

Abstract
Recent studies reveal that multifunctional protein β-arrestin 2 (Arrb2) modulates cell apoptosis. Survival and various aspects of liver injury were investigated in WT and Arrb2 KO mice after bile duct ligation (BDL). We found that deficiency of Arrb2 enhances survival and attenuates hepatic injury and fibrosis. Following BDL, Arrb2-deficient mice as compared with WT controls displayed a significant reduction of hepatocyte apoptosis as demonstrated by the TUNEL assay. Following BDL, the levels of phospho-Akt and phospho-glycogen synthase kinase 3β (GSK3β) in the livers were significantly increased in Arrb2 KO compared with WT mice, although p-p38 increased in WT but not in Arrb2-deficient mice. Inhibition of GSK3β following BDL decreases hepatic apoptosis and decreased p-p38 in WT mice but not in Arrb2 KO mice. Activation of Fas receptor with Jo2 reduces phospho-Akt and increases apoptosis in WT cells and WT mice but not in Arrb2-deficient cells and Arrb2-deficient mice. Consistent with direct interaction of Arrb2 with and regulating Akt phosphorylation, the expression of a full-length or N terminus but not the C terminus of Arrb2 reduces Akt phosphorylation and coimmunoprecipates with Akt. These results reveal that the protective effect of deficiency of Arrb2 is due to loss of negative regulation of Akt due to BDL and decreased downstream GSK3β and p38 MAPK signaling pathways.
Keywords: Akt PKB, apoptosis, gene regulation, liver injury, signal transduction, β-arrestin 2, BDL
Introduction
Cholestasis is defined as an impairment of bile flow and is usually seen in certain human liver diseases such as primary biliary cirrhosis and primary sclerosing cholangitis (1, 2). With liver injury, hepatocytes and cholangiocytes are damaged as a consequence of the buildup of bile acids, toxic metabolites, reactive oxygen species, and cytokines that cause cell death, apoptosis, and fibrosis (1, 3). Some signaling pathways antagonize cell death following liver injury, thereby influencing the balance between pro- and anti-apoptotic signals. These survival pathways belong to the MAPK- and PI3K-signaling pathways. The treatment of cholestatic liver injury is currently limited (4), and the discovery of new mechanisms of cellular injury may provide the opportunity to develop novel therapeutic strategies.
Increasing evidence reveals that β-arrestin 2 (Arrb2),3 a multifunctional adaptor, may have either a pro- or anti-apoptotic effect (5–8). The most thoroughly studied Arrb-dependent signaling system is ERK1/2 activation (8–10). The importance of Arrb2 in liver injury, where it is abundantly expressed, is not known. It has been shown that Arrb2 modulates activation of serine/threonine kinase Akt (11, 12). Akt modulates cell survival and apoptosis (13–16). Activated Akt in turn phosphorylates a variety of proteins involved in survival and apoptotic pathways leading to diminished cell apoptosis (14, 17, 18). Akt is activated following bile duct ligation (BDL), and inhibition of the activation markedly increases liver injury indicating that Akt activation is a survival factor after liver injury (19). Activated Akt phosphorylates several downstream targets of the PI3K pathway, including glycogen synthase kinase 3β (GSK3β) (20, 21). GSK3β is a constitutively active enzyme that is inactivated by Akt through phosphorylation of serine 9 (20, 21). GSK3β is a crucial regulator of cell survival and apoptosis and has recently been shown to be involved in liver ischemia reperfusion injury (20, 22). Accumulating evidence indicates that GSK3 modulates key steps in each of the two major apoptotic signaling pathways (the mitochondrial intrinsic apoptotic and the death receptor-mediated extrinsic apoptotic signaling pathways).
The overall objective of this study was to investigate the role of Arrb2 in cholestatic liver injury by using a loss of function in vivo model. We defined whether a deficiency of Arrb2 affects overall survival, liver injury, and Arrb2-mediated signaling pathways using the BDL mouse as an established model of extrahepatic cholestasis. We found that deficiency of Arrb2 significantly enhances animal survival and reduced hepatic fibrosis and hepatocyte apoptosis. The protective effect of deficiency of Arrb2 was associated with increased phospho-Akt and inactivation of GSK3β, p38 MAPK, and Fas signaling pathways and inhibition of Fas-induced apoptosis. Together, these data suggest that Arrb2 plays a critical role in obstructive cholestasis and significantly contributes to cholestatic liver injury and fibrosis.
Experimental Procedures
Animals and BDL Mice
Arrb2 knock-out mice (Arrb2 KO) on a C57BL/6 background were kindly provided by Dr. Robert Lefkowitz and were bred at East Tennessee State University (7, 8). Wild type (WT) C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained at the Division of Laboratory Animal Resources at East Tennessee State University, a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All procedures were approved by the East Tennessee State University Committee on Animal Care. Male mice aged 6–8 weeks were used in all experiments. BDL was performed as described in our previous study (1). Briefly, mice were anesthetized by inhalation of 1–5% isoflurane driven by 100% oxygen flow with the use of EZ-Anesthesia system. Under deep anesthesia, the peritoneal cavity was opened and the common bile duct was double-ligated and cut between the ligatures (BDL mouse). Control animals underwent a sham operation that consisted of exposure but not ligation of the common bile duct. After the sham operation or BDL at different times, mice were euthanized to obtain livers.
In Situ Apoptosis Assay
The frozen liver sections from wild type and Arrb2 KO mice were harvested for terminal deoxynucleotidyltransferase biotin-dUTP nick end labeling (TUNEL) assay. Nucleosomal DNA fragmentation in cells was determined by the TUNEL assay using an in situ apoptosis detection kit according to the manufacturer's instructions. The number of apoptotic cells was counted in randomly selected fields to calculate the ratio of apoptotic cells and total 500 cells.
Histological and Immunohistochemical Analysis
Hematoxylin and eosin (H&E) was performed to assess general histology. Sirius red was used to evaluate liver fibrosis. Negative control sections were incubated with non-immune immunoglobulin under the same conditions. 3–3′-Diaminobenzidine chromogen solution was used for visualization by light microscopy. The tissue sections were then counterstained with hematoxylin for 3 min. Three slides from each block were evaluated with bright field microscopy.
Transfections
Arrb2 full-length GFP vector and control GFP vector were kindly provided by Dr. Gang Pei, Shanghai Institutes for Biological Sciences, China. WT mouse embryonic fibroblasts (MEFs) and Arrb2 KO MEFs were kindly provided by Dr. Robert Lefkowitz (Duke University Medical Center, Durham, NC). MEFs were seeded in culture plates 24 h before transfection. All transfection was performed using Lipofectamine 2000 reagent (Invitrogen) (7).
Generation of EGFP and Arrb2 Mutant Constructs
To generate Arrb2 with EGFP fusion expression constructs, the N terminus of the EGFP(1–180) and the C terminus of the Arrb2(188–418) DNA fragments were amplified by PCR using pIRES2-EGFP vector and full-length Arrb2 cDNA construct as template and then cloned into the pBluescript SK vector. After sequence confirmation, the EGFP-Arrb2 fragment was first subcloned into the pCDNA3 vector through HindIII and BamHI restriction sites, and the N- and C-terminal fragments of Arrb2 were subcloned into the EGFP fragment containing pCDNA3 vector by BamHI and EcoRI sites. Expression constructs were confirmed by sequencing.
Western Blots
Briefly, protein concentration was measured using the bicinchoninic acid kit (7). Equal amounts of protein were separated by 12% SDS-PAGE then transferred to a nitrocellulose membrane (Bio-Rad). The membrane was then incubated at room temperature in a blocking solution composed of 5% skim milk powder dissolved in 1×TBS for 1 h. The membrane was then incubated overnight at 4 °C with the blocking solution containing first antibody. The signals were detected with the ECL system (Amersham Biosciences). The signals were quantified by scanning densitometry and computer-assisted image analysis. The primary antibodies used in this study were Akt, GSK3β, p38, ERK1/2, phospho-Akt, phospho-GSK3β, phospho-p38, phospho-ERK1/2, and Arrb2 (Cell Signaling Technology, Beverly, MA). Antibody against GAPDH was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Coimmunoprecipitation (Co-IP)
Arrb2 KO MEFs were transfected with Arrb2 full-length GFP vector, C- or N-terminal truncated Arrb2 GFP vectors, or FLAG-Akt vector for 16 h. The cells were treated with serum deprivation (SD) for 6 h. Co-IP was performed with Pierce® coimmunoprecipitation kit (Pierce) as described by us (7). Briefly, cells were incubated with IP Lysis/Wash Buffer on ice for 5 min to obtain cell lysates. Anti-GFP antibody was immobilized using AminoLink Plus coupling resin. The antibody-coupled resin was incubated with cell lysates overnight at 4 °C. Protein complexes bound to the antibody were then eluted with elution buffer and analyzed by Western blot as described before.
Statistical Analysis
All data were represented as means ± S.E. The data were analyzed using one-way analysis of variance followed by Bonferroni tests to determine where differences existed among the groups. Differences were considered statistically significant for values of p < 0.05.
Results
Deficiency of Arrb2 Enhances Animal Survival Following BDL
We investigated the role of the multifunctional protein Arrb2 (7, 23, 24) in BDL-induced liver damage. Arrb2 KO mice and WT mice were subjected to BDL, and mortality was monitored for 5–30 days. As shown in Fig. 1A, BDL-induced mortality was significantly decreased in Arrb2 KO mice compared with WT mice. There were no deaths in sham control mice. BDL caused a significant reduction in the body weight of the WT mice compared with the Arrb2 KO mice (data not shown). The expression of Arrb2 in the liver was significantly enhanced after BDL as detected by Western blot (Fig. 1B).
FIGURE 1.

Arrb2 deficiency enhances animal survival after BDL. A, survival of wild type (WT) and Arrb2 knock-out (KO) mice (n = 25–30 per group) was observed from 5 to 30 days after BDL. *, p < 0.01, Arrb2 KO mice versus WT mice. B, livers were harvested from WT mice after 3 days of sham operation or BDL, and Arrb2 expression was determined by Western blot. *, p < 0.01 compared with WT sham control group.
Arrb2 Deficiency Attenuates Hepatic Apoptosis and Hepatic Fibrosis after BDL
To evaluate whether Arrb2 plays a role in BDL-induced hepatic apoptosis, we subjected WT mice and Arrb2 KO mice to BDL for different time periods. The percentage of liver area occupied with bile infarcts, which correlates with the degree of liver apoptosis (25), was determined by H&E staining. We observed that BDL-induced areas of bile infarcts in WT mice appeared more prominent compared with Arrb2 KO mice, and the areas increased in size over time (Fig. 2A). Furthermore, morphometric evaluation of the percentage of liver with bile infarcts showed that deficiency of Arrb2 significantly reduces BDL-induced bile infarct area compared with WT mice (Fig. 2B). The percentage of liver area occupied with trichrome-stained areas was less in Arrb2 KO mice compared with WT mice following BDL suggesting that the early fibrousness in the BDL model is reduced by Arrb2 deficiency (Fig. 2, C and D).
FIGURE 2.

Hepatocyte apoptosis is reduced in Arrb2 KO mice following BDL. Hepatic morphology was determined by H&E staining in WT mice and Arrb2 KO mice following sham operation or BDL at different times (A and B). A, areas of bile infarcts in WT mice are more prominent compared with Arrb2 KO mice (×400). B, morphometric evaluation of the percentage of liver area with bile infarcts in WT mice and Arrb2 KO mice by Image Pro Plus Version 6.0. *, p < 0.05, Arrb2 KO mice versus WT mice. Hepatic morphology was examined by sirius red staining in WT mice and Arrb2 KO mice at different time points after BDL for C and D. C, sirius red staining showed significantly decreased hepatic fibrosis (red) in Arrb2 KO mice compared with WT mice (×400). D, morphometric evaluation of percentage of liver with hepatic fibrosis using Image Pro Plus Version 6.0. *, p < 0.01, Arrb2 KO mice versus WT mice. E, 3 days after BDL in WT and Arrb2 KO mice, the mice were sacrificed to obtain the livers. Apoptotic cells (dark cells) were determined by TUNEL assay. Magnification ×40. The bar graph at right shows the percentage of TUNEL-positive cells. *, p < 0.01.
Arrb2 Loss of Function Reduces BDL-induced Apoptosis
Growing evidence indicates that Arrb2 plays a critical role in regulating apoptosis (6, 26), and liver cell apoptosis is an important mechanism in the liver injury following BDL. We therefore determined whether Arrb2 is involved in BDL-mediated apoptosis. We examined hepatocyte apoptosis in WT and Arrb2 KO mice following BDL using the TUNEL assay. Fig. 2E shows that a significant number of cells in the liver of WT mice were undergoing apoptosis, whereas only a few apoptotic cells were detected in the liver of Arrb2 KO mice following BDL.
Arrb2 Deficiency Increases the Levels of Phospho-Akt and Phospho-GSK3β in the Liver Following BDL
Recent evidence has shown that Arrbs modulate Akt signaling (11, 12), and Akt reduction is associated with hepatocyte apoptosis in BDL (19). To investigate whether Arrb2 regulates the activation of Akt signaling in the liver following BDL, we determined the levels of phospho-Akt. As shown in Fig. 3A, the following BDL levels of phospho-Akt in the liver were significantly increased in Arrb2 KO compared WT mice. GSK3β is an important downstream target from Akt (21). Phosphorylation of GSK3β on the inactivating residue serine 9 by Akt results in GSK3β inactivation (21, 28). The levels of phospho-GSK3β (Ser-9) in the liver were significantly higher in the Arrb2 KO than in the WT mice following BDL (Fig. 3B). These data suggest that Arrb2 deficiency enhances BDL activation of Akt and decreases (by increased phosphorylation) GSK3β activity. Another survival pathway, MAPK/ERK1/2 (29), was not altered by Arrb2 deficiency because ERK1/2 MAPK phosphorylation in both WT mice and Arrb2 KO mice was found to be similar (Fig. 3C).
FIGURE 3.

Inhibition of GSK3β reduces BDL-induced apoptosis. Three days after BDL, livers were harvested for A and B. A, levels of total and p-Akt were determined by Western blot. B, levels of total and p-GSK3β were examined as in A. *, p < 0.01. GSK3β inhibitor SB216763 (0.6 mg/kg body weight, i.v.) or vehicle (10% dimethyl sulfoxide) was administrated 30 min before BDL and then administrated every 24 h until mice were euthanized. On day 3, livers were harvested for C and D. C, total and p-ERK1/2 levels were determined by Western blot. D, apoptotic cells (dark cells) were determined by TUNEL assay. Magnification ×40. The bar graph at right shows the percentage of apoptotic cells. *, p < 0.01.
GSK3β Inhibition Decreases BDL-induced Hepatic Apoptosis
To determine whether the observed decreased GSK3β activity due to Arrb2 deficiency is responsible for decreased liver injury following BDL, we administered in vivo the pharmacological GSK3β inhibitor SB216763 that has been widely used to study the role of GSK3β both in vitro and in vivo (28, 30). We observed that inhibition of GSK3β with SB216763 resulted in significant decreases in hepatic apoptosis in WT mice in response to BDL. However, inhibition of GSK3β did not alter hepatic apoptosis in Arrb2 KO mice following BDL (Fig. 3D), a finding consistent with GSK3β participating in series but not in parallel to Arrb2 on the inhibition of hepatic apoptosis.
Effect of GSK3β Inhibition on the Levels of Phospho-p38 MAPK Following BDL
Recently, it has been reported that GSK3β modulates p38 MAPK signaling (31). Activation of p38 induces hepatocyte apoptosis in models of cholestasis (33). We evaluated whether GSK3β plays a role in BDL-mediated phosphorylation of p38 MAPK. Both BDL and sham-treated WT mice and Arrb2 KO mice were administered GSK3β inhibitor SB216763. BDL significantly increased the levels of phospho-p38 MAPK in WT mice but did not alter the levels of phospho-p38 MAPK in Arrb2 KO mice compared with the sham control mice (Fig. 4). The ratio of phospho-p38 to total p38 was 3-fold higher in sham Arrb2 KO mice compared with WT sham mice. Interestingly, inhibition of GSK3β in WT mice decreased the levels of phospho-p38 MAPK induced by BDL. However, inhibition of GSK3β in Arrb2 KO mice did not change the levels of phospho-p38 MAPK perhaps due to GSK3β already being completely inactivated (phosphorylated) in Arrb2 KO mice (Fig. 4). These data suggest that Arrb2 plays a role in GSK3β-mediated activation of p38 MAPK following BDL.
FIGURE 4.
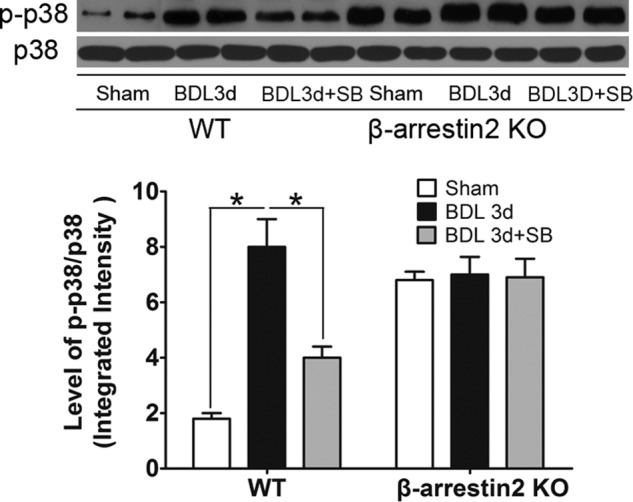
Effects of GSK3β on levels of p38 MAPK following BDL. GSK3β inhibitor SB216763 (0.6 mg/kg body weight, i.v.) or vehicle (10% dimethyl sulfoxide) was administrated 30 min before sham operation or BDL and then administered every 24 h until mice were euthanized. On day 3, mice were euthanized to obtain the livers. The levels of total and p-p38 MAPK were determined by Western blot. *, p < 0.01.
Arrb2 N Terminus but Not the C Terminus Directly Inactivates Akt
Previous studies have shown Arrb2 directly binds to Akt and alters Akt function, either up-regulating or down-regulating, depending on the signaling source. Previous studies have not disclosed the site where Arrb2 binds to Akt (33). We created both full-length and N- or C-terminal Arrb2 expression vectors, and we determined whether these will alter the activation of Akt. MEF cells lacking Arrb2 were serum-deprived (SD) for 6 h to decrease Akt, and then Arrb2 expression was induced by the above expression vectors (Fig. 5A). There was similar transfection of Arrb2 in all transfection studies (data not shown). SD deceased p-Akt but not total Akt in cells expressing full-length and N-terminal Arrb2 expression vectors but did not alter p-Akt in cells expressing empty or C-terminal Arrb2 expression vectors (Fig. 5A), suggesting that the N terminus but not the C terminus of Arrb2 is responsible for dephosphorylation of Akt. The potential interaction between the full-length or N or C terminus of Arrb2 and Akt was assessed by coimmunoprecipitation. Arrb2 KO MEF cells were transfected with Arrb2 full-length GFP or FLAG-tagged Akt plasmids following SD for 6 h. As shown in Fig. 5B, full-length Arrb2 coimmunoprecipitated with Akt. We then transfected Arrb2 KO MEF cells with GFP-tagged C- or N-terminal Arrb2 and FLAG-tagged Akt plasmids following SD for 6 h. Notably, the N-terminal Arrb2 but not the C-terminal Arrb2 coimmunoprecipitated with Akt (Fig. 5C). Taken together, these results suggest that the interaction of the N-terminal Arrb2 with Akt promotes Akt dephosphorylation.
FIGURE 5.

Arrb2 C terminus but not N terminus directly inactivates Akt. A, Arrb2 KO MEFs were transfected with Arrb2 full-length (FL) GFP vector or N- or C-terminal truncated Arrb2 GFP vectors or their GFP control vector. After 16 h of transfection, the cells were treated with SD for 6 h. The levels of total Akt, p-Akt, and GFP were determined by immunoblot. *, p < 0.05; **, p < 0.01. B, we transfected Arrb2 KO MEFs with Arrb2 full-length GFP vector or FLAG-Akt vector for 16 h. The cells were treated with SD for 6 h. Cell extracts were immunoprecipitated (IP)with anti-FLAG antibody and then analyzed together with cell lysates by immunoblotting (IB) with indicated primary antibodies. C, Arrb2 KO MEFs were transfected with C- or N-terminal truncated Arrb2 GFP vectors or FLAG-Akt vector. After 16 h of transfection, the cells were treated with SD for 6 h. Immunoprecipitates for Arrb2 C or N terminus and Akt were determined as in B. All of the results are representatives of three independent experiments.
Arrb2 Deficiency Inhibits Fas-induced Hepatocyte Apoptosis and Fas-reduced Phospho-Akt
Previous studies have shown that activation of Fas receptor is required for hepatocyte apoptosis following BDL (34). We determined whether Arrb2 deficiency prevents Fas-induced hepatocyte apoptosis by treating WT mice and Arrb2 KO mice with anti-Fas antibody Jo2 that activates the Fas receptor. We found that administration of Jo2 significantly increased the percentage of apoptotic cells in the livers in WT mice but not Arrb2 KO mice (Fig. 6A). Furthermore, we also found that treatment with Jo2 dramatically attenuated the levels of phospho-Akt in WT mice but not in Arrb2 KO mice (Fig. 6B). Therefore, these data reveal that Arrb2 deficiency reduces Fas-dependent increase in apoptosis and reduction of phospho-Akt.
FIGURE 6.

Deficiency of Arrb2 inhibits hepatocyte apoptosis through Fas. A, WT and Arrb2 KO mice were administered Jo2 at two different doses (0.15 and 0.3 μg/g body weight, i.p.). After 6 h of treatment, livers were harvested and fixed in 10% buffered formalin. Apoptotic cells (dark cells) were examined by TUNEL assay. **, p < 0.01; ***, p < 0.001. B, mice were injected as in A. At 6 h of injection, the expression of total Akt and p-Akt in livers was examined by immunoblot. C, WT and Arrb2 KO MEFs were transfected with Arrb2 full-length vector or control (Con) vector. After transfection for 24 h, the cells were treated with Jo2 (200 ng/ml), and the levels of total Akt and p-Akt were measured by Western blot. *, p < 0.01. D, WT, Arrb1 KO, Arrb2 KO, and Arrb1/2 double KO MEFs were treated with Jo2 (200 ng/ml) for 24 h. Apoptosis was determined by TUNEL assay. *, p < 0.05; **, p < 0.01.
Reduction of Phospho-Akt Due to Activation of Fas Receptor Requires Arrb2
To determine whether Arrb2 is required for inhibition of phospho-Akt following Fas activation, WT MEFs and Arrb2-deficient MEFs were transfected with Arrb2 full-length plasmid or control plasmid and then were incubated with or without Jo2, which activates the Fas receptor. As shown in Fig. 6C, Jo2 treatment decreases p-Akt but not total Akt in WT cells. In contrast, in Arrb2-deficient cells, Jo2 did not decrease p-Akt levels. Consistent with the effect of Arrb2 on p-Akt, which is an anti-apoptotic protein, the Jo2-dependent increase in TUNEL-positive MEF cells was markedly reduced in Arrb1 or -2 and in Arrb1- and -2-deficient cells compared with WT cells (Fig. 6D).
Discussion
Previous studies have shown that either Arrb1 or Arrb2 KO mice appear healthy and function normally unless challenged (24). This study specifically addressed the role of Arrb2 in the liver injury due to BDL. Our results demonstrate that Arrb2 deficiency enhanced animal survival, decreased hepatocyte apoptosis and liver fibrosis, increased Akt, inactivated GSK3β, and reduced p38 MAPK signaling, the latter being previously demonstrated to be required for cholestatic liver injury (32). Administration of a GSK3β inhibitor to BDL mice reproduced the effect of Arrb2 KO on liver cell apoptosis, Akt activation, and p38 inactivation. In vitro, activation of the Fas receptor, previously shown to be required for apoptosis in BDL (34) in the presence but not absence of Arrb2, decreases Akt and increases apoptosis. Finally, we demonstrate that the Arrb2-dependent decrease in Akt phosphorylation requires the interaction of the N terminus but not the C terminus of Arrb2. The results raise the likelihood of a novel role of Arrb2 in the modulation of liver injury.
The scaffolding proteins Arrb1 and Arrb2 have been conventionally associated with receptor internalization. However, new roles of Arrbs in receptor trafficking and signal transduction have been discovered in recent years. Arrbs can scaffold different sets of molecules that determine different and even opposite effects on the same signaling cascade dependent on the receptor activated (24). In the case of Akt, which we focused on in these studies, previous studies by Beaulieu et al. (12) have shown Arrb2 can deactivate Akt by the formation of an Arrb-Akt complex upon stimulation of the D2-class dopamine receptors. Our studies show that in the liver injury model, the presence of Arrb2 similarly reduces p-Akt, which is associated with increased liver injury. Both in vitro and in vivo studies where we decreased phosphorylated Akt levels employing activation of Fas receptor by Jo2 antibody showed that Arrb2 is required for Fas receptor-dependent decreases in Akt phosphorylation and apoptosis. Other studies by Povsic et al. (11) have also shown that Arrbs have opposite effects, with insulin-like growth factor 1 activation of PI3K and Akt, which cause inhibition of apoptosis. Our studies did not determine the specific membrane receptors that are involved in the modulation of Arrb2 inactivation of Akt. This is a reasonable omission because previous studies have shown a large number of G-protein-coupled receptors to be involved in cholestatic liver injury (4); nevertheless, reductions of Akt due to Fas is one that we know is very relevant to cholestatic liver injury (19). We reveal for the first time that dephosphorylation of Akt by Arrb2 requires the N terminus of Arrb2, and we demonstrated direct protein-protein interactions between Akt and Arrb2 by co-immunoprecipitation studies and that this interaction also depends on the N terminus of Arrb2. Additional studies will be needed to demonstrate the specific subdomains where Akt and Arrb2 interact, because this additional information may allow the development of more specific inhibitors. Although drugs that nonspecifically disrupt Akt/Arrb2 scaffolding (e.g. lithium) may have therapeutic effects in the brain, the potential for these classes of drugs to modify liver injury has not been explored.
In our studies we found that the degree of Akt phosphorylation was inversely related to GSK3β activity. GSK3β is an important downstream target from Akt (21). Phosphorylation of GSK3β on the inactivating residue serine 9 by Akt results in GSK3β inactivation (21, 28). Previous studies have shown a reduction of GSK3β is associated with reduced liver injury in BDL mice (27). We extended these previous observations by demonstrating that GSK3β inhibition reduces liver injury due to BDL. The GSK3β inhibitor SB216763 has been widely used to study the role of GSK3β in vivo, and our studies showed it significantly reduced both liver cell apoptosis and phosphorylation of p38. In models of cholestatic liver cell injury, previous studies showed that the only MAPK to increase liver cell apoptosis was p38 (32). Although the GSK3β inhibitor did not decrease p38 phosphorylation in Arrb2 KO mice with BDL, we postulate that deficiency reduces p38 phosphorylation to a level that cannot be further decreased. Baseline p-p38 was increased in sham Arrb2 KO mice compared with WT mice, which is likely a phenotype of the KO. Because the increased baseline p-p38 was not associated with liver injury in Arrb2 KO mice, it is likely other counterbalancing influences come into play to prevent p38-dependent liver injury. Previous studies have shown that GSK3β regulates the stability of Arrb2 complex formations. GSK3 inhibitors or heterozygosity for GSK3β expression reduces Arrb2-Akt complex formation and increases Akt activity (35). Consistent with GSK3β regulating the Arrb2-Akt complex in liver injury is our finding of the ability of the GSK3β inhibitor to reproduce the effect of Arrb2 KO on reduction of liver injury and decreased p38 phosphorylation. Further studies are needed to determine whether GSK3β regulates Arrb2-Akt complexes, and their regulation of cholestatic liver injury.
In summary, this study is the first to elucidate the molecular mechanisms for Arrb2 regulation of hepatocyte apoptosis induced by BDL. Activation of Akt and inhibition of GSK3β, which are both negatively regulated by Arrb2, promote hepatocyte survival and inhibit hepatocyte apoptosis in Arrb2 KO mice. These findings suggest that targeted inhibition of Arrb2 might provide a novel therapeutic approach to prevent liver injury.
Author Contributions
X. Y., H. L., X. Z., Y. F., D. H., Y. C., and N. X. performed the experiments. D. Y., H. F., C. S., Z. L., and G. L. supervised experiments. X. Y., D. Y., and G. L. wrote the manuscript.
Acknowledgments
We thank Dr. Robert Lefkowitz, Duke University Medical School (Durham, NC), for providing Arrb2 KO mice and WT MEFs, Arrb1 KO, Arrb2 KO, and Arrb1/2 double KO MEFs. We also thank Dr. Gang Pei, Shanghai Institutes for Biological Sciences, China, for GFP Arrb2 full-length vector and control vector.
This work was supported in part by National Institutes of Health Grant RO1 DK54208 from NIDDK (to G. L.), Grant 020120 from NIDA, Grants 094740 and 114716 from NIGMS (to D. Y.), and Grant C06RR0306551 and by National Natural Science Foundation of China Grants 81570454 and 81370433. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Arrb2
- β-Arrestin 2
- BDL
- bile duct ligation
- GSK3β
- glycogen synthase kinase 3β
- MEF
- mouse embryonic fibroblast
- EGFP
- enhanced GFP
- SD
- serum deprivation.
References
- 1.LeSage G. D., Alvaro D., Glaser S., Francis H., Marucci L., Roskams T., Phinizy J. L., Marzioni M., Benedetti A., Taffetani S., Barbaro B., Fava G., Ueno Y., and Alpini G. (2004) α-1 adrenergic receptor agonists modulate ductal secretion of BDL rats via Ca2+- and PKC-dependent stimulation of cAMP. Hepatology 40, 1116–1127 [DOI] [PubMed] [Google Scholar]
- 2.Osawa Y., Seki E., Adachi M., Suetsugu A., Ito H., Moriwaki H., Seishima M., and Nagaki M. (2010) Role of acid sphingomyelinase of Kupffer cells in cholestatic liver injury in mice. Hepatology 51, 237–245 [DOI] [PubMed] [Google Scholar]
- 3.Nalapareddy Pd., Schüngel S., Hong J. Y., Manns M. P., Jaeschke H., and Vogel A. (2009) The BH3-only protein bid does not mediate death-receptor-induced liver injury in obstructive cholestasis. Am. J. Pathol. 175, 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoemaker M. H., and Moshage H. (2004) Defying death: the hepatocyte's survival kit. Clin. Sci. 107, 13–25 [DOI] [PubMed] [Google Scholar]
- 5.Gao H., Sun Y., Wu Y., Luan B., Wang Y., Qu B., and Pei G. (2004) Identification of βarrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 14, 303–317 [DOI] [PubMed] [Google Scholar]
- 6.Wang P., Gao H., Ni Y., Wang B., Wu Y., Ji L., Qin L., Ma L., and Pei G. (2003) β-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J. Biol. Chem. 278, 6363–6370 [DOI] [PubMed] [Google Scholar]
- 7.Li H., Hu D., Fan H., Zhang Y., LeSage G. D., Caudle Y., Stuart C., Liu Z., and Yin D. (2014) β-Arrestin 2 negatively regulates toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10. J. Biol. Chem. 289, 23075–23085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowitz R. J., and Shenoy S. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 9.Buchanan F. G., and DuBois R. N. (2006) Emerging roles of β-arrestins. Cell Cycle 5, 2060–2063 [DOI] [PubMed] [Google Scholar]
- 10.DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., and Bunnett N. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Povsic T. J., Kohout T. A., and Lefkowitz R. J. (2003) β-Arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. J. Biol. Chem. 278, 51334–51339 [DOI] [PubMed] [Google Scholar]
- 12.Beaulieu J. M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., and Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 13.Fruman D. A., and Cantley L. C. (2002) Phosphoinositide 3-kinase in immunological systems. Semin. Immunol. 14, 7–18 [DOI] [PubMed] [Google Scholar]
- 14.Cantley L. C. (2002) The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y., Foster R., Sun X., Yin Q., Li Y., Hanley G., Stuart C., Gan Y., Li C., Zhang Z., and Yin D. (2008) Restraint stress induces lymphocyte reduction through p53 and PI3K/NF-κB pathways. J. Neuroimmunol. 200, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y., Feng Y., Kang J., Liu C., Li Z., Li D., Cao W., Qiu J., Guo Z., Bi E., Zang L., Lu C., Zhang J. Z., and Pei G. (2007) Critical regulation of CD4+ T cell survival and autoimmunity by β-arrestin 1. Nat. Immunol. 8, 817–824 [DOI] [PubMed] [Google Scholar]
- 17.Fukao T., and Koyasu S. (2003) PI3K and negative regulation of TLR signaling. Trends Immunol. 24, 358–363 [DOI] [PubMed] [Google Scholar]
- 18.Fukao T., Tanabe M., Terauchi Y., Ota T., Matsuda S., Asano T., Kadowaki T., Takeuchi T., and Koyasu S. (2002) PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881 [DOI] [PubMed] [Google Scholar]
- 19.Osawa Y., Hannun Y. A., Proia R. L., and Brenner D. A. (2005) Roles of AKT and sphingosine kinase in the antiapoptotic effects of bile duct ligation in mouse liver. Hepatology 42, 1320–1328 [DOI] [PubMed] [Google Scholar]
- 20.Jope R. S., and Johnson G. V. (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102 [DOI] [PubMed] [Google Scholar]
- 21.Martin M., Rehani K., Jope R. S., and Michalek S. M. (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren F., Duan Z., Cheng Q., Shen X., Gao F., Bai L., Liu J., Busuttil R. W., Kupiec-Weglinski J. W., and Zhai Y. (2011) Inhibition of glycogen synthase kinase 3β ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 54, 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefkowitz R. J. (1998) G protein-coupled receptors. III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 273, 18677–18680 [DOI] [PubMed] [Google Scholar]
- 24.Ma L., and Pei G. (2007) β-Arrestin signaling and regulation of transcription. J. Cell Sci. 120, 213–218 [DOI] [PubMed] [Google Scholar]
- 25.Kahraman A., Barreyro F. J., Bronk S. F., Werneburg N. W., Mott J. L., Akazawa Y., Masuoka H. C., Howe C. L., and Gores G. J. (2008) TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology 47, 1317–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moorman J., Zhang Y., Liu B., LeSage G., Chen Y., Stuart C., Prayther D., and Yin D. (2009) HIV-1 gp120 primes lymphocytes for opioid-induced, β-arrestin 2-dependent apoptosis. Biochim. Biophys. Acta 1793, 1366–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pi-Chieh Wang K., Lee L. M., Lin T. J., Sheen-Chen S. M., Lin J. W., Chiu W. T., Wang C. C., and Hung K. S. (2011) Gene transfer of IGF1 attenuates hepatocellular apoptosis after bile duct ligation. J. Surg. Res. 167, 237–244 [DOI] [PubMed] [Google Scholar]
- 28.Hu X., Paik P. K., Chen J., Yarilina A., Kockeritz L., Lu T. T., Woodgett J. R., and Ivashkiv L. B. (2006) IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 24, 563–574 [DOI] [PubMed] [Google Scholar]
- 29.Li J. G., Chen C., and Liu-Chen L. Y. (2002) Ezrin-radixin-moesin-binding phosphoprotein-50/Na+/H+ exchanger regulatory factor (EBP50/NHERF) blocks U50,488H-induced down-regulation of the human kappa opioid receptor by enhancing its recycling rate. J. Biol. Chem. 277, 27545–27552 [DOI] [PubMed] [Google Scholar]
- 30.Menon B., Johnson J. N., Ross R. S., Singh M., and Singh K. (2007) Glycogen synthase kinase-3β plays a pro-apoptotic role in β-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes: Role of β1 integrins. J. Mol. Cell. Cardiol. 42, 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manson M. M. (2003) Cancer prevention–the potential for diet to modulate molecular signalling. Trends Mol. Med. 9, 11–18 [DOI] [PubMed] [Google Scholar]
- 32.Grambihler A., Higuchi H., Bronk S. F., and Gores G. J. (2003) cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J. Biol. Chem. 278, 26831–26837 [DOI] [PubMed] [Google Scholar]
- 33.DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 34.Miyoshi H., Rust C., Roberts P. J., Burgart L. J., and Gores G. J. (1999) Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 117, 669–677 [DOI] [PubMed] [Google Scholar]
- 35.O'Brien W. T., Huang J., Buccafusca R., Garskof J., Valvezan A. J., Berry G. T., and Klein P. S. (2011) Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 121, 3756–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]