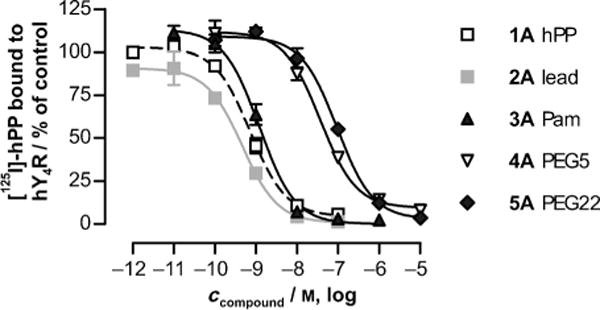
Mean [125I]-hPP displacement curves at the hY4R.
Fluorescence microscopy was used to characterize the uptake of the TAMRA-labeled hPP variants by receptor-mediated endocytosis in HEK293 cells stably expressing the hY4R–EYFP fusion protein (Figures 3a and 4a, see also Figure S3 in the Supporting Information). In response to ligand stimulation, intracellular accumulation of TAMRA fluorescence was seen for hPP (1B) and lead peptide (2B), while palmitoylation (3B: ca. 150%) facilitated internalization significantly. Surprisingly, PEGylated variants (4B, 5B#: < 2%) showed markedly decreased endocytosis with no detectable peptide uptake. Specific [125I]-hPP binding to cell-surface receptors after 1 h stimulation (Figure 4c) confirmed that the palmitoylated variant strongly increased internalization of the ligand–receptor complex, thereby eliminating virtually all the [125I]-hPP binding sites from the cell surface (3A: < 5%). PEGylated peptides did not induce internalization or loss of cell-surface receptor binding (4A: 80%, 5A: 120%). Importantly, these effects were specific because they required covalent attachment of the modification to the peptide and its binding and activation of the cognate receptor (see Figure S4 in the Supporting Information).
Figure 3.
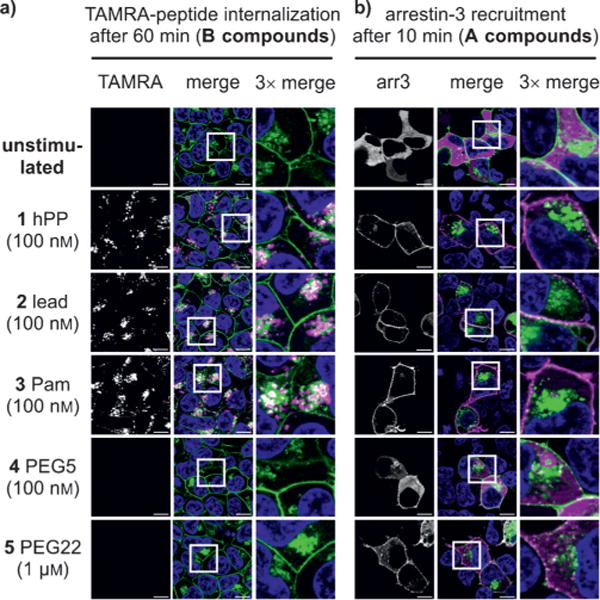
a) hY4R-mediated uptake of TAMRA-modified peptide analogues and b) arr3 recruitment. White vesicles in the overlayed channel indicate co-localization of hY4R (green) and peptide or arr3 (magenta). Scale bar: 10 μm.
Figure 4.
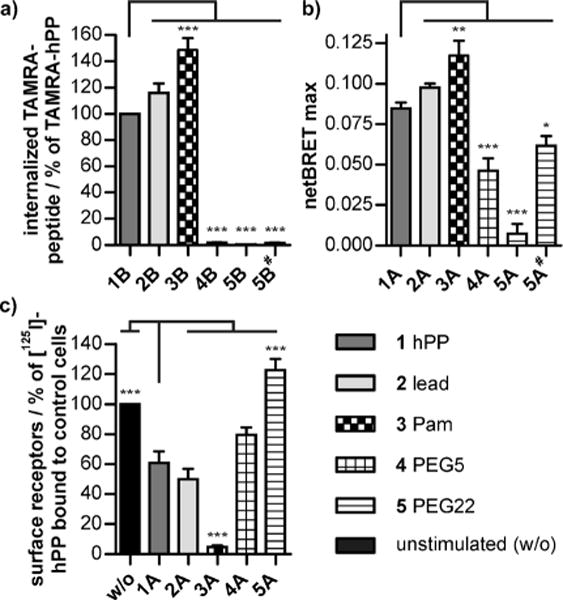
a) Internalization of TAMRA-labeled peptides after 60 min stimulation. b) Agonist-induced arr3 association after 10 min measured by BRET. c) Cell-surface receptors before (w/o) and after 60 min exposure to the agonist. The stimulation concentration was 100 nm for all compounds except for 5# (1 μM).
Arrestin recruitment was visualized using HEK293 cells transiently expressing EYFP-tagged hY4R and mCherry-fused arrestin-3 (arr3; Figure 3b). Rapid arr3 redistribution was seen after stimulation by all the analogues, which is in full agreement with previous demonstrations of arr3 involvement[9] in Y4R internalization.[10] Only the PEGylated variants (4A, 5A#) showed reduced arr3 recruitment. Arr3–receptor interactions were further quantified by bioluminescence resonance energy transfer (BRET; Figure 4b). The high maximal netBRET (see Figure S5 in the Supporting Information) showed robust arr3 recruitment by hPP (1A) and lead peptide (2A). Stimulation with the palmitoylated analogue (3A) increased arr3 recruitment, whereas PEGylated peptides (4A, 5A) showed considerably reduced BRETmax values in a concentration-dependent manner. Thus, ligand palmitoylation enhanced internalization and arr3 recruitment to the receptor without showing increased potency and affinity. This is in contrast to other receptors, where lipidation of the ligand led to enhanced receptor activity.[11]
However, the effects of lipidation critically depend on the length of the fatty acid chain, as peptides modified with shorter fatty acids (see Figure S6 in the Supporting Information) did not enhance arr3 recruitment or internalization (see Figure S7 in the Supporting Information).
PEGylated peptides, in contrast, showed diminished arr3 recruitment and no receptor internalization. PEGylation of various molecules has resulted either in enhanced[12] or hindered internalization,[13] but the exact sizes of the attached PEG were inconsistent.[14] In the present study, a low-molecular-weight PEG moiety (PEG5) attached to the peptide resulted in high activity, despite impaired uptake, arr3 recruitment, and receptor internalization. Clearly, the receptor conformation is biased towards the G protein for lower molecular weight PEG moieties.
To investigate the behavior of the peptide analogues in a more translatable system, the antisecretory responses (Isc)[15] were measured in Col-24 epithelial layers, where the lead peptide showed a rapid and transient activity with maximum effects at about 5 min (Figure 5a), as observed previously for hPP.(15) In contrast, the response onset to palmitoylated (3A) and PEGylated (4A) hPP variants was delayed, and both analogues showed prolonged reductions in Isc, with 3A exhibiting a higher maximum than the parental peptide (2A). Interestingly, 5A did not alter the Isc value during 45 min exposure. Analogous time courses were obtained in human colon mucosa (which expresses hY2R and hY4R; see Figure S8a in the Supporting Information).[16]
Figure 5.
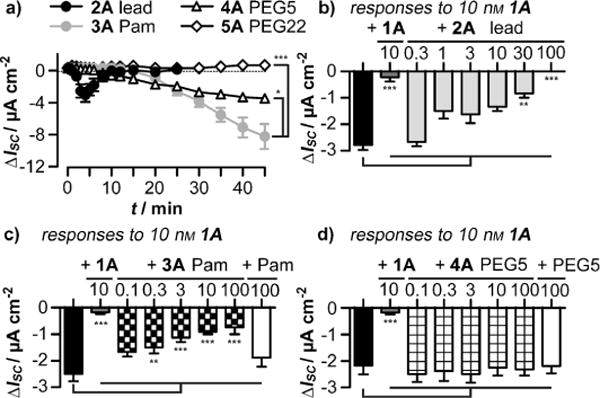
Responses of human Col-24 epithelial layers after treatment with a) 100 nm 2A, 3A, 4A, or 5A. Desensitization of responses to 10 nm hPP after the indicated concentrations of 1A (b–d), or 2A (b), 3A (c), or 4A (d) in nM. The black bars show responses to hPP without prestimulation. The white bars in (c,d) reflect hPP responses after the addition of 100 nM unconjugated Pam or PEG5.
Concentration–response curves in Col-24 cells revealed good activity and increased efficacy for 3A, and one order of magnitude enhanced potency (but unaltered efficacy) for 4A (see Figure S8b in the Supporting Information). Prestimulation with 2A or 3A, but not with 4A, resulted in a concentration-dependent desensitization of hY4R (Figure 5b–d).
In Col-24 cells, additionally transfected with hY4R-EYFP, we also observed rapid internalization of native (1B), lead (2B), and palmitoylated (3B) peptides, whereas the PEGylated derivative (5B) showed impaired cellular uptake (Figure 6a,b).
Figure 6.
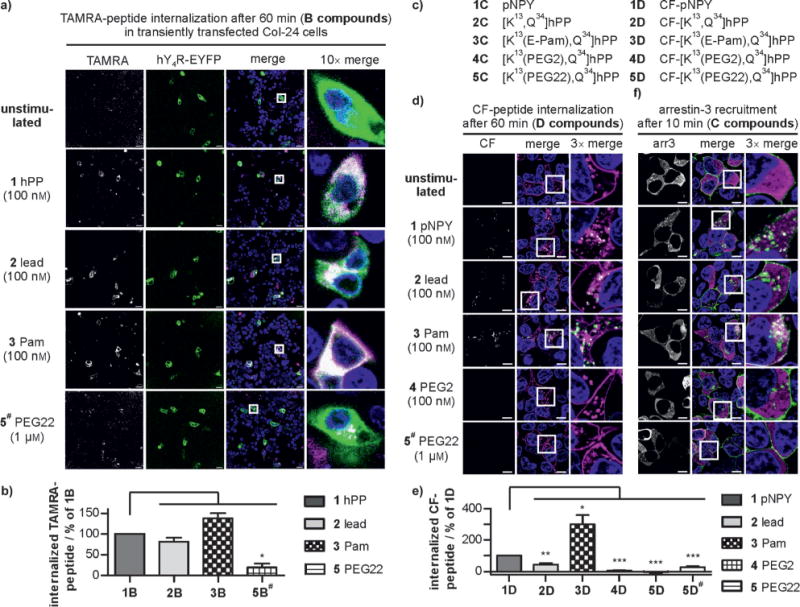
a) Live-cell images and b) quantification of ligand uptake (magenta) after 60 min exposure in Col-24 cells transiently transfected with hY4R (green). Scale bar: 20 μm. c) Porcine NPY (pNPY) and modified [K13,Q34]hPP analogues. Imaging (d) and quantification (e) of the hY2R-mediated (magenta) internalization of CF compounds (green) after 60 min stimulation. f) Arr3 distribution (magenta) after 10 min stimulation of hY2R (green) in transiently transfected HEK293 cells. Scale bar: 10 μm. Peptide concentration: 100 nM, except for 5B# and 5D# (1 μm).
The impact of peptide modification on hY4R activation and internalization were also transferable to hY2R, despite displaying a distinct binding mode with respect to hY4R (see Figure S9 in the Supporting Information). Moreover, we investigated a different set of chemically modified peptides, namely [K13,Q34]hPP conjugates, which we had fully characterized previously,[6] bearing Pam as well as PEGs of different sizes at position 13 and another fluorophore (5(6)-carboxyfluorescein (CF)) at the N-terminus (Figure 6c). These derivatives were tested for internalization and arr3 recruitment in HEK293 cells expressing hY2R (Figure 6d–f). Increased receptor-mediated uptake of the peptide upon palmitoylation (3D) and significantly reduced internalization of PEGylated derivatives (4D, 5D#) was confirmed by live-cell microscopy. Enhanced arr3 recruitment for 3C compared to porcine NPY (1C) and the lead peptide (2C) was observed, while minimal arr3 recruitment was detected with 4C or 5C. These results are in good agreement with the data obtained for [K22,Q34]hPP peptides, thus showing that the impact of different modifications depends only on their nature and is transferable to different receptor and peptide combinations.
In conclusion, our data show that different chemical modifications, used to improve the metabolic stability of ligands, can diametrically bias peptide agonists towards either G proteins or arrestins (Figure 7). Long fatty acids bias the agonist towards arrestins, which results in enhanced internalization of the receptor. In contrast, PEGylation allows G-protein activation but disrupts arr3 recruitment, thereby blocking internalization and receptor desensitization. These new findings provide powerful tools to control ligand uptake and receptor response. Accordingly, the type of modification to improve the half-life of peptide drugs deserves careful consideration in the future.
Figure 7.
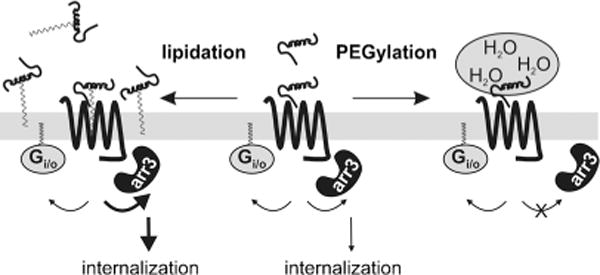
Impact of peptide modifications on agonist bias.
Supplementary Material
Footnotes
We acknowledge financial support from the EU (FP7, No. 223057 GIPIO), the DFG (SFB610, SFB1052, SPP1623), the NIH (GM077561), and the graduate school BuildMoNa. S.B. kindly acknowledges the financial contribution from the Fonds der Chemischen Industrie. We thank R. Reppich-Sacher, K. Löbner, C. Dammann, B. Goettgens, and B. Gill-Barman for excellent assistance.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201403750.
Contributor Information
Veronika Mäde, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany).
Stefanie Babilon, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany).
Dr. Navjeet Jolly, King’s College London, Wolfson Centre for Age-Related Diseases, Guy’s Campus, London SE1 1UL (UK)
Lizzy Wanka, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany).
Dr. Kathrin Bellmann-Sickert, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany)
Dr. Luis E. Diaz Gimenez, Vanderbilt University, Department of Pharmacology, Nashville, TN 37232 (USA)
Dr. Karin Mörl, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany)
Prof. Dr. Helen M. Cox, King’s College London, Wolfson Centre for Age-Related Diseases, Guy’s Campus, London SE1 1UL (UK)
Prof. Dr. Vsevolod V. Gurevich, Vanderbilt University, Department of Pharmacology, Nashville, TN 37232 (USA)
Dr. Annette G. Beck-Sickinger, Universität Leipzig, Faculty of Biosciences, Pharmacy and Psychology, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig (Germany).
References
- 1.Bellmann-Sickert K, Beck-Sickinger AG. Trends Pharmacol Sci. 2010;31:434–441. doi: 10.1016/j.tips.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Pedragosa-Badia X, Stichel J, Beck-Sickinger AG. Front Endocrinol. 2013;4:5. doi: 10.3389/fendo.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babilon S, Mörl K, Beck-Sickinger AG. Biol Chem. 2013;394:921–936. doi: 10.1515/hsz-2013-0123. [DOI] [PubMed] [Google Scholar]
- 4.Yulyaningsih E, Zhang L, Herzog H, Sainsbury A. Br J Pharmacol. 2011;163:1170–1202. doi: 10.1111/j.1476-5381.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz T. Vol. US20080261871 A1, 23.10.2008 [Google Scholar]
- 6.Bellmann-Sickert K, Elling CE, Madsen AN, Little PB, Lundgren K, Gerlach LO, Bergmann R, Holst B, Schwartz TW, Beck-Sickinger AG. J Med Chem. 2011;54:2658–2667. doi: 10.1021/jm101357e. [DOI] [PubMed] [Google Scholar]
- 7.a) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]; b) Ahrens VM, Bell-mann-Sickert K, Beck-Sickinger AG. Future Med Chem. 2012;4:1567–1586. doi: 10.4155/fmc.12.76. [DOI] [PubMed] [Google Scholar]
- 8.Kubetzko S, Sarkar CA, Pluckthun A. Mol Pharmacol. 2005;68:1439–1454. doi: 10.1124/mol.105.014910. [DOI] [PubMed] [Google Scholar]
- 9.Berglund MM, Schober DA, Statnick MA, McDonald PH, Gehlert DR. J Pharmacol Exp Ther. 2003;306:147–156. doi: 10.1124/jpet.103.051227. [DOI] [PubMed] [Google Scholar]
- 10.a) Parker SL, Kane JK, Parker MS, Berglund MM, Lundell IA, Li MD. Eur J Biochem. 2001;268:877–886. doi: 10.1046/j.1432-1327.2001.01966.x. [DOI] [PubMed] [Google Scholar]; b) Böhme I, Stichel J, Walther C, Mörl K, Beck-Sickinger AG. Cell Signalling. 2008;20:1740–1749. doi: 10.1016/j.cellsig.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 11.Todorovic A, Holder JR, Bauzo RM, Scott JW, Kavanagh R, Abdel-Malek Z, Haskell-Luevano C. J Med Chem. 2005;48:3328–3336. doi: 10.1021/jm0490843. [DOI] [PubMed] [Google Scholar]
- 12.Pawar SK, Badhwar AJ, Kharas F, Khandare JJ, Vavia PR. Int J Pharm. 2012;436:183–193. doi: 10.1016/j.ijpharm.2012.05.078. [DOI] [PubMed] [Google Scholar]
- 13.a) Sen P, Ghosh S, Ezban M, Pendurthi UR, Vijaya Mo-han Rao L. Haemophilia. 2010;16:339–348. doi: 10.1111/j.1365-2516.2009.02121.x. [DOI] [PubMed] [Google Scholar]; b) Mickler FM, Vachutinsky Y, Oba M, Miyata K, Nishiyama N, Kataoka K, Brauchle C, Ruthardt N. J Controlled Release. 2011;156:364–373. doi: 10.1016/j.jconrel.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Feng SS. Biomaterials. 2014;35:3340–3347. doi: 10.1016/j.biomaterials.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 15.a) Cox HM, Tough IR, Zandvliet DW, Holliday ND. Br J Pharmacol. 2001;132:345–353. doi: 10.1038/sj.bjp.0703815. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cox HM, Tough IR. Br J Pharmacol. 1995;116:2673–2678. doi: 10.1111/j.1476-5381.1995.tb17225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox HM, Tough IR. Br J Pharmacol. 2002;135:1505–1512. doi: 10.1038/sj.bjp.0704604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.