

Abstract
Plasma membrane proteins play critical roles in cell-to-cell recognition, signal transduction and material transport. Because of their accessibility, membrane proteins constitute the major targets for protein-based drugs. Here, we described an approach, which included stable isotope labeling by amino acids in cell culture (SILAC), cell surface biotinylation, affinity peptide purification and LC-MS/MS for the identification and quantification of cell surface membrane proteins. We applied the strategy for the quantitative analysis of membrane proteins expressed by a pair of human melanoma cell lines, WM-115 and WM-266-4, which were derived initially from the primary and metastatic tumor sites of the same individual. We were able to identify more than 100 membrane and membrane-associated proteins from these two cell lines, including cell surface histones. We further confirmed the surface localization of histone H2B and three other proteins by immunocytochemical analysis with confocal microscopy. The contamination from cytoplasmic and other nonmembrane-related sources is greatly reduced by using cell surface biotinylation and affinity purification of biotinylated peptides. We also quantified the relative expression of 62 identified proteins in the two types of melanoma cells. The application to quantitative analysis of membrane proteins of primary and metastatic melanoma cells revealed great potential of the method in the comprehensive identification of tumor progression markers as well as in the discovery of new protein-based therapeutic targets.
Keywords: Quantitative proteomics, LC-MS/MS, SILAC, membrane proteins, cell surface biotinylation, immunocytochemistry, malignant melanoma, biomarkers
Introduction
Membrane proteins represent about one-third of the proteins encoded by the human genome and assume a variety of key biological functions, such as cell-to-cell recognition, signal relay, and transport of ions and solutes.1 Because of their accessibility, membrane proteins also constitute more than two-thirds of the current protein-based drug targets.2 Despite their importance in biological processes and drug discovery, membrane proteins are usually challenging to study because of the difficulty in preparing clean plasma membrane (PM) fractions. In this context, PM proteins are hydrophobic and they have to be transferred into aqueous solution to be available for subsequent analysis. In addition, the relatively low abundances of most membrane proteins impose further challenges for determining their identities and functions.
Traditional two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) is useful for the separation and relative quantification of proteins at large scale, but this technique does not resolve membrane proteins very well, mainly because of their poor solubility in the buffers used for protein separation in the first dimension.3 Recently, advances in MS-based proteomics have facilitated the identification of a large number of membrane proteins and their post-translational modifications by using multidimensional LC coupled with MS/MS, which could overcome the disadvantages associated with conventional 2D-PAGE.4–9 These multidimensional LC-based techniques improved the separation power and could facilitate the identification of more proteins from complex mixtures. However, it is difficult to isolate and enrich PMs to reduce the sample complexity, and the majority of the proteins identified are not membrane-related. In addition, the PM protein fractions prepared by the typical ultracentrifugation method are usually contaminated with other cellular components.10 Therefore, selective enrichment is frequently essential for the study of low-abundance membrane and membrane-associated proteins.
Biotinylation and affinity purification of cell-surface proteins, when coupled with MS, can improve the throughput and selectivity for the detection of membrane proteins in low abundance.11–23 For example, relative enrichments of 1600- and 400-fold of PM proteins versus mitochondria and endoplasmic reticulum (ER), two major sources of contaminations in PM fractions prepared by ultracentrifugation methods, were achieved by using a commercial biotinylation reagent followed by 1D-SDS-PAGE separation and LC-MS/MS analysis.11 In previous studies, the biotin-conjugated proteins were frequently purified and enriched after the biotinylation reaction and cell lysis. The affinity-enriched membrane fractions, however, are still a complex mixture and contain significant contamination of cytoplasmic proteins. Further separation of the enriched membrane proteins by using SDS-PAGE, 2-D gel or HPLC is often necessary to reduce sample complexity.14,19,24
Different MS-based strategies have been developed for the quantitative profiling of proteins.25 However, quantification of membrane proteins at large scale seems to be more challenging than that of soluble proteins. For instance, isotope-coded affinity tag (ICAT),26 a widely used chemical tagging reagent for protein quantification, has been rarely employed for interrogating membrane proteins, probably as a result of the poor compatibility of ICAT with the high concentration of detergent required for dissolving membrane proteins.27 Recently, an optimized strategy for ICAT was published, and it was demonstrated that ICAT could be used for the quantitative analysis of hydrophobic proteins.28 Nevertheless, a relatively small number of papers have been published for the quantitative assessment of membrane proteins.19,29–33 Among the various isotope-labeling approaches, stable isotope labeling by amino acids in cell culture (SILAC) is simple and accurate,34 and SILAC has been demonstrated to be suitable for the quantitative analysis of cell membrane proteins using SDS-PAGE and LC-MS/MS.29
To improve the purification efficiency and to minimize contamination, in this report, we describe an approach for the selective quantification of surface membrane proteins. In our strategy, intact cells cultured in SILAC light and heavy media were labeled in situ with a membrane impermeable biotin reagent, sulfo-NHS-LC-biotin, which can conjugate with the N-terminus or the side chain amino group of lysine residues in a protein to afford an amide linkage (Schemes 1 and 2). The light and heavy cell lysates were mixed and digested with trypsin. The biotinylated peptides of membrane proteins were subsequently enriched via affinity purification and subjected to LC-MS/MS analysis.
Scheme 1. Strategy for the Relative Quantification of Surface Membrane Proteins in Primary and Metastatic Human Melanoma Cells.
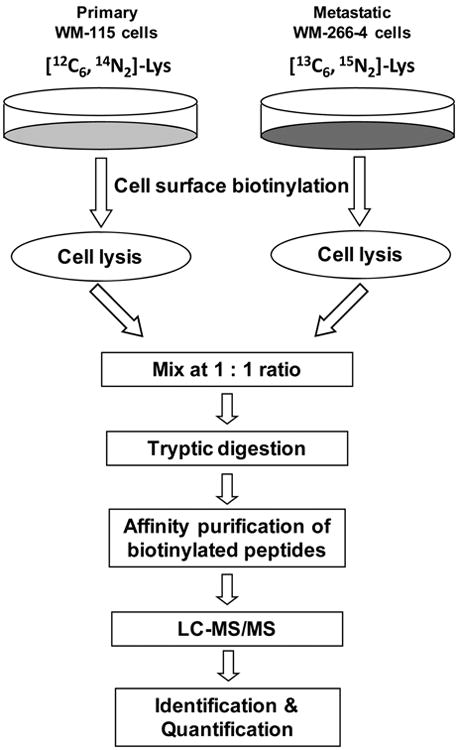
Scheme 2. Cell Surface Protein Biotinylation with Sulfo-NHS-LC-biotina.
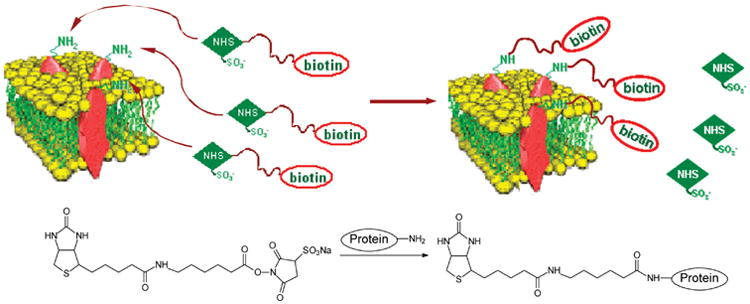
aThe chemical reaction between the sulfo-NHS-LC-biotin and amino groups in proteins is also shown.
We employed the above strategy and assessed the expression of cell surface membrane proteins in two human melanoma cell lines, WM-115 and WM-266-4. We were able to identify more than 100 membrane and membrane-associated proteins and quantified the relative expression of 62 identified proteins in these two cell lines. To the best of our knowledge, this is the first characterization of the differential expression of cell surface membrane proteins of primary and metastatic tumor cells using MS-based quantitative proteomic techniques.
Materials and Methods
Cell Culture
A pair of human malignant melanoma cell lines, WM-115 and WM-266-4, which were derived from the primary and metastatic tumor tissues of the same patient, were obtained from the American type Culture Collection (ATCC, Manassas, VA). The cells were cultured under ATCC-recommended conditions in Eagle's Minimum Essential Medium (EMEM) supplied with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 100 IU/mL of penicillin, 100 μg/mL of streptomycin and 1.5 g/L of sodium bicarbonate. For SILAC experiments, the EMEM medium in the absence of l-lysine was custom prepared by AthenaES (Baltimore, MD). [13C6,15N2]-l-lysine (heavy lysine) and unlabeled l-lysine (light lysine) were purchased from Cambridge Isotope Laboratories (Andover, MA) and Sigma (St. Louis, MO), respectively. Light and heavy EMEM media containing either light or heavy lysine were prepared according to the standard formulation of EMEM except with the supplement of dialyzed FBS (Invitrogen). The WM-115 cells were grown in light medium, and the metastatic WM-266-4 cells were cultured in heavy medium for at least 6 days to allow the complete incorporation of the isotope-labeled amino acid.
Biotinylation of Cell-Surface Membrane Proteins
Cells, which were attached to the culturing flasks, were washed gently with prewarmed PBS (pH 7.4) at 37 °C three times to remove amine-containing culture media and FBS. A 1-mL solution of 0.5 mg/mL sulfo-NHS-LC-biotin (Pierce, Rockford, IL) in PBS was added to each flask and the cells were incubated at 37 °C for 10 min. Following biotinylation, 1 mL of 100 mM Tris-HCl (pH 7.4) was added to each flask to quench the reaction by incubation at room temperature for 5 min, and the cells were washed three times with ice-cold PBS to remove excess reagent and byproducts.
Preparation of Cell-Surface Membrane Proteins
After a 10-min labeling reaction, biotinylated WM-115 and WM-266-4 cells from four 75-cm2 culturing flasks (∼1 × 107 cells in total) were harvested into ice-cold PBS by scraping and pelleted by centrifugation at 800g at 4 ° C for 5 min. The WM-115 and WM-266-4 cells were lysed separately in lysis buffer on ice for 30 min with gentle shaking. The lysis buffer contained 50 mM HEPES (pH 7.4), 2% NP-40 (v/v), 0.5% SDS (w/v), 100 mM NaCl, 0.5 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1.5 mM MgCl2, 1 mM PMSF and a protease inhibitor cocktail. Cell lysates were centrifuged at 16 000g and at 4 °C for 30 min, and the resulting supernatants were collected. The cell lysates were diluted by at least 30-fold, and the protein concentrations of these diluted lysates were determined by using Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA). The WM-115 and WM-266-4 cell lysates were then combined at a ratio of 1/1 (w/w).
To remove large amounts of salts and detergent, proteins were precipitated by adding trichloroacetic acid (TCA) to a final concentration of 20% and incubated on ice for at least 1 h. After incubation, the precipitates were pelleted by centrifugation at 10 000g for 5 min and washed with an ice-cold solution of 10 mM HCl/90% acetone three times. The resultant protein pellets were dried under vacuum.
Enzymatic Digestion
The TCA precipitates were resuspended in a 100-mM NH4HCO3 solution (pH 8.5) containing 0.2% SDS at room temperature for 30 min, and then heated at 95 °C for 10 min. Urea was then added to the solution to a final concentration of 8 M to improve further the solubility of membrane proteins. These procedures were adapted from previously published procedures for enhancing the solubility of membrane proteins.19,35 It is worth noting that heating protein in the presence of urea may lead to the carbamylation of proteins.36 Nevertheless, database search based on the LC-MS/MS results revealed the absence of peptides with the N-terminus or the side chain of lysine residues being carbamylated.
The solution was cooled to room temperature, and the proteins were reduced with dithiothreitol (DTT) at a final concentration of 10 mM and alkylated in dark with iodoacetamide (IAA) at a final concentration of 50 mM for 30 min. The resulting protein solutions were diluted 4 times with 100 mM NH4HCO3 (pH 8.5) and digested with mass spectrometry-grade trypsin (Promega, Madison, WI) at an enzyme-to-substrate ratio of 1/100 (w/w) at 37 °C overnight. The peptide mixtures were dried in a SpeedVac.
Affinity Purification
Biotinylated peptides were purified and enriched by using an affinity column packed with avidin-agarose resin (Sigma-Aldrich) as previously described.37 Briefly, the packed avidin-agarose resin was washed with 10 column volumes of binding buffer (20 mM potassium phosphate, 0.15 M NaCl, pH 7.5) prior to loading the tryptic digests. After a 30-min incubation, unbound peptides were removed by washing the resin thoroughly with the binding buffer and pure water. The bound peptides were eluted with an equi-volume mixture of CH3CN and H2O at 65 °C. The elution process was repeated, and the eluates were combined, dried in a SpeedVac, and stored at −20 °C for further analysis.
LC-MS/MS
The affinity-purified peptide mixtures were analyzed by LC-MS/MS in triplicate on an LTQ linear ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an electrospray ionization (ESI) source. An Agilent 1100 capillary HPLC system with a Zorbax SB-C18 capillary column (5 μm in particle size, 0.5 × 250 mm, Agilent Technologies, Santa Clara, CA) was employed for peptide separation. The chromatographic separation was performed with a linear gradient of 2–15% CH3CN in 0.6% aqueous solution of acetic acid in the first 13 min, followed by a 70-min gradient of 15–50% CH3CN in 0.6% acetic acid; the flow rate was 6 μL/min.
The capillary temperature for the electrospray source was maintained at 275 °C and the source voltage was set at 4 kV. The maximum ion injection time was 50 ms with automatic gain control (AGC) values of 3 × 103, 1 × 103, and 1 × 103 ions for full-scan MS, MS in selected ion monitoring (SIM) mode, and MS/MS, respectively. The product-ion spectra were acquired in a data-dependent scan mode, where the eight most abundant ions observed in MS were chosen for collision-induced dissociation (CID). The normalized collision energy was 35% and the activation Q was 0.25. In addition, the dynamic exclusion feature was activated to maximize the detection of peptide ions of low abundance.
Quantitative Analysis and Data Processing
Raw data of product-ion spectra from the LC-MS/MS analysis were searched against NCBI human RefSeq protein database using TurboSE-QUEST in Bioworks 3.2 (Thermo Electron, San Jose, CA). Variable modifications on lysine residues were allowed with a mass increase of 8.01 Da to recognize heavy lysine residues, and increases of 339.14 and 347.15 Da for biotinylated light and heavy lysine residues, respectively. The static carbamidomethylation (CAM) of cysteine residues was used with a mass increase of 57.01 Da. Peptide filters with cross-correlation (Xcorr) via charge states (≥1.9, ≥ 2.4, ≥ 3.5 for peptide ions that are singly, doubly, and triply charged) and delta correlation (ΔCn ≥ 0.1) were used to sort the search results. All the tandem mass spectra of the identified peptides with biotin labeling were manually inspected. The cellular localization and function of each identified protein were further investigated based on NCBI Gene Ontology (GO) annotation and Gene Ontology Annotation (GOA) database (http://www.ebi.ac.uk/GOA).
Protein quantification was based on the biotinylated peptides by comparing manually the relative ion abundances of unlabeled and isotope-labeled peptides with the average of at least three MS scans with a mass tolerance of ±0.5 Da and an S/N ratio greater than 3. We also quantified some of the identified proteins by monitoring selectively the fragmentations of the precursor ions for the biotinylated peptides carrying either light- or heavy-lysine residue(s). The ratios of the unlabeled and isotope-labeled proteins were determined using the peak areas found in the corresponding selected-ion chromatograms (SICs) of the monitored peptide pairs. For those ions of low relative abundance, SICs for monitoring the formation of the three most abundant fragment ions in MS/MS were employed for the quantification. All the quantification results were based on LC-MS/MS measurements of samples from three independent mixings of the cell lysates.
Immunocytochemistry and Confocal Microscopy
Four identified proteins, including histone H2B and three membrane proteins, MCAM (melanoma cell adhesion molecule, or CD146), ALCAM (activated leukocyte cell adhesion molecule, or CD166), and ITGA3 (integrin alpha 3, or CD49C), were assessed by immunofluorescence staining using confocal microscopy. To stain the three membrane proteins, WM-115 and WM-266-4 cells grown on coverslips were washed with PBS and fixed with 4% paraformaldehyde in PBS for 12 min. The fixed cells were treated with a blocking buffer (3% BSA in PBS) at room temperature for 30 min. After blocking, cells were stained with primary mouse monoclonal antibodies (mAbs) recognizing human MCAM, ALCAM, and ITGA3, respectively (Abcam, Cambridge, MA). After incubation in a humidified chamber at 37 °C for 1 h, cells were washed with PBS and blocking buffer, and then stained with an Alexa Fluor 488-conjugated rabbit secondary antibody to mouse IgG (Invitrogen) at 37 °C for 30 min. For histone H2B, cells were stained without fixation. In this case, all operations were performed on ice, and ice-cold PBS and blocking buffers were also used. Briefly, cells were first stained with mouse monoclonal anti-human H2B antibody (MBL International, Woburn, MA) on ice and then with the same secondary antibody described above. Control experiments were also carried out for unfixed WM-115 and WM-266-4 cells stained with the secondary antibody alone. After staining, cells were fixed with 4% paraformaldehyde.
Following washing with PBS, coverslips with the antibody-stained WM-115 and WM-266-4 cells were mounted onto microscope slides with 10 μL of Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing DAPI (4′,6-diamidino-2-phenylindole). Images were collected with a Leica TCS SP2/UV confocal microscope (Heidelberg, Germany) at a magnification factor of 63×. Images of WM-115 and WM-266-4 cells stained with the same antibody were captured with the same parameters to compare the staining of these two types of cells. Relative intensities of fluorescence staining were analyzed using ImageJ (http://rsb.info.nih.gov/ij/).
Flow Cytometric Analysis
After the biotinylation reaction, the WM-115 and WM-266-4 cells were detached from culturing flasks into trypsin-EDTA solution and collected by centrifugation at 800g at 4 °C for 5 min. The cells were washed with ice-cold PBS (Ca2+/Mg2+-free, 5 mM EDTA) and binding buffer (Ca2+/Mg2+-free PBS, 2% FBS, 1% sodium azide, 5 mM EDTA, 20 mM HEPES, pH 7.4). Before staining, the number of cells in each sample was determined by using a hemocytometer. An excess amount of streptavidin-R-phycoerythrin conjugate (SAPE, Invitrogen) was added to each cell sample (5 × 105 cells), followed by incubation at 4 °C in the dark for 30 min. The cells were subsequently washed twice with binding buffer and three times with PBS to remove the free SAPE. The cells were then fixed with 2% paraformaldehyde and kept at 4 °C. Flow cytometric measurements were carried out on a BD FACSAria Cell Sorting System (BD Biosciences) and the data were analyzed using FlowJo (Tree Star, Inc.)
Results and Discussion
General Strategy for the Comparative Studies of Cell Surface Proteins
Our general strategy encompassed SILAC for the metabolic incorporation of isotope-labeled amino acids, in situ biotinylation of surface proteins of intact cells using the membrane-impermeable sulfo-NHS-LC-biotin, cell lysis, enzymatic digestion, affinity purification of biotinylated peptides, and LC-MS/MS for protein identification and quantification (Schemes 1 and 2). Since its first introduction, SILAC has been widely used for MS-based protein quantification and it has also been employed to quantify membrane and membrane-bound proteins.29,38,39
The strategy of labeling and selective enrichment of membrane proteins by biotinylation and subsequent affinity purification has been employed to improve the selectivity and throughput for profiling membrane proteins.11,12,14–23 After affinity enrichment and purification, the resultant membrane fractions, however, are still contaminated with cytoplasmic proteins. The contamination could arise from the nonspecific binding of the cytoplasmic proteins to avidin resin, from noncovalent protein–protein interactions and/or from the biotinylation of proteins in nonviable cells.18,24 Washing with buffers at high-salt concentration and high pH during affinity purification could reduce contaminations to some extent. However, cytosolic proteins could still account for up to 15% of all the identified proteins.11 In addition, the affinity-enriched membrane protein fractions from cell lysates are still a complex mixture. Further separation of the enriched membrane proteins by using 1-D SDS-PAGE, 2-D gel, or HPLC is usually necessary to reduce the sample complexity prior to enzymatic digestion.14,19,24
To improve the efficiency of purification and to minimize contamination, we decided to digest the protein mixtures and enrich the biotinylated peptides using biotin/avidin-based affinity purification. After a single step of affinity purification, the biotinylated peptides of membrane proteins are subjected to LC-MS/MS analysis. Because biotinylation occurs only at lysine residues exposed on the cell surface and only the labeled peptides are selectively enriched, the sample complexity is markedly reduced.
A potential drawback for the purification of biotinylated peptides rather than biotinylated proteins is that the protein representation of the peptides is only limited to the biotinylated peptides.13,18,21 However, our previous work,37 together with the successful implementation of a similar approach for the selective enrichment of ICAT-conjugated cysteine-containing peptides,26 revealed that protein identification and quantification based on the biotinylated peptides are reliable.
SILAC with [13C6,15N2]-l-lysine will render each lysine residue in the proteins from WM-266-4 cells being replaced with a heavy lysine, which results in a mass increase of 8.01 Da. After biotinylation, the lysine residues are no longer susceptible to trypsin cleavage. Therefore, these biotinylated peptides contain at least one internal lysine residue. As a result, the biotinylated light- and heavy-lysine residues exhibit mass increases of 339.14 and 347.15 Da, respectively. Relative quantification could be readily achieved by using the relative abundances of ions corresponding to the biotinylated light- and heavy-lysine-bearing peptide pairs.
To evaluate the accuracy of quantification based on SILAC and LC-MS/MS analysis of biotinylated peptides, we carried out control experiments by mixing the light and heavy cell lysates from WM-266-4 cells at ratios of 1/1, 3/1, and 10/1 (w/w). The SICs for monitoring the biotinylated peptides from five proteins identified in WM-266-4 cells were plotted, and the resulting ratios for these five proteins recovered from LC-MS analysis are shown in Table 1. These results demonstrated that the relative ratios of light and heavy peptides are consistent with the expected ratios (Table 1). The ratios of multiple isotopic peptide pairs from the same protein were also in good agreement with each other and with the expected ratios (Figure 1). Moreover, we mixed the light and heavy cell lysates at 1/1 (w/w) ratio and analyzed, for three times, the resulting tryptic digestion mixtures by using LC-MS/MS in data-dependent scan mode. The LC-MS/MS results allowed us to quantify 20 proteins, and the average ratio determined by LC-MS/MS was 1.01 with a relative standard deviation of 11% (Table S1).
Table 1. A List of Biotinylated Proteins and Their Peptides Identified from WM-266-4 Cells Used To Evaluate the Quantification Strategy Based on Cell Surface Biotinylation and Affinity-Enriched Peptidesa.
| NCBI accession | protein name | peptide used for quantification | charge | m/z | ratio (L/H) 1:1 | ratio (L/H) 3:1 | ratio (L/H) 10:1 |
|---|---|---|---|---|---|---|---|
| NP_006491 | melanoma cell adhesion molecule | R.IFLCQGK@R.P | 2 | 684.9 | 1.12 | 3.14 | 10.35 |
| R.IFLCQGK#R.P | 2 | 680.9 | |||||
| R.LPSGNHMK@ESR.E | 3 | 534.9 | 0.99 | 3.18 | 9.91 | ||
| R.LPSGNHMK#ESR.E | 3 | 532.3 | |||||
| NP_001001390 | CD44 antigen isoform 3 precursor | R.YVQK@GEYR.T | 2 | 695.3 | 0.75 | 2.35 | 8.85 |
| R.YVQK#GEYR.T | 2 | 691.3 | |||||
| NP_002194 | integrin alpha 2 precursor | R.K@YAYSAASGGR.R | 2 | 739.3 | 0.98 | 2.95 | 10.02 |
| R.K#YAYSAASGGR.R | 2 | 735.3 | |||||
| NP_001670 | Na+/K+ -ATPase beta 3 subunit | R.IIGLK@PEGVPR.I | 2 | 763.4 | 1.04 | 3.07 | 10.11 |
| R.IIGLK#PEGVPR.I | 2 | 759.4 | |||||
| NP_005492 | integrin alpha 3 isoform b precursor | R.TGAVYLCPLTAHK@DDCER.M | 3 | 818.4 | 1.18 | 2.91 | 10.67 |
| R.TGAVYLCPLTAHK#DDCER.M | 3 | 815.7 | |||||
| Mean | 1.01 | 2.93 | 9.99 | ||||
| SD | 0.15 | 0.30 | 0.62 | ||||
| RSD% | 14.74 | 10.38 | 6.20 |
Note: #, biotinylated light (L) lysine; @, biotinylated heavy (H) lysine; cysteine is CAM-modified.
Figure 1.

Evaluation of the quality of quantification based on SILAC and biotinylated peptides. Selected-ion chromatograms (SICs) of two pairs of light (L) and heavy (H) biotinylated peptides of MCAM from WM-266-4 cells. The expected ratios of peak area of light and heavy peptides are 1:1, 3:1, and 10:1 from top panel to bottom panel. The biotinylated light lysine ([12C6,14N2]-l-lysine) and heavy lysine ([13C6,15N2]-l-lysine) are labeled with “#” and “@”, respectively. (A) SICs of the [M + 3H]3+ ions of LPSGNHMK#ESR (m/z 532.4) and LPSGNHMK@ESR (m/z 535.1); (B) SICs of the [M + 2H]2+ ions of IFLCQGK#R (m/z 681.0) and IFLCQGK@R (m/z 684.9), in which the cysteine residue is CAM-modified. The calculated ratios of peak area of these peptides are listed in Table 1.
A typical pair of peptides, which were used for quantification based on MS, as well as their product-ion spectra acquired in data-dependent scan mode, are depicted in Figure 2. The ratios of isotopic peptide pairs of different charge states (in this case, 2+ and 3+) were also examined, and they gave consistent results (data not shown). On the basis of the results from the above-mentioned control quantification experiments, we concluded that our strategy, which included SILAC, biotinylation reaction, one-step affinity purification of biotinylated peptides and LC-MS/MS analysis, can afford accurate and reliable quantification of protein expression.
Figure 2.

A typical isotopic pair of biotinylated peptides from WM-115 (light, “L”) and WM-266-4 cells (heavy, “H”) used for quantification by ESI-MS. The identities of peptides were confirmed by MS/MS acquired in data-dependent scan mode. (A) ESI mass spectra of an isotopic peptide pair, IGDLQAFQGHGAGN-LAGLK#GR ([M + 3H]3+ ion, m/z 807.2) and IGDLQAFQGHGA-GNLAGLK@GR ([M + 3H]3+ ion, m/z 810.0) of the solute carrier family 3 protein from WM-115 and WM-266-4 cells; (B and C) the product-ion spectra of the biotinylated peptides carrying a light (B) and heavy (C) lysine.
It is worth noting that not all identified biotinylated peptides can be employed for quantifying the expression of their corresponding proteins. For instance, some peptides were identified based on their tandem mass spectra, but their molecular ions exhibit relatively low abundances or the backgrounds of the full-scan MS were relatively high. Under such circumstances, we cannot quantify these isotopic peptide pairs based on the intensities of peaks found in ESI-MS. Instead we acquired MS/MS for these peptide ions in the SIM mode, which offered improved selectivity, sensitivity and accuracy for the quantification. However, even with the SIM mode, a small portion of identified peptides, because of their low abundances, still could not be quantified (data not shown).
Proteomic Analysis of Membrane Proteins of Primary and Metastatic Melanoma Cells
To examine the differential expression of surface membrane proteins of melanoma cells at different tumor progression stages, we first labeled the cell- surface proteins of the WM-115 and WM-266-4 cells, which were cultured in media containing light and heavy lysines, respectively, with sulfo-NHS-LC-biotin in situ. After removing the excess labeling reagent, we lysed the cells, mixed the light and heavy cell lysates at a ratio of 1/1 (w/w), digested the proteins in the lysates with trypsin, and subjected the digestion mixture to LC-MS/MS analysis. From the LC-MS/MS results acquired in the data-dependent scan mode, we were able to identify 109 proteins (Figure 3 and Table S3). A majority (78%, Figure 3) of these proteins were localized on the cell surface and PM, as classified by GO database annotations. These include integral membrane, transmembrane and peripheral membrane proteins, such as a large set of integrins, cell adhesive molecules, CD antigens and receptors. All the identified proteins carry at least one peptide with internal lysine residue(s) tagged with a biotin moiety.
Figure 3.
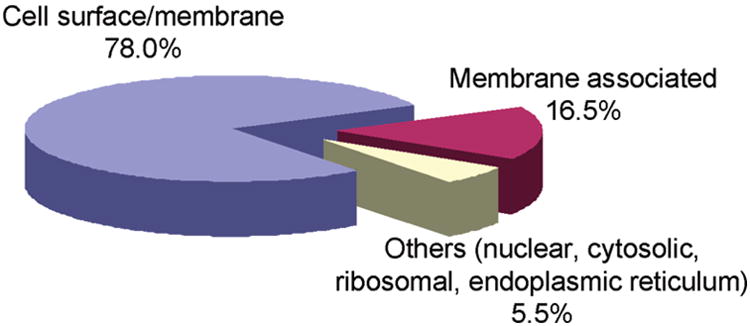
Pie chart of subcellular classification of the 109 proteins identified from WM-115 and WM-266-4 cells, based on GO database annotations and previous publications. See text and Table S3 for details.
The rest (22%) of the identified proteins were not classified as cell surface and PM proteins by the GO database annotations. However, the membrane localization of the majority of these proteins was reported previously (Table S2 in the Supporting Information). These proteins contributed to 16.5% of all the identified proteins. For instance, several proteins with chaperone functions, including heat shock proteins (HSPs) and protein disulfide isomerases (PDIs), were identified.
HSPs and PDIs are generally considered to be cytoplasmic or ER proteins and the majority of them do not contain transmembrane domains. Several recent studies, however, revealed the cell membrane localization of these proteins.15,20,21,40,41 In this respect, a previous comprehensive profiling of cell surface proteins revealed that HSPs have relatively high abundance, compared with other cell surface proteins.15,20,21 Little is known about the cellular pathways through which the HSPs reach the cell surface, and it remains unclear what the functions are for these proteins to be present on the cell surface. In this context, it was suggested that the HSPs might be associated with misfolded proteins or peptide fragments and are transferred out of the cytosol via a nonclassic pathway.20 In addition, the presence of these proteins on the cell surface may help maintain the structural integrity of various receptor complexes.20
Other than the chaperone proteins, histones H1, H2A, H2B, H3 and H4 were also found to be present on the surface of melanoma cells in our experiments. Histones are the major protein components of chromatin and play important roles in DNA packaging, chromosome stabilization and gene expression. 42,43 However, histones were also shown to exist on the cell surface.44–49
As discussed above, a majority of the proteins identified were determined to be membrane and membrane-associated proteins by GO database annotations and recent publications. However, we also observed a few peptides arising from nonmembrane-related proteins, including those from cytosol, nucleus, ER and ribosome, which in total accounted for only 5.5% of all the proteins identified. It remains unexplored whether these proteins are also localized on the cell surface or membrane. Nevertheless, compared with the classical membrane enrichment techniques, such as ultracentrifugation and fractionation,50 the proportion of nonmembrane-related proteins identified in our experiments is relatively small.
Because of the low abundances, some identified proteins were not quantified. In this regard, we were able to quantify the relative expression of 62 proteins in these two melanoma cell lines (Table 2 and Table S3). Among the quantified proteins, the majority of them exhibited only slight differences in expression between these two cell lines. Only about one-third of the proteins (22 out of 62, as diagramed in Figure 4) are down- or up-regulated in the metastatic melanoma cells, relative to the primary melanoma cells, if the ratio of 2 or above is considered as a significant change. With this criterion, the number of proteins that are up-regulated in WM-266-4 cells is comparable to the number of proteins that are down-regulated. For instance, several proteins, such as ALCAM, CD9, EphA2, erbB-2, histone H4, histone H2B and immunoglobulin super-family member 4D isoform 1 (IGSF4), are down-regulated. On the other hand, butyrophilin, CD109, major histocompatibility complex class II DR alpha precursor (HLA-DRA), ITGB3, MCAM and several other proteins are up-regulated in WM-266-4 cells.
Table 2. A List of Quantified Proteins and Their Corresponding Ratios of Expression in WM-266-4 Cells with Respect to WM-115 Cells.
| NCBI accession | protein name | surface/membrane GOa | Ratio WM-266–4/115 mean ± SD (n = 3) | WM266-4 hitsb (H)c | WM115 hits (L)d |
|---|---|---|---|---|---|
| Up-Regulated Proteins | |||||
| NP_853509 | butyrophilin, subfamily 3, member A1 | Y | 2.37 ± 0.59 | 3 | 3 |
| NP_598000 | CD109 | Y | 3.65 ± 0.04 | 4 | 1 |
| NP_000102 | down-regulated in adenoma protein | Y | 5.97 ± 0.55 | 2 | 0 |
| NP_000203 | integrin beta chain, beta 3 precursor | Y | 2.38 ± 0.07 | 19 | 9 |
| NP_061984 | major histocompatibility complex, class II, DR alpha precursor | Y | >5 | 2 | 0 |
| NP_004986 | matrix metalloproteinase 14 preproprotein | Y | 2.18 ± 0.48 | 6 | 0 |
| NP_006491 | melanoma cell adhesion molecule | Y | 3.13 ± 0.69 | 19 | 6 |
| NP_005920 | melanoma-associated antigen p97 isoform 1, precursor | Y | 3.60 ± 1.05 | 2 | 0 |
| XP_933500 | PREDICTED: similar to immunoglobulin superfamily, member 3 isoform 1 | Y | 2.62 ± 0.10 | 2 | 0 |
| NP_005496 | scavenger receptor class B, member 1 isoform 1 | Y | 3.25 ± 0.35 | 6 | 0 |
| NP_001143 | solute carrier family 25, member 5 | Y | >20 | 3 | 0 |
| NP_001012679 | solute carrier family 3 | Y | 2.53 ± 0.18 | 9 | 6 |
| Down-Regulated Proteins | |||||
| NP_001618 | activated leukocyte cell adhesion molecule | Y | 0.07 ± 0.03 | 0 | 15 |
| NP_001760 | CD9 antigen | Y | 0.31 ± 0.02 | 1 | 4 |
| NP_004422 | ephrin receptor EphA2 | Y | 0.36 ± 0.06 | 0 | 9 |
| NP_001005862 | erbB-2 isoform b | Y | 0.29 ± 0.07 | 0 | 3 |
| NP_003536 | H4 histone family, member J | R | 0.45 ± 0.26 | 19 | 20 |
| NP_001019770 | histone 2, H2bf | R | 0.38 ± 0.19 | 1 | 9 |
| NP_055148 | immunoglobulin superfamily, member 4D isoform 1 | Y | 0.47 ± 0.11 | 3 | 4 |
| NP_005388 | podocalyxin-like precursor isoform 2 | Y | <0.05 | 0 | 2 |
| NP_543006 | signal-regulatory protein beta 2 isoform 2 | Y | 0.29 ± 0.07 | 1 | 2 |
| NP_003371 | vimentin | R | 0.43 ± 0.03 | 1 | 4 |
GO database annotations, Y, cell surface/membrane protein; R, membrane-associated protein based on previous publications (see Supporting Information for details).
Hits: total number of peptides (including same peptides with multiple identification and different m/z) identified with SEQUEST search based on LC-MS/MS data acquired in data-dependent scan mode. Although no peptides were identified for some of the listed proteins from LC-MS/MS data acquired in the data-dependent scan mode, biotinylated peptide(s) was (were) identified from LC-MS/MS results obtained in the SIM mode.
H, heavy.
L, light.
Figure 4.
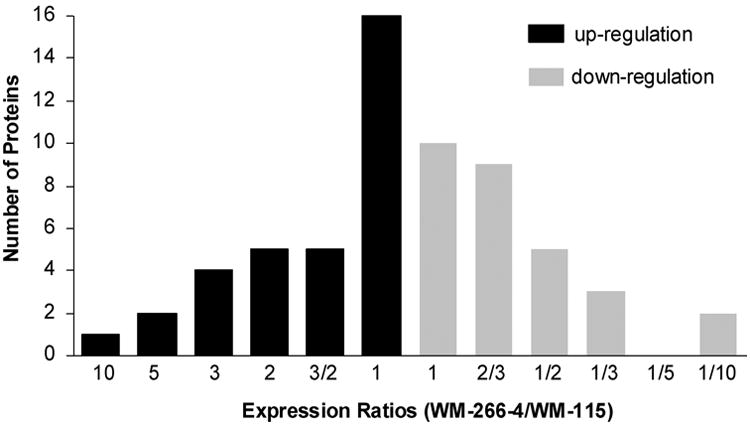
A summary of the quantified proteins, which were up-(black bar) and down-(gray bar) regulated in WM-266-4 with respect to WM-115 cells.
Immunocytochemistry for the Validation of Surface Localization and Quantification
A potential pitfall of the above quantification strategy lies in that the biotinylation step involves two different types of cells, which may result in different labeling efficiency with sulfo-NHS-LC-biotin, thereby giving rise to the observation of different levels of membrane proteins. Thus, it is important to assess the relative expression of some membrane proteins in these two cell lines by using an independent method.
To evaluate the reliability of the above MS-based quantification results and to confirm the surface localization of the identified membrane proteins, we carried out immunocytochemistry experiments for four identified proteins, including H2B, MCAM, ALCAM, and ITGA3. As depicted in Figure 5, the confocal microscopic images confirmed the cell surface localization of these proteins (The unfixed WM-115 and WM-266-4 cells were also stained with the secondary antibody alone to serve as negative control, see Figure S1). Particularly, the expression of histone H2B, an abundant nuclear protein, was detected on the surface of most WM-115 cells with a punctuate distribution (Figure 5A, left panel). However, H2B showed a lower level of expression on the surface of most WM-266-4 cells (Figure 5A, right panel). It is worth noting that, to visualize the cell-surface distribution of histone H2B, we stained the WM-115 and WM-266-4 cells without fixation so that the integrity of PM can be maintained.
Figure 5.
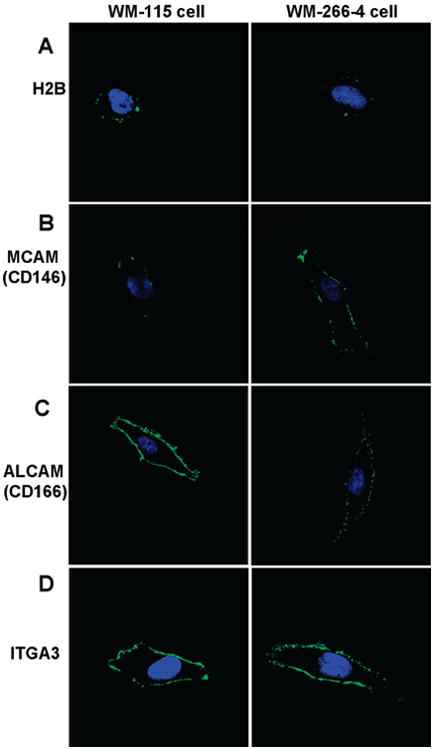
Immunocytochemical detection of cell surface proteins. The left and right panels represent the staining of WM-115 and WM-266-4 cells, respectively. Cells were stained with primary mAbs to (A) H2B, (B) MCAM, (C) ALCAM, and (D) ITGA3 followed by staining with Alexa Fluor 488-labeled secondary antibody (green). Cells were also stained with DAPI (blue) to identify nuclei. Images of WM-115 and WM-266-4 cells stained with same primary mAbs were captured with identical parameters to compare the staining of the two cell lines.
To date, the mechanism by which the histone proteins are expressed on PM has not been elucidated. A recent study showed that histone proteins are able to transverse directly PMs and mediate penetration of macromolecules covalently attached to them.47 This process was believed to occur by direct translocation and not by typical endocytic pathways. It has also been shown that all major histone proteins, including H2A, H2B, H3, H4 and H1, could be located on the PM of activated T-cells.45,46 All these cell-surface histones are in the form of nucleosomes, which appear to be anchored to the cell surface through an interaction with sulfated polysaccharides; however, the function of the cell surface nucleosomes remains unclear. Considering the identification of almost all major histones and their nonuniform distribution on the cell membrane (Figure 5A), our MS and immunofluorescence results are in line with a previous report.45
We next compared the relative intensities of immunofluorescence for these two cell lines and correlated these findings with the MS-based quantitative data (Table 3). The ratios calculated from the immunofluorescence measurements are generally in keeping with the quantification results obtained from the MS-based analysis. Therefore, we may conclude that the WM-115 and WM-266-4 cells exhibit similar efficiency in the labeling of the amino group of surface proteins with the sulfo-NHS-LC-biotin.
Table 3. Comparison of Quantitative Results from Immunofluorescence Staining and MS-Based Proteomics.
| protein name | Immunofluorescence ratio (WM-266-4/115) mean ± SD (n = 5) | MS ratio (WM-266-4/115) mean ± SD (n = 3) |
|---|---|---|
| H2B | 0.29 ± 0.06 | 0.38 ± 0.19 |
| MCAM | 5.08 ± 1.23 | 3.13 ± 0.69 |
| ALCAM | 0.04 ± 0.01 | 0.07 ± 0.03 |
| ITGA3 | 1.19 ± 0.35 | 0.96 ± 0.11 |
To gain further insights into the labeling of the two types of cells, we stained the biotin-labeled cells with streptavidin-R-phycoerythrin and followed the labeling reaction by flow cytometric analysis. While it is difficult to calculate the labeling efficiency based on this assay, the results showed that the two cell lines exhibit similar kinetics in the labeling reaction (Figure S2). In this respect, we normalized the fluorescent intensities found at other time intervals against the highest fluorescent intensity observed. It is worth noting that the fluorescent intensity does not reach a plateau at 10 min; we, however, chose to label the cells for a relatively short period of time (i.e., 10 min) to minimize the potential effects of biotinylation on damaging the integrity of plasma membrane.
Characterization of the Differential Expression of Cell Surface Proteins
The identification and characterization of the differential expression of cell surface proteins are potentially useful for screening candidates for biomarkers. Membrane proteins account for more than two-thirds of protein-based drug targets.2 The above MS-based quantitative analysis facilitates the analysis of differential protein expression at large scale and holds great potential for screening purpose. In addition, understanding the difference of primary and metastatic cancers at proteomic level may potentially benefit cancer treatment. Next we will discuss the membrane and membrane-associated proteins that we quantified in this study.
CD (Cluster of Differentiation) molecules are protein or carbohydrate antigens recognized by specific antibodies, which are used to distinguish particular cell types. CD molecules often serve as receptors or ligands that are important to a cell. For example, MCAM (CD146), previously known as MUC18 or Mel-CAM, is an integral membrane glycoprotein functioning as Ca2+-independent cell adhesive molecule involved in heterophilic cell-cell interactions.51 Although an earlier study found no direct correlation between its expression and metastatic behavior of melanoma,52 an increasing body of work has shed light on the functions of MCAM, particularly about its role in tumor growth and the progression of human malignant melanoma.51,53–55 Our MS-based quantitative proteomic data and immunostaining results (Figure 5B) consistently demonstrated the overexpression of MCAM in the metastatic over the primary melanoma cells. These results support that MCAM may serve as a diagnostic marker for monitoring the progression of melanoma.
ALCAM (CD166) was found to be significantly down-regulated in WM-266-4 cells. ALCAM is a type-I transmembrane glycoprotein of the immunoglobin superfamily.56,57 It has been well-documented that ALCAM is the ligand for the lymphocyte cell-surface receptor CD6.58 Several reports have claimed that ALCAM is associated with cancer development.59–65 However, the expression pattern of ALCAM does not exhibit simple correlation with tumor development.60,62,64 Although up-regulation of ALCAM was found in metastatic melanoma,64 our MS data revealed a significant down-regulation of ALCAM in WM-266-4 cells, which was also confirmed by the immunofluorecence measurements (Figure 5C). Other CD molecules, such as lymphocyte function-associated antigen 3 (CD58), CD59 antigen p18–20 and CD9, were expressed at a slightly lower level in WM-266-4 cells (Table 2 and Table S3).
The CD9 antigen was described originally as a surface protein of non-T acute lymphoblastic leukemia cells and developing B-lymphocytes.66 It plays a pivotal role in regulating cell adhesion, motility, and proliferation and has been considered as an important inhibitory factor for metastasis of various human cancers. It has been reported that CD9 is expressed predominantly on primary rather than on metastatic tumors, and the down-regulation of CD9 in metastatic melanoma might have prognostic significance with respect to the potential for metastasis.67 Recent studies showed that CD9 may reduce the metastatic spread of human small-cell lung carcinoma via the inhibition of cell proliferation and motility,68 and that it could also be a potential cell membrane marker in human breast cancer cell lines.69
In this study, several integrins, such as ITGAV (integrin alpha V or CD51) and ITGB3 (integrin beta chain beta 3, or CD61), were shown to be up-regulated in metastatic melanoma cells. In this context, the expression of these integrins (CD51/CD61) was found to be associated with tumor progression and metastasis formation in human malignant melanoma.70,71 They are well-recognized as molecular markers correlating with the change from radial to vertical growth in primary melanoma.70 Changes in the expression or function of these cell adhesion molecules can therefore contribute to tumor progression by altering the adhesion status of cells and modulating cell signaling pathways.72,73
We were also able to quantify several proteins that have potential roles in a variety of cell signaling pathways, i.e., erbB-2 (HER2), ephrin receptor (EPHA2), and signal-regulatory protein beta 2 (SIRPG). In this respect, erbB-2, a transmembrane tyrosine kinase receptor, is known to be associated with aggressive breast cancers, making this receptor an attractive therapeutic target.74 However, conflicting results have been published regarding the expression pattern of erbB-2 in primary and metastatic melanoma.75–78 Our proteomic data supported the down-regulation of this receptor in WM-266-4 cells. Other molecules, such as melanoma-associated antigen p97, scavenger receptor and several solute carrier family proteins, also displayed different expression between these two cell lines. Further assessment of the roles of these proteins in melanoma progression is necessary.
Conclusions
We demonstrated that combining SILAC, cell surface biotinylation, affinity peptide purification and LC-MS/MS is very useful for the large-scale quantitative analysis of surface membrane proteins. Contaminations from cytoplasm and other nonmembrane related sources were greatly reduced by using in situ cell surface biotinylation and the subsequent affinity purification of biotinylated peptides; therefore, high selectivity and specificity were achieved for the identification of membrane proteins. Additionally, metabolic labeling with SILAC, together with LC-MS/MS, facilitates the reliable quantification of surface membrane proteins expressed on two human melanoma cell lines derived from the same individual at different stages of tumor development. Integrins, cell adhesive molecules, CD antigens and receptors, which are essential for tumor development, were quantified in our study. Other proteins, such as histones, were also detected on the surface of WM-115 and WM-266-4 cells, and the cell surface localization of histone H2B was further confirmed by immunocytochemistry. The identification of aberrantly expressed proteins on the surface of metastatic melanoma cells sets the stage for the future investigation on the implications of these proteins in the progression of human melanoma. Moreover, the application to quantitative analysis of membrane proteins of primary and metastatic melanoma cells using this strategy has demonstrated its great potential in the comprehensive identification of tumor progression biomarkers in pathology and in the discovery of new protein-based drug targets.
Supplementary Material
Acknowledgments
This work was supported by NIH (Grant No. R01 CA 116522). We thank Dr. David Carter from CEPCEB Microscopy and Imaging Core Facility of UC Riverside for help with confocal microscope, Dr. Liyan Ping for help on image analysis, and Dr. Barbara Walter from IIGB Core Instrument Facility of UC Riverside for flow cytometric measurements.
Footnotes
Supporting Information Available: The proteins identified with unique peptides, the ratios of cell-surface proteins expressed on WM-115 and WM-266-4 cells, the control SILAC quantification results, the confocal microscope images for cells stained with the secondary antibody alone, and flow cytometry results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: Un amour impossible. Electrophoresis. 2000;21:1054–1070. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1054::AID-ELPS1054>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Wu CC, Yates JR. The application of mass spectrometry to membrane proteomics. Nat Biotechnol. 2003;21:262–267. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]
- 5.Wu CC, MacCoss MJ, Howell KE, Yates JR. A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 6.Xiang R, Shi Y, Dillon DA, Negin B, Horvath C, Wilkins JA. 2D LC/MS analysis of membrane proteins from breast cancer cell lines MCF7 and BT474. J Proteome Res. 2004;3:1278–1283. doi: 10.1021/pr049852e. [DOI] [PubMed] [Google Scholar]
- 7.Fischer F, Wolters D, Rogner M, Poetsch A. Toward the complete membrane proteome–High coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics. 2006;5:444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Lohaus C, Nolte A, Bluggel M, Scheer C, Klose J, Gobom J, Schuller A, Wiebringhaus T, Meyer HE, Marcus K. Multidimensional chromatography: A powerful tool for the analysis of membrane proteins in mouse brain. J Proteome Res. 2007;6:105–113. doi: 10.1021/pr060247g. [DOI] [PubMed] [Google Scholar]
- 9.Speers AE, Wu CC. Proteomics of integral membrane proteins-theory and application. Chem Rev. 2007;107:3687–3714. doi: 10.1021/cr068286z. [DOI] [PubMed] [Google Scholar]
- 10.Pasquali C, Fialka I, Huber LA. Subcellular fractionation, electromigration analysis and mapping of organelles. J Chromatogr, B. 1999;722:89–102. doi: 10.1016/s0378-4347(98)00314-4. [DOI] [PubMed] [Google Scholar]
- 11.Zhao YX, Zhang W, Kho YJ, Zhao YM. Proteomic analysis of integral plasma membrane proteins. Anal Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- 12.Goshe MB, Blonder J, Smith RD. Affinity labeling of highly hydrophobic integral membrane proteins for proteome-wide analysis. J Proteome Res. 2003;2:153–161. doi: 10.1021/pr0255607. [DOI] [PubMed] [Google Scholar]
- 13.Chen WNU, Yu LR, Strittmatter EF, Thrall BD, Camp DG, Smith RD. Detection of in situ labeled cell surface proteins by mass spectrometry: Application to the membrane subproteome of human mammary epithelial cells. Proteomics. 2003;3:1647–1651. doi: 10.1002/pmic.200300468. [DOI] [PubMed] [Google Scholar]
- 14.Hastie C, Saxton M, Akpan A, Cramer R, Masters JR, Naaby-Hansen S. Combined affinity labelling and mass spectrometry analysis of differential cell surface protein expression in normal and prostate cancer cells. Oncogene. 2005;24:5905–5913. doi: 10.1038/sj.onc.1208747. [DOI] [PubMed] [Google Scholar]
- 15.Jang JH, Hanash S. Profiling of the cell surface proteome. Proteomics. 2003;3:1947–1954. doi: 10.1002/pmic.200300563. [DOI] [PubMed] [Google Scholar]
- 16.Lee SJ, Kim KH, Park JS, Jung JW, Kim YH, Kim SK, Kim WS, Goh HG, Kim SH, Yoo JS, Kim DW, Kim KP. Comparative analysis of cell surface proteins in chronic and acute leukemia cell lines. Biochem Biophys Res Commun. 2007;357:620–626. doi: 10.1016/j.bbrc.2007.03.191. [DOI] [PubMed] [Google Scholar]
- 17.Mojica W, Sun JL. Plasma membrane protein identification by cell surface biotinylation and tandem mass spectrometry of clinical specimens. Mod Pathol. 2006;19:162–162. [Google Scholar]
- 18.Nunomura K, Nagano K, Itagaki C, Taoka M, Okamura N, Yamauchi Y, Sugano S, Takahashi N, Izumi T, Isobe T. Cell surface labeling and mass spectrometry reveal diversity of cell surface markers and signaling molecules expressed in undifferentiated mouse embryonic stem cells. Mol Cell Proteomics. 2005;4:1968–1976. doi: 10.1074/mcp.M500216-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Scheurer SB, Rybak JN, Roesli C, Brunisholz RA, Potthast F, Schlapbach R, Neri D, Elia G. Identification and relative quantification of membrane proteins by surface biotinylation and two-dimensional peptide mapping. Proteomics. 2005;5:2718–2728. doi: 10.1002/pmic.200401163. [DOI] [PubMed] [Google Scholar]
- 20.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW, Misek DE, Hanash SM. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–7616. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 21.Sostaric E, Georgiou AS, Wong CH, Watson PF, Holt WV, Fazeli A. Global profiling of surface plasma membrane proteome of oviductal epithelial cells. J Proteome Res. 2006;5:3029–3037. doi: 10.1021/pr060366w. [DOI] [PubMed] [Google Scholar]
- 22.Tang XT, Yi W, Munske GR, Adhikari DP, Zakharova NL, Bruce JE. Profiling the membrane proteome of Shewanella oneidensis MR-1 with new affinity labeling probes. J Proteome Res. 2007;6:724–734. doi: 10.1021/pr060480e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Zhou G, Zhao YX, White MA, Zhao YM. Affinity enrichment of plasma membrane for proteomics analysis. Electrophoresis. 2003;24:2855–2863. doi: 10.1002/elps.200305569. [DOI] [PubMed] [Google Scholar]
- 24.Zhao YX, Zhang W, White MA, Zhao YM. Capillary high-performance liquid chromatography/mass spectrometric analysis of proteins from affinity-purified plasma membrane. Anal Chem. 2003;75:3751–3757. doi: 10.1021/ac034184m. [DOI] [PubMed] [Google Scholar]
- 25.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 26.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 27.Smolka MB, Zhou HL, Purkayastha S, Aebersold R. Optimization of the isotope-coded affinity tag-labeling procedure for quantitative proteome analysis. Anal Biochem. 2001;297:25–31. doi: 10.1006/abio.2001.5318. [DOI] [PubMed] [Google Scholar]
- 28.Ramus C, de Peredo AG, Dahout C, Gallagher M, Garin J. An optimized strategy for ICAT quantification of membrane proteins. Mol Cell Proteomics. 2006;5:68–78. doi: 10.1074/mcp.M500205-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Liang XQ, Zhao J, Hajivandi M, Wu R, Tao J, Amshey JW, Pope RM. Quantification of membrane and membrane-bound proteins in normal and malignant breast cancer cells isolated from the same patient with primary breast carcinoma. J Proteome Res. 2006;5:2632–2641. doi: 10.1021/pr060125o. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Xiang R, Horvath C, Wilkins JA. Quantitative analysis of membrane proteins from breast cancer cell lines BT474 and MCF7 using multistep solid phase mass tagging and 2D LC/MS. J Proteome Res. 2005;4:1427–1433. doi: 10.1021/pr050100+. [DOI] [PubMed] [Google Scholar]
- 31.Foster LJ, Zeemann PA, Li C, Mann M, Jensen ON, Kassem M. Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation. Stem Cells. 2005;23:1367–1377. doi: 10.1634/stemcells.2004-0372. [DOI] [PubMed] [Google Scholar]
- 32.Stockwin LH, Blonder J, Bumke MA, Lucas DA, Chan KC, Conrads TP, Issaq HJ, Veenstra TD, Newton DL, Rybak SM. Proteomic analysis of plasma membrane from hypoxiaadapted malignant melanoma. J Proteome Res. 2006;5:2996–3007. doi: 10.1021/pr0601739. [DOI] [PubMed] [Google Scholar]
- 33.Bisle B, Schmidt A, Scheibe B, Klein C, Tebbe A, Kellermann J, Siedler F, Pfeiffer F, Lottspeich F, Oesterhelt D. Quantitative profiling of the membrane proteome in a halophilic archaeon. Mol Cell Proteomics. 2006;5:1543–1558. doi: 10.1074/mcp.M600106-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 35.Simpson RJ. Proteins and Proteomics: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. [Google Scholar]
- 36.McCarthy J, Hopwood F, Oxley D, Laver M, Castagna A, Righetti PG, Williams K, Herbert B. Carbamylation of proteins in 2-D electrophoresis–Myth or reality. J Proteome Res. 2003;2:239–242. doi: 10.1021/pr025564b. [DOI] [PubMed] [Google Scholar]
- 37.Qiu H, Wang Y. Probing adenosine nucleotide-binding proteins with an affinity-labeled nucleotide probe and mass spectrometry. Anal Chem. 2007;79:5547–5556. doi: 10.1021/ac0622375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loyet KM, Ouyang WJ, Eaton DL, Stults JT. Proteomic profiling of surface proteins on Th1 and Th2 cells. J Proteome Res. 2005;4:400–409. doi: 10.1021/pr049810q. [DOI] [PubMed] [Google Scholar]
- 39.Zhang G, Spellman DS, Skolnik EY, Neubert TA. Quantitative phosphotyrosine proteomics of EphB2 signaling by stable isotope labeling with amino acids in cell culture (SILAC) J Proteome Res. 2006;5:581–588. doi: 10.1021/pr050362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Multhoff G, Hightower LE. Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones. 1996;1:167–176. doi: 10.1379/1466-1268(1996)001<0167:cseohs>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Botzler C, Issels R, Multhoff G. Heat-shock protein 72 cell-surface expression on human lung carcinoma cells in associated with an increased sensitivity to lysis mediated by adherent natural killer cells. Cancer Immunol Immunother. 1996;43:226–230. doi: 10.1007/s002620050326. [DOI] [PubMed] [Google Scholar]
- 42.Van Holde KE. Chromatin. Springer-Verlag; New York: 1989. [Google Scholar]
- 43.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 44.Ojcius DM, Muller S, Hasselkus-Light CS, Young JD, Jiang S. Plasma membrane-associated proteins with the ability to partially inhibit perforin-mediated lysis. Immunol Lett. 1991;28:101–108. doi: 10.1016/0165-2478(91)90106-k. [DOI] [PubMed] [Google Scholar]
- 45.Watson K, Gooderham NJ, Davies DS, Edwards RJ. Nucleosomes bind to cell surface proteoglycans. J Biol Chem. 1999;274:21707–21713. doi: 10.1074/jbc.274.31.21707. [DOI] [PubMed] [Google Scholar]
- 46.Watson K, Edwards RJ, Shaunak S, Parmelee DC, Sarraf C, Gooderham NJ, Davies DS. Extra-nuclear location of histones in activated human peripheral blood lymphocytes and cultured T-cells. Biochem Pharmacol. 1995;50:299–309. doi: 10.1016/0006-2952(95)00142-m. [DOI] [PubMed] [Google Scholar]
- 47.Hariton-Gazal E, Rosenbluh J, Graessmann A, Gilon C, Loyter A. Direct translocation of histone molecules across cell membranes. J Cell Sci. 2003;116:4577–4586. doi: 10.1242/jcs.00757. [DOI] [PubMed] [Google Scholar]
- 48.Herren T, Burke TA, Das R, Plow EF. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry. 2006;45:9463–9474. doi: 10.1021/bi060756w. [DOI] [PubMed] [Google Scholar]
- 49.Parseghian MH, Luhrs KA. Beyond the walls of the nucleus: the role of histones in cellular signaling and innate immunity. Biochem Cell Biol. 2006;84:589–604. doi: 10.1139/o06-082. [DOI] [PubMed] [Google Scholar]
- 50.Ruth MC, Old WM, Emrick MA, Meyer-Arendt K, Aveline-Wolf LD, Pierce KG, Mendoza AM, Sevinsky JR, Hamady M, Knight RD, Resing KA, Ahn NG. Analysis of membrane proteins from human chronic myelogenous leukemia cells: comparison of extraction methods for multidimensional LC-MS/MS. J Proteome Res. 2006;5:709–719. doi: 10.1021/pr050313z. [DOI] [PubMed] [Google Scholar]
- 51.Shih IM. The role of CD146 (Mel-CAM) in biology and pathology. J Pathol. 1999;189:4–11. doi: 10.1002/(SICI)1096-9896(199909)189:1<4::AID-PATH332>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 52.Denton KJ, Stretch JR, Gatter KC, Harris AL. A study of adhesion molecules as markers of progression in malignant melanoma. J Pathol. 1992;167:187–191. doi: 10.1002/path.1711670205. [DOI] [PubMed] [Google Scholar]
- 53.Luca M, Hunt B, Bucana CD, Johnson JP, Fidler IJ, Bar-Eli M. Direct correlation between MUC18 expression and metastatic potential of human melanoma cells. Melanoma Res. 1993;3:35–41. doi: 10.1097/00008390-199304000-00006. [DOI] [PubMed] [Google Scholar]
- 54.Xie S, Luca M, Huang S, Gutman M, Reich R, Johnson JP, Bar-Eli M. Expression of MCAM/MUC18 by human melanoma cells leads to increased tumor growth and metastasis. Cancer Res. 1997;57:2295–2303. [PubMed] [Google Scholar]
- 55.Leslie MC, Zhao YJ, Lachman LB, Hwu P, Wu GJ, Bar-Eli M. Immunization against MUC18/MCAM, a novel antigen that drives melanoma invasion and metastasis. Gene Ther. 2007;14:316–323. doi: 10.1038/sj.gt.3302864. [DOI] [PubMed] [Google Scholar]
- 56.Abidi SM, Saifullah MK, Zafiropulos MD, Kaput C, Bowen MA, Cotton C, Singer NG. CD166 expression, characterization, and localization in salivary epithelium: implications for function during sialoadenitis. J Clin Immunol. 2006;26:12–21. doi: 10.1007/s10875-006-7119-6. [DOI] [PubMed] [Google Scholar]
- 57.Bowen MA, Bajorath J, D'Egidio M, Whitney GS, Palmer D, Kobarg J, Starling GC, Siadak AW, Aruffo A. Characterization of mouse ALCAM (CD166): the CD6-binding domain is conserved in different homologs and mediates cross-species binding. Eur J Immunol. 1997;27:1469–1478. doi: 10.1002/eji.1830270625. [DOI] [PubMed] [Google Scholar]
- 58.Aruffo A, Bowen MA, Patel DD, Haynes BF, Starling GC, Gebe JA, Bajorath J. CD6-ligand interactions: a paradigm for SRCR domain function. Immunol Today. 1997;18:498–504. doi: 10.1016/s0167-5699(97)01130-4. [DOI] [PubMed] [Google Scholar]
- 59.Burkhardt M, Mayordomo E, Winzer KJ, Fritzsche F, Gansukh T, Pahl S, Weichert W, Denkert C, Guski H, Dietel M, Kristiansen G. Cytoplasmic overexpression of ALCAM is prognostic of disease progression in breast cancer. J Clin Pathol. 2006;59:403–409. doi: 10.1136/jcp.2005.028209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jezierska A, Matysiak W, Motyl T. ALCAM/CD166 protects breast cancer cells against apoptosis and autophagy. Med Sci Monit. 2006;12:263–273. [PubMed] [Google Scholar]
- 61.Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol. 2007;20:102–107. doi: 10.1038/modpathol.3800720. [DOI] [PubMed] [Google Scholar]
- 62.Kristiansen G, Pilarsky C, Wissmann C, Stephan C, Weissbach L, Loy V, Loening S, Dietel M, Rosenthal A. ALCAM/CD166 is up-regulated in low-grade prostate cancer and progressively lost in high-grade lesions. Prostate. 2003;54:34–43. doi: 10.1002/pros.10161. [DOI] [PubMed] [Google Scholar]
- 63.Lunter PC, van Kilsdonk JW, van Beek H, Cornelissen IM, Bergers M, Willems PH, van Muijen GN, Swart GW. Activated leukocyte cell adhesion molecule (ALCAM/CD166/MEMD), a novel actor in invasive growth, controls matrix metalloproteinase activity. Cancer Res. 2005;65:8801–8808. doi: 10.1158/0008-5472.CAN-05-0378. [DOI] [PubMed] [Google Scholar]
- 64.van Kempen LC, van den Oord JJ, van Muijen GN, Weidle UH, Bloemers HP, Swart GW. Activated leukocyte cell adhesion molecule/CD166, a marker of tumor progression in primary malignant melanoma of the skin. Am J Pathol. 2000;156:769–774. doi: 10.1016/S0002-9440(10)64943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Degen WG, van Kempen LC, Gijzen EG, van Groningen JJ, van Kooyk Y, Bloemers HP, Swart GW. MEMD, a new cell adhesion molecule in metastasizing human melanoma cell lines, is identical to ALCAM (activated leukocyte cell adhesion molecule) Am J Pathol. 1998;152:805–813. [PMC free article] [PubMed] [Google Scholar]
- 66.Kersey JH, LeBien TW, Abramson CS, Newman R, Sutherland R, Greaves M. P-24: a human leukemia-associated and lymphohemopoietic progenitor cell surface structure identified with monoclonal antibody. J Exp Med. 1981;153:726–731. doi: 10.1084/jem.153.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Si Z, Hersey P. Expression of the neuroglandular antigen and analogues in melanoma. CD9 expression appears inversely related to metastatic potential of melanoma. Int J Cancer. 1993;54:37–43. doi: 10.1002/ijc.2910540107. [DOI] [PubMed] [Google Scholar]
- 68.Zheng R, Yano S, Zhang H, Nakataki E, Tachibana I, Kawase I, Hayashi S, Sone S. CD9 overexpression suppressed the liver metastasis and malignant ascites via inhibition of proliferation and motility of small-cell lung cancer cells in NK cell-depleted SCID mice. Oncol Res. 2005;15:365–372. doi: 10.3727/096504005776449699. [DOI] [PubMed] [Google Scholar]
- 69.Huang H, Groth J, Sossey-Alaoui K, Hawthorn L, Beall S, Geradts J. Aberrant expression of novel and previously described cell membrane markers in human breast cancer cell lines and tumors. Clin Cancer Res. 2005;11:4357–4364. doi: 10.1158/1078-0432.CCR-04-2107. [DOI] [PubMed] [Google Scholar]
- 70.Johnson JP. Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Rev. 1999;18:345–357. doi: 10.1023/a:1006304806799. [DOI] [PubMed] [Google Scholar]
- 71.Saalbach A, Wetzel A, Haustein UF, Sticherling M, Simon JC, Anderegg U. Interaction of human Thy-1 (CD 90) with the integrin alphavbeta3 (CD51/CD61): an important mechanism mediating melanoma cell adhesion to activated endothelium. Oncogene. 2005;24:4710–4720. doi: 10.1038/sj.onc.1208559. [DOI] [PubMed] [Google Scholar]
- 72.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 73.Kuphal S, Bauer R, Bosserhoff AK. Integrin signaling in malignant melanoma. Cancer Metastasis Rev. 2005;24:195–222. doi: 10.1007/s10555-005-1572-1. [DOI] [PubMed] [Google Scholar]
- 74.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 75.Natali PG, Nicotra MR, Digiesi G, Cavaliere R, Bigotti A, Trizio D, Segatto O. Expression of gp185HER-2 in human cutaneous melanoma: implications for experimental immunotherapeutics. Int J Cancer. 1994;56:341–346. doi: 10.1002/ijc.2910560308. [DOI] [PubMed] [Google Scholar]
- 76.Persons DL, Arber DA, Sosman JA, Borelli KA, Slovak ML. Amplification and overexpression of HER-2/neu are uncommon in advanced stage melanoma. Anticancer Res. 2000;20:1965–1968. [PubMed] [Google Scholar]
- 77.Eliopoulos P, Mohammed MQ, Henry K, Retsas S. Overexpression of HER-2 in thick melanoma. Melanoma Res. 2002;12:139–145. doi: 10.1097/00008390-200204000-00006. [DOI] [PubMed] [Google Scholar]
- 78.Kluger HM, DiVito K, Berger AJ, Halaban R, Ariyan S, Camp RL, Rimm DL. Her2/neu is not a commonly expressed therapeutic target in melanoma—a large cohort tissue microarray study. Melanoma Res. 2004;14:207–210. doi: 10.1097/01.cmr.0000130874.33504.2f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.