

Abstract
Breast cancer is a leading cause of morbidity and mortality among women. Metastasis is initiated after epithelial-mesenchymal-transition (EMT). We have found a connection between EMT markers and the expression of four microRNAs (miRs) mediated by the signaling enzyme phospholipase D (PLD). Low aggressive MCF-7 breast cancer cells have low endogenous PLD enzymatic activity and cell invasion, concomitant with high expression of miR-203, -887, and -3619 (that decrease PLD2 translation and a luciferase reporter) and miR-182 (targeting PLD1) that are, therefore, “tumor-suppressor-like” miRs. The combination miR-887+miR-3619 abolished >90% of PLD enzymatic activity. Conversely, post-EMT MDA-MB-231 cells have low miR expression, high levels of PLD1/2, and high aggressiveness. The latter was reversed by ectopically transfecting the miRs, which was negated by silencing miRs with specific siRNAs. We determined that the molecular mechanism is that E-cadherin triggers expression of the miRs in pre-EMT cells, whereas vimentin dampens expression of the miRs in post-EMT invasive cells. This novel work identifies for the first time a set of miRs that are activated by a major pre-EMT marker and deactivated by a post-EMT marker, boosting the transition from low invasion to high invasion, as mediated by the key phospholipid metabolism enzyme PLD.
Keywords: chemotaxis, microRNA (miRNA), phospholipid, phosphorylation, signal transduction
Introduction
Breast cancer is a leading cause of morbidity and mortality among women. It is estimated that 1 in 8 women in America will develop invasive breast cancer in their lifetime (1). The 5-year survival rate of breast cancer dramatically decreases from 100% in Stage I, non-metastatic disease, to a mere 22% in Stage IV, advanced metastatic disease (1). Metastasis is a complicated process involving detachment from the primary tumor, cellular invasion into surrounding tissue, entering and exiting the vascular system, and establishment of a novel tumor in a secondary site. Widely used breast cancer cell lines for research are MCF-7 and MDA-MB-231. The molecular classification of these widely used cancer cell lines is as follows. MCF-7 cells are anatomically luminal A/B with the phenotype ER+/PR+/−/HER−, and patients with these carcinomasare endocrine-responsive. MDA-MB-231 cells are anatomically claudin/E-cadherin-low with the phenotype ER−/PR−/HER−, and patients with these carcinomas are endocrine non-responsive.
Phospholipase D (PLD)2 is a vital enzyme in many tissue types, including healthy breast tissue, and its enzymatic products exert influence over an abundance of cellular processes, including cytoskeletal rearrangement, membrane trafficking, and cell migration (2). The role of PLD role in these signaling events also makes it integral to cancer cell metastasis (3–5). PLD itself plays an important role in maintaining the integrity of cellular membranes by hydrolyzing phosphatidylcholine (PC) producing phosphatidic acid, a critical secondary messenger in cellular processes. Furthermore, increased PLD protein expression and enzymatic activity have been reported in multiple cancer types, including breast cancer (5–8). However, the post-transcriptional regulation of PLD remains widely underexplored.
Here, we focus on microRNAs (miRs), small ∼22-nucleotide molecules of non-coding RNA that function to inhibit protein translation (9–11). After transcription and processing, microRNAs associate with the Argonaute (Ago) proteins and the RNA-induced silencing complex (RISC) (12–14). The active miR is then able to associate with the 3′-UTR of target mRNA molecules with partial complementarity (15). It is estimated more than half of the protein coding genes can be regulated by miRs, thus allowing miR regulation to affect most cellular processes (16). Expression profiles of various miR molecules have been shown to vary greatly in cancer when compared with healthy tissue (17–19). Therefore, it would seem inherent that miRs could play a major role in various aspects of cancer, and recent studies have found that expression profiles of several miR molecules varied greatly in cancer when compared with healthy tissue (17, 18). Furthermore, multiple miRs have been shown to play a role in breast cancer metastasis (18, 20).
Although it has previously been reported that microRNA-203 can inhibit the proliferation and invasion of U251 glioblastoma cells by directly targeting PLD2 (22), insights into the molecular mechanisms of how this is accomplished are still needed. Likewise, it is unlikely that only one miR will be enough to act on regulating mammalian isoforms PLD1 and PLD2, as it appears to be the norm that several miRs will cooperate to impede translation of a particular protein.
Knowing the importance of both PLD and miRs in healthy and cancer cells, we investigated if a novel regulation of PLD translation by miR molecules existed. We report for the first time a repertoire of such miRs that declined in invasive breast cancer cells compared with normal breast cells. We also report that as the formation of the EMT progressed and breast cancer cells became more mesenchymal, the miRs driven by EMT markers E-cadherin and vimentin were down-regulated, and thus, PLD protein expression and cell invasion were increased.
Materials and Methods
Reagents
miR mimics specific to PLD1 and PLD2 were Dharmacon products purchased from GE Healthcare. The miR specific to PLD1 was 182–5p (TargetScan Human accession number MIMAT0000259). The miRs specific to PLD2 were 203a, 3619–5p, 887–5p (accession numbers MIMAT0000264, MIMAT0017999, and MIMAT0004951 respectively). For specific experiments (as indicated in the text and figures) we used miR plasmids that were tagged to the GFP coding sequence. miExpress Precursor miRNA Expression Clones were from GeneCopoeia (Rockville, MD) and include these catalogue numbers: HmiR0249-M04-B, HmiR0957-MR04-B,and HmiR0512-MR04-B, which are specific for these miRs in PLD2: hsa-mir-203, hsa-mir-3619, and hsa-mir-887. Additionally, one Precursor miRNA Expression Clone for PLD1 was from GeneCopoeia, that of hsa-mir-182 (catalogue #HmiR0115). Transit2020 transfection reagent was from Mirus (Houston, TX).
Cell Culture and Transfection of Cells
MDA-MB-231, BT-474, and BT-549 human breast cancer cells were obtained from ATCC (Manassas, VA). MCF-7 cells were from Dr. Steven J. Berberich (Wright State University). MCF-7 and MDA-MB-231 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). BT-474 cells were cultured in Hybri-Care Media (ATCC) supplemented with 1.5 g/liter NaHCO3 + 10% FBS. BT-549 cells were cultured in RPMI 1640 media (ATCC) supplemented with 0.023 units/ml of insulin + 10% FBS. Cells were maintained at 37 °C in an incubator with a humidified atmosphere of 5% CO2. Cells were seeded in 6-well plates with an equal number of cells per well. Cells were then allowed to grow for 12–24 h before transfection. Plasmid transfection reactions included 1–2 μg of DNA plasmid and 1 μg of DNA/2-μl volume of Transit2020 transfection reagent in 200 μl of Opti-MEM serum-free medium. The above transfection reaction was incubated at room temperature for 20 min and was subsequently added to the appropriate well in 2 ml of complete media. Mimic transfection reactions included adequate Opti-MEM serum-free media, 50 nm concentrations of the appropriate miR mimic, and the appropriate volume of DharmaFECT 2 transfection reagent based on the cell line and number of cells seeded as recommended by the manufacturer. The above transfection reaction was incubated at room temperature for 20 min and was subsequently added to the appropriate well in 2 ml of complete media. Cell transfections were 36–48 h in duration.
SDS-PAGE and Western Blot Analyses
Samples of equal volume and equal protein amount were prepared with SDS buffer and boiled for 5 min. Samples were run on 4–20% acrylamide pre-cast gels purchased from ThermoScientific at 100 V for 1 h. Protein was transferred onto PVDF Transfer Membrane purchased from ThermoScientific at ∼420 mA for 1 h. Blots were incubated in primary antibody in 5% BSA in TBS-Tween buffer overnight at 4 °C. Blots were washed 3 times with TBS-T for 5 min each wash. Blots were subsequently incubated in the appropriate secondary antibody in 5% BSA in TBS-Tween buffer for 1 h. Blots were washed 3 times with TBS-Tween for 5 min each wash. ECL Western blotting detection reagents (GE Healthcare) were used for detection. Antibodies used include: Abgent PLD2 mouse monoclonal antibody (catalog no. AT3337a), Cell Signaling β-actin rabbit monoclonal antibody (catalog no. 4970S), Santa Cruz Biotechnology GFP rabbit polyclonal antibody (catalog no. sc-8334), Cell Signaling anti-rabbit IgG HRP-linked antibody (catalog no. 7074S), and Cell Signaling anti-mouse IgG HRP-linked antibody (catalog no. 7076S).
Luciferase Assay
Luciferase reporter assays were performed using the LightSwitch Luciferase Assay kit from Active Motif (catalog no. 32031). The reporter vectors contain a 3′-UTR region and a downstream RenSP luciferase region. Cells were co-transfected as previously described earlier herein with either a miR mimic or plasmid DNA and a LightSwitch 3′-UTR positive control vector (catalog no. 32022) or PLD2 3′-UTR vector (catalog no. 32011) in a 96-well plate for 36 h. LightSwitch Luciferase Assay Reagent was added to the wells, and the signal was read on a luminometer. The signal knockdown from the control 3′-UTR to the PLD2 3′-UTR for each miRNA was calculated and normalized to a negative control mimic.
Quantitative Polymerase Chain Reaction Assay
Cells that were transfected with the PLD miExpress Precursor miRNA Expression Clones were used for RNA lysates 48 h post-transfection using the Taqman microRNA Cells-to-CT kit according to the manufacturer's protocol (Life Technologies; catalog no. 4391848). RNA concentrations were determined using a NanoDrop spectrophotometer, and samples were normalized to ∼66 ng/μl RNA. Reverse transcription was performed in a 15-μl reaction volume with 1 μg of RNA, 1.5 μl of 10T Buffer (included in the Taqman microRNA Cells-to-Ct Kit purchased from Ambion), 1 mm dNTPs, 3.8 units of RNase inhibitor, and 1 μl of Multiscribe Reverse Transcriptase and incubated for 1 cycle at 16 °C for 30 min, 42 °C for 30 min, and then 85 °C for 5 min. Quantitative PCR reactions were run in a 20-μl reaction volume using 10 μl of Taqman Master Mix, ∼88 ng of total input RNA, and 1 μl of the relevant microRNA gene expression assay (6-carboxyfluorescein-labeled) multiplexed with the housekeeping gene (U6). Taqman miR primers and fluorescent probes were from Life Technologies. Quantitative PCR conditions for the Stratagene cycler were 95 °C for 10 min and then 40 cycles of 15 s at 95 °C and then 1 min at 60 °C. The cycle threshold Ct values were chosen from the linear part of the PCR amplification curve where an increase in fluorescence can be detected at >10 S.E. above the background signal. ΔCt was calculated as: ΔCt = average PLD Ct × average housekeeping Ct, and gene -fold expression was calculated as 2−(ΔΔCt) = 2−(experimental condition ΔCt − control ΔCt).
PLD Activity Assay
Cell lysates were processed for PLD activity in PC8 liposomes and n-[3H]butanol beginning with the addition of the following reagents (final concentrations): 3.5 mm PC8 phospholipid, 45 mm HEPES (pH 7.8), and 1.0 μCi of n-[3H]butanol in a liposome form to accomplish the transphosphatidylation reaction of PLD and were incubated for 20 min at 30 °C with continuous shaking. Reactions were stopped with the addition of 0.3 ml of ice-cold chloroform/methanol (1:2), and lipids were isolated and resolved by thin layer chromatography. The amount of [3H]phosphatidylbutanol ([3H]PBut) that co-migrated with phosphatidylbutanol (PBut) standards (Rf = 0.45–0.50) was measured by scintillation spectrometry.
Immunofluorescence Microscopy
COS-7 cells overexpressing GFP-tagged miRs were plated on sterile coverslips in a 6-well plate and transfected and cultured similar to MDA-MB-231 and MCF-7 cells as described above. Cells were fixed onto coverslips with 4% paraformaldehyde for at least 10 min. Cells were then permeabilized with 0.5% Triton X-100 in PBS (PBS-T) for 10 min. 10% FBS in PBS-T (IF blocking buffer) was used to block each coverslip for 4 h at room temperature. Coverslips were incubated in primary antibody (Santa Cruz 1:200 rabbit anti-PLD2 (H-133) IgG or Santa Cruz 1:200 mouse-anti-PLD1 (F-12) IgG in IF blocking buffer) for 1 h at room temperature. Subsequently, coverslips were thoroughly washed with PBS. The secondary antibody incubation was with Santa Cruz 1:200 goat-anti rabbit or (anti-mouse) TRITC IgG in IF blocking buffer for 1 h at 4 °C. Again, coverslips were extensively washed with PBS. Coverslips were then incubated with 1:2000 DAPI in PBS for 5 min and immediately washed thoroughly with PBS. Once sufficiently dry, coverslips were mounted onto glass slides with VectaShield Mounting Media. Cells were visualized using 100× objective of Nikon Eclipse immunofluorescence microscope. Images were obtained of green (EGFP), red (PLD2 or PLD1), and blue (nuclei) from multiple fields of cells. Infinity Analyze or Adobe Photoshop software was used to create overlay images.
Cell Invasion Assay
MDA-MB-231 human breast cancer cells were serum-starved for 2 h and resuspended at a concentration 1.5 × 106 cells/ml in chemotaxis buffer (DMEM + 0.5% bovine serum albumin). Next, ∼3 × 105 cells were applied to the upper chambers of 8-μm PET Matrigels (24-well format) with a 6.5-μm diameter membrane, and cells were allowed to invade for 6 h at 37 °C in a humidified 5% CO2 cell culture incubator. The final concentration of chemoattractant used was 0 or 30 nm EGF in 500 μl of chemotaxis buffer placed in the lower wells of 24-well plates. Cells were scraped from the Matrigel insert and then stained for 1 h with hematoxylin. Six separate fields of cells were counted for each invasion assay and expressed in terms of total number of invading cells ±S.E.
PLD-targeting miRs
Knowing the 3′-UTR sequences of both PLD isoforms, PLD1 and PLD2, we found five putative miRs that align with specific regions by using the TargetScanHuman bioinformatics analysis. Additionally, we found that the predicted binding site of these five microRNA to PLD mRNA is widely conserved among several mammalian species as shown in Table 1. We used several bioinformatic algorithms to determine the favorability of miRNA:mRNA binding pairs, which are also reported in Table 1. First we calculated the Gibbs free energy of the miRNA seed sequence:mRNA binding using mfold web server (The RNA Institute, University At Albany). Second, we reported mirSVR score as obtained from the miRanda database. Finally, we reported the weighted context ++ score as reported by TargetScanHuman and calculated according to Agarwal et al. (23). The mirSVR score and weighted context ++ score represent different algorithms in miRNA binding prediction that incorporate multiple miRNA:mRNA binding factors. In all, negative scores represent favorable binding, with greater favorability with increased score magnitude. We derived DNA plasmids from GeneCopoeia with the relevant miRs sequences cloned in or a scrambled negative control clone (catalog nos. HmiR0249-MR04-B, HmiR05120MR0–04-B, Hmi-R0957-MR04-B, and CmiR0001-MR04-B) as well as RNA mimics from GE Healthcare-Dharmacon (catalog nos. C-300605-05-0002, C-300557-07-0002, C-300562-03-0002, C-301245-01-0002, C-301515-00-0002, or CN-001000-01-05). Additionally, we cloned the 3′-UTR sequence of PLD2 downstream to a luciferase ORF and co-transfected mammalian cells with this construct and the putative miRs, looking for a potential decreased luciferase activity if the miR-3′-UTR match occurred in living cells.
TABLE 1.
Predicted binding sites of various microRNAs on PLD mRNA are conserved among species and are energetically favorable
The bold font represents 100% conservation of the PLD mRNA nucleotide sequence. miRNA sequences listed are the seed sequence of each miRNA, which represents the miRNA region that associates with mRNA. Species conservation information was taken from TargetScanHuman. ΔG values were calculated for the human binding pair of the miRNA seed sequence and corresponding PLD mRNA sequence using mform web server software (The RNA Institute, University at Albany). mirSVR scores are as reported in the miRanda-mirSVR database. A negative score denotes a favorable match with scores <−1.0 representing the top 7% of predictions. Data for miR-3619–5p were not yet available on miRanda-mirSVR at time of publication. Weighted context ++ scores were obtained from TargetScanHuman based on the algorithm described in Agarwal et al. (23). Again, a negative score denotes a favorable reaction with greater magnitude indicating higher favorability.
| Species | miR-182 | miR-203 | miR-887 | miR-3619 |
|---|---|---|---|---|
| 3′- … AGAUGGUAACGGUU | 3′- … GUAAAGU | 3′- … GGCAAGUG | 3′- … GGACGACU 3′- … GGACGACU | |
| Human | 5′- … UCUAGG--UUGCCAA … | 5′- … CAUUUCA … | 5′- … CCGUUCAA … | 5′- … 227CCUGCUGC … 5′- … 501CCUGCUGA … |
| Chimp | 5′- … UCUAGG--UUGCCAA … | 5′- … CAUUUCA … | 5′- … CCGUUCAA … | 5′- … 227CCUGCUGC … 5′- … 501CCUGCUGA … |
| Mouse | 5′- … UCCAUACAAGUUGCCAA … | 5′- … CAUUUCA … | 5′- … UGGUUCAU … | 5′- … 227CCUGUCAC … 5′- … 501CCUGCUGA … |
| Rat | 5′ … UCCGGG--UUGCCAAA … | 5′- … CAUUUCA … | 5′- … CGGUUCAC … | 5′- … 227CUUGUCAC … 5′- … 501CCUGCUGA … |
| Pig | 5′- … UCUACA--UUGCCAAA … | 5′- … CAAGCGUUUCA … | 5′- … CAGUUCAA … | 5′- … 227CUCGAUGGAUGC 5′- … 501GCUGCUAC … |
| ΔG (human) | −13.60 | −11.70 | −13.40 | −13.30–19.00 |
| mirSVR score | −0.4703 | −1.2145 | −1.3069 | Not available |
| Weighted context ++ score | −0.07 | −0.08 | −0.66 | −0.08–0.27 |
Statistics
Data are presented as the mean ± S.E. The difference between means was assessed by the single factor analysis of variance test. Probability of p < 0.05 indicated a significant difference.
Results
PLD Is Associated with Increased Invasiveness in Breast Cancer Cells
Due to its positive effect on cancer cell migration, we looked at the levels of PLD gene expression in three breast cell lines after increasing time of incubation with epidermal growth factor (EGF). Fig. 1A documents significantly increased PLD gene expression in the highly aggressive MDA-MB-231 cells compared with HMEC cells after EGF stimulation, suggesting a role for PLD in breast cancer cell invasion. To establish the importance of MCF-7 or MDA-MB-231 cancer cells to PLD-mediated processes as relates to metastasis, we performed cell invasion assays through Matrigel. As shown in Fig. 1B, both low-invasive MCF-7 and high-invasive MDA-MB-231 breast cancer cells increasingly migrated through the Matrigel matrix in a significant response to EGF as a function of time, whereas control HMEC cells did not invade to any great extent during the same timeframe. Fig. 1C shows actual photographs of representative fields for invaded cells (after 6 h) in Matrigel membranes.
FIGURE 1.

PLD is important for breast cancer cell invasion. A, PLD gene expression was measured by quantitative RT-PCR in three cell lines after stimulation with 30 nm EGF for 0, 3, and 6 h. B, cell invasion was measured in HMEC, MCF-7, and MDA-MB-231 cells for 3 and 6 h in response to 30 nm EGF as chemoattractant. Results are expressed in terms of the mean total number of cells invading the Matrigel insert ±S.E. for each time point. Cell invasion assays were performed in duplicate, and ∼six fields were counted per insert. C, actual photographs (20× magnification) of representative fields for invaded cells (after 6 h) in Matrigel. Green arrows point at round, empty circles that are the membrane pores. Blue arrows point at irregularly shaped, black spots, which are the stained cells that had invaded the Matrigel. D, enzymatic activity of endogenous PLD was measured in HMEC, MCF-7, and MDA-MB-231 cell lines. Results are expressed in terms of the mean % of total PLD activity compared with the mock control samples ±S.E. Duplicate sample were assayed with n = 3 total assays performed. E, endogenous protein expression of PLD1 and PLD2 was detected by Western blot (W.B.) analysis. β-Actin was used as a readout for equal protein loading control. F, both endogenous and overexpressed PLD enzymatic activity was measured in the presence of increasing amounts of PLD inhibitor, FIPI or NFOT. G, cell invasion was measured in MDA-MB-231 cells and overexpression of PLD2 and incubation with PLD inhibitor, FIPI or NFOT.
Additionally, cell lysates from MCF-7 and MDA-MB-231 breast cancer cells similarly stimulated with EGF for similar times were used for the PLD activity measurement. MDA-MB-231 cells had more PLD activity than that of MCF-7 cells, which was also greater than that of the control HMEC cells (Fig. 1D). The increased extent of both cell invasion and PLD activity in MDA-MB-231 cells was also correlated to the increased protein expression of both PLD1 and PLD2 in these cells compared with either the MCF-7 or HMEC cells (Fig. 1E), as detected using Western blot analyses of lysates used in the PLD activity assay. PLD enzymatic inhibitors (FIPI and NFOT) were used to assess the importance of active PLD in breast cancer cell invasion. Fig. 1F illustrates the inhibitory effect of FIPI and NFOT on PLD enzymatic activity, implying their reliability as PLD inhibitors. Furthermore, we saw a dramatic increase in cell invasion with PLD2 overexpression that was subsequently negated by the addition of PLD inhibitors (Fig. 1G).
Endogenous Gene Expression of Putative PLD Regulating miRs Is Down-regulated in Highly Aggressive Breast Cancer Cells
Other groups and our laboratory have established that PLD is involved in cancer cell invasion and metastasis (3, 4, 21, 22). Although it has previously been reported that microRNA-203 can inhibit the proliferation and invasion of U251 glioblastoma cells by directly targeting PLD2 (22), the study did not provide insights into the molecular mechanisms of how this is accomplished. Likewise, it is unlikely that only one miR will be enough to act on regulating PLD2, as it appears to be the norm that several miRs will cooperate to impeding translation of a particular protein. As shown in the schematic presented in Fig. 2, the human PLD2 gene is located on human chromosome 17. Knowing the 3′-UTR sequence of both PLD1 and PLD2, we found four putative microRNAs that target PLD by using bioinformatics scan software (TargetScanHuman), as indicated under “Materials and Methods.”
FIGURE 2.

Prediction of PLD associating microRNA molecule. Bioinformatic analysis was performed using TargetScanHuman or EBI Search (The European Bioinformatics Institute) and yielded regions where they could bind. Two miRs were predicted to have two targets on the PLD2–3′-UTR. Inset at bottom left, PLD1–3′-UTR.
The C-terminal 3′-UTR of PLD2 spans 568 bp in which potential alignment with miR-203, miR-887, and miR-3619 can be found. Note that there are two potential regions of alignment for miR-3619 to the PLD2 3′-UTR, whereas the remaining miRs tested herein (miR-203 and miR-887) have only one region of alignment with the PLD2 3′-UTR. We have found that in MCF-7 and MDA-MB-231 cells, the following repertoire of miRs targeted PLD2: miR-203, miR-887, miR-3619. Additionally, one miR targeted PLD1: 182.
We hypothesized that if one or several microRNA exist that could impair translation of an mRNA for PLD, thus acting as a tumor suppressor microRNA, then one or more of these potential PLD-specific miRs listed could be of interest to this study. Using miR primer/probe sets specific for miR-182, -203, -887, and -3619-5p, we determined the endogenous levels of expression of each miR in the two different human breast cancer cell lines, MCF-7 and MDA-MB-231, relative to the control cell line, HMEC. As shown in Fig. 3A, we found four miRs that had decreased endogenous miR expression in the highly invasive MDA-MB-231 cancer cells compared with the lowly invasive MCF-7 cells and have termed miR-182, -203, -887, and -3619 as tumor suppressor-like miRs. We also present data in Fig. 3, B–E, indicating the endogenous microRNA at normal expression levels and demonstrate that gene expression can indeed be detected in vivo (Fig. 3, B–E) in absolute terms (i.e. not related to the housekeeping gene U6, as in Fig. 3A).
FIGURE 3.

Endogenous and overexpressed gene expression of PLD targeting microRNA in breast cancer cells. A, quantitative RT-PCR was performed to determine the relative endogenous gene expression levels of certain miRs that were potential targets of PLD (miR-203, miR-887, miR-3619, and miR182) in HMEC, MCF-7, and MDA-MB-231 cells. Error bars represent results from three independent experiments performed in duplicate. The symbols * or # denote statistically significant (p < 0.05) increases or decreases, respectively, between samples and controls (HMEC cells). B–E, amplification plots of endogenous miR gene expression (miR-203, -887, -3619, and -182, respectively) in MCF-7 cells and MDA-MB-231 cells measured by quantitative RT-PCR. Each line is from triplicate samples of isolated RNA/back-transcribed to cDNA; blue shades are triplicates from MCF-7 cells; red shades are from MDA-MB-231 cells. F–I, gene expression of exogenous miRs plasmids after transfection (miR-203, miR-887, miR-3619, and miR-182, respectively) was measured by quantitative RT-PCR in MCF-7 and MDA-MB-231 cells. miR gene expression in mock-transfected cells was used as a control.
Overexpression of MicroRNAs Decreased PLD Protein Expression and Enzymatic Activity
The previous experiment measured endogenous expression of these particular miRs in these two breast cancer cell lines. Next, we used commercially available miR plasmids (full information is included under “Material and Methods”) specific for miR-182, -203, -887, and -3619 that contained the microRNA sequence that putatively targeted PLD1 or PLD2 to overexpress each miR in MCF-7 and MDA-MB-231 cells and performed quantitative PCR. We successfully overexpressed miR-containing plasmids in MCF7 and MDA-MB-231 cells (Fig. 3, F–I) as shown by an increase in gene expression of miR-203, -887, and -3619 (that potentially could target PLD2) and miR-182 (that could potentially target PLD1) in transfected cells compared with non-transfected cells. Overexpression of miR203 yielded a large increase in gene expression in MDA-MB-231 cells (Fig. 3F) due to the fact that its endogenous are extremely low compared with MCF7 cells (Fig. 3B).
Whether or not the effects of miR overexpression could be manifested on PLD1 or PLD2 protein expression or even PLD enzymatic activity remained to be seen. Therefore, to address these possibilities, we next overexpressed these miR plasmid DNAs in cells for 48 h and then prepared whole cell lysates that were then used for SDS-PAGE and subsequent Western blotting or for PLD enzymatic assays. As shown in Fig. 4, A and B, the PLD2-specific miRs each decreased PLD2 protein expression in MCF7 and MDA-MB-231 cells, respectively, which also yielded a dose-dependent negative effect on PLD2 protein levels (Fig. 4C).
FIGURE 4.

Effect of miRNA overexpression on PLD protein expression and activity. A–B, PLD2 protein expression in MCF-7 and MDA-MB-231 cells, respectively, in the presence of miR overexpression was detected by Western blot (W.B.). β-Actin was used as a control. C, PLD2 protein expression was decreased with increasing amounts of overexpressed microRNA-3619. β-Actin was used as a control. D, PLD enzymatic activity was measured in HMEC, MCF-7, and MDA-MB-231 cell lines in the presence of microRNA overexpression. Non-transfected cells were used as a control. The symbols * or # denote statistically significant (p < 0.05) increases or decreases, respectively, between samples and controls. E, effect of different combination of miRs on PLD enzymatic activity.
Additionally, overexpression of each of the four miRs tested significantly reduced PLD enzymatic activity in the highly invasive MDA-MB-231 cells compared with non-invasive/non-cancerous HMEC cells (Fig. 4D); a combination of miRs produced the maximum effect on enzymatic activity (Fig. 4E). These data indicate that miR overexpression can modulate PLD2 protein and enzyme activity in the highly invasive MDA-MB-231 cells versus the less invasive MCF-7 cells.
Next, we performed immunofluorescence microscopy on COS7 cells overexpressing miR plasmids (Fig. 5A) in which we used commercially available miR plasmids (full information is included under “Materials and Methods”). The reason we used miR plasmids is that they were obtained with a GFP coding sequence that we used as a proxy to detect the expression of miR in whole cells by fluorescence. The plasmids were specific for miR-182, -203, -887, and -3619 containing the microRNA sequence that putatively targeted PLD1 or PLD2 to overexpress each miR in MCF-7 and MDA-MB-231 cells and performed quantitative PCR.
FIGURE 5.

Overexpression of miR plasmids abrogated PLD protein expression in cells resulting in decreased cell invasion. A, cells were overexpressed with GFP-tagged miR plasmids. Immunofluorescence indicates decreased PLD protein expression in the presence of miR overexpression. Bars = 5 μm. B, Matrigel invasion assay showed increased cell invasion with PLD overexpression. Invasion returned to basal level with the addition of PLD-targeting miRNA. C, Matrigel invasion assay on cells previously transfected with siRNAs specific for the indicated miRs. D and E, overexpression of select miR plasmids or mimics, respectively, decreased cell invasion in MCF-7 and MDA-MB-231 cells. F, representative images of cell invasion assay showing a decrease in invaded cell in the presence of miR-203 using 10× magnification. In B–E, * and # represent significance (p < 0.05) by ANOVA over the respective controls, either increases (*) or decreases (#).
Breast Cancer Cells Adopted a Less Invasive Phenotype When Overexpressed with Tumor Suppressor-like miRs
The overall objective of this study was to investigate the role of miRs as putative factors responsible for down-regulating PLD in healthy breast cells, and the lack there of resulted in elevated PLD in highly aggressive cancer cells. Invasive properties of highly aggressive MDA-MB-231 cells were further increased by the overexpression of PLD1 and PLD2, which was then negated by overexpression of PLD isoform specific miR (Fig. 5B). miR-203 was chosen to represent PLD2-specific miRNAs due to its consistent inhibition of cell invasion shown in Fig. 5, D and E. Selective manipulation of gene expression of these miRs could be used to alter/control the severity of the invasive phenotype of these two human breast cancer cell lines, which is a function of both endogenous PLD enzymatic activity (Fig. 1C) and the ability of the cell to invade through tissue/appropriate matrix by ectopically transfecting particular miRs into each of the two different cell lines. We accomplished this objective by transfecting the tumor suppressor-like miRs into both the highly aggressive MDA-MB-231 cells and the less aggressive MCF-7 cells and measured the change in invasive properties of each cell line.
Specific siRNAs that target miRs were also used, and results are in Fig. 5C, indicating that cell invasion was enhanced over controls in cells that had been transfected with siRNAs. To further test for biological responses as read-outs for the corrected phenotypes, we assayed cancer cell invasion through Matrigel matrix. As shown in Fig. 5, D and E, overexpression of the tumor suppressor-like miR plasmids and mimics, respectively, in the more invasive MDA-MB-231 cells resulted in significantly reduced EGF-mediated cell invasion compared with the control cells, which phenocopied less invasive breast cancer. Representative images are shown in Fig. 5F. These data further implicate these tumor suppressor-like miRs and their regulation of PLD in the role of breast cancer metastasis.
Next, we used a GoClone reporter plasmid containing the 3′-UTR sequence of PLD2 or PLD1 downstream to a GFP ORF and microRNA mimics that putatively targeted PLD as a way to co-transfect breast cancer cells (Fig. 6A). These transfections with the 3′-UTR of PLD were under the control of a marine (Renilla) luciferase reporter gene (RenSP) that would allow for the production of luciferase after successful co-transfection and subsequent co-overexpression of the PLD 3′-UTR and the putative PLD-targeted miR mimics, which could then be measured using a luciferase-based fluorometric assay. We found that all three miRs tested significantly decreased luciferase activity in the presence of the PLD2–3′-UTR in COS7 cells (miR-203, miR-887, and miR-3619) compared with mock-transfected or negative control cells (Fig. 6B). This reinforced the concept above of the three miRs as tumor suppressor-like miRs, as PLD expression was significantly inhibited.
FIGURE 6.

MicroRNA gene expression responds to EMT markers. A, schematic depicting the setup of Renilla luciferase that was performed to assess miR binding to PLD2–3′-UTR. Normal cells were co-transfected with a reporter vector, containing the PLD2–3′-UTR region linked to luciferase, and the respective miR mimic. B, miR-203, -887, -3619, and -182 significantly decreased luminescence of the transfected cells, which represents inhibition of PLD2–3′-UTR reporter translation. C, summary of published data depicting over- or under-expression of pre- and post-EMT markers in MCF-7 and MDA-MB-231 cells. EGFR, epidermal growth factor receptor, D, Western blot analysis of pre- and post-EMT markers (E-cadherin and vimentin, respectively) in MCF-7 and MDA-MB-231 cells. E, transfection of different plasmid expressing EMT differentiation markers on miR expression. F and G, Western blot analysis shows protein expression silencing of E-cadherin in MCF-7 cells or vimentin in MDA-MB-231 cells, respectively. H and I, gene expression of miR-3619 and -182 were measured in the presence of silencing of EMT markers (E-cadherin and vimentin, respectively) by quantitative RT-PCR. The symbols * or # denote statistically significant (p < 0.05) increases or decreases, respectively, between samples and controls.
Molecular Mechanism: MicroRNA Gene Expression Respondsto EMT Markers
We have shown that PLD-targeting miR gene expression decreased in more metastatically invasive breast cancer cells (Fig. 3A), but we needed to demonstrate the molecular mechanism on how this is accomplished. Given the role of PLD on metastasis that our laboratory has reported (3), we hypothesized that this change was a function of the epithelial-mesenchymal transition. We used E-cadherin as a pre-EMT marker and vimentin as a post-EMT marker to investigate this hypothesis. The relative levels of EMT marker expression in MCF-7 and MDA-MB-231 were listed in Fig. 6C. We confirmed the protein expression of E-cadherin (pre-EMT) and vimentin (post-EMT) in MCF-7 and MDA-MB-231 cells (Fig. 6D, Western blots). Plasmids expressing EMT differentiation markers had effects on miR expression (Fig. 6E). Conversely, by silencing E-cadherin in MCF-7 cells, we were able to replicate the decreased miR-3619 gene expression characteristic of MDA-MB-231 cells (Fig. 6, F and G). Conversely, by silencing vimentin in MDA-MB-231 cells, we were able to increase miR-182 expression (Fig. 6, H and I). These data indicate PLD-targeting miR expression is regulated by EMT processes. The molecular mechanism is dependent on EMT markers as follows: E-cadherin depletion correlates with increased miR expression, and the opposite is true for vimentin depletion.
We are proposing a model (Fig. 7) where PLD protein expression is down-regulated by a repertoire of microRNAs in healthy breast cells. In cancerous breast cells expression of these miRs was suppressed as EMT progressed (particularly with the appearance of vimentin and the decrease of E-cadherin), leading to increased PLD protein production. This surplus of PLD protein subsequently facilitated cell invasion and thereby metastasis.
FIGURE 7.
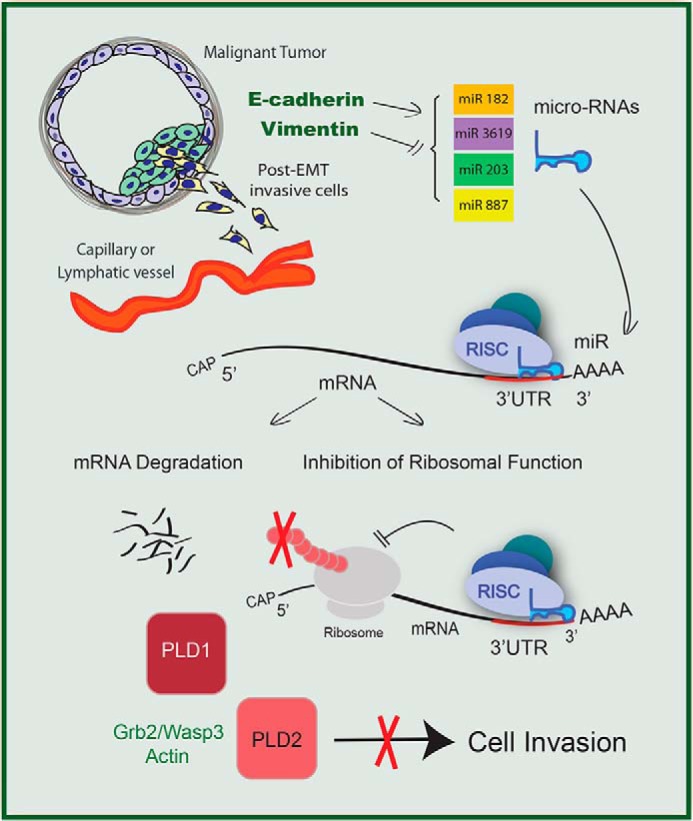
Proposed model of microRNA activity on PLD2–3′-UTR inhibiting cell invasion. MicroRNA molecules targeting PLD serve as tumor suppressor molecules by down-regulating PLD signaling pathways. The absence of these microRNA molecules in breast cancer cells correlates with increased cell invasion. RISC, RNA-induced silencing complex.
Discussion
We have found a connection between EMT markers and the expression of four miRs mediated by the signaling enzyme PLD. Low aggressive MCF-7 and BT-474 breast cancer cells have low endogenous PLD enzymatic activity and cell invasion concomitant with high expression of miR-203, -887, and -3619 (that decrease PLD2 translation and a luciferase reporter) and miR-182 (targeting PLD1). The combination of miRs abolished >90% PLD enzymatic activity. Conversely, post-EMT MDA-MB-231 and BT-549 cells have low levels of expression of the same miRs and high levels of PLD1/2 expression and high aggressiveness. The latter was reversed by ectopically transfecting the miRs, which was negated by silencing miRs with specific siRNAs. In this study we have also determined that the molecular mechanism is that E-cadherin triggers expression of the miRs in pre-EMT cells, whereas vimentin dampens expression of the miRs in post-EMT, invasive cells. This novel work identifies for the first time that a set of miRs that is activated by a major pre-EMT marker and deactivated by a post-EMT marker, boosts the transition from low invasion to high invasion, as mediated by the key phospholipid metabolism enzyme PLD.
The present data identify the mechanism of action of pre- and post-EMT drivers on breast cancer cell invasion, through a discrete set of micro-RNAs that directly regulate the lipid signaling enzyme PLD. Here we propose a model by which regulation of PLD protein expression was disrupted in highly aggressive MDA-MB-231 breast cancer cells. We have found four microRNAs that regulated either PLD1 or PLD2 mammalian isoforms by directly binding to the respective PLD-3′-UTR and thereby inhibiting protein translation. The tumor suppressor-like miRs had endogenously decreased levels of gene expression in highly aggressive MDA-MB-231 breast cancer cells compared with HMEC non-cancerous breast cells. This decrease in miR gene expression correlated with increased PLD protein expression. By overexpressing the tumor suppressor-like miRs, miR-203, -887, and -3619, targeting PLD2, and miR-182 targeting PLD1, we were able to decrease the respective PLD protein expression as well as PLD enzymatic activity in MCF-7 and MDA-MB-231 breast cancer cells. These experiments suggest a post-transcriptional regulation of PLD by these tumor suppressor-like miRs. Based on these data, we propose a mechanism of direct binding of these miRs to their respective PLD mRNA and subsequent inhibition of protein expression. Additionally, we propose this mechanism naturally occurs in non-cancerous HMEC breast cells, which is dysregulated in highly invasive MDA-MB-231 breast cancer cells.
Phospholipase D is a critical membrane protein at the center of complex signaling networks increasing both cell movement and cell proliferation. PLD acts through protein-protein interactions with signaling molecules including Grb2, Rac2, WASP, S6K, JAK3, etc. and also through the enzymatic production of a vital lipid secondary messenger, phosphatidic acid (3). PLD has been implicated in perpetuating cancer progression by serving as a cell survival signal and by facilitating invasion, the first step in metastasis (3–5). Elevated PLD protein expression and activity have been associated with more advanced cancer cell lines (5–8).
Furthermore, by overexpressing these miRs we confirmed the hypothesis that the tumor suppressor-like miR-203, miR-887, miR-3619, and miR-182 were able to diminish the invasively aggressive properties of MDA-MB-231 cells. We propose a mechanism for this whereby suppressing the PLD-mediated effect on cell invasion was accomplished through inhibition of PLD2 translation. As a consequence the well known effects of PLD on phosphatidic acid-mediated cell migration were inhibited as well as the formation of protein-protein interactions of PLD and proteins of the cell motility machinery (namely, Grb2, Wasp, Arp3, and actin). Additionally, by silencing these miRs, we were able to reverse their inhibitory effect on cell invasion. Thus, we report for the first time specific miRNAs that play a major role in breast cell cancer biological activity to explain the role of PLD.
We have found that as EMT progresses and breast cancer cells became more mesenchymal, a set of miRs specific for PLD is down-regulated and thus the PLD gene, and protein expression is increased. By silencing a pre-EMT marker protein (E-cadherin) we were able to diminish the gene expression of the tumor suppressor-like miR-3619 and miR-182. Conversely, we increased gene expression of these miRs by silencing the post-EMT marker vimentin. Therefore, we report that these tumor suppressor-like miRs were under regulatory control of the EMT status of the cell, and thus cells undergoing EMT prepared for invasion by accumulating PLD protein via down-regulation of these miRs.
Thus, we report a novel regulatory mechanism for the observed changes in PLD levels in MDA-MB-231 breast cancer cells via the vimentin-triggered silencing of miR-203, miR-887, miR-3619, and miR-182. The long term application of this study could be realized as a potential clinical application in assessing the efficacy of tumor suppressor-like miR-mediated down-regulation of PLD in combination with inhibitors of PLD activity directed toward inactivating the aggressive cell invasion phenotype.
Author Contributions
K. F. performed the experiments, analyzed the data, and edited the manuscript. J. G.-C. planned and directed this research Project, analyzed data, and composed the manuscript.
Acknowledgment
We thank Dr. Labib Rouhana (Department of Biological Sciences, Wright State University) for his assistance in creating Table 1 of miR free energies and also Karen M. Henkels for invaluable editorial contributions.
This work was supported, in whole or in part, by National Institutes of Health Grant HL056653-14 (to J. G.-C.) and American Heart Association Grant 13GRNT17230097. The authors declare that they have no conflicts of interest with the contents of this article.
- PLD
- phospholipase D
- PC
- phosphatidylcholine
- miR and miRNA
- microRNA
- EMT
- epithelial-to-mesenchymal transition
- TRITC
- tetramethylrhodamine isothiocyanate
- FIPI
- 5-fluoro-2-indolyl des-chlorohalopemide
- NFOT
- N-[2-[1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-8-yl]ethyl]-2-naphthalenecarboxamide.
References
- 1.DeSantis C. E., Fedewa S. A., Goding Sauer A., Kramer J. L., Smith R. A., Jemal A. (2015) Breast cancer statistics (2015): convergence of incidence rates between black and white women. CA Cancer J. Clin. 10.3322/caac.21320 [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Cambronero J. (2014) Phospholipase D in cell signaling: from a myriad of cell functions to cancer growth and metastasis. J. Biol. Chem. 289, 22557–22566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henkels K. M., Boivin G. P., Dudley E. S., Berberich S. J., and Gomez-Cambronero J. (2013) Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene 32, 5551–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knoepp S. M., Chahal M. S., Xie Y., Zhang Z., Brauner D. J., Hallman M. A., Robinson S. A., Han S., Imai M., Tomlinson S., and Meier K. E. (2008) Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol. Pharmacol. 74, 574–584 [DOI] [PubMed] [Google Scholar]
- 5.Uchida N., Okamura S., and Kuwano H. (1999) Phospholipase D activity in human gastric carcinoma. Anticancer Res. 19, 671–675 [PubMed] [Google Scholar]
- 6.Uchida N., Okamura S., Nagamachi Y., and Yamashita S. (1997) Increased phospholipase D activity in human breast cancer. J. Cancer Res. Clin. Oncol 123, 280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada Y., Hamajima N., Kato T., Iwata H., Yamamura Y., Shinoda M., Suyama M., Mitsudomi T., Tajima K., Kusakabe S., Yoshida H., Banno Y., Akao Y., Tanaka M., and Nozawa Y. (2003) Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J. Mol. Med. 81, 126–131 [DOI] [PubMed] [Google Scholar]
- 8.Levin W. J., Press M. F., Gaynor R. B., Sukhatme V. P., Boone T. C., Reissmann P. T., Figlin R. A., Holmes E. C., Souza L. M., and Slamon D. J. (1995) Expression patterns of immediate early transcription factors in human non-small cell lung cancer: The Lung Cancer Study Group. Oncogene 11, 1261–1269 [PubMed] [Google Scholar]
- 9.Carrington J. C., and Ambros V. (2003) Role of microRNAs in plant and animal development. Science 301, 336–338 [DOI] [PubMed] [Google Scholar]
- 10.Baehrecke E. H. (2003) MiRNAs: micro managers of programmed cell death. Curr. Biol. 13, R473–R475 [DOI] [PubMed] [Google Scholar]
- 11.Ambros V. (2001) MicroRNAs: tiny regulators with great potential. Cell 107, 823–826 [DOI] [PubMed] [Google Scholar]
- 12.Meister G., Landthaler M., Patkaniowska A., Dorsett Y., Teng G., and Tuschl T. (2004) Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 15, 185–197 [DOI] [PubMed] [Google Scholar]
- 13.Okamura K., Ishizuka A., Siomi H., and Siomi M. C. (2004) Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 18, 1655–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaucheret H., Vazquez F., Crété P., and Bartel D. P. (2004) The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 16.Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nygaard S., Jacobsen A., Lindow M., Eriksen J., Balslev E., Flyger H., Tolstrup N., Møller S., Krogh A., and Litman T. (2009) Identification and analysis of miRNAs in human breast cancer and teratoma samples using deep sequencing. BMC Med. Genomics 2, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang J., Ahmad A., and Sarkar F. H. (2012) The role of microRNAs in breast cancer migration, invasion and metastasis. Int. J. Mol. Sci. 13, 13414–13437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eroles P., Tormo E., Pineda B., Espin E., and Lluch A. (2015) MicroRNAs in breast cancer: one More Turn in Regulation. Curr. Drug Targets, in press [DOI] [PubMed] [Google Scholar]
- 20.Scott G. K., Goga A., Bhaumik D., Berger C. E., Sullivan C. S., and Benz C. C. (2007) Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J. Biol. Chem. 282, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 21.Henkels K. M., Farkaly T., Mahankali M., Segall J. E., and Gomez-Cambronero J. (2011) Cell invasion of highly metastatic MTLn3 cancer cells is dependent on phospholipase D2 (PLD2) and Janus kinase 3 (JAK3) J. Mol. Biol. 408, 850–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z., Li D., Cheng Q., Ma Z., Jiang B., Peng R., Chen R., Cao Y., and Wan X. (2014) MicroRNA-203 inhibits the proliferation and invasion of U251 glioblastoma cells by directly targeting PLD2. Mol. Med. Rep. 9, 503–508 [DOI] [PubMed] [Google Scholar]
- 23.Agarwal V., Bell G. W., Nam J. W., Bartel D. P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]