

Abstract
Glycoprotein hormones (GPHs) are the main regulators of the pituitary-thyroid and pituitary-gonadal axes. Selective interaction between GPHs and their cognate G protein-coupled receptors ensure specificity in GPH signaling. The mechanisms of how these hormones activate glycoprotein hormone receptors (GPHRs) or how mutations and autoantibodies can alter receptor function were unclear. Based on the hypothesis that GPHRs contain an internal agonist, we systematically screened peptide libraries derived from the ectodomain for agonistic activity on the receptors. We show that a peptide (p10) derived from a conserved sequence in the C-terminal part of the extracellular N terminus can activate all GPHRs in vitro and in GPHR-expressing tissues. Inactivating mutations in this conserved region or in p10 can inhibit activation of the thyroid-stimulating hormone receptor by autoantibodies. Our data suggest an activation mechanism where, upon extracellular ligand binding, this intramolecular agonist isomerizes and induces structural changes in the 7-transmembrane helix domain, triggering G protein activation. This mechanism can explain the pathophysiology of activating autoantibodies and several mutations causing endocrine dysfunctions such as Graves disease and hypo- and hyperthyroidism. Our findings highlight an evolutionarily conserved activation mechanism of GPHRs and will further promote the development of specific ligands useful to treat Graves disease and other dysfunctions of GPHRs.
Keywords: follicle-stimulating hormone (FSH), G protein-coupled receptor (GPCR), hormone receptor, signal transduction, signaling, thyroid, luteinizing hormone, thyroid-stimulating hormone
Introduction
Glycoprotein hormone receptors (GPHRs)2 were cloned about 25 years ago (1–3) and, since then, the glycoprotein hormones (GPHs) TSH, LH/hCG, and FSH are considered as the agonists for their respective receptors. In contrast to other rhodopsin-like G protein-coupled receptors (GPCRs), they possess a large extracellular leucine-rich repeat (LRR) hormone-binding domain, which is linked via a hinge region (HR) to the 7-transmembrane helix domain (7TM) (see Fig. 1A). Based on the crystal structure of human FSH bound to the receptor's extracellular domain (ECD), it has been suggested that the FSHR grabs the FSH in a “hand-clasp” mode, orienting the hormone to interact with the extracellular loops and juxtamembrane regions of the 7TM to induce signaling (4). However, to date, all attempts to identify agonistic parts of GPHs have failed, and even extreme high concentrations of hCG did not activate the 7TM of LHR lacking the ECD (5).
FIGURE 1.

Peptides derived from the ECD C terminus are agonists at GPHRs. A, the structural architecture of a prototypical GPHR is shown. An ECD consisting of a signal peptide (SP), an LRR domain, the HR, and the 7TM domain can be distinguished. All constructs were epitope-tagged with an N-terminal HA epitope (yellow) and a C-terminal FLAG epitope (green). Positions of mutations studied are given for LHR and TSHR. B, the ECD C terminus contains a block of 9 amino acids that is conserved among GPHRs. C–E, peptides (1 mm) of different lengths derived from the C-terminal part of the LHR (C), TSHR (D), and FSHR (E) hinge region were tested on LHR-, TSHR- and FSHR-transfected COS-7 cells, respectively. cAMP levels of transfected COS-7 cells were determined as described under “Experimental Procedures.” All peptides were controlled on empty vector-transfected cells and showed no cAMP signal above basal (cAMP level pcDps: 3.7 ± 0.3 nm/well; cAMP level LHR with 1 μm hCG: 56.8 ± 8.5 nm/well). **, p < 0.01, ***, p < 0.001, as compared with basal (without peptide), paired Student's t test. All data are means ± S.E. of three independent experiments performed in triplicate.
In addition to TSH and thyrostimulin (6), the TSHR can be activated by mutations in the ECD and by ECD-directed autoantibodies, which cause hyperthyroidism and Graves disease, respectively (7). The detailed mechanism of how the ECD integrates the activating actions of TSH, thyrostimulin, mutations, and autoantibodies to trigger GPHR signal transduction is unsolved, although there are numerous mutagenesis studies (5, 8) and crystallographic data of the liganded GPHR ECD (4, 9–11).
Based on findings that proteolytic cleavage of the ECD by trypsin, or artificially generated ECD deletions and truncations, can increase TSHR activity, it was proposed that the ECD functions as an internal inverse agonist inhibiting 7TM signaling until ligand binding at the ECD (8, 12–17). We have shown that deletion of the entire ECD did not activate the LHR, which provokes an alternative hypothesis of an “intramolecular agonistic unit” where an internal agonist within the ECD is exposed upon ligand binding at the ECD (5, 8, 18). The latter hypothesis is supported by LHR studies showing that parts of the ECD are necessary to stabilize active state conformations of the 7TM (19, 20).
Here we show that all GPHRs are activated by an internal peptide sequence, which is located in the C-terminal part of the ECD and integrates the diverse stimuli into one activating signal. This provides an explanation of how Graves disease autoantibodies and several mutations cause diseases of the thyroid and gonads.
Experimental Procedures
DNA Constructs and Functional Assays
Full-length GPHR sequences were cloned into the mammalian expression vector pcDps (5, 21, 22). A hemagglutinin (HA) epitope and a FLAG epitope were inserted directly downstream of the signal peptide and at the very C terminus of the GPHRs, respectively. All mutant constructs were generated by a PCR-based site-directed mutagenesis and fragment replacement strategy (details given in Table 1) and confirmed by sequencing.
TABLE 1.
Description of human GPHRs constructs used in this study
| Construct | GPHR amino acid residues in the construct | M3 amino acid residues in the construct | HA tag | FLAG tag | Modifications |
|---|---|---|---|---|---|
| LHR WT | 1–699 | − | + | + | − |
| LHR S277N | 1–699 | − | + | + | S277N |
| LHR E354K | 1–699 | − | + | + | E354K |
| LHR S277N/E354K | 1–699 | − | + | + | S277N; E354K |
| LHR D578H | 1–699 | − | + | + | D578H |
| LHR E354K/D578H | 1–699 | − | + | + | E354K; D578H |
| TSHR WT | 1–764 | − | + | + | − |
| TSHR S281N | 1–764 | − | + | + | S281N |
| TSHR E409K | 1–764 | − | + | + | E409K |
| FSHR WT | 1–669 | − | + | + | − |
| M3-LHR | 361–699 | 1–66 | + | + | − |
| SP-p10-LHR | 1–26; 350–699 | − | − | + | − |
| SP-p10E354K-LHR | 1–26; 350–699 | − | − | + | E354K |
For functional assays, GPHR constructs were expressed heterologously in COS-7 cells upon transient transfection and grown in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2 in a humidified atmosphere. For the cAMP assay, cells were split into 96-well plates (1.5 × 104 cells/well) and transfected with 250 ng of vector constructs using MACSfectinTM (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's protocol. Wild type receptors served as positive controls and empty vector served as negative control, respectively. 72 h after transfection, cells were incubated with DMEM containing 1 mm 3-isobutyl-methyl-xanthine (IBMX) and the indicated concentrations of peptides and/or hormones for 1 h at 37 °C. For cAMP measurements, cells were lysed in 25 μl of lysis buffer (5 mm HEPES; 0.1% BSA; 0.3% Tween 20; 1 mm, pH 7.4) and kept frozen at −20 °C until measurement. To measure cAMP concentration, the AlphaScreen cAMP assay kit (PerkinElmer Life Sciences) was used according to the manufacturer's protocol. The accumulated cAMP was measured in 384-well white OptiPlate microplates (PerkinElmer Life Sciences) with the Fusion AlphaScreen multilabel reader (PerkinElmer Life Sciences). To estimate cell surface expression of receptors carrying an N-terminal HA tag, an indirect ELISA was used (23).
Tissue Preparation
Activation of endogenously expressed GPHRs with p10 peptide was tested on male C57/Bl6 wild type mice. Testis and thyroid gland were isolated and stored in PBS on ice. Testis was homogenized in 1 ml of PBS. After centrifugation for 5 min at 300 × g, the pellet was dissolved in DMEM with 1 mm IBMX. Thyroids were disrupted with a scalpel blade and homogenized through a needle in 100 μl of DMEM with 1 mm IBMX. 15 μl of these cell solutions were transferred to a 96-well plate, and 15 μl of 2-fold concentrated agonist in DMEM with IBMX were added. Suspensions were incubated for 1 h at 37 °C followed by centrifugation for 5 min at 1,000 × g. The supernatant was discarded, and the pellets were subjected to lysis by adding 25 μl of lysis buffer and kept frozen at −20 °C until measurement. Measurement of cAMP content was done as described above.
Intact thyroid follicles were isolated from mice expressing the cAMP FRET sensor Epac1-camps and plated in glass-bottom Petri dishes coated with a collagen gel as described previously (24). Imaging was performed in a buffer containing 144 mm NaCl, 5.4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, pH 7.3. Ratiometric FRET measurements were performed on a Zeiss Axiovert 200 inverted microscope equipped with an oil-immersion 63× objective, a polychrome V light source (Till Photonics), a 505 DCXR beam splitter, and an iXon Ultra 897 EMCCD camera (Andor). Images were acquired every 5 s with 5-ms illumination time and monitored with the MetaFluor 5.0 software (Molecular Devices) as the ratio between emission at 535 ± 20 nm (YFP) and emission 480 ± 15 nm (CFP), upon excitation at 436 ± 10 nm. The YFP emission was corrected for direct excitation of YFP at 436 nm and the bleed-through of CFP emission into the YFP channel as described previously (25).
For testosterone ELISA, testis were isolated and homogenized in 1 ml of ice-cold PBS. After centrifugation for 5 min at 300 × g, the pellet was dissolved in Hanks' balanced salt solution (100 mg of tissue/1 ml). 60 μl of this solution were transferred to a 96-well plate, and the indicated concentrations of agonists were added. Cells were incubated for 24 h at 37 °C. After two freeze-thaw cycles to break down cell membranes, samples were centrifuged for 5 min at 1,500 × g. 50 μl of the supernatant were assayed with Testosterone Saliva ELISA (IBL International GmbH, Hamburg, Germany) to determine the testosterone concentration.
Peptide Synthesis
Solid phase peptide synthesis was performed on an automated peptide synthesizer MultiPep from Intavis AG (Köln, Germany) using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry. The final side chain deprotection and cleavage from the solid support employed a mixture of TFA, water, and triisopropylsilane (95:2.5:2.5, volume %) for the peptides. The peptides were purified to >95% purity using preparative RP-HPLC (Shimadzu LC-8, Duisburg, Germany) equipped with a PLRP-S column (300 × 25 mm, Agilent, Waldbronn, Germany). For both analytical and preparative use, the mobile phases were water (A) and acetonitrile (B), respectively, each containing 0.1% TFA. Samples were eluted with a linear gradient from 5% B to 90% B in 30 min for analytical runs and in 90 min for preparative runs. Finally, all peptides were characterized by analytical HPLC (Agilent 1100) and MALDI-MS (Bruker Microflex LT, Bremen, Germany), which gave the expected [M+H]+ mass peaks.
RT-qPCR
For expression analysis RNA from Nthy-ori 3-1 cells was isolated using TRIzol (Sigma-Aldrich) according to manufacturer's instructions. For quantitative real-time PCR analysis (qPCR), 1 μg of total RNA was reverse-transcribed (Omniscript; Qiagen) using a mixture of oligo(dT) and random hexamer primers. qPCR was performed by GoTaq® qPCR Master Mix (Promega, Madison, WI). cDNA from 25 ng of total RNA and from 0.2 μm of the forward and reverse primers were used. Oligonucleotide primers were as follows: 5′gttgccccatgtcatccag-3′and 5′-catcgatcacaggacccca-3′, thyroglobulin (TG); 5′-catctacggatcggcctgt-3′ and 5′-cccaagatgaaggctccca-3′, sodium/iodide symporter (NIS). They were designed using the Primer3 software (26) to flank intron sequences. PCR was performed in an MX 3000P instrument (Stratagene, La Jolla, CA) using the following protocol: 5 min at 50 °C, 2 min at 95 °C, and 40 cycles of 15 s at 95 °C and 30 s at 60 °C. To confirm the presence of a single amplicon, product melting curves were recorded. Threshold cycle (Ct) values were set within the exponential phase of the PCR. Data were normalized to β2-microglobulin, and ΔCT values were used to calculate the relative expression levels. Gene regulation was statistically evaluated by subjecting the 2−ΔΔCt method (27).
Homology Modeling of Structural Conformations and TSHR Model Assembling
For the generation of the GPHR models, the crystal structure of the ECD was taken from Protein Data Bank (PDB) code 3G04 (9), and the 7TM domain was generated by homology modeling using the crystal structure of different GPCR. In detail, an advanced TSHR homology model was prepared by using the following structural templates from homologous family A GPCRs. The LRR domain (with 11 repeats) complexed with the bound hormone was modeled based on the previously determined FSHR-FSH structure (PDB code 4AY0 (11)). This FSHR crystal structure comprises an N-terminal (TSHR positions Gln289–Ser304) and a C-terminal fragment of the hinge region (TSHR positions Ser383–Ile411). Disulfide bridges between cysteines of the entire ECD were kept as observed in this FSHR crystal structure. Details of the modeling protocol for the extracellular N-terminal part were described previously in Ref. 18 and were refined in this current study.
For modeling of the TSHR 7TM domain, composed of the transmembrane helices (TMs) and the three connecting extracellular loops (ECL) and three intracellular loops, in an active state conformation, the solved structural complex of the β2-adrenergic receptor (ADRB2)/Gs was used (28). Besides common modifications for template preparations such as loop length adjustment (29), the structural modifications on this template were: (a) deletion of the G protein (Gs); (b) deletion of the extracellularly fused T4 lysozyme; (c) removal of the ligand from the ligand-binding pocket; (d) substitution of the slightly kinked TM5 by a regular α-helix (because the GPHRs do not have a proline that causes a kink in this helix in contrast to most other family A GPCRs (for details, see Refs. 30 and 31); and (e) substitution of the ADRB2 ECL2 by the ECL2 of opsin. This substitution was necessary according to the favored conformation and localization of the TSHR ECL2 as suggested by the rhodopsin/opsin structures (32), which is different from the ADRB2 ECL2 conformation (β-sheet versus helical) and localization. For this purpose, the structures of opsin (PDB code 3CAP) and the ADRB2 were superimposed and the entire ADRB2 ECL2 was replaced by the opsin loop (from the transitions to transmembrane helices TM4 and TM5). Amino acids of this chimeric receptor template were than mutated with residues of the TSHR, followed by molecular dynamics (3 ns) and energetic minimizations of the side chains with constrained backbone atoms (until converging at a termination gradient of 0.05 kcal/mol × Å). This system was than minimized without any constraint.
For comparison between TSHR and LHR or FSHR, the amino acids of the TSHR 7TM were mutated to the specific residues of LHR and FSHR, respectively. By fixing the backbone, the FSHR and LHR models were minimized. Moreover, in a further step, the extra- and intracellular loops of all three receptors were subjected to molecule dynamics of 0.5 ns by fixing the backbone atoms of the transmembrane helices, followed by energetic optimization.
Finally, the 7TM of the TSHR and the ECD in complex with TSH were assembled. The N terminus of the 7TM model (at position 411) and the ECD/TSH model (C terminus at position 410) were superimposed and fused, whereby the orientation or spatial distance of the extracellular part relative to the ECLs is not known so far without a determined crystal structure of the entire complex, but is restricted due to the fact that the transition between the ECD and TM1 is simultaneously connected via a cysteine bridge between Cys284 (C terminus of the LRR) and Cys408 (C terminus of the hinge region close to TM1). This cysteine bridge functions as a structural constraint, together with a second cysteine bridge between Cys398 and Cys283. In consequence, the ECLs tightly surround the Pro400–Asp410 fragment that is adjacent to TM1 and above the helical bundle. The assembled complex was than minimized by constraining the backbone first, followed by a minimization of the entire model without constraints.
To ensure the structural dimension of TM1 (N and C terminus) and by this the transition to the ECD in the TSHR model to the best resolution, 15 available GPCR crystal structures from the β2-adrenergic receptor (PDB code 2RH1), metarhodopsin II (PDB code 3PQR), δ-opioid receptor (PDB code 4EJ4), or the neurotensin receptor (PDB code 4GRV) were superimposed and analyzed to define the common number of residues constituting TM1. Of note, the TM1 structures can be slightly different with respect to the adjustment relative to helices in spatial proximity, but the dimension at the extracellular side is highly conserved with only minimal deviations (33). Therefore, we defined on the basis of determined GPCR crystal structures the most common N terminus of the TM1 at position 1.28 (Ballesteros and Weinstein numbering system (34)), which corresponds to the TSHR Asp410.
Structural modifications to generate homology models were performed with the software Sybyl X2.0 (Certara, Princeton, NJ). The AMBER F99 force field was used for energy minimization and dynamics. Structure images were produced using the PyMOL software (Schrödinger, LLC, New York).
Results and Discussion
Identification of an Internal Agonist in GPHRs
To identify the region of the potential internal agonist, we tested peptides derived from the very C-terminal portion of the ECD, which is highly conserved among GPHRs (Fig. 1B) and functionally relevant (35). Thus, peptide libraries with different lengths covering this 25-amino acid region were synthesized. Functional testing of these libraries revealed two peptides (each 10 amino acids long) with agonistic activity on the LHR (Fig. 1C). The two 10-amino acid-long peptides contained Phe350–Tyr359 and Asn351–Asp360 (Fig. 1C) and covered the highly conserved region (Fig. 1B, blue box). All other peptides showed no function at the LHR.
Similarly, screening of peptides derived from the same region of the TSHR and FSHR showed significant activity for the peptides covering the conserved segment (Fig. 1, D and E). The most efficient peptide at all GPHRs, FNPCEDIMGY (referred to as p10), showed no response on mock-transfected cells (Fig. 2A) and cells transfected with the human β2 adrenergic receptor or V2 vasopressin receptor (Fig. 2B). Concentration-response studies revealed low potency for p10 (EC50 value >2 mm) at GPHRs (Fig. 2, C and D). To investigate whether the high EC50 value of p10 is due to competition with the potentially internal agonist sequence, we used a chimeric construct where the entire ECD of the LHR was replaced by the N terminus of the M3 muscarinic acetylcholine receptor (M3-LHR) (5). As shown in Fig. 2D, the EC50 value of p10 at the internal agonist-free M3-LHR remained unchanged, indicating only a low affinity interaction between the internal agonistic sequence and the 7TM. The apparently very low affinity of the peptide did not allow for radioligand binding studies. However, high affinity ligand binding is not required because the peptide is part of the receptor's N terminus and, therefore, covalently bound to the 7TM in a 1:1 stoichiometry. Consequently, we did not reach saturation of receptor activation with the p10 concentrations used (Fig. 2, C and D), which explains the lower Emax value (efficacy) of p10 as compared with GPH (see Fig. 5A).
FIGURE 2.

p10 specifically activates GPHRs. A, p10 (1 mm) was tested on COS-7 cells transfected with the LHR, FSHR, and TSHR. B, COS-7 cells were transfected with the human ADRB2 and the human V2 vasopressin receptor (AVPR2) and incubated with isoprenaline (isopre: 1 μm), arginine-vasopressin (AVP: 1 μm) and p10 (2 mm). Empty pcDps vector (mock) served as negative control (pcDps; cAMP level was 2.9 ± 0.4 nm/well). C and D, concentration-response curves of p10 on LHR, M3-LHR, and TSHR revealed an EC50 value >2 mm. Basal pcDps levels were 3.6 ± 0.3 nm/well. Data are means ± S.E. of three independent experiments performed in triplicate.
FIGURE 5.

Specificity of p10-mediated activation of GPHRs. A, COS-7 cells transfected with LHR and TSHR and their inactive mutants E354K and E409K, respectively, were incubated with their respective glycoprotein hormone (LHR: 500 nm hCG; TSHR: 100 milliunits/ml bTSH) and p10 and cAMP levels were determined. Basal cAMP level for pcDps was 1.8 ± 0.8 nm/well. B, COS-7 cells transfected with TSHR, LHR, and FSHR were incubated with their GPH (100 milliunits/ml bTSH, 500 nm hCG, 5 milliunits/ml FSH) and p10 E5K as indicated. Basal cAMP level for pcDps (empty vector) was 1.7 ± 0.5 nm/well. cAMP levels were determined (see “Experimental Procedures”). C and D, Nthy-ori 3-1 cells (40) were incubated with TSH, p10, and p10 E5K. In C and D, cAMP levels (basal cAMP levels: 1.1 ± 0.3 nm/well) (C) and mRNA expression levels (D) of the NIS and TG prior to and after stimulation were determined (see “Experimental Procedures”). Expression of NIS and TG was normalized to β2-microglobulin (Ct 15.7 ± 0.2). Gene regulation values are given as means of 2−ΔΔCt ± S.E. Data are means ± S.E. of three independent experiments performed in triplicate. *, p < 0.05, **, p < 0.01, ***, p < 0.001 (paired Student's t test).
To constrain the 1:1 stoichiometry of p10 and the 7TM, we generated an N-terminally truncated LHR construct where the signal peptide was directly fused upstream of the p10 region (SP-p10-LHR). SP-p10-LHR, which still contained p10 but no additional upstream sequence, displayed an increased basal activity as compared with the full-length receptors (Fig. 3). This is consistent with previous studies showing an increased basal activity after deletion of the ECD (see above). Introduction of the inactivating mutation E354K into the p10 region (see below) completely abolished basal activity of this receptor construct, but p10 was able to activate SP-p10 E354K-LHR (Fig. 3). This again excludes that the ECD functions as inverse agonist because one would also expect increased basal activity in SP-p10 E354K-LHR. Our data instead support the scenario that direct attachment of the p10 sequence to the 7TM is sufficient for p10-mediated 7TM activation. However, the efficacy of cAMP formation in SP-p10-LHR-transfected cells was similar to p10, indicating that the active conformation of p10 is most optimal when p10 is imbedded in the ECD.
FIGURE 3.
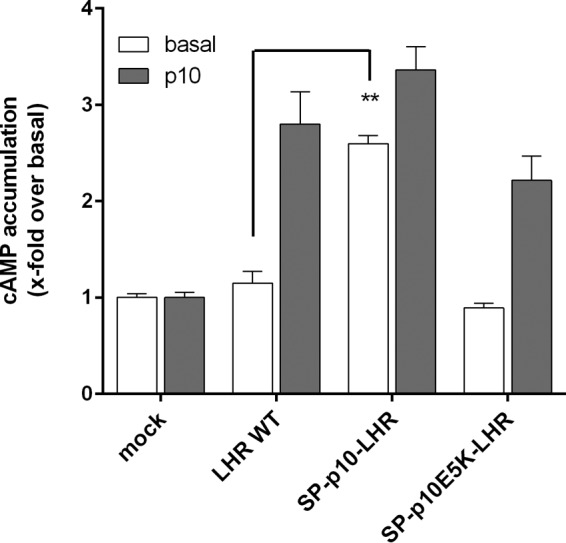
N-terminal truncation activates the LHR. COS-7 cells were transfected with the indicated LHR constructs, and cAMP levels were determined with p10 (2 mm) and without. Empty pcDps vector (mock) served as negative control (pcDps; cAMP level was 1.2 ± 0.2). Data are means ± S.E. of three independent experiments performed in triplicate.
Structure-Function Relationship Analysis of p10
To study the relevance of the individual positions in the p10 region, we performed systematic alanine-scanning mutagenesis in the LHR (Fig. 4A) and the p10 peptide (Fig. 4C). Seven of 10 alanine LHR mutants displayed significantly reduced activity upon hCG stimulation (Fig. 4A). For example, mutation of Phe350 and Asp355 did not alter cell surface expression, suggesting direct participation in agonistic function of the p10 region (Fig. 4B). This is consistent with studies at the TSHR where mutations of the corresponding positions of Phe350 and Asp355 led to reduced receptor activation (36). This is most probably due to misfolding or disorientation of the p10 sequence within its 7TM-binding pocket. Interestingly, alanine substitutions of positions Asn351, Pro352, and Gly358 in LHR induced increased basal activity (Fig. 4A). This suggests that the amino acids at these positions most probably do not specifically interact with the 7TM but mutations of these residues change the fold of the p10 region into more active states. Mutations of the corresponding positions in the TSHR also resulted in constitutive activity (37).
FIGURE 4.

Alanine-scanning mutagenesis of the p10 region in GPHRs and the p10 peptide. A, COS-7 cells were transfected with the indicated LHR constructs, and cAMP levels were determined with hCG (500 nm) and without. Empty pcDps vector (mock) served as negative control (pcDps; cAMP level was 2.6 ± 0.8). Data are means ± S.E. of three independent experiments performed in triplicate. B, cell surface expression of HA epitope-tagged receptors transiently expressed in COS-7 cells was determined by cell surface ELISA (see “Experimental Procedures”). Optical density (OD) is given as the percentage of LHR WT minus the OD of mock-transfected cells. The OD values for mock-transfected cells and LHR WT were 0.02 ± 0.007 and 0.642 ± 0.092, respectively. Data are given as means ± S.E. of three independent experiments performed in triplicate. C, alanine-scanning mutagenesis was performed on p10 and tested on COS-7 cells transfected with WT LHR and TSHR. GPH served as positive control (GPH; 100 milliunits/ml bTSH for TSHR and 500 nm hCG for LHR). Peptides (2 mm) were also tested on cells transfected with the empty vector (mock), and data are given as -fold over mock treated with the indicated peptide. Basal cAMP level of mock-transfected cells was 1.7 ± 0.3 nm/well. Means ± S.E. of three independent experiments performed in triplicate are presented.
Consistent with the mutagenesis, data at the LHR alanine substitution in the p10 peptide revealed abolished or reduced agonistic activity at the peptide positions 1 and 4–8 (corresponding to Phe350 and from Cys353 to Met357) when tested on the LHR and TSHR (Fig. 4C). There was no major difference of the peptide mutants acting on the TSHR and LHR. None of the alanine substitutions in p10 increased efficacy at 2 mm. This all indicates functional equivalence between the p10 region and in the 7TM peptide-binding side of GPHRs.
Mutations in the Internal Agonist Cause Endocrine Diseases
There are inactivating mutations in the p10 region identified in patients with inherited pseudohermaphroditism (LHR, E354K) (38) and hypothyroidism (TSHR, D410N) (39). We speculated that receptor inactivation is due to loss of agonistic properties of the internal sequence. E354K in LHR showed no activity in cAMP assays after hCG stimulation but this mutant receptor was activated by p10 (Fig. 5A). The corresponding mutation in the TSHR (E409K) could also be activated by p10 (Fig. 5A). Both mutants were detectable at the cell surface (Fig. 6). Consistently, p10 containing this Glu to Lys substitution (p10 E5K) had no agonistic activity and significantly reduced GPH-induced cAMP formation on all three GPHRs (Fig. 5B). This shows that mutations in the internal agonistic sequence of GPHRs are functionally relevant in vivo and responsible for cases of hypogonadism and hypothyroidism in humans.
FIGURE 6.

Expression of WT and mutant GPHRs. COS-7-cells were transfected with either WT or mutant GPHRs, and expression of receptors was measured by cell surface ELISA (see “Experimental Procedures”). OD is given as the percentage of WT GPHR minus OD of mock-transfected cells. For surface ELISA, the OD value for mock-transfected cells, LHR WT, and TSHR WT was 0.025 ± 0.006, 0.895 ± 0.069, and 0.834 ± 0.072, respectively. Data are given as means ± S.E. of three independent experiments each performed in triplicate.
p10 Activates GPHRs in a Thyroid Cell Line and Tissues
We tested whether the TSHR, endogenously expressed in immortalized human primary thyroid follicular epithelial cells (Nthy-ori 3-1 (40)), can be activated by p10. Incubation of the Nthy-ori 3-1 cells with TSH and p10 resulted in robust elevations of cAMP (Fig. 5C). Induction of NIS mRNA and TG mRNA expression is a prominent biological effect of TSHR stimulation in thyrocytes (41, 42). Both TSH and p10 significantly increased NIS and TG mRNA levels in Nthy-ori 3-1 cells, whereas p10 E5K blocked TSH-induced mRNA levels of both transcripts (Fig. 5D). Next, cell suspensions from mouse thyroid (TSHR) and mouse testis (FSHR and LHR) were treated with p10 and the inactive p10 E5K. p10 induced a robust cAMP response in cells from thyroid (Fig. 7A) and testis (Fig. 7B), whereas p10 E5K did not but blocked GPH-induced receptor activation. In testicular Leydig cells, testosterone synthesis is stimulated by LHR activation. As shown in Fig. 7C, p10 increased testosterone levels and p10 E5K blocked hCG-induced testosterone production.
FIGURE 7.

p10 activates GPHRs in tissues. A and B, cell suspensions from mouse thyroid (A) and mouse testis (B) were incubated with TSH and hCG, respectively, and with p10, p10 E5K, and a control peptide (scrambled amino acids of p10). Basal cAMP levels for thyroid and testis were 5.8 ± 2.3 and 30.1 ± 10.5 nm/well, respectively. C, cell suspension from testis was incubated with the indicated compounds, and testosterone levels were determined with an ELISA (see “Experimental Procedures”). Data are means ± S.E. of three (A and C) and five (B) independent experiments performed in triplicate. *, p < 0.05, **, p < 0.01, ***, p < 0.001 (paired Student's t test).
To generate live image of the time course of p10-induced cAMP formation, we isolated intact thyroid follicles from mice expressing the cAMP FRET sensor Epac1-camps. In this system, TSH produces a robust cAMP signal in thyrocytes within minutes (24). Similarly, p10 (0.5 mm) induced cAMP formation, whereas p10 E5K or a scrambled peptide did not (Fig. 8).
FIGURE 8.

Real-time monitoring of cAMP levels in thyroid follicles isolated from the cAMP reporter mice. Thyroid follicles isolated from Epac1-camps mice were visualized by time-lapse fluorescence microscopy. A, CFP and corrected YFP images were recorded to determine YFP/CFP ratio images. B and C, the graphs show normalized YFP/CFP ratio values calculated from CFP and YFP images. D, FRET values were normalized to the basal level (set to 1) and the response to stimulation with 10 μm forskolin (set to 0). Data are means ± S.E. of three (scrambled peptide) and five (p10, p10 E5K) experiments. ***, p < 0.001 versus p10 (one-way analysis of variance followed by Bonferroni's post hoc test).
Mutant p10 Reduces GPHR Activation by Mutations and Antibodies
Next, we tested whether p10 E5K has an inhibitory effect on mutation-induced constitutive activity. The constitutive activity of S281N, an ECD mutation of the TSHR found in patients with congenital hyperthyroidism (43–45), was suppressed by p10 E5K (Fig. 9A). A similar effect was seen with the corresponding mutation in the LHR (S277N) (Fig. 9A). This suggests that constitutive activity of S281N and S277N is most probably due to the mutation-induced conformational changes in the ECD exposing the internal agonist sequence to the 7TM. To support this hypothesis, we introduced the inactivating mutation E354K (located in the p10 region) into the constitutive active S277N. S277N/E354K was not basally active but was activated by p10 (Fig. 9B). Cell surface expression of this double mutant was reduced (20% of the WT LHR; Fig. 6).
FIGURE 9.

The mutant p10 E5K blocks mutation and antibody-induced GPHR activation. A–D, COS-7 cells were transfected with WT and the indicated mutant LHR and TSHR and incubated with 500 nm hCG or 100 milliunits/ml bTSH and p10 and inactivating p10 E5K (A–C) or 500 ng/ml of the monoclonal autoantibody M22 (D). cAMP levels were determined as described (see “Experimental Procedures”). cAMP level of mock-transfected cells was 2.3 ± 0.9 nm/well. Data are means ± S.E. of three experiments performed in triplicate. *, p < 0.05, **, p < 0.01, ***, p < 0.001 (paired Student's t test).
Most activating GPHR mutations were found in the 7TM domain (46). Based on previous studies (5), we can rule out that the constitutive activity of those 7TM mutations is p10 sequence-mediated. We showed that an LHR construct lacking the ECD including the p10 region and harboring an activating mutation in TM6 (D578H) has high basal activity (5). Therefore, D578H must promote an active conformation of the 7TM. Because p10 E5K most probably interacts with the 7TM, we asked whether p10 E5K has an effect on an activating mutation within the 7TM (D578H). Unexpectedly, p10 E5K functions as an inverse agonist at D578H, reducing the high basal activity of this mutant (Fig. 9C). This suggests that p10 E5K binds to the mutant 7TM and shifts its conformation into the inactive state. Consistently, the double mutant E354K/D578H had an almost abolished basal activity (Fig. 9C), most probably because the mutant p10 region (E354K) within this construct acts as an artificial inverse agonist. Incubation with hCG further decreased the basal activity of E354K/D578H (Fig. 9C), indicating that the mutant p10 region undergoes structural changes upon GPH binding, which further promotes the inverse agonistic action of the mutated p10 region.
Graves disease is caused by persistent stimulation of thyrocyte TSHR by autoantibodies. These autoantibodies usually bind to the LRR domain of the TSHR (47, 48). As expected, the activating human autoantibody M22 (49) induced a robust cAMP signal at the TSHR, but not at the mutant receptor E409K, suggesting that the antibody requires the functional internal agonist sequence to trigger the 7TM activation (Fig. 9D). The activating properties of the autoantibody could be reduced by the inactive peptide p10 E5K (Fig. 9D). This shows that autoantibodies in Graves disease utilize the same activation mechanism as GPHs, exposing the internal agonist.
Potential Mechanism of GPHR Activation through the Internal Agonist
We identified an intramolecular agonistic unit for GPHRs that comprises a linear amino acid sequence between the HR and TM1. Currently, the molecular mechanism unmasking this internal agonistic unit is speculative. However, obviously, GPHs, activating autoantibodies, and mutations in the ECD trigger structural changes such that the internal agonist sequence adopts an active conformation. Unfortunately, all crystal structures of GPHR ECDs (4, 9–11) provide no direct hint for such a 7TM activation mechanism because the internal agonist sequence is not entirely included in these structural models. It was previously speculated that the HR conveys hormone binding at LRR domain to an intramolecular agonistic unit (18). In agreement with this scenario, many activating mutations in GPHRs were found in the HR (5, 43, 50).
To gain further insights into a potential activating scenario, we took advantage of the ECD and 7TM crystal structures of GPHRs and other rhodopsin-like GPCRs, respectively, and generated an advanced homology model of the TSHR. Based on this model, the C-terminal half of the p10 region is part of the TM1 (Fig. 10A). The N-terminal half of the p10 region is localized closely above the 7TM, embedded in a cleft between the ECLs (Fig. 10A), suggesting a preoccupied binding pocket mainly formed by determinants of the ECLs. The accuracy of this model is supported by a number of experimental observations. First, based on our model, only the N-terminal part of the p10 region takes part in ECL interactions and may therefore act as agonist (Fig. 10A). Indeed, peptides lacking the four C-terminal amino acids of p10 (proposed to be the N terminus of TM1) showed a reduced but still significant activity at the TSHR and LHR (Fig. 10B). Second, the conserved Glu409 in the TSHR p10 region is a functional key player (see above) and, according to this model, likely interacts with Lys660 at the N-terminal part of TM7. Consistent with this model, mutation of Lys660 leads to receptor inactivation (51). There are also other residues in the ECL part of GPHRs that are important for receptor signaling (Fig. 10, C and D). Consistently, many peptide receptors, such as the chemokine receptors, have their ligand-binding pocket in the ECL part. Even the β2 adrenergic receptor, having its agonist-binding pocket buried in the helical bundle, shows significant structural changes of the ECL part upon receptor activation (52) highlighting the ECL region as crucial for receptor activation.
FIGURE 10.

Structural homology model of the TSHR serpentine domain including the p10 region. A, the 7TM structure of the TSHR with the p10 region (red, backbone ribbon tube, side chains from Phe405–Tyr414), which equates in the TSHR exactly the transition between the extracellular HR and the TM1, is represented as a model based on crystal structures of homologous GPCRs (see “Experimental Procedures”). Ntt, N-terminal tail; Ctt, C-terminal tail. Inset: the surfaces of the 7TM and the p10 region are highlighted, showing that the flexibility of this fragment between the ECLs is spatially restricted by steric constraints. B, COS-7 cells transfected with TSHR and LHR were incubated with shortened p10 (1 mm) and p10 as positive control. cAMP levels of mock-transfected cells were 2.1 ± 1.1 nm/well. Data are means ± S.E. of three experiments performed in triplicate. *, p < 0.05, **, p < 0.01, ***, p < 0.001 (paired Student's t test). C, the positioning of the p10 region is shown in the overlay of the TSHR, LHR, and FSHR. Based on this model, the amino acid interface of p10 and its binding pocket is shown in D. Several positions of this interface were mutated and functionally tested. Positions marked with + lead to receptor activation, and positions marked with − lead to receptor inactivation. For references, see the TSHR database Sequence-Structure-Function-Analysis of Glycoprotein Hormone Receptors (46). E, the complex model of TSHR (white, backbone ribbon) with bound TSH and the activating antibody M22 visualizes a potential arrangement and the principle mechanism of GPHR activation. For comparison of binding sites, the LRR domain of TSHR complexed with the activating antibody M22 (9) was superimposed with the TSHR LRR domain/TSH model. The entire complex is derived by the arrangement of available structural information on GPHRs and GPCRs (see “Experimental Procedures”). ICL, intracellular loop.
Our current concept of GPHR activation suggests an isomerization of the p10 region pre-bound in the ECL-binding pocket (Fig. 10E). This could be very similar to the 11-cis/all-trans isomerization of retinal in opsins where distinct structural changes of a covalently bound ligand can trigger 7TM activation and does not require high affinity ligand/7TM interactions. In GPHR, the ECD ensures specific ligand binding and the HR integrates this extracellular signal into structural changes, exposing or structuring the internal agonist sequence to activate the 7TM. The transduction of these structural changes likely involves the disulfide bridges (Cys283/Cys398; Cys284/Cys408) physically linking the LRR domain via the HR directly to the p10 region (which contains Cys408). The importance of these disulfide bridges in receptor activation is supported by activating mutations at Cys283 and Cys284 in the TSHR (53).
Activation by an internal sequence appears to be a common signaling mechanism in many GPCRs. For example, protease-activated receptors expose an agonistic, short sequence (TRAP-6) after N-terminal cleavage by thrombin (54). We recently showed for adhesion GPCRs that a short sequence in the C-terminal part of the ectodomain functions as a tethered agonist activating G protein-signaling cascades (55).
Conclusion
GPHRs contain an internal agonist within their ECD. Peptides derived from this sequence can directly activate GPHRs, and modified peptides can reduce GPHR activation induced by GPHs, mutations, and antibodies. These results are compatible with an activation scenario of GPHRs in which an intramolecular agonistic unit is switched into an active state upon structural changes of the ECD and subsequently triggers 7TM-mediated activation of G protein-signaling cascades. Due to its peptide nature, the high concentration needed for stimulation, and the loss of specificity to the various GPHRs, in vivo experiments essentially require the development of more potent and specific small molecule ligands, which has already been started (56–58). The identification of the internal agonist sequence for GPHRs now allows for characterization of its agonist-binding pocket followed by rational ligand design. Such ligands could be therapeutically useful for blocking TSHR activation due to autoantibodies in Graves disease and activating mutations in GPHRs. The proof of concept that non-peptidic, high affinity compounds can block autoantibody-induced TSHR activation has been recently documented (58).
Author Contributions
A. B., A. S., A. R., and D. C. performed the experiments. G. K. performed the modeling studies. A. B. and T. S. analyzed the data. S. R. synthesized the peptides. A. B. and T. S. designed the study and wrote the paper with contributions from all authors.
Acknowledgments
We thank Thomas S. Heard for critical reading of the manuscript and for suggestions. We also acknowledge Carola Rintisch and Katja Ettig for technical assistance. We thank Mariam Gamaleldin for help with FRET experiments.
The work was supported by the Deutsche Forschungsgemeinschaft (SSP ThyroidTransAct; BR 5108/1-1 AO 619225 and SFB/Transregio 166, project C1), the BMBF (IFB AdipositasDiseases Leipzig, FZ: 01EO1001), Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Würzburg (B-281) and the University of Leipzig (Junior research grant by the Medical Faculty). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
- GPHR
- glycoprotein hormone receptor
- GPH
- glycoprotein hormone
- TSH
- thyroid-stimulating hormone
- TSHR
- TSH receptor
- hCG
- hCG, human chorionic gonadotropin
- LH
- luteinizing hormone
- LHR
- luteinizing hormone receptor
- FSHR
- follicle-stimulating hormone receptor
- bTSH
- bovine TSH
- 7TM
- seven-transmembrane spanning domain
- ECD
- extracellular domain
- ECL
- extracellular loop
- GPCR
- G protein-coupled receptors
- HR
- hinge region
- IBMX
- 3-isobutyl-methyl-xanthine
- LRR
- leucine-rich repeat
- NIS
- sodium/iodide symporter
- qPCR
- quantitative PCR
- TG
- thyroglobulin
- SP
- signal peptide
- TM
- transmembrane helix
- OD
- optical density.
References
- 1.Parmentier M., Libert F., Maenhaut C., Lefort A., Gérard C., Perret J., Van Sande J., Dumont J. E., and Vassart G. (1989) Molecular cloning of the thyrotropin receptor. Science 246, 1620–1622 [DOI] [PubMed] [Google Scholar]
- 2.McFarland K. C., Sprengel R., Phillips H. S., Köhler M., Rosemblit N., Nikolics K., Segaloff D. L., and Seeburg P. H. (1989) Lutropin-choriogonadotropin receptor: an unusual member of the G protein-coupled receptor family. Science 245, 494–499 [DOI] [PubMed] [Google Scholar]
- 3.Sprengel R., Braun T., Nikolics K., Segaloff D. L., and Seeburg P. H. (1990) The testicular receptor for follicle stimulating hormone: structure and functional expression of cloned cDNA. Mol. Endocrinol. 4, 525–530 [DOI] [PubMed] [Google Scholar]
- 4.Fan Q. R., and Hendrickson W. A. (2005) Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sangkuhl K., Schulz A., Schultz G., and Schöneberg T. (2002) Structural requirements for mutational lutropin/choriogonadotropin receptor activation. J. Biol. Chem. 277, 47748–47755 [DOI] [PubMed] [Google Scholar]
- 6.Nakabayashi K., Matsumi H., Bhalla A., Bae J., Mosselman S., Hsu S. Y., and Hsueh A. J. (2002) Thyrostimulin, a heterodimer of two new human glycoprotein hormone subunits, activates the thyroid-stimulating hormone receptor. J. Clin. Invest. 109, 1445–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies T. F., Ando T., Lin R. Y., Tomer Y., and Latif R. (2005) Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J. Clin. Invest. 115, 1972–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vlaeminck-Guillem V., Ho S. C., Rodien P., Vassart G., and Costagliola S. (2002) Activation of the cAMP pathway by the TSH receptor involves switching of the ectodomain from a tethered inverse agonist to an agonist. Mol. Endocrinol. 16, 736–746 [DOI] [PubMed] [Google Scholar]
- 9.Sanders J., Chirgadze D. Y., Sanders P., Baker S., Sullivan A., Bhardwaja A., Bolton J., Reeve M., Nakatake N., Evans M., Richards T., Powell M., Miguel R. N., Blundell T. L., Furmaniak J., and Smith B. R. (2007) Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid 17, 395–410 [DOI] [PubMed] [Google Scholar]
- 10.Sanders P., Young S., Sanders J., Kabelis K., Baker S., Sullivan A., Evans M., Clark J., Wilmot J., Hu X., Roberts E., Powell M., Núñez Miguel R., Furmaniak J., and Rees Smith B. (2011) Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J. Mol. Endocrinol. 46, 81–99 [DOI] [PubMed] [Google Scholar]
- 11.Jiang X., Liu H., Chen X., Chen P. H., Fischer D., Sriraman V., Yu H. N., Arkinstall S., and He X. (2012) Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 12491–12496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Sande J., Massart C., Costagliola S., Allgeier A., Cetani F., Vassart G., and Dumont J. E. (1996) Specific activation of the thyrotropin receptor by trypsin. Mol. Cell. Endocrinol. 119, 161–168 [DOI] [PubMed] [Google Scholar]
- 13.Mizutori Y., Chen C. R., McLachlan S. M., and Rapoport B. (2008) The thyrotropin receptor hinge region is not simply a scaffold for the leucine-rich domain but contributes to ligand binding and signal transduction. Mol. Endocrinol. 22, 1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majumdar R., and Dighe R. R. (2012) The hinge region of human thyroid-stimulating hormone (TSH) receptor operates as a tunable switch between hormone binding and receptor activation. PLoS ONE 7, e40291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agrawal G., and Dighe R. R. (2009) Critical involvement of the hinge region of the follicle-stimulating hormone receptor in the activation of the receptor. J. Biol. Chem. 284, 2636–2647 [DOI] [PubMed] [Google Scholar]
- 16.Chen C. R., Chazenbalk G. D., McLachlan S. M., and Rapoport B. (2003) Targeted restoration of cleavage in a noncleaving thyrotropin receptor demonstrates that cleavage is insufficient to enhance ligand-independent activity. Endocrinology 144, 1324–1330 [DOI] [PubMed] [Google Scholar]
- 17.Zhang M., Tong K. P., Fremont V., Chen J., Narayan P., Puett D., Weintraub B. D., and Szkudlinski M. W. (2000) The extracellular domain suppresses constitutive activity of the transmembrane domain of the human TSH receptor: implications for hormone-receptor interaction and antagonist design. Endocrinology 141, 3514–3517 [DOI] [PubMed] [Google Scholar]
- 18.Krause G., Kreuchwig A., and Kleinau G. (2012) Extended and structurally supported insights into extracellular hormone binding, signal transduction and organization of the thyrotropin receptor. PLoS ONE 7, e52920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karges B., Gidenne S., Aumas C., Haddad F., Kelly P. A., Milgrom E., and de Roux N. (2005) Zero-length cross-linking reveals that tight interactions between the extracellular and transmembrane domains of the luteinizing hormone receptor persist during receptor activation. Mol. Endocrinol. 19, 2086–2098 [DOI] [PubMed] [Google Scholar]
- 20.Nurwakagari P., Breit A., Hess C., Salman-Livny H., Ben-Menahem D., and Gudermann T. (2007) A conformational contribution of the luteinizing hormone-receptor ectodomain to receptor activation. J. Mol. Endocrinol. 38, 259–275 [DOI] [PubMed] [Google Scholar]
- 21.Schulz A., Schöneberg T., Paschke R., Schultz G., and Gudermann T. (1999) Role of the third intracellular loop for the activation of gonadotropin receptors. Mol. Endocrinol. 13, 181–190 [DOI] [PubMed] [Google Scholar]
- 22.Biebermann H., Schöneberg T., Hess C., Germak J., Gudermann T., and Grüters A. (2001) The first activating TSH receptor mutation in transmembrane domain 1 identified in a family with nonautoimmune hyperthyroidism. J. Clin. Endocrinol. Metab. 86, 4429–4433 [DOI] [PubMed] [Google Scholar]
- 23.Schöneberg T., Schulz A., Biebermann H., Grüters A., Grimm T., Hübschmann K., Filler G., Gudermann T., and Schultz G. (1998) V2 vasopressin receptor dysfunction in nephrogenic diabetes insipidus caused by different molecular mechanisms. Hum. Mutat. 12, 196–205 [DOI] [PubMed] [Google Scholar]
- 24.Calebiro D., Nikolaev V. O., Gagliani M. C., de Filippis T., Dees C., Tacchetti C., Persani L., and Lohse M. J. (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLos Biol. 7, e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Börner S., Schwede F., Schlipp A., Berisha F., Calebiro D., Lohse M. J., and Nikolaev V. O. (2011) FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 6, 427–438 [DOI] [PubMed] [Google Scholar]
- 26.Rozen S., and Skaletsky H. (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386 [DOI] [PubMed] [Google Scholar]
- 27.Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 28.Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., and Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costanzi S. (2012) Homology modeling of class a G protein-coupled receptors. Methods Mol. Biol. 857, 259–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinau G., Hoyer I., Kreuchwig A., Haas A. K., Rutz C., Furkert J., Worth C. L., Krause G., and Schülein R. (2011) From molecular details of the interplay between transmembrane helices of the thyrotropin receptor to general aspects of signal transduction in family a G-protein-coupled receptors (GPCRs). J. Biol. Chem. 286, 25859–25871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinau G., Neumann S., Grüters A., Krude H., and Biebermann H. (2013) Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr. Rev. 34, 691–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleinau G., Claus M., Jaeschke H., Mueller S., Neumann S., Paschke R., and Krause G. (2007) Contacts between extracellular loop two and transmembrane helix six determine basal activity of the thyroid-stimulating hormone receptor. J. Biol. Chem. 282, 518–525 [DOI] [PubMed] [Google Scholar]
- 33.Worth C. L., Kleinau G., and Krause G. (2009) Comparative sequence and structural analyses of G-protein-coupled receptor crystal structures and implications for molecular models. PLoS ONE 4, e7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballesteros J. A., and Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 35.Alvarez C. A., Narayan P., Huang J., and Puett D. (1999) Characterization of a region of the lutropin receptor extracellular domain near transmembrane helix 1 that is important in ligand-mediated signaling. Endocrinology 140, 1775–1782 [DOI] [PubMed] [Google Scholar]
- 36.Mueller S., Kleinau G., Jaeschke H., Neumann S., Krause G., and Paschke R. (2006) Significance of ectodomain cysteine boxes 2 and 3 for the activation mechanism of the thyroid-stimulating hormone receptor. J. Biol. Chem. 281, 31638–31646 [DOI] [PubMed] [Google Scholar]
- 37.Kleinau G., Jäschke H., Neumann S., Lättig J., Paschke R., and Krause G. (2004) Identification of a novel epitope in the thyroid-stimulating hormone receptor ectodomain acting as intramolecular signaling interface. J. Biol. Chem. 279, 51590–51600 [DOI] [PubMed] [Google Scholar]
- 38.Stavrou S. S., Zhu Y. S., Cai L. Q., Katz M. D., Herrera C., Defillo-Ricart M., and Imperato-McGinley J. (1998) A novel mutation of the human luteinizing hormone receptor in 46XY and 46XX sisters. J. Clin. Endocrinol. Metab. 83, 2091–2098 [DOI] [PubMed] [Google Scholar]
- 39.de Roux N., Misrahi M., Brauner R., Houang M., Carel J. C., Granier M., Le Bouc Y., Ghinea N., Boumedienne A., Toublanc J. E., and Milgrom E. (1996) Four families with loss of function mutations of the thyrotropin receptor. J. Clin. Endocrinol. Metab. 81, 4229–4235 [DOI] [PubMed] [Google Scholar]
- 40.Lemoine N. R., Mayall E. S., Jones T., Sheer D., McDermid S., Kendall-Taylor P., and Wynford-Thomas D. (1989) Characterisation of human thyroid epithelial cells immortalised in vitro by simian virus 40 DNA transfection. Br. J. Cancer 60, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmutzler C., and Köhrle J. (1998) Implications of the molecular characterization of the sodium-iodide symporter (NIS). Exp. Clin. Endocrinol. Diabetes 106, Suppl. 3, S1–S10 [DOI] [PubMed] [Google Scholar]
- 42.Dumont J. E., Lamy F., Roger P., and Maenhaut C. (1992) Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol. Rev. 72, 667–697 [DOI] [PubMed] [Google Scholar]
- 43.Grüters A., Schöneberg T., Biebermann H., Krude H., Krohn H. P., Dralle H., and Gudermann T. (1998) Severe congenital hyperthyroidism caused by a germ-line neo mutation in the extracellular portion of the thyrotropin receptor. J. Clin. Endocrinol. Metab. 83, 1431–1436 [DOI] [PubMed] [Google Scholar]
- 44.Kopp P., Muirhead S., Jourdain N., Gu W. X., Jameson J. L., and Rodd C. (1997) Congenital hyperthyroidism caused by a solitary toxic adenoma harboring a novel somatic mutation (serine281 → isoleucine) in the extracellular domain of the thyrotropin receptor. J. Clin. Invest. 100, 1634–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duprez L., Parma J., Costagliola S., Hermans J., Van Sande J., Dumont J. E., and Vassart G. (1997) Constitutive activation of the TSH receptor by spontaneous mutations affecting the N-terminal extracellular domain. FEBS Lett. 409, 469–474 [DOI] [PubMed] [Google Scholar]
- 46.Kleinau G., Kreuchwig A., Worth C. L., and Krause G. (2010) An interactive web-tool for molecular analyses links naturally occurring mutation data with three-dimensional structures of the rhodopsin-like glycoprotein hormone receptors. Hum. Mutat. 31, E1519–1525 [DOI] [PubMed] [Google Scholar]
- 47.Costagliola S., Bonomi M., Morgenthaler N. G., Van Durme J., Panneels V., Refetoff S., and Vassart G. (2004) Delineation of the discontinuous-conformational epitope of a monoclonal antibody displaying full in vitro and in vivo thyrotropin activity. Mol. Endocrinol. 18, 3020–3034 [DOI] [PubMed] [Google Scholar]
- 48.Smith B. R., Sanders J., and Furmaniak J. (2007) TSH receptor antibodies. Thyroid 17, 923–938 [DOI] [PubMed] [Google Scholar]
- 49.Sanders J., Jeffreys J., Depraetere H., Evans M., Richards T., Kiddie A., Brereton K., Premawardhana L. D., Chirgadze D. Y., Núñez Miguel R., Blundell T. L., Furmaniak J., and Rees Smith B. (2004) Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: sequence structure and function. Thyroid 14, 560–570 [DOI] [PubMed] [Google Scholar]
- 50.Nakabayashi K., Kudo M., Kobilka B., and Hsueh A. J. (2000) Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. J. Biol. Chem. 275, 30264–30271 [DOI] [PubMed] [Google Scholar]
- 51.Claus M., Jaeschke H., Kleinau G., Neumann S., Krause G., and Paschke R. (2005) A hydrophobic cluster in the center of the third extracellular loop is important for thyrotropin receptor signaling. Endocrinology 146, 5197–5203 [DOI] [PubMed] [Google Scholar]
- 52.Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., and Kobilka B. K. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho S. C., Van Sande J., Lefort A., Vassart G., and Costagliola S. (2001) Effects of mutations involving the highly conserved S281HCC motif in the extracellular domain of the thyrotropin (TSH) receptor on TSH binding and constitutive activity. Endocrinology 142, 2760–2767 [DOI] [PubMed] [Google Scholar]
- 54.Vu T. K., Hung D. T., Wheaton V. I., and Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 55.Liebscher I., Schön J., Petersen S. C., Fischer L., Auerbach N., Demberg L. M., Mogha A., Cöster M., Simon K. U., Rothemund S., Monk K. R., and Schöneberg T. (2014) A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 9, 2018–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heitman L. H., Oosterom J., Bonger K. M., Timmers C. M., Wiegerinck P. H., and Ijzerman A. P. (2008) [3H]Org 43553, the first low-molecular-weight agonistic and allosteric radioligand for the human luteinizing hormone receptor. Mol. Pharmacol. 73, 518–524 [DOI] [PubMed] [Google Scholar]
- 57.Neumann S., Huang W., Titus S., Krause G., Kleinau G., Alberobello A. T., Zheng W., Southall N. T., Inglese J., Austin C. P., Celi F. S., Gavrilova O., Thomas C. J., Raaka B. M., and Gershengorn M. C. (2009) Small-molecule agonists for the thyrotropin receptor stimulate thyroid function in human thyrocytes and mice. Proc. Natl. Acad. Sci. U.S.A. 106, 12471–12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neumann S., Nir E. A., Eliseeva E., Huang W., Marugan J., Xiao J., Dulcey A. E., and Gershengorn M. C. (2014) A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology 155, 310–314 [DOI] [PMC free article] [PubMed] [Google Scholar]