

Background: AS160 regulates insulin-sensitive glucose transport in adipocytes.
Results: Knockdown of the AS160 substrate Rab10 differentially affected Glut4 and LRP1 in adipocytes and fibroblasts.
Conclusion: Glut4 and LRP1 are sorted from a Rab10-independent constitutive trafficking pathway into a highly regulated Rab10-dependent pathway in adipocytes.
Significance: Rab10 is a marker for the specialized insulin-sensitive pathway in adipocytes and an AS160 substrate that limits exocytosis through this pathway.
Keywords: adipocyte, endocytosis, exocytosis, glucose, glucose transporter type 4 (GLUT4), insulin, LRP1, Rab10, as160/TBC1D4
Abstract
The RabGAP AS160/TBC1D4 controls exocytosis of the insulin-sensitive glucose transporter Glut4 in adipocytes. Glut4 is internalized and recycled through a highly regulated secretory pathway in these cells. Glut4 also cycles through a slow constitutive endosomal pathway distinct from the fast transferrin (Tf) receptor recycling pathway. This slow constitutive pathway is the only Glut4 cycling pathway in undifferentiated fibroblasts. The α2-macroglobulin receptor LRP1 cycles with Glut4 and the Tf receptor through all three exocytic pathways. To further characterize these pathways, the effects of knockdown of AS160 substrates on the trafficking kinetics of Glut4, LRP1, and the Tf receptor were measured in adipocytes and fibroblasts. Rab10 knockdown decreased cell surface Glut4 in insulin-stimulated adipocytes by 65%, but not in basal adipocytes or in fibroblasts. This decrease was due primarily to a 62% decrease in the rate constant of Glut4 exocytosis (kex), although Rab10 knockdown also caused a 1.4-fold increase in the rate constant of Glut4 endocytosis (ken). Rab10 knockdown in adipocytes also decreased cell surface LRP1 by 30% by decreasing kex 30–40%. There was no effect on LRP1 trafficking in fibroblasts or on Tf receptor trafficking in either cell type. These data confirm that Rab10 is an AS160 substrate that limits exocytosis through the highly insulin-responsive specialized secretory pathway in adipocytes. They further show that the slow constitutive endosomal (fibroblast) recycling pathway is Rab10-independent. Thus, Rab10 is a marker for the specialized pathway in adipocytes. Interestingly, mathematical modeling shows that Glut4 traffics predominantly through the specialized Rab10-dependent pathway both before and after insulin stimulation.
Introduction
In response to postprandial hyperglycemia, insulin increases glucose uptake into muscle and adipose tissue. Glucose uptake into muscle and fat is rate-limited by the number of facilitative glucose transporters present in the plasma membranes of cells. Insulin increases glucose uptake predominantly by causing a change in the steady-state distribution of glucose transporter 4 (Glut4), from intracellular compartments to the plasma membrane, where it can facilitate glucose diffusion into the cell (1–4).
Under basal conditions, less than 5% of the total cellular Glut4 in adipocytes is in the plasma membrane (2, 3). In this state, ∼10% of the Glut4 is found co-localized with endocytic markers, predominantly in the sorting tubules associated with “early” (cathepsin-negative) and “late” (cathepsin-positive) endosomes. The majority of the Glut4 (85–90%) is found in small, specialized tubulo-vesicular compartments known as Glut4 storage vesicles (GSVs),2 and in the trans-Golgi region. Consistent with this subcellular distribution, intracellular Glut4 exists in two kinetically distinct pools in basal cells. There is a small pool (10–20%) that is continuously exchanging with the plasma membrane with a rate constant of exocytosis (kex) consistent with endosomal recycling (kex = 0.03 min−1; t½ = 20 min)3 and a large pool (70–90%) that is in highly regulated, very slowly cycling compartments (kex = 0.0007 min−1; t½ >16 h) (5–11). After insulin stimulation, 20–40% of the Glut4 is present in the plasma membrane. Insulin increases surface Glut4 by at least two mechanisms as follows: 1) insulin decreases the amount of Glut4 in the GSVs and trans-Golgi region 2–3-fold (2, 3), and 2) insulin increases the overall kex of the cycling Glut4 5–6-fold (from ∼0.005 to ∼0.03 min−1) (5–10). Insulin has little effect on Glut4 endocytosis in adipocytes. How insulin exerts these effects on Glut4 exocytosis has not been fully elucidated.
A signal transduction pathway linking activation of the insulin receptor directly to proteins on GSVs has been described previously (12, 13). The activated insulin receptor tyrosine kinase phosphorylates downstream substrates, including IRS1. Phospho-IRS1 binds to and activates type I phosphatidylinositol 3-kinase (PI 3-kinase). PI 3-kinase generates phosphatidylinositol 3,4,5-trisphosphate, which recruits and activates kinases that in turn activate Akt (protein kinase B). Activations of PI 3-kinase and Akt are both necessary and largely sufficient for Glut4 translocation. Akt phosphorylates the Akt substrate of 160 kDa (AS160/TBC1D4), which localizes to Glut4 vesicles. When AS160 is phosphorylated by Akt in response to insulin, it is bound by 14-3-3, and it dissociates from the Glut4 vesicles. AS160 is an inhibitor of Glut4 exocytosis; phosphorylation and release of AS160 stimulates Glut4 exocytosis. However, the precise mechanism(s) of this inhibition are incompletely understood.
AS160 is a Rab GTPase-activating protein (Rab GAP), and this Rab GAP activity is required to inhibit Glut4 exocytosis in basal cells (14). Rab GAPs function by enhancing the intrinsic GTPase activity of Rabs, which are small G-proteins that regulate many steps in vesicular trafficking. Rabs are molecular switches that cycle between a GTP-bound form, which can bind downstream effector proteins, and a GDP-bound form, which cannot bind effectors. Thus, the likely function of AS160 on GSVs is to prevent the accumulation of the GTP-bound form of a Rab (or Rabs) required for efficient exocytosis of these compartments. In the basal state, this Rab protein would be maintained predominantly in the GDP-bound inactive form due to active AS160 on GSVs. After insulin stimulation, AS160 is phosphorylated, inactivated by 14-3-3 binding, and removed from GSVs, allowing the Rab to exchange GDP for GTP, form an active conformation, and bind effectors required for Glut4 translocation. Thus, phosphorylation of AS160 would allow efficient GTP loading of a Rab substrate required for the conversion of GSVs to a primed state that is competent to fuse to the plasma membrane. The fusion of these primed vesicles with the plasma membrane is regulated by insulin through an additional AS160-independent mechanism, possibly through direct regulation of Rab effector proteins (11, 15, 16).
Many Rabs are found on Glut4-containing vesicles, including Rab1–8, -10, -11, -14, -18, and -35 (17–19). Of these, AS160 has in vitro Rab GAP activity against Rab2, -8, -10, and -14 (18, 19). Depletion of Rab10 in adipocytes inhibits Glut4 translocation, whereas expression of a Rab10 mutant that is resistant to AS160 elevates basal cell surface Glut4 (20–23). Loss of Rab14 also attenuates insulin-stimulated Glut4 translocation in adipocytes, whereas expression of an AS160-resistant mutant acts as a dominant negative inhibitor (22–24). Likewise, depletion of Rab8 and -14 inhibits Glut4 translocation in muscle cells (25–27). These observations are consistent with Rab8, -10, or -14 being targets of AS160 that control the release of sequestered GSVs. However, Glut4 trafficking through the cell involves many Rab-dependent steps. In addition, Rab8, -10, and -14 have been implicated in the trafficking of proteins in many cell types, including undifferentiated fibroblasts, although accumulation of Glut4 in the highly regulated GSVs is seen only in fully differentiated adipocytes and muscle cells. Therefore, Rab8, -10, or -14 could affect Glut4 trafficking through steps other than the highly regulated rate-limiting step of GSV release/priming. Consistent with this, additional effects of AS160 have been observed at the plasma membrane and in endosomes, suggesting it may regulate multiple steps in Glut4 trafficking (10, 11, 16, 28, 29).
We have developed a series of highly reproducible, sensitive, and quantitative assays to measure Glut4 trafficking kinetics (8–11). These assays allow us to distinguish the effects of treatments on several different steps in Glut4 trafficking. We have also developed mathematical models that replicate the unique trafficking of Glut4, including accurately simulating all of the observed effects of AS160 knockdown. Using these models, we can make predictions about the kinetic phenotypes expected for Glut4 trafficking in adipocytes in which the AS160 Rab substrate(s) required for GSV priming has been knocked down. Fitting the observed kinetics data with these models allows us to test hypotheses about the specific site of action of the Rabs in the Glut4 trafficking pathway. Therefore, to determine which Rab proteins regulate GSV priming, the effects of Rab depletion on the trafficking kinetics of Glut4 were measured in both adipocytes and fibroblasts. The effects of knockdown of four AS160 substrates, Rab10, -14, -8a, and -8b were examined. We also examined the effects of knockdown on the trafficking of LRP1, an α2-macroglobulin (α2-M) receptor that traffics with Glut4 through GSVs (30), and the Tf receptor, an endosomal protein that does not traffic through GSVs. These phenotypes were compared with those predicted by our models. In this study, we describe our results in the Rab10 knockdown cells. The phenotype of the Rab10 knockdown cells is exactly the phenotype predicted for knockdown of the AS160-regulated Rab protein that controls GSV exocytosis. In a future paper, we will describe the results from the Rab14, -8a, and -8b knockdown cells.4 In contrast to Rab10, the phenotypes of the other Rab knockdown cells are inconsistent with a role in GSV exocytosis, supporting the idea that there are additional steps regulated by AS160 through its Rab substrates (8, 10, 11).
Experimental Procedures
Tissue Culture
3T3-L1 cells were obtained from the ATCC. Cells were passaged as fibroblasts, plated into 96-well plates, and differentiated into adipocytes as described previously (8). For experiments on fibroblasts, cells were plated into 96-well plates and used 3 days later after reaching confluence.
Viral Infections
3T3-L1 fibroblasts were infected with lentivirus encoding HA-Glut4/GFP as described previously (8). At the titers used, 70–80% of cells were infected and expressed the reporter protein; under these conditions, most of the infected cells will be infected with only one virion. The uninfected cells in each sample were used as internal controls to measure background fluorescence from nonspecific antibody labeling and autofluorescence. After recovery from lentiviral infection, cells were infected with retroviruses expressing shRNAs targeting Rabs (20, 21) or a control sequence not targeting any mouse gene (31) as described previously (11). These viruses contained the pSiren-RetroQ backbone and were gifts from Dr. Gustav Lienhard. After recovery from retroviral infection, stable cell lines were generated by selection in DMEM, 10% calf serum containing 2.5 μg/ml puromycin (Sigma) (11). All assays were repeated multiple times (see Table 1) on cells from at least three retroviral infections for each knockdown construct. Under the conditions used in our experiments, there were no observable cytopathic effects from infection with any of the viruses. Furthermore, all cell types responded as uniform populations in GFP and antibody staining (11).
TABLE 1.
Comparison of the observed concentration of surface Glut4 to the expected values calculated from the measured rate constants
Surface Glut4 (PMobs) was directly measured in surface binding experiments (Fig. 1). The rate constants of exocytosis (kex) and endocytosis (ken) and the size of the cycling pool (Ymax) were determined from anti-HA uptake and +LYi transition experiments (Figs. 2, A and B). The expected values for surface Glut4 (PMcalc) were calculated from the measured rate constants using the following equations: 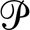 = (kex)/(ken); PMcalc cyc
= ()/(1 + ); PMcalc total
= ()/(1 + ) Ymax. The relative surface Glut4, PMrel = (PM)/(PMcontrol, insulin); n, number of independent experiments; *, p value = 0.01 relative to control; **, p value = 0.001–0.0030 relative to control; ***, p value < 0.0002 relative to control.
= (kex)/(ken); PMcalc cyc
= ()/(1 + ); PMcalc total
= ()/(1 + ) Ymax. The relative surface Glut4, PMrel = (PM)/(PMcontrol, insulin); n, number of independent experiments; *, p value = 0.01 relative to control; **, p value = 0.001–0.0030 relative to control; ***, p value < 0.0002 relative to control.
| PMobs ± S.D. (n) | PMrel | kex min−1 ± S.D. (n) | ken min−1 ±S.D. (n) | |
PMcalc cyc | Ymax | PMcalc total | PMrel | |
|---|---|---|---|---|---|---|---|---|---|
| Insulin | |||||||||
| Control | 19.8% ± 1.39 (15) | 1.00 | 0.034 ± 0.003 (9) | 0.13 ± 0.007 (14) | 0.261 | 20.7% × | 1.00 | 20.7% | 1.00 |
| Rab10 KD | 6.9% (±0.99; 9)** | 0.35 | 0.013 ± 0.001 (7)*** | 0.18 ± 0.013 (8)*** | 0.072 | 6.7% × | 0.97 | 6.5% | 0.31 |
| Basal | |||||||||
| Control | 1.5% (±0.50; 9) | 0.08 | 0.0053 (±0.001; 5) | 0.13 | 0.041 | 4.0% × | 0.30 | 1.2% | 0.06 |
| Rab10 KD | 1.3% (±0.70; 9) | 0.07 | 0.0053 | 0.18 | 0.029 | 2.9% × | 0.26* | 0.7% | 0.04 |
Antibodies and Reagents
HA.11 monoclonal antibody (α-HA; Covance) was purchased as ascites. Antibody was purified using protein A-Sepharose as described previously (8). Purified α-HA was labeled with an AlexaFluor647 (AF647) protein labeling kit (Invitrogen) according to the manufacturer's instructions, and unincorporated dye was removed using 2 × 10 ml of Zeba desalting spin columns in series (7000 molecular weight cutoff, Pierce). α2-Macroglobulin was purchased from AssayPro, desalted using a 5-ml HiTrap desalting column (GE Healthcare) to remove glycine, and labeled using an AF647 monoclonal antibody labeling kit (Invitrogen). It was then activated using methylamine (Sigma), and then quenched dye and excess methylamine were removed by desalting into PBS as described previously (9). Antibody and α2-M concentrations and labeling efficiencies were determined by absorption spectroscopy (8).
Glut4 Kinetic Assays
Kinetic experiments were performed essentially as described previously (9–11). For all experiments, cells were serum-starved for 2 h at 37 °C in low serum media (LSM; 0.5% FBS in DMEM). Insulin (100 nm) was added to the LSM for the final 45 min of starvation in some samples, as indicated.
Surface Labeling
To label surface Glut4, cells were placed on ice and incubated with 50 μg/ml AF647-α-HA antibody for 1 h. Cells were then washed and analyzed by flow cytometry (see below).
Basal to Insulin Transition Kinetics
Basal serum-starved cells were incubated for increasing amounts of time with insulin (100 nm). At the end of the time course, cells were placed on an ice slurry to halt trafficking, and surface Glut4 was labeled as described above, and cells were analyzed by flow cytometry.
Insulin + LYi Transition Kinetics
Cells were serum-starved in LSM, and insulin was added for the final 45 min. The incubation media were then replaced with pre-warmed LSM supplemented with insulin and the PI 3-kinase inhibitor LY294002 (LYi, 50 μm; EMD Chemicals), and the cells were incubated for increasing amounts of time. At the end of the time course, cells were placed in an ice slurry to halt trafficking; surface Glut4 was labeled as described above, and cells were analyzed by flow cytometry. The data were fit with single exponents to estimate the overall endocytic rate constant (ken), which is approximately equal to the observed relaxation rate constant (ken ≈ kobs). This method for measuring ken has been validated using two additional independent measurement techniques (9).
α-HA Uptake
Cells were serum-starved in LSM ± insulin and then incubated at 37 °C in the continuous presence of labeled antibody (50 μg/ml AF647-α-HA) in LSM ± insulin for increasing amounts of time. At the end of the time course, cells were placed in an ice-water slurry to halt uptake, washed, and analyzed by flow cytometry. The data were fit with single exponents as shown in Equation 1,
to estimate the overall exocytic rate constant (kex = kobs) and the relative amount of Glut4 in the actively cycling pool (Ymax). Ymin = labeling at 0 min ≈ cell surface Glut4 (labeling is essentially instantaneous at the antibody concentration used in these experiments (8)). To compare and average data from multiple experiments, the data were standardized by dividing by the maximum fluorescence measured in control insulin-stimulated cells in each experiment. To calculate the fraction of total cycling Glut4 in the plasma membrane from the observed surface binding data, the average surface α-HA fluorescence was divided by the average maximal fluorescence measured in the uptake experiments in insulin-stimulated cells.
α2-Macroglobulin Uptake and Surface Labeling
Experiments were performed as described previously (9). Cells were serum-starved for 2 h in serum-free DMEM and then incubated with or without 100 nm insulin for 30 min. Cells were then incubated with 4 μg/ml AF647-α2-M (± insulin) for increasing times, placed on ice, and additional cells on the same plate labeled for 90 min to label cell surface receptors. Cells were gently washed with ice-cold PBS, collagenase digested at 37 °C for 10 min, and then placed on ice. This warm incubation allows surface-bound α2-M to internalize to prevent loss during analysis. Cells were analyzed by flow cytometry as described below, and uptake data were fitted with linear equations. To measure the rate constant of α2-M endocytosis (ken), the internal to surface ratio was calculated for each time point; the slope of the In/Sur versus time plot equals ken (32).
Tf Efflux and Tf Receptor Surface Labeling
Experiments were performed as described previously (9). Cells were preincubated for 2 h in LSM. The incubation media were removed and replaced with LSM containing Alexa647-conjugated iron-loaded (holo) Tf (5 μg/ml), and incubation was continued at 37 °C for 30 min. The cells were rapidly washed, then incubated at 37 °C in LSM with excess unlabeled holo-Tf (500 μg/ml), and then placed on ice. Incubation in media with excess unlabeled holo-Tf was sufficient to remove all surface Tf. Cells were then analyzed by flow cytometry. The data were fitted by equations for a single exponential decay. In these experiments kobs = kex. To determine the relative amounts of cell surface Tf receptor, cells were treated with or without insulin for the last 30 min of the preincubation and then labeled with biotinylated α-Tf receptor antibody (anti-mouse CD71-Biotin, Leinco Technologies) and AF647-streptavidin (Invitrogen/Molecular Probes) at 4 °C.
Flow Cytometry
Flow cytometry, gating, and analysis were performed essentially as described previously (8–11). Briefly, labeled cells in 96-well plates on ice were washed three times with 200 μl of ice-cold PBS. Cells were incubated with 20 μl of collagenase (type III; 1 mg/ml in 2% BSA/PBS; Worthington) for 5 min at 37 °C and then resuspended in 180 μl of PBS. For experiments with non-differentiated fibroblasts, cells were resuspended in 180 μl of PBS, 0.5 mm EDTA. Cells were gently filtered through a 100-μm cell strainer to remove clumps of cells and analyzed on an Accuri C6 cytometer. Detection thresholds for adipocytes were set at 1,000,000 for FSC-H (forward scatter-height) and 500,000 for SSC-H (side scatter-height). The only threshold for fibroblasts was 750,000 for FSC-H. Log intensities of scattered light and fluorescence (FL1, 488 nm excitation/533 nm emission; FL2, 488 nm excitation/585 nm emission; FL3, 488 nm excitation/>670 nm emission; FL4, 640 nm excitation/675 nm emission) were collected for each cell. For analysis of adipocytes, selective gating using CFlow Plus software (Accuri) was used to analyze only the adipocytes in the sample, excluding any residual fibroblasts and cellular debris in the sample, as described previously (8), except that on this instrument adipocytes could be identified using gates in a single two-dimensional histogram of FSC-H versus FL3-H. For analysis of non-differentiated fibroblasts, cells were identified in the two-dimensional histogram of FSC-H versus SSC-H. For either cell type, cells infected with HA-Glut4/GFP and uninfected cells in the same sample were separately analyzed using gates in a two-dimensional histogram of FL1-H versus FL3-H. The geometric means of the fluorescence values of the gated populations were determined using FCS Express (De Novo Software; the distribution of fluorescence values of individual cells in a sample population is approximately log normal, thus standard statistical analysis is not appropriate; however, the sample to sample variation of the geometric means has a normal distribution, and standard statistical analysis is then appropriate; data not shown). The mean fluorescence values of uninfected cells are a measure of cellular autofluorescence (FL1) and nonspecific antibody labeling (FL4). These values were subtracted from the mean values of FL1 and FL4 measured for the infected cell population within the same sample (11).
Data Analysis
Glut4
The geometric mean fluorescence values for AF647-α-HA labeling (FL4) and GFP (FL1) were determined for each sample (∼1000 cells/sample after gating for adipocytes or fibroblasts) and corrected for background fluorescence as described above. In most experiments, the mean fluorescence ratio (MFR) was calculated for each individual sample to correct for differences in expression of the HA-Glut4/GFP reporter construct (MFR = corrected geometric mean FL4 (α-HA)/corrected geometric mean FL1 (GFP)), and the average mean of n = 2–3 replicates was calculated for each time point/experimental condition. All data in each experiment were standardized to the average control values (basal or insulin, as indicated). The data points in each figure are the average ± error (S.E. or standard deviation, S.D., as indicated) of the average standardized values for n = 4–15 replicate experiments (2–3 replicates/experiment, 1000 cells/replicate).
α2-M or Tf
Adipocytes or fibroblasts were gated as described above, and the geometric mean fluorescence for AF647-labeling (α2-M, Tf, or α-Tf receptor; FL4) was determined for each sample. To correct for autofluorescence, the average geometric mean FL4 fluorescence for n = 2–4 samples of unlabeled cells was subtracted from the geometric mean fluorescence for the samples of labeled cells.
Fits of the data were performed using GraphPad Prism version 5.0 for Windows/Mac (GraphPad Software, San Diego) as well as custom code implemented in Matlab (MATLAB and Statistics Toolbox Release 2013a, The MathWorks, Inc., Natick, MA) (10). All fits were done on combined data sets, including data points from each of n = 4–15 individual experiments. The rate constants obtained from the fits of the combined data were compared with the average of the rate constants obtained from fits of each independent experiment and were always in good agreement. The statistical significance of observed differences between steady-state distributions under different experimental conditions as well as calculated exocytic rate constants (kex calc) was assessed by either t test or two-way analysis of variance (Figs. 1, A and B, 4, A and B, 5, A and D, 6, A and D, 7, B and C, and 8, B and C), whereas differences between experimental conditions for the kinetics data were assessed by comparing fits of the data sets using either single exponents (Figs. 1C, 2, A and B, 3, A and B, 4, C and D, 7A, and 8A) or lines (Figs. 5, B and C, and 6, B and C) and calculating the p value for the null hypothesis that both sets of data were best fit by the same function (***, p ≤ 0.0001; **, p ≤ 0.001; and *, p ≤ 0.015).
FIGURE 1.
Knockdown of Rab10 decreases cell surface Glut4 in adipocytes. 3T3-L1 adipocytes expressing a Glut4 reporter construct (HA-Glut4/GFP) and either a scrambled (control; white) or Rab-targeting shRNA (Rab10 KD; gray) were incubated with 100 nm insulin for increasing amounts of time and placed on ice. Surface-exposed HA-Glut4/GFP was labeled with AlexaFluor647-conjugated α-HA antibody (AF647-α-HA). A, surface Glut4 (AF647-α-HA). B, total HA-Glut4/GFP expression (GFP). C, basal to insulin transition, MFR (mean AF647/mean GFP). Lines, single exponential fits of the data. All data are standardized to control samples as indicated and are the mean ± S.D. (A) or the average MFR ± S.D. (C) of n = 7–15 independent experiments (Table 1) or mean ± S.D. (B) of n = 96 samples from three independent experiments. ***, p ≤ 0.0001 that values are the same in control and knockdown cells.
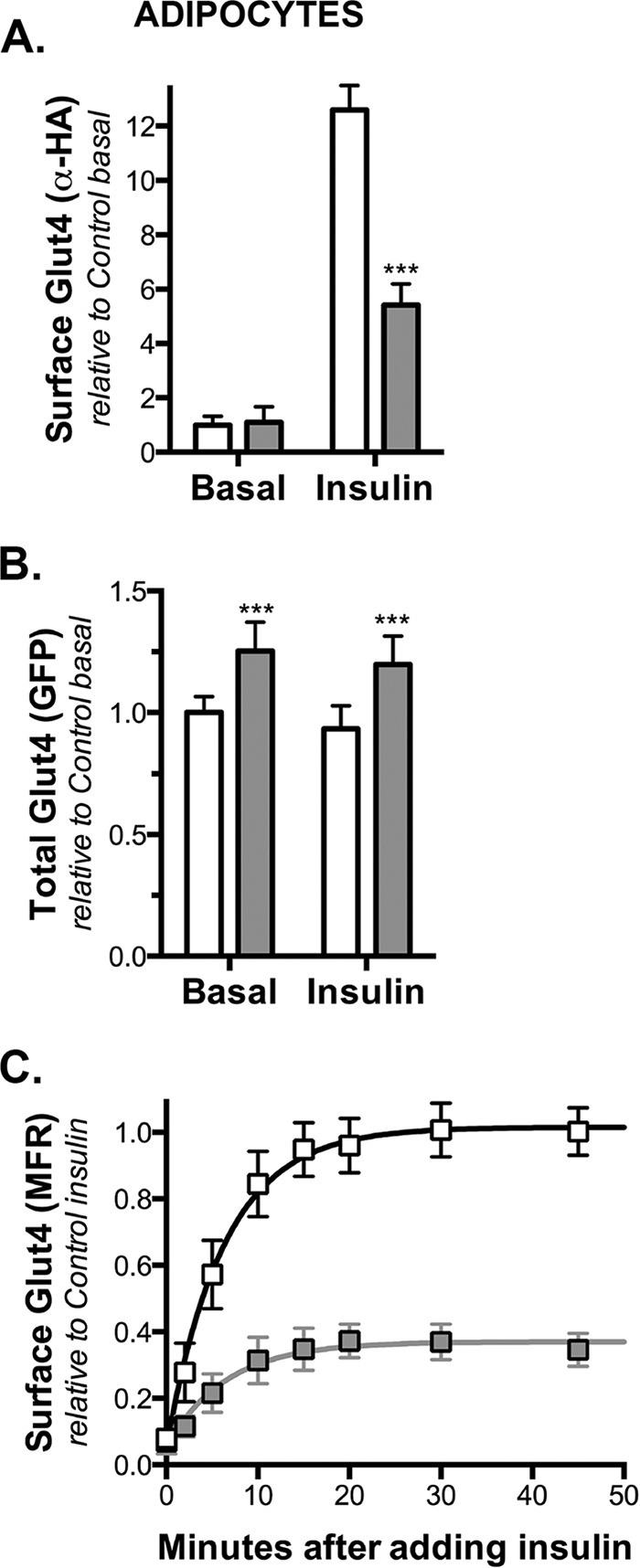
FIGURE 4.
Knockdown of Rab10 has no effect on Glut4 trafficking in fibroblasts. Fibroblasts expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were treated as described in Figs. 1–3, and surface HA-Glut4/GFP was labeled. A, basal to insulin transition, average MFR ± S.D. of n = 4–10 independent experiments. Lines, simulations (Table 3). B, total HA-Glut4/GFP expression (GFP), mean ± S.D. of n = 64 samples from two independent experiments. ***, p ≤ 0.0001, and values are the same in control and knockdown cells. C, ken ± S.D. determined from single-exponential decay fits of D. Insulin + LYi transition data average MFR ± S.D. of n = 5–6 independent experiments. Lines, single exponential fits of the data. All data are standardized to control samples as indicated.
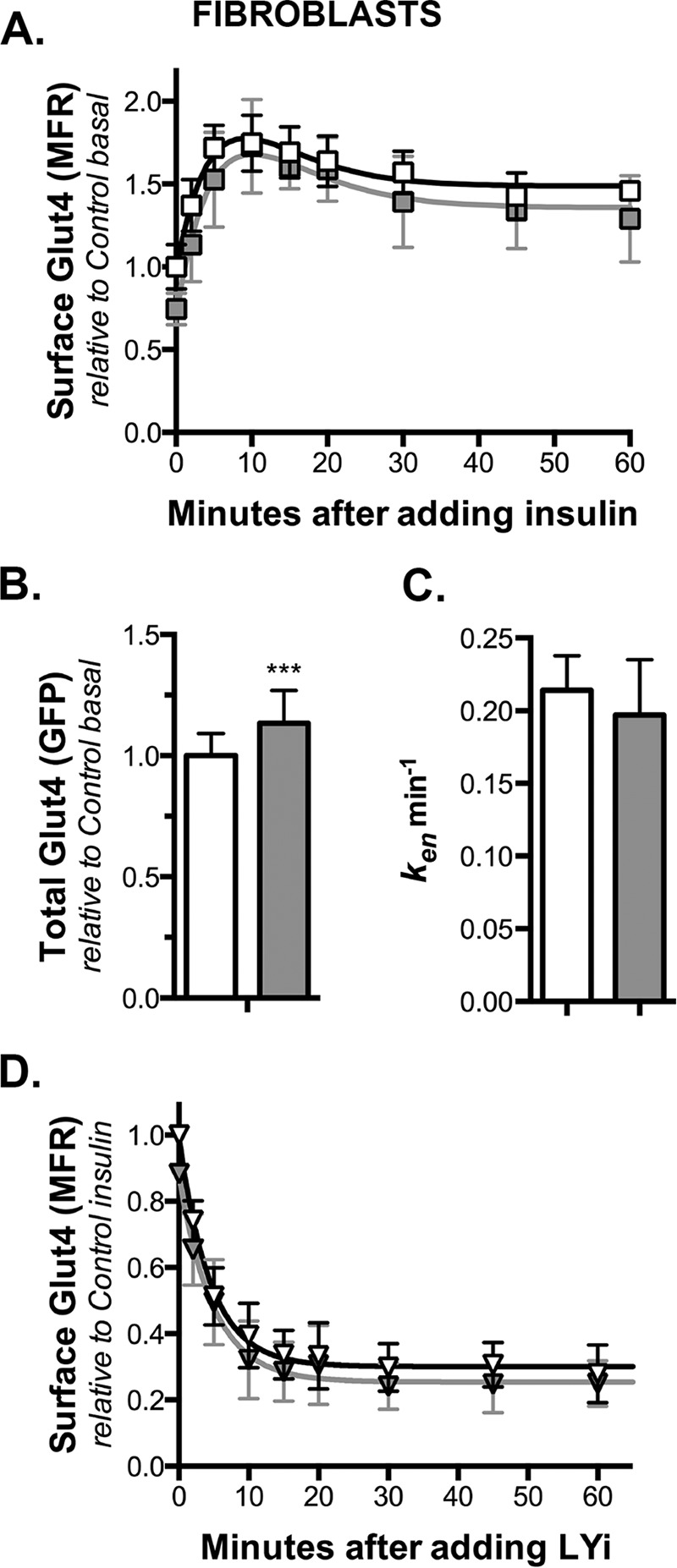
FIGURE 5.
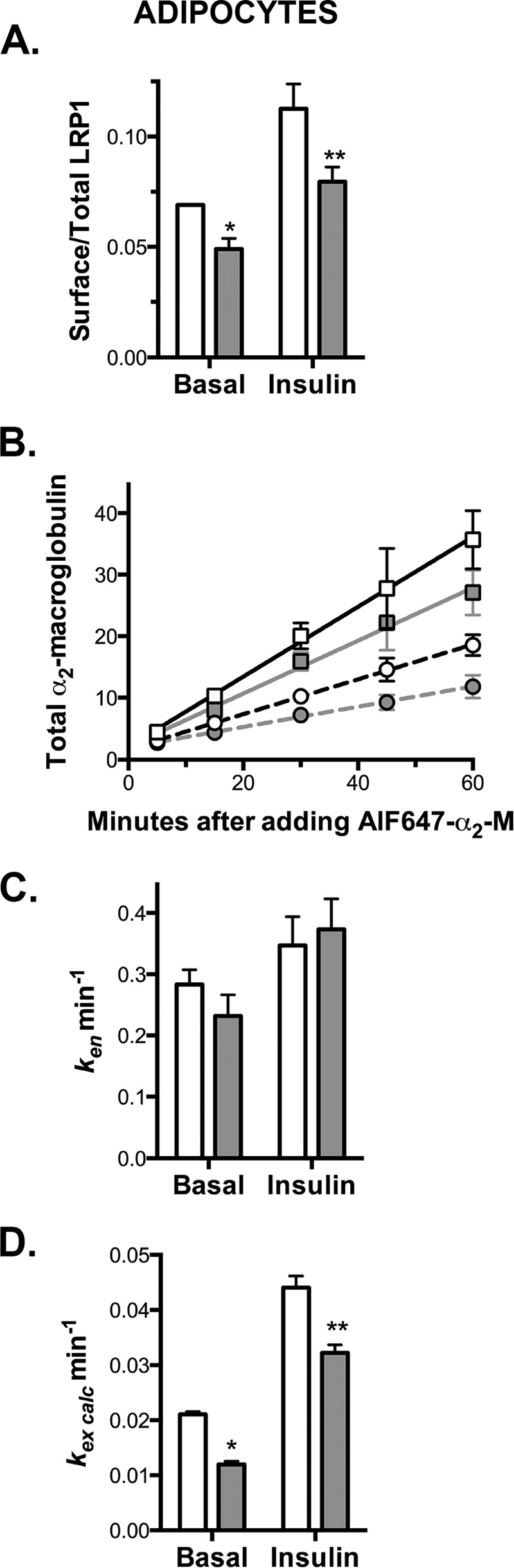
Knockdown of Rab10 inhibits LRP1 exocytosis in adipocytes. Adipocytes expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were incubated ± 100 nm insulin for 30 min (circle, basal; squares, insulin), then incubated at 37 °C for increasing times with AF647, α2-M ± insulin (4 μg/ml), and placed on ice. Additional cells on the same plate were then labeled for 90 min on ice to label cell surface receptors. A, surface/total LRP1; data are standardized to Ymax from cells incubated with chloroquine (data not shown (9)). B, AF647, α2-M uptake; arbitrary units, data are standardized to control basal uptake at t = 15 min in fibroblasts (Fig. 6). Lines, linear fits of the data. C, ken determined from the slope of the In/Sur versus time plot (In/Sur = uptake/surface; data not shown). D, kex calculated using surface/total Glut4 (PM) and ken (kex calc = (PM/(1 − PM)) ken). Data are mean ± S.D. (A and C) or mean ± S.E. (B and D) of n = 5–7 independent experiments. **, p ≤ 0.001, or *, p ≤ 0.015, that values are the same in control and knockdown cells.
FIGURE 6.
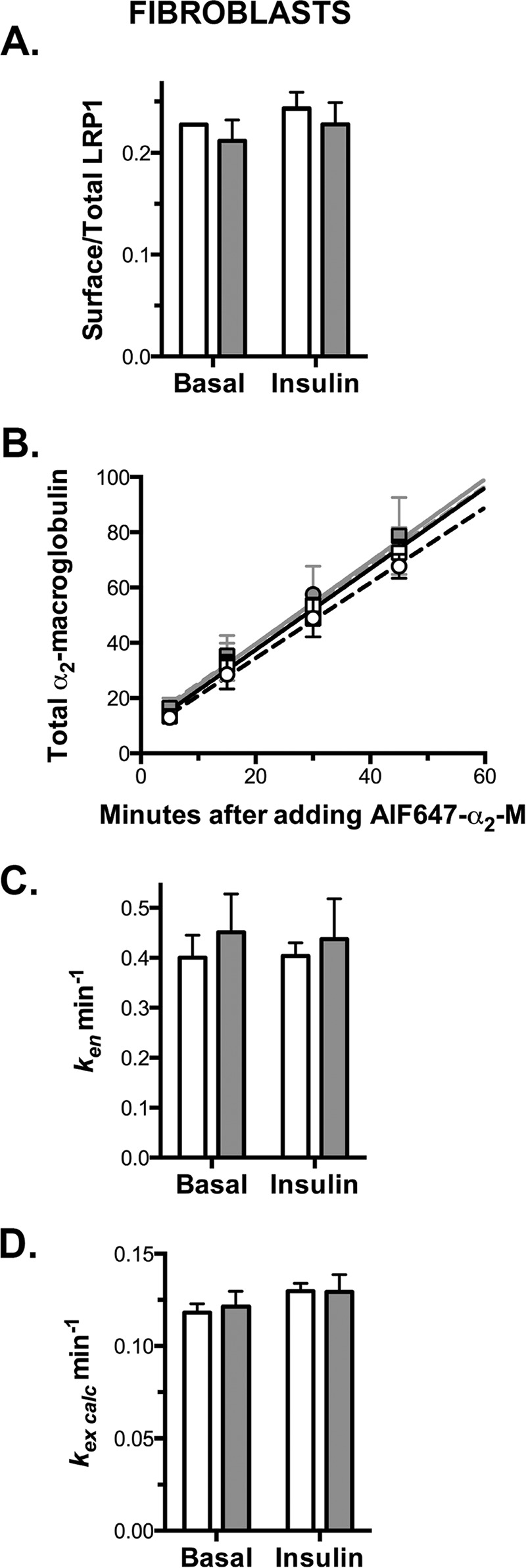
Knockdown of Rab10 has no effect on LRP1 exocytosis in fibroblasts. Fibroblasts expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were treated as described in Fig. 5. A, surface/total LRP1. B, AF647, α2-M uptake. Lines, linear fits of the data. C, ken determined from the slope of the In/Sur versus time plot (data not shown). D, kex calculated using surface/total Glut4 (PM) and ken (kex calc = (PM/(1 − PM)) ken). Data are mean ± S.D. (A and C) or mean ± S.E. (B and D) of n = 4 independent experiments.
FIGURE 7.
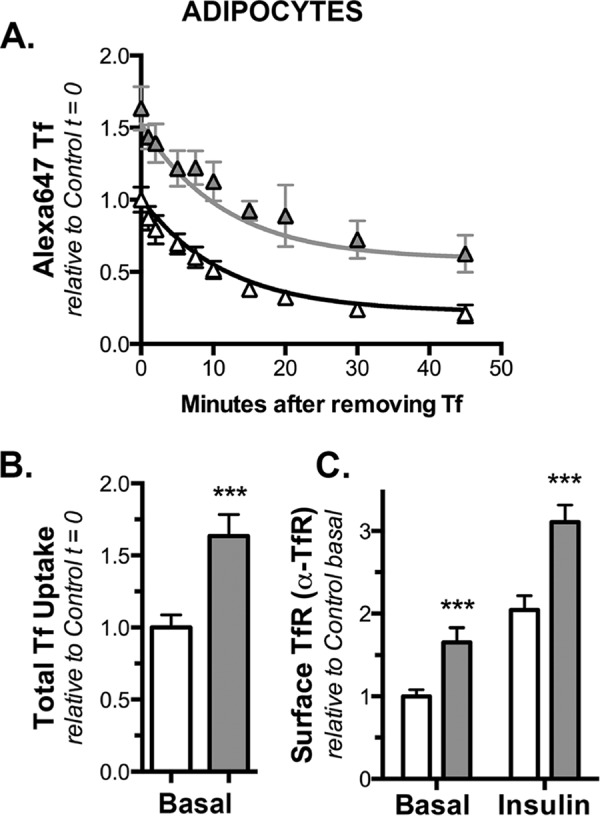
Knockdown of Rab10 has no effect on Tf receptor exocytosis in adipocytes. Adipocytes expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were incubated at 37 °C for 30 min with AF647-holo-Tf, then for increasing time in media with excess unlabeled holo-Tf and placed on ice. To label cell surface receptors, additional cells on the same plate were incubated ± insulin at 37 °C and then labeled with biotinylated α-Tf receptor and AF647-streptavidin on ice. A, AF647-Tf efflux; data are mean ± S.D. from n = 3 independent experiments standardized to Y0 from control cells. Lines, single-exponential decay fits of the data (kobs ≈ kex). There was no significant difference in kex in the two cell types (kex = 0.09 ± 0.008 min−1; p = 0.94), and that k is the same for both data sets. B, AF647, Tf uptake (Y0); mean ± S.D. from n = 9 samples from three independent experiments, data are standardized to control basal. C, surface TfR; mean ± S.D. of n = 6 samples from three independent experiments, data are standardized to control basal. ***, p ≤ 0.0001, that values are the same in control and knockdown cells.
FIGURE 8.
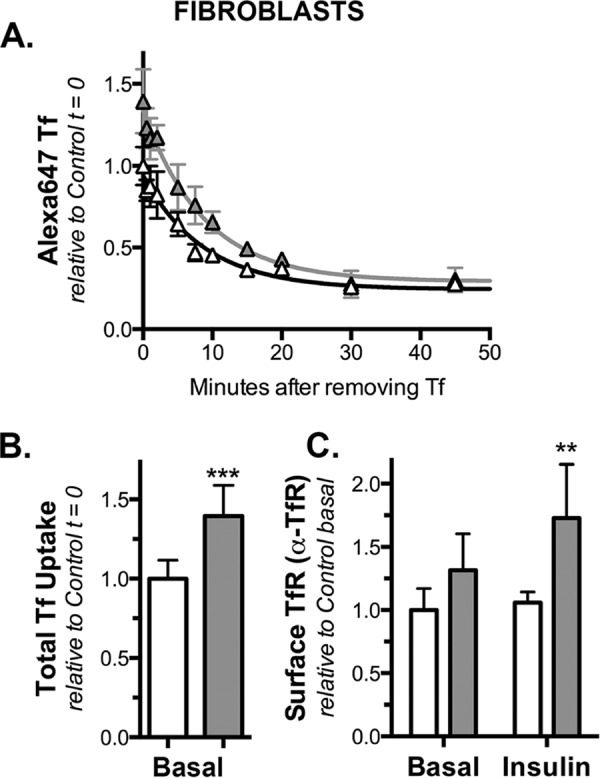
Knockdown of Rab10 has no effect on Tf receptor exocytosis in fibroblasts. Fibroblasts expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were treated as described in Fig. 7. A, AF647-Tf efflux (mean ± S.D. from n = 2 independent experiments). There was no significant difference in kex in the two cell types (kex = 0.12 ± 0.01 min−1; p = 0.092, that k is the same for both data sets). B, AF647, Tf uptake (Y0); mean ± S.D. from n = 6 samples from two independent experiments. C, surface Tf receptor; mean ± S.D. of n = 12 samples from two independent experiments. ***, p ≤ 0.0001, or **, p ≤ 0.001, that values are the same in control and knockdown cells.
FIGURE 2.
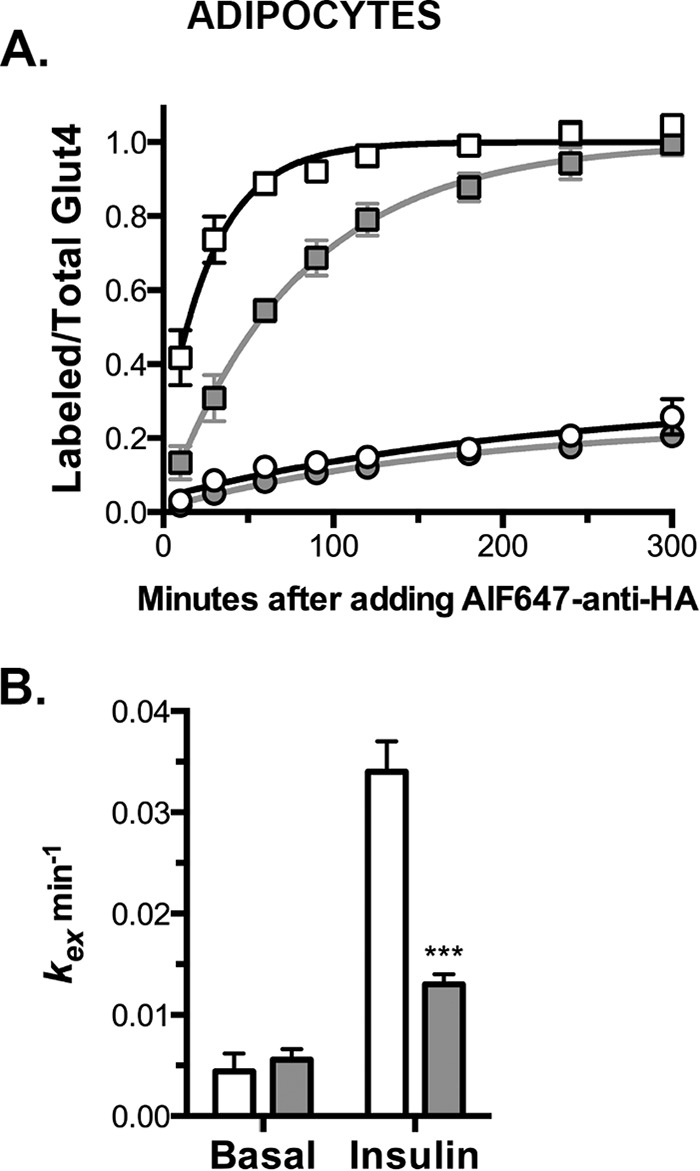
Knockdown of Rab10 inhibits exocytosis of Glut4 in adipocytes. A, 3T3-L1 adipocytes expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were pretreated ± 100 nm insulin for 45 min and then incubated at 37 °C for increasing times with AF647-α-HA ± insulin. Circles, basal; squares, insulin. Data are standardized to Ymax control insulin and are the average MFR ± S.D. (uptake) of n = 4–9 experiments (Table 1). Lines, single-exponential fits of the data (kobs = kex; Ymax = total cycling pool size). B, kex ± S.D. determined from single exponential fits of the combined data. ***, p ≤ 0.0001, that control and Rab10 knockdown cells are best fit by the same function.
FIGURE 3.
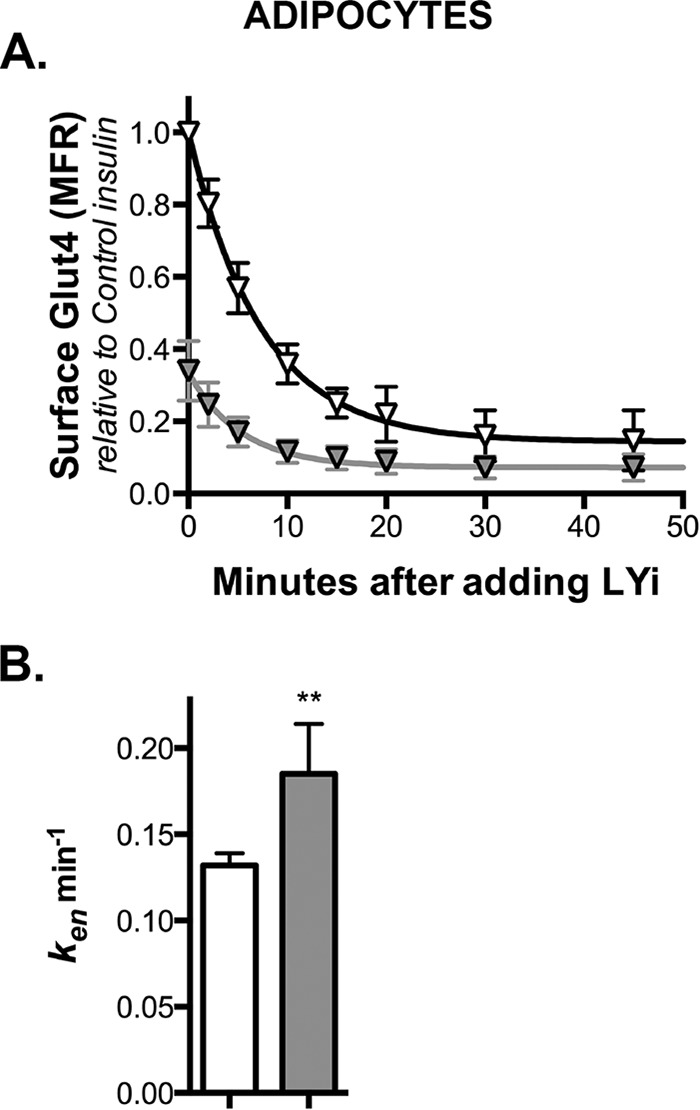
Knockdown of Rab10 accelerates endocytosis of Glut4 in adipocytes. A, 3T3-L1 adipocytes expressing HA-Glut4/GFP and shRNA (control, white; Rab10, gray) were pretreated with 100 nm insulin for 45 min and then incubated at 37 °C for increasing times with LY294002 (LYi). Cells were then placed on ice, and surface HA-Glut4/GFP was labeled. Lines, single-exponential decay fits of the data (kobs ≈ ken + 0.004). Data are standardized to control insulin at t = 0, and are the average MFR ± S.D. (transition) of n = 6–12 independent experiments (Table 1). B, ken± S.D. determined from single exponential fits of the combined data. **, p ≤ 0.001, that control and knockdown cells are best fit by the same function.
To fit the data with more complicated mathematical models (Fig. 9), simultaneous fits of multiple data sets were done. A system of coupled ordinary differential equations was identified that explicitly represents the output of each type of experiment. These were then fitted using a combination of numerical integration and nonlinear least squares regression to optimize the parameter values simultaneously over the entire range of experiments and experimental data as described previously (10).
FIGURE 9.

Modeling and simulations. A, Glut4 trafficking itinerary in fibroblasts and adipocytes. 1) Glut4 is internalized from the PM via a slow pathway distinct from the fast TfR pathway and delivered to SE. Proteins in the SE are sorted into different exocytic pathways, including the fast recycling pathway followed by the TfR (thin dotted lines). 2 and 3) The slow constitutive recycling pathway through endosomal recycling intermediate compartments (ERC; thin solid lines). AS160 regulates 4) trafficking from the SE into GSVs, and 5) release/priming of sequestered GSVs for fusion to the PM through its Rab substrates. 6) An AS160-independent Akt and PI3K-dependent step regulates the tethering/docking/fusion of primed GSVs to the PM. Rab10 is rate-limiting for exocytosis from the GSV, whereas an unidentified Rab (Rab-?) is required for recycling from the ERC. B, differential equations describing the transfer of Glut4 between four compartments, with a single rate constant for each of six steps: ken, endocytosis (PM to SE); ksort, sorting (SE to ERC); kfuseE, endosomal fusion (ERC to PM); kseq, sequestration (SE to GSV); kfuseG, GSV fusion (GSV to PM); and krec, recycling (SE to PM; LRP and TfR). To determine the values of the rate constants to use in simulations (Fig. 10), the experimental data in Figs. 1–6 were fitted with a system of coupled ordinary differential equations derived from this model that explicitly represents the output of these experiments (10).
Modeling and Simulations
The Glut4 trafficking itinerary depicted in Fig. 9A was modeled mathematically as a series of simple differential equations that describe the transfer of Glut4 between four compartments, with a single rate constant for each of the five steps (Fig. 9B). These rate constants/steps are as follows: ken, endocytosis from the plasma membrane (PM) to sorting endosomes (SE); ksort, sorting from endosomes to the endosomal recycling intermediate compartment (ERC) in the constitutive exocytic pathway; kfuseE, transport from the ERC and fusion to the plasma membrane; kseq, sorting from endosomes into the sequestered GSVs; and kfuseG, release of sequestered GSVs and fusion to the plasma membrane. LRP1 has an additional route of recycling from the sorting endosomes to the plasma membrane that it shares with the transferrin receptor (TfR), with a single rate constant (krec). Glut4 cannot efficiently exit via this fast recycling pathway (krec, Glut4 = 0). Sorting endosomes (SE) are defined as the compartment where Glut4, LRP1, and the TfR are sorted into three separate exocytic pathways as follows: the fast TfR recycling pathway, the slower constitutive ERC pathway, and the highly regulated, specialized GSV pathway. The ERC is defined functionally as a recycling intermediate between sorting endosomes and the plasma membrane that accumulates behind a rate-limiting step in exocytosis via the slow constitutive pathway. The equations were used to simulate the movement of Glut4 by solving them numerically using computing software as described previously (8, 10, 11).
Glut4
The rate constants for Glut4 used in the modeling (ken, ksort, kfuseE, kseq, and kfuseG) were determined by simultaneous free fits of the control fibroblast, control adipocyte, and Rab10 knockdown adipocyte kinetics data (Figs. 1C, 2A, 3A, and 4, A and D; Table 2). Insulin has little effect on ken in either fibroblasts or adipocytes (9). In addition, there was no significant change in ksort in fits where this rate constant was allowed to vary between basal and insulin-stimulated conditions in the fibroblast cells (Ref. 10 and data not shown). Therefore, both ken and ksort were held constant between the basal and insulin-stimulated conditions in the fits. The other rate constants (kfuseE, kseq, and kfuseG) were allowed to vary between basal and insulin-stimulated cells.
TABLE 2.
Summary of rate constants (k) and plasma membrane distributions (PM) for LRP1 and the TfR (Figs. 5–8)
Values ± S.D. of n replicate experiments: LRP1, PM and ken; fibroblasts n= 4; adipocytes n= 7 or 5; TfR, PM and kex, fibroblasts n= 6 or 2; adipocytes n= 6 or 3. ***, p ± 0.0001; **, p ± 0.001; *, p ± 0.015 that Rab10 KD and control values are the same. All values are mean ± S.D.

LRP1
The rate constants for LRP1 endocytosis used in the modeling (ken) were calculated from the slope of the internal/ surface data from the α2-M uptake experiments in adipocytes and fibroblasts for each cell type (Figs. 5 and 6). The rate constants for LRP1 recycling via the fast Tf receptor pathway used in the modeling (krec) were determined from simulations of cell surface LRP1 in control cells, using Glut4 values for all other rate constants (krec, Glut4 = 0).
Replicates
The observed effects were consistent across three separate retroviral infections and with multiple experiments using each batch of cells and multiple replicate samples in each experiment (as described).
Results
Cell Surface Glut4 in Insulin-stimulated Adipocytes Was Decreased by Rab10 Knockdown
Consistent with previous reports, knockdown of Rab10 in differentiated adipocytes caused a 57% decrease in surface Glut4 levels relative to control adipocytes after insulin stimulation (Fig. 1A). Depletion of Rab10 did not significantly alter basal surface levels of Glut4. Cell surface Glut4 levels are proportional to the total amount of Glut4 expressed in cells. This can be measured as the total HA-Glut4/GFP expressed per cell (Fig. 1B). There was a small (1.25-fold) increase in total HA-Glut4/GFP in Rab10 knockdown cells relative to control cells. This increase is likely due to an increase in stability and not to an increase in expression of the reporter construct (10). To correct for this difference in protein levels, the mean fluorescence ratios (MFR; mean fluorescence AF647-α-HA/mean fluorescence GFP) were calculated for each sample. Using these corrected values, knockdown of Rab10 in differentiated adipocytes decreased the proportion of total Glut4 at the cell surface in insulin-stimulated cells 65% relative to control adipocytes (Fig. 1C and Table 1).
The decrease in cell surface Glut4 in the Rab10 knockdown cell lines was not due to a decrease in the rate of response to insulin stimulation (Fig. 1C). Surface Glut4 increased rapidly and remained elevated for at least 3 h in both control and Rab10 knockdown cells (Ref. 8 and data not shown). The rate of transition from the basal to the insulin-stimulated steady-state levels of surface Glut4 were the same in the Rab10 knockdown and control adipocytes (kobs = 0.15 min−1; t½ = 4.5 min).
Exocytosis of Glut4 in Adipocytes Was Inhibited by Rab10 Knockdown
The proportion of total Glut4 at the cell surface is dependent on the relative rates of insertion of Glut4 into the plasma membrane (exocytosis) and clearance from the plasma membrane (endocytosis). It is also dependent on the amount of total Glut4 that is actively cycling.5 To measure the intrinsic rate constant of exocytosis (kex) and the amount of cycling Glut4, α-HA uptake assays were performed (Fig. 2). In these experiments, cells were stimulated with insulin until the steady-state distribution of Glut4 is reached, and then cells were incubated with fluorescently labeled α-HA antibody at 37 °C for increasing amounts of time. Antibody labeling of cell surface Glut4 is essentially instantaneous at the concentrations of antibody used, and the antibody remains bound to HA-Glut4/GFP as it is internalized and trafficked within the cell (8). As unlabeled intracellular HA-Glut4/GFP exchanges with the labeled Glut4 in the plasma membrane, it is rapidly labeled with antibody until maximum fluorescence is reached. The Glut4 in insulin-stimulated adipocytes behaves as a single cycling pool, and the data were fit using single exponential equations. In these fits, the observed relaxation rate constant (kobs) is equal to kex, and the maximum mean fluorescence ratio (Ymax) is the amount of total Glut4 that is cycling and can be labeled with antibody.
In Rab10 knockdown (KD) cells, the cycling pool size (proportion of total Glut4 that is actively cycling) in insulin-stimulated cells was not significantly different from control cells (Fig. 2 and Table 1). Thus, Rab10 knockdown did not cause a significant fraction of the total Glut4 to accumulate in non-cycling compartments (e.g. degradative or biosynthetic compartments or in defective GSVs that are unable to fuse). In insulin-stimulated cells, knockdown of Rab10 significantly decreased kex relative to the control cells (kex = 0.034 min−1 control, 0.013 min−1 Rab10 KD; Fig. 2 and Table 1). There was no significant difference in the rate constant of exocytosis between the control and Rab knockdown cells under basal conditions (Table 1). However, there was a small but significant difference in the distribution of Glut4 between the constitutive and GSV cycling pathways in Rab10 knockdown and control cells under these conditions. Rab10 knockdown decreased the concentration of Glut4 in the endosomal pool (decreased Ymax) and increased the concentration of Glut4 sequestrated in GSVs in basal cells (Table 1; p < 0.015 that the Rab10 KD and control data are best fit by the same exponential function).
Endocytosis of Glut4 in Adipocytes Was Accelerated by Rab10 Knockdown
A change in the proportion of total Glut4 at the cell surface can also be caused by a change in the intrinsic rate constant of endocytosis (ken). To measure this, transition kinetic assays were performed using the PI 3-kinase inhibitor LY294002 (LYi; Fig. 3). In this experiment, cells were stimulated with insulin until the steady-state distribution of Glut4 was reached. LYi was then added, the cells were incubated for increasing amounts of time, and surface Glut4 was measured. LYi inhibits exocytosis of Glut4, without affecting endocytosis (9). Therefore, addition of LYi causes an exponential decay in surface Glut4. The observed relaxation rate constant determined by a single exponential fit of this decay is approximately equal to ken (9). Unexpectedly, ken was significantly increased in the Rab10 knockdown cells relative to the control cells (ken = 0.13 min−1 control, 0.18 min−1 Rab10 KD; Fig. 3 and Table 1). This increase is not an artifact of shRNA expression or viral infection, because it is not observed in cells expressing shRNAs specific for AS160, insulin-regulated aminopeptidase, sortilin, or CDP138 (Ref. 11 and data not shown).
Effects of Rab10 Knockdown on kex and ken Account for the Observed Changes in Cell Surface Glut4
To examine whether the measured changes in the kinetic parameters were sufficient to account for the decrease in Glut4 at the cell surface observed in Rab10 knockdown cells, the kinetics data were analyzed mathematically using a simple two-step/three-compartment model (“static retention”) (6, 8–10). In this model, Glut4 is found in the following three pools: in the PM; in actively cycling sorting endosomal compartments (SE); and in non-cycling (sequestered) compartments (GSVs). The amount of Glut4 in the plasma membrane pool is dependent on the partition coefficient (), which is the ratio of the rate constant of insertion into the plasma membrane (exocytosis from the sorting endosomes, kex) and the rate constant of internalization from the plasma membrane (endocytosis to the sorting endosomes, ken). It is also dependent on the size of the actively cycling pool (Ymax = PM + SE; GSV = 1 − Ymax). This model is described by Equations 2 and 3,
 |
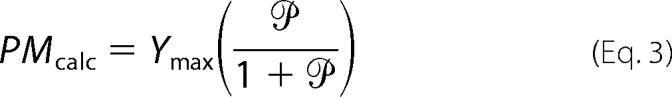 |
Using Equations 2 and 3, the fraction of total Glut4 expected at the plasma membrane (PMcalc) can be calculated using the values of kex, Ymax, and ken determined from the uptake and LYi transition experiments (Figs. 2 and 3). The actual amount of Glut4 found at the plasma membrane was measured in the basal to insulin transition experiments, with total Glut4 defined as Ymax in insulin-stimulated cells (PMobs = surface/total Glut4; Table 1). As reported previously, ∼20% of the total pool of cycling Glut4 (Glut4 that can be labeled with α-HA) was found at the plasma membrane in control adipocytes after insulin stimulation (10). This was reduced to 6.9% in the Rab10 knockdown cells. There is excellent agreement between PMobs and PMcalc for both the control and Rab10 knockdown cells. These calculations show that the effects of knockdown of Rab10 on cell surface Glut4 are due to effects on both exocytosis and endocytosis of Glut4. They also offer internal confirmation of the accuracy of the rate constants and subcellular distributions determined from the three independent experiments (9–11).
Glut4 Trafficking in Fibroblasts Was Unaffected by Rab10 Knockdown
Rab10 has been implicated in protein trafficking in many cell types, including undifferentiated cells that do not exhibit regulated exocytosis. Glut4 traffics through both a constitutive endocytic pathway as well as through a highly regulated specialized trafficking pathway in adipocytes. It has been proposed in many models that Glut4 cycles predominantly through the constitutive pathway after insulin stimulation (33, 34). Therefore, it is possible that Rab10 functions in the general endocytic pathway and that the inhibition of Glut4 translocation by knockdown of Rab10 involves the constitutive and not the specialized highly insulin-responsive pathway. Glut4 is expressed only after adipocyte differentiation, and the specialized trafficking of Glut4 through GSVs develops during differentiation at the same time as Glut4 expression increases (10). It is not observed in pre-adipocytes (fibroblasts) that express exogenous Glut4. Therefore, to investigate whether Rab knockdown affects the constitutive Glut4 endosomal recycling pathway, the trafficking kinetics of expressed Glut4 were measured in fibroblasts (Fig. 4). Knockdown of a protein that limits exocytosis from the highly regulated sequestered GSVs but not from the constitutive pathway will affect Glut4 trafficking in adipocytes but not in fibroblasts.
As expected for a protein that regulates GSV exocytosis, knockdown of Rab10 in fibroblasts had no significant effect on cell surface Glut4 relative to control cells (Fig. 4A). As observed in adipocytes, there was a small increase in HA-Glut4/GFP expression in Rab10 knockdown cells relative to control cells (1.13-fold; Fig. 4B). As reported previously, GFP expression was lower in fibroblasts than in adipocytes for both control and Rab10 knockdown cells (51% less in control cells and 57% less in Rab10 knockdown cells ((10); data not shown).
Glut4 endocytosis in fibroblasts was also not significantly affected by Rab10 knockdown relative to control cells (ken = 0.22 min−1 control, 0.20 min−1 Rab10 KD; Fig. 4, C and D). Thus, the increase in Glut4 ken observed in the Rab knockdown cells is specific for adipocytes and is not observed in fibroblasts. Differentiation decreases the rate constant of endocytosis of Glut4 in control cells by 40% (0.13 min−1 in adipocytes versus 0.22 min−1 in fibroblasts). We have proposed that this decrease in ken is due to redistribution of Glut4 between a fast endocytic pathway (ken = 0.6 min−1, the pathway followed by the Tf receptor) and a slow endocytic pathway (ken = 0.05 min−1) (10). We hypothesize that Rab10 knockdown affects this redistribution, as ken remains unchanged in the Rab10 knockdown cells after differentiation. We speculate that this increase may be the result of altering the proportion of Glut4 recycling via the GSV versus the constitutive pathway (Rab10 knockdown increases the proportion of ERC-derived vesicles relative to GSVs that fuse to the plasma membrane by decreasing the rate of exocytosis of GSVs).
These data show that Rab10 only affects Glut4 trafficking through the specialized trafficking pathway in adipocytes. Thus, differentiation redirects Glut4 from a Rab10-independent, constitutive recycling pathway into a Rab10-dependent highly regulated pathway through GSVs.
Exocytosis of LRP1 Was Inhibited by Rab10 Knockdown in Adipocytes but Not in Fibroblasts
LRP1 is an α2-M receptor expressed in fibroblasts and adipocytes. LRP1 traffics with Glut4 in both cell types and is co-localized with Glut4 in GSVs after differentiation (30). Thus, it is expected that proteins that limit exocytosis from the highly regulated, sequestered GSVs, but not from the constitutive pathway, will inhibit LRP1 exocytosis in adipocytes but not in fibroblasts.
To study the trafficking of LRP1, we examined the binding and uptake of its ligand α2-M (Figs. 5 and 6). α2-M binds with high affinity to LRP1 at the cell surface, but it dissociates from the LRP1 when it reaches the low pH of the endosomes. LRP1 recycles back to the plasma membrane where it binds additional ligand and is re-internalized. The labeled α2-M is retained within the cell and is eventually delivered to the lysosomes. Thus, AF647-α2-M labeling increases linearly with time. The rate of uptake is dependent on both the total amount of LRP1 at the cell surface and the rate constant of endocytosis of LRP1. The slope of the internal α2-M/surface α2-M versus time plot equals ken (32). The rate constant of exocytosis can be calculated from the surface binding data and ken (9, 10).
Consistent with its effects on Glut4 trafficking, Rab10 knockdown decreased cell surface LRP1 and the rate of AF647-α2-M uptake by 30–40% in adipocytes but not in fibroblasts (Figs. 5, A and B, 6, A and B, and Table 2). Rab10 knockdown had no significant effect on LRP1 ken (Figs. 5C, 6C, and Table 2). The decrease in α2-M uptake in adipocytes is the result of inhibition of LRP1 exocytosis in Rab10 knockdown cells. Rab10 knockdown decreased kex calc 30–40% relative to control cells in adipocytes but not fibroblasts (Figs. 5D, 6D, and Table 2).
Exocytosis of the Tf Receptor Was Not Affected by Rab10 Knockdown in Either Adipocytes or Fibroblasts
Glut4 is both internalized and recycled through pathways that are distinct from those followed by the Tf receptor (10). This occurs in both fibroblasts and adipocytes. Thus, there are at least two endocytic cycling pathways in fibroblasts and three pathways in adipocytes. The two constitutive cycling pathways (the “fast” Tf pathway, kex = 0.12–0.2 min−1, ken = 0.6 min−1; and the slower Glut4 pathway, kex = 0.025–0.04 min−1, ken = 0.12–0.2 min−1) have very similar kinetics in fibroblasts and adipocytes, strongly suggesting that both of these pathways are conserved after differentiation. Differentiation leads to the expression of a third cycling pathway, the GSV pathway that is highly regulated and is insulin-responsive (kex = 0.0007 min−1 basal, 0.03 min−1 after insulin stimulation). While Glut4 and LRP1 traffic through the slow constitutive recycling pathway as well as through regulated GSVs, the Tf receptor is largely excluded from both of these pathways. Thus, it is expected that knockdown of a Rab protein required for GSV exocytosis should have no effect on Tf receptor cycling.
To examine Tf receptor trafficking, we examined the rate of efflux of Tf from preloaded endosomes as well as the cell surface levels of the Tf receptor in both adipocytes and fibroblasts (Figs. 7 and 8). In these experiments, cells are incubated for 30 min with AF647-holo-Tf. The cells are then washed to remove the excess labeled Tf, and the rate of loss of intracellular Tf is measured at increasing times. The observed relaxation rate constant is equal to kex for the Tf receptor. Consistent with a role in GSV exocytosis, Rab10 knockdown had no effect on Tf receptor trafficking kinetics in either adipocytes or fibroblasts (Figs. 7, 8, and Table 2). Interestingly, Rab10 knockdown increased the total amount of AF647-Tf taken up relative to control cells in both adipocytes and fibroblasts (1.6- and 1.4-fold). However, kex was not significantly affected by Rab10 knockdown (kex = 0.09 min−1 in adipocytes, 0.12 min−1 in fibroblasts; p = 0.92–0.94 that the data from Rab10 knockdown and control cells is best fit by exponential functions that share the same relaxation rate constants). The increase in uptake is due to an increase in the total number of Tf receptors in Rab10 knockdown cells; Rab10 knockdown cells also increased cell surface Tf receptor 1.6–1.7-fold in adipocytes and 1.3–1.7-fold in fibroblasts (as determined by α-Tf receptor antibody binding). Thus, although Rab10 knockdown did not affect the endocytic cycling of the Tf receptor, it affected the total amount of Tf receptor expressed in both adipocytes and fibroblasts. We suggest that this increase in total Tf receptor expression may be through effects on the degradation of the Tf receptor, as observed for HA-Glut4/GFP (Figs. 1 and 4) (10).
Modeling and Simulations, Insulin Stimulates Both Sorting into and Exocytosis from Specialized, Regulated Secretory Compartments in Control Adipocytes
Although the two-step/three-compartment model (static retention) is useful for data analysis, it is insufficient to account for the complex kinetics behaviors observed for Glut4. We have previously shown that a number of independent steps contribute to the unique trafficking of Glut4 relative to the transferrin receptor (TfR) in adipocytes (Fig. 9A (10)). These steps include the following: 1) endocytosis from the PM into SE through a slow internalization pathway; 2) sorting from endosomes into and 3) recycling from a slow constitutive pathway through ERC, a pathway distinct from the fast (TfR) recycling pathway; 4) sorting from endosomes into a specialized, highly regulated sequestration pathway through GSVs; 5) release of GSVs from sequestration/GSV priming; 6) tethering/docking/fusion of primed GSVs; and 7) retrieval of Glut4 from lysosomal degradation (pathway not shown). Release/priming involves GTP loading and activation of a Rab protein (or proteins) bound to the sequestered GSVs. This Rab protein is inhibited by the AS160-Rab GAP. Sorting from the endosomes into the sequestered GSVs is also regulated by AS160, through an unknown mechanism (10). The tethering/docking/fusion step is regulated by Akt and PI 3-kinase through an AS160-independent process (11, 16). The regulated GSV pathway (steps 4–6) is observed only in differentiated adipocytes and not in fibroblasts. LRP1 cycles through both of the pathways followed by Glut4, as well as through the fast TfR pathway (10).
This model can be described mathematically as a series of ordinary differential equations that describe the transfer of Glut4 between four compartments (PM, SE, ERC, and GSV), with a single rate constant for each of the five steps (Fig. 9B). These rate constants are as follows: ken, endocytosis from plasma membrane to sorting endosomes; ksort, sorting from endosomes into the ERC; kfuseE, transport from the ERC and fusion to the plasma membrane; kseq, sorting from endosomes into the sequestered GSVs, and kfuseG, release and fusion of sequestered GSVs to the plasma membrane (in the mathematical model, step 5, priming, and step 6, tethering/docking/fusion, were combined to simplify the data fitting and simulations). In fibroblasts, this model reduces to a three-step/three-compartment model (kseq and kfuseG = 0; Fig. 9B, black dotted line). LRP1 has an additional route of recycling from the sorting endosomes to the plasma membrane that it shares with the TfR, with a single rate constant, krec (Fig. 9B, gray dotted line). Glut4 cannot efficiently exit via this fast recycling pathway (Fig. 9B, krec, Glut4 = 0). Sorting endosomes are defined functionally as the compartment(s) where Glut4, LRP1, and the TfR are sorted into three exocytic pathways as follows: the fast TfR recycling pathway; the slower constitutive endosomal ERC pathway; and the highly regulated, specialized GSV pathway. The ERC is defined functionally as a recycling intermediate between sorting endosomes and the plasma membrane that accumulates behind a rate-limiting step in the slow constitutive pathway. All of the trafficking assays for both Glut4 and LRP1 can be accurately simulated using these differential equations (10).
To estimate the values of the rate constants, simultaneous free fits were done for the fibroblast data (Fig. 4, A and B) or the adipocyte data (Figs. 1C, 2, A and B, and 3) for control cells and the Rab10 knockdown cells. To test the hypothesis that Rab10 knockdown affects one or more of these rate constants, ken, ksort, kfuseE, kfuseG, and kseq were either modeled as being shared between the control and Rab10 knockdown cells or as unique for each cell type. Fits of fibroblast data were done by setting kfuseG and kseq to 0 (Glut4 does not traffic through the GSV pathway in fibroblasts). Software was developed to allow testing of every possible combination of hypotheses (8 alternative fits in fibroblasts, 32 alternative fits in adipocytes; see supplemental material).
Free unconstrained fits of the data from control 3T3-L1 fibroblasts yielded the following estimates of the rate constants: ken = 0.18 min−1; ksort = 0.053 min−1; kfuseE, basal = 0.03 min−1; kfuseE, insulin = 0.08 min−1 (Table 3). Although there was no statistically significant differences measured in fibroblasts, the best fit of the data included a small (20%) decrease in ksort in Rab10 knockdown versus control cells. These rate constants yielded excellent simulations of the transition data in fibroblasts in both cell types (Fig. 4A).
TABLE 3.

* kfuseE constrained to the Fibroblast values. PM inf, inferred values.
† Simultaneous fits to test hypotheses that all rate constants are shared between Control and Rab10 knockdown cells except those listed.
Free unconstrained fits of the data from control adipocytes yielded the following estimates for the rate constants: ken = 0.12 min−1; ksort = 0.014 min−1; kfuseE, basal = 0.43 min−1; kfuseE, insulin =0.07 min−1; kfuseG, basal = 0.0005 min−1; kfuseG, insulin = 0.033 min−1; kseq, basal = 0.003 min−1; and kseq, insulin = 0.35 min−1. With the exceptions of ken and kfuseE, basal, these rate constants are very similar to those calculated from kinetics and subcellular distribution data for primary adipocytes reported previously (10). The measured differences in ken between primary adipocytes and 3T3-L1 cells have been previously described. In these unconstrained fits there are two steps that are highly regulated by insulin as follows: exocytosis from GSVs (kfuseG) and sorting from the endosomes into the sequestered GSVs (kseq) (insulin increases both >60-fold). Regulation of both of these steps was observed in all 32 of the unconstrained simultaneous fits of data from control and Rab10 knockdown cells (Table 3 and supplemental material).
In the unconstrained fits, kfuseE is highly variable. This reflects the fact that this rate constant is not rate-limiting for Glut4 trafficking and is therefore not well defined (it can vary significantly with no effect on the overall kinetics in the simulations). In the free fits of the adipocyte data, the constitutive pathway in basal cells is essentially reduced to a single step (kfuseE, basal ≫ ksort; 0.5 versus 0.01–0.014 min−1). However, constraining the fits by requiring adipocytes and fibroblasts to share the same kfuseE (kfuseE, basal = 0.03 min−1 and kfuseE, insulin =0.08 min−1) did not significantly alter the values for any of the other estimated rate constants, and the simulations from the two fits were indistinguishable (Table 3 and data not shown). Thus, the unconstrained simultaneous fits of the data from control and Rab10 knockdown adipocytes were used in all simulations (Fig. 10).
FIGURE 10.
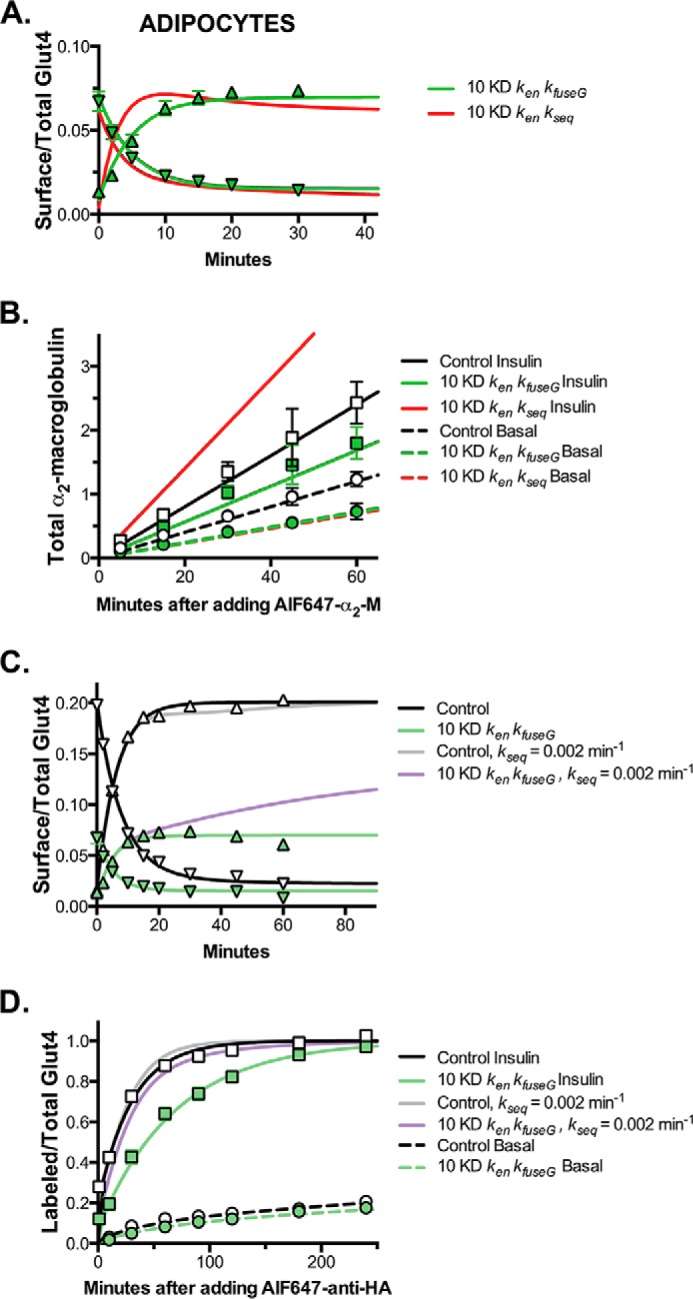
Modeling and simulations, Rab10 knockdown inhibits exocytosis from GSVs and Glut4 continues to cycle through the Rab10-dependent GSVs after insulin stimulation in adipocytes. A and C, basal to insulin transitions, surface/total Glut4 (data from Fig. 1C; up triangles) and insulin + LYi transitions (data from Fig. 3; down triangles). B, α2-M uptake (data from Fig. 5B; circles, basal; squares, insulin). D, anti-HA uptake (data from Fig. 2; circles, basal; squares, insulin). Two hypotheses were compared for Rab10 knockdown. Rab10 knockdown affects the following: 1) ken and kfuseG (green lines), and 2) ken and kseq (red lines). The control experiments were simulated with the best overall fit (black lines). We also tested the hypothesis that 3) Glut4 and LRP1 redistribute from the GSVs into the endosomal recycling pathway after insulin simulation (kseq, basal = kseq, insulin = 0.002 min−1; control, gray lines; Rab10 knockdown, purple lines). The only hypothesis tested that accurately simulates the effects of Rab10 knockdown on Glut4 and LRP1 trafficking is hypothesis 1, Rab10 knockdown affects ken and kfuseG.
Modeling and Simulations, Rab10 Limits Exocytosis from Specialized, Regulated Secretory Compartments in Adipocytes
Rab10 knockdown inhibited exocytosis of Glut4 and LRP1 in adipocytes but not in fibroblasts. Thus, although Rab10 affects protein trafficking in many cell types, it only affects the trafficking of Glut4 and LRP1 through the highly regulated, specialized GSV pathway. It does not affect the slow constitutive recycling pathway. Therefore, Rab10 knockdown must affect either release and fusion of GSVs (kfuseG) or trafficking from endosomes into the sequestered GSVs (kseq). Rab10 knockdown also accelerated Glut4 endocytosis in adipocytes. Therefore, to model the effect of Rab10 knockdown on Glut4 trafficking, three hypotheses were compared as follows: Rab10 KD affects 1) ken and kfuseG; 2) ken and kseq; or 3) ken, kfuseG, and kseq (Fig. 10, A and B, and Table 2). In the fit testing hypothesis 1, ken and kfuseG were significantly different in Rab10 KD adipocytes compared with control cells (Table 3). These rate constants accurately simulated all of the Glut4 data as well as the α2-M uptake data in Rab10 knockdown cells (Fig. 10, A and B, green lines). In contrast, the fit of hypothesis 2 did not yield good simulations of the kinetics of the Glut4 transitions (Fig. 10A, red lines). Both the basal to insulin and the insulin + LYi transitions were too fast (ken > 0.3 min−1), and there was an overshoot that was not observed in the data. Hypothesis 2 also did not yield good simulations of the α2-M uptake data in Rab10 knockdown cells (Fig. 10B, red lines). Inhibition of kseq in insulin-stimulated cells would cause an increase, not an inhibition, of LRP1 exocytosis and α2-M uptake. Inhibition of kseq would cause a 10-fold increase in the amount of LRP1 in endosomes at steady state after insulin stimulation (supplemental material; control SE = 5%; Rab10 KD S.E. = 45%). This redistribution of LRP1 increases its exocytosis through the fast TfR recycling pathway (Glut4 cannot exit via this pathway). In contrast, inhibition of kfuseG causes a 2-fold decrease in the amount of Glut4 in the endosomes after insulin stimulation. The fit testing hypothesis 3 that allows both kseq and kfuseG to vary between cell types was not significantly different from the fit of hypothesis 1 that allows only kfuseG to vary (Table 3). These results show that effects on endocytosis (ken) and GSV release and fusion (kfuseG)are sufficient to accurately simulate all of the phenotypes observed in the Rab10 knockdown adipocytes.
Discussion
Modeling and simulations show that inhibition of the release and fusion of GSVs is sufficient to account for all of the trafficking phenotypes observed in Rab10 knockdown cells (Fig. 9). Placement of Rab10 at the release step regulated by AS160 is consistent with previous studies, which observed an attenuation of Glut4 translocation in Rab10 knockdown cells, and an increase in surface Glut4 in basal cells when a GTP-hydrolysis resistant dominant active mutant was expressed in adipocytes (20, 22). Additionally, knockdown of Rab10 partially rescued the increase in basal surface Glut4 observed in AS160 knockdown cells, and overexpression of Rab10 enhanced the effect of AS160 knockdown in basal cells, suggesting the proteins work at the same step (20, 23). Finally, greater than 90% of Glut4 and IRAP-containing vesicles fusing to the plasma membrane immediately after stimulation by insulin (and release of GSVs from sequestration) are decorated with Rab10 (22). Very few of these vesicles also contained the Tf receptor.
Rab10 is expressed in both differentiated and undifferentiated cells, whereas the Rab GAP AS160 is expressed only after adipocyte differentiation. Therefore, one simple mechanism for inhibition of Glut4 exocytosis in adipocytes, and generation of the GSV pathway, would be to put a break on a constitutive Rab10-dependent pathway by expression of AS160 (Fig. 9A). In this model, expression and targeting of AS160 to vesicles in a Rab10-dependent constitutive exocytic pathway would be sufficient to induce the formation of the insulin-responsive GSVs. Inconsistent with this hypothesis, however, expression of AS160, even the insulin-resistant mutant form, AS160–4P, in fibroblasts is insufficient to inhibit Glut4 trafficking (data not shown). Furthermore, knockdown of Rab10 did not inhibit Glut4 exocytosis in fibroblasts (Fig. 4), although Rab10 is expressed and functional in these cells (Fig. 8). Thus, differentiation induces the expression of proteins that cause Glut4 to be sorted from a constitutive Rab10-independent pathway into a Rab10-dependent secretory pathway. This pathway is highly regulated in adipocytes (inhibited in basal cells and insulin responsive). Therefore, Rab10 is a functional marker of the specialized GSV pathway.
It has been suggested that Glut4 cycles predominantly through the GSV pathway in basal cells and that it shifts into the endosomal pathway after insulin stimulation (33, 34). However, although the majority of Glut4 is found in GSVs in basal cells (80–90%), and very little is found in the ERC (≤ 5%), the majority of the Glut4 that is actively cycling in basal cells is moving through the constitutive and not the GSV pathway. This is due to the difference in the rates of exocytosis from these two compartments. Our models indicate that the majority of vesicles (3 out of 4) fusing to the plasma membrane under basal conditions come from the constitutive ERC and not from the regulated GSV pathway. Consistent with this, the Glut4 and IRAP-containing vesicles that are fusing with the plasma membrane under basal conditions contain Rab14 but not Rab10 (22). Immediately after insulin stimulation, there is a burst of GSV exocytosis, as the sequestered vesicles are released and fused. Under these conditions, the majority of the Glut4 and IRAP-containing vesicles fusing to the plasma membrane are GSVs that contain Rab10. But what happens after the cells reach a new steady state? The static retention model proposes that Glut4 would cycle through the constitutive pathway until insulin is withdrawn (34). In contrast, in our model Glut4 will continue to cycle predominantly through the GSV pathway. Even after the cells have reached a new steady-state distribution, exocytosis from the GSV pathway will be higher than exocytosis from the ERC pathway (with a ratio of >10:1).
In order for there to be a pool of Glut4 cycling through the constitutive ERC pathway in basal adipocytes, the rate constant for trafficking of Glut4 into the GSVs from the endosomes (kseq) must be low relative to the rate constant for sorting into the ERC pathway in basal cells (ksort; unconstrained simultaneous free fits of the data yield the estimates kseq = 0.002 min−1; ksort =0.014 min−1). Otherwise, all of the Glut4 would accumulate in the very slowly cycling sequestered GSVs under basal conditions (kfuseG ≈0.0005 min−1; in the extreme form of this model static retention, kseq = 0). This is what was observed in unconstrained simultaneous free fits of the adipocyte data (Table 3 and supplemental material). In response to insulin, there is a large increase in both kseq and kfuseG (kseq≈ 0.35 min−1; kfuseG ≈ 0.033 min−1) in fits of all of the tested models. The increase in kseq is consistent with electron microscopy data in primary adipocytes that show that the rate of flux out of endosomes (kseq + ksort) must increase at least 8-fold after insulin stimulation (10). This increase in kseq drives Glut4 through the GSVs rather than ERC after insulin stimulation.
To test the hypothesis that Glut4 cycles through the GSVs and not the constitutive ERC pathway after insulin stimulation, our model was modified to drive exocytosis through the ERC after insulin stimulation by not allowing the increase in kseq (kseq, basal = kseq, insulin = 0.002 min−1). This model can accurately simulate the behavior of Glut4 in control adipocytes (Fig. 10, C and D, gray line). However, it does not accurately simulate the behaviors observed in the Rab10 knockdown cells. Although Rab10 knockdown does decrease cell surface Glut4 in the basal to insulin transition at early times in this model (Fig. 10C, purple line), there would be a slow increase in Glut4 at the cell surface in the Rab10 knockdown cells if Glut4 predominantly cycled through the ERC after insulin stimulation. This increase would occur because cycling through the ERC is not affected by Rab10 knockdown; thus, there would be an increase in Glut4 kex as Glut4 is internalized and redistributed into the Rab10-independent pathway. In contrast to this prediction, we observed that cell surface Glut4 rapidly reaches a new steady state after insulin stimulation in the Rab10 knockdown cells (t½ <5 min), then the levels remain stable or decrease with increasing incubation time (Fig. 10C, green triangles). The ERC cycling model also predicts that after the cells reach their new steady-state distribution of Glut4 between the ERC and GSV compartments, there would be very little difference in the α-HA uptake experiments in the two cell lines after insulin stimulation (Fig. 10D, purple line). Again, this would occur because Rab10 knockdown does not affect recycling through the constitutive ERC pathway. This prediction is also inconsistent with our data. Even after a pre-incubation of 45 min to several hours, the defect in exocytosis is still observed in the Rab10 KD cells (Fig. 10D, green squares, and data not shown). Thus, our simulations indicate that Glut4 continues to cycle predominantly through the Rab10-dependent GSV pathway after insulin stimulation and that Rab10 dependence is a key distinguishing feature of the specialized Glut4 sequestration pathway versus the constitutive recycling pathways in adipocytes. They also support the idea that both trafficking into GSVs (kseq) and the fusion of GSVs to the plasma membrane (kfuseG) are regulated by insulin (10).
Rab10 has a variety of effectors that may be required for steps downstream of Rab10 activation in Glut4 trafficking. These effectors play roles in almost every step of the vesicle trafficking cycle, including sorting, vesicle formation (budding, coating), motility, and tethering. Rab10 effectors with likely roles in sorting/budding of vesicles include AP1σ1, a clathrin adaptor subunit (35), and EHBP1 (36), a protein that interacts with the dynamin-like protein EHD1; both EHBP1 and EHD1 are required for Glut4 translocation (37). Another Rab10 effector in adipocytes with a possible role in budding is TBC1D13, a Rab GAP for Rab35 (38), which in neurons can recruit the Arf6 GAP ACAP2 (39). These interactions suggest a model where Rab10 activation would inactivate Rab35, allowing activation of Arf6, a GTPase involved in vesicle budding. Interestingly, the ACAP1 isoform acts as a clathrin adaptor in endosomes, interacts with the central loop of Glut4, and is required for sorting into the specialized GSV compartment (40). Our LRP1 data (Fig. 10B) suggest that any sorting functions of Rab10 would occur in compartments such as the trans-Golgi network that are in communication with, but are distinct from, the constitutive recycling pathways.
Rab10 is also likely to be involved in insulin-regulated transport of GSVs through a direct interaction with myosin Va (41). Myosin Va is required for insulin-stimulated Glut4 translocation, is phosphorylated by Akt in response to insulin, and localizes to the Glut4 vesicles that fuse with the plasma membrane (15, 22). Myosin Va and Rab10 also are likely to participate in the tethering/docking of vesicles to the plasma membrane as these proteins interact with the Sec15/Exo6/6b subunits of the exocyst, a tethering complex that is required for insulin-stimulated Glut4 translocation (42–46). GSVs are also tethered to the exocyst by the small GTPase RalA (47, 48); Rab10 was recently shown to activate RalA by recruiting the RalA GEF Rlf/Rgl2 (49). Therefore, there may be multiple functional interactions between Rab10 and effectors that take place during the rate-limiting vesicle priming/fusion step in Glut4 exocytosis. Consistent with the known function of these Rab10 effector proteins, Rab10 knockdown decreased the accumulation of Glut4 in and near the plasma membrane after insulin stimulation (22–24). However, it did not inhibit the insulin-stimulated increase in Glut4 insertion efficiency, indicating that fusion itself is regulated through an additional mechanism downstream of the GTP-loading/activation of Rab10, perhaps through direct regulation of the Rab10 effectors (11, 15, 16) or through other elements of the downstream fusion machinery (50).
Author Contributions
C. C. M. conceived and coordinated the study, analyzed the data, and wrote the paper. C. C. M. and A. C. F. C. designed, performed, and analyzed the mathematical modeling and simulations. P. D. B. designed, performed and analyzed the experiments shown in Figs. 1–4. E. N. H. designed, performed, and analyzed the experiments shown in Figs. 5 and 6. I. R. designed, performed, and analyzed the experiments shown in Figs. 7 and 8. I. R. provided technical assistance and contributed to the preparation of all figures. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by grants from the American Diabetes Association. The authors declare that they have no conflicts of interest with the contents of this article.

This article contans supplemental Table S1.
Glut4 cycles through a slow endosomal pathway in fibroblasts and other undifferentiated cells that is distinct from the fast recycling pathway followed by the TfR (kex = 0.12–0.2 min−1, t½ of 3–6 min).
P. D. Brewer, E. N. Habtemichael, I. Romenskaia, C. C. Mastick, and A. C. F. Coster, submitted for publication.
Glut4 that is not cycling, e.g. that is accumulated in biosynthetic or degradative compartments or sequestered in non-cycling GSVs does not traffic to the plasma membrane and does not contribute to cell surface Glut4.
- GSV
- Glut4 storage vesicle
- α2-M
- α2-macroglobulin
- α-HA
- anti-HA antibody
- AF647
- AlexaFluor647
- ERC
- endosomal recycling intermediate compartment
- LSM
- low serum media
- In/Sur
- internal/surface
- LYi
- PI 3-kinase inhibitor LY294002
- PM
- plasma membrane
- SE
- sorting or “early” endosome(s)
- Tf
- transferrin
- PI
- phosphatidylinositol
- GAP
- GTPase-activating protein
- MFR
- mean fluorescence ratio
- TfR
- Tf receptor.
References
- 1.James D. E., Brown R., Navarro J., and Pilch P. F. (1988) Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 333, 183–185 [DOI] [PubMed] [Google Scholar]
- 2.Slot J. W., Geuze H. J., Gigengack S., Lienhard G. E., and James D. E. (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113, 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malide D., Ramm G., Cushman S. W., and Slot J. W. (2000) Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells. J. Cell Sci. 113, 4203–4210 [DOI] [PubMed] [Google Scholar]
- 4.Muretta J. M., and Mastick C. C. (2009) How insulin regulates glucose transport in adipocytes. Vitam. Horm. 80, 245–286 [DOI] [PubMed] [Google Scholar]
- 5.Govers R., Coster A. C., and James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coster A. C., Govers R., and James D. E. (2004) Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic 5, 763–771 [DOI] [PubMed] [Google Scholar]
- 7.Martin O. J., Lee A., and McGraw T. E. (2006) GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J. Biol. Chem. 281, 484–490 [DOI] [PubMed] [Google Scholar]
- 8.Muretta J. M., Romenskaia I., and Mastick C. C. (2008) Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J. Biol. Chem. 283, 311–323 [DOI] [PubMed] [Google Scholar]
- 9.Habtemichael E. N., Brewer P. D., Romenskaia I., and Mastick C. C. (2011) Kinetic evidence that Glut4 follows different endocytic pathways than the receptors for transferrin and α-2-macroglobulin. J. Biol. Chem. 286, 10115–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brewer P. D., Habtemichael E. N., Romenskaia I., Mastick C. C., and Coster A. C. (2014) Insulin-regulated Glut4 translocation: membrane protein trafficking with six distinctive steps. J. Biol. Chem. 289, 17280–17298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brewer P. D., Romenskaia I., Kanow M. A., and Mastick C. C. (2011) Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J. Biol. Chem. 286, 26287–26297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 13.Klip A., Sun Y., Chiu T. T., and Foley K. P. (2014) Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 306, C879–C886 [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto K., and Holman G. D. (2008) Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 295, E29–E37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshizaki T., Imamura T., Babendure J. L., Lu J. C., Sonoda N., and Olefsky J. M. (2007) Myosin 5a is an insulin-stimulated Akt2 (protein kinase Bbeta) substrate modulating GLUT4 vesicle translocation. Mol. Cell. Biol. 27, 5172–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang L., Fan J., Bai L., Wang Y., Chen Y., Yang L., Chen L., and Xu T. (2008) Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J. Biol. Chem. 283, 8508–8516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cormont M., Tanti J. F., Zahraoui A., Van Obberghen E., Tavitian A., and Le Marchand-Brustel Y. (1993) Insulin and okadaic acid induce Rab4 redistribution in adipocytes. J. Biol. Chem. 268, 19491–19497 [PubMed] [Google Scholar]
- 18.Larance M., Ramm G., Stöckli J., van Dam E. M., Winata S., Wasinger V., Simpson F., Graham M., Junutula J. R., Guilhaus M., and James D. E. (2005) Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 280, 37803–37813 [DOI] [PubMed] [Google Scholar]
- 19.Mîinea C. P., Sano H., Kane S., Sano E., Fukuda M., Peränen J., Lane W. S., and Lienhard G. E. (2005) AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 391, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sano H., Eguez L., Teruel M. N., Fukuda M., Chuang T. D., Chavez J. A., Lienhard G. E., and McGraw T. E. (2007) Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303 [DOI] [PubMed] [Google Scholar]
- 21.Sano H., Roach W. G., Peck G. R., Fukuda M., and Lienhard G. E. (2008) Rab10 in insulin-stimulated GLUT4 translocation. Biochem. J. 411, 89–95 [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Wang Y., Zhang J., Deng Y., Jiang L., Song E., Wu X. S., Hammer J. A., Xu T., and Lippincott-Schwartz J. (2012) Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 198, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadacca L. A., Bruno J., Wen J., Xiong W., and McGraw T. E. (2013) Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol. Biol. Cell 24, 2544–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed S. E., Hodgson L. R., Song S., May M. T., Mastick C. C., Verkade P., and Tavare J. M. (2013) A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. J. Cell Sci. 126, 1931–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishikura S., Bilan P. J., and Klip A. (2007) Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem. Biophys. Res. Commun. 353, 1074–1079 [DOI] [PubMed] [Google Scholar]
- 26.Ishikura S., and Klip A. (2008) Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am. J. Physiol. Cell Physiol. 295, C1016–C1025 [DOI] [PubMed] [Google Scholar]
- 27.Sun Y., Bilan P. J., Liu Z., and Klip A. (2010) Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 107, 19909–19914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan S. X., Ng Y., Burchfield J. G., Ramm G., Lambright D. G., Stöckli J., and James D. E. (2012) The Rab GTPase-activating protein TBC1D4/AS160 contains an atypical phosphotyrosine-binding domain that interacts with plasma membrane phospholipids to facilitate GLUT4 trafficking in adipocytes. Mol. Cell. Biol. 32, 4946–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koumanov F., Richardson J. D., Murrow B. A., and Holman G. D. (2011) AS160 Phosphotyrosine-binding domain constructs inhibit insulin-stimulated GLUT4 vesicle fusion with the plasma membrane. J. Biol. Chem. 286, 16574–16582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jedrychowski M. P., Gartner C. A., Gygi S. P., Zhou L., Herz J., Kandror K. V., and Pilch P. F. (2010) Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J. Biol. Chem. 285, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., and McGraw T. E. (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2, 263–272 [DOI] [PubMed] [Google Scholar]
- 32.Wiley H. S., and Cunningham D. D. (1982) The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 257, 4222–4229 [PubMed] [Google Scholar]
- 33.Xu Y., Rubin B. R., Orme C. M., Karpikov A., Yu C., Bogan J. S., and Toomre D. K. (2011) Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J. Cell Biol. 193, 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stöckli J., Fazakerley D. J., and James D. E. (2011) GLUT4 exocytosis. J. Cell Sci. 124, 4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ewing R. M., Chu P., Elisma F., Li H., Taylor P., Climie S., McBroom-Cerajewski L., Robinson M. D., O'Connor L., Li M., Taylor R., Dharsee M., Ho Y., Heilbut A., Moore L., et al. (2007) Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi A., Chen C. C., Banerjee R., Glodowski D., Audhya A., Rongo C., and Grant B. D. (2010) EHBP-1 functions with RAB-10 during endocytic recycling in Caenorhabditis elegans. Mol. Biol. Cell 21, 2930–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guilherme A., Soriano N. A., Furcinitti P. S., and Czech M. P. (2004) Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J. Biol. Chem. 279, 40062–40075 [DOI] [PubMed] [Google Scholar]
- 38.Davey J. R., Humphrey S. J., Junutula J. R., Mishra A. K., Lambright D. G., James D. E., and Stöckli J. (2012) TBC1D13 is a RAB35 specific GAP that plays an important role in GLUT4 trafficking in adipocytes. Traffic 13, 1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi H., and Fukuda M. (2012) Rab35 regulates Arf6 activity through centaurin-β2 (ACAP2) during neurite outgrowth. J. Cell Sci. 125, 2235–2243 [DOI] [PubMed] [Google Scholar]
- 40.Li J., Peters P. J., Bai M., Dai J., Bos E., Kirchhausen T., Kandror K. V., and Hsu V. W. (2007) An ACAP1-containing clathrin coat complex for endocytic recycling. J. Cell Biol. 178, 453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roland J. T., Lapierre L. A., and Goldenring J. R. (2009) Alternative splicing in class V myosins determines association with Rab10. J. Biol. Chem. 284, 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Babbey C. M., Bacallao R. L., and Dunn K. W. (2010) Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am. J. Physiol. Renal Physiol. 299, F495–F506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X. W., and Saltiel A. R. (2011) Ral's engagement with the exocyst: breaking up is hard to do. Cell Cycle 10, 2299–2304 [DOI] [PubMed] [Google Scholar]
- 44.Inoue M., Chang L., Hwang J., Chiang S. H., and Saltiel A. R. (2003) The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature 422, 629–633 [DOI] [PubMed] [Google Scholar]
- 45.Jin Y., Sultana A., Gandhi P., Franklin E., Hamamoto S., Khan A. R., Munson M., Schekman R., and Weisman L. S. (2011) Myosin V transports secretory vesicles via a Rab GTPase cascade and interaction with the exocyst complex. Dev. Cell 21, 1156–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sano H., Peck G. R., Blachon S., and Lienhard G. E. (2015) A potential link between insulin signaling and GLUT4 translocation: association of Rab10-GTP with the exocyst subunit Exoc6/6b. Biochem. Biophys. Res. Commun. 465, 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X. W., Leto D., Chiang S. H., Wang Q., and Saltiel A. R. (2007) Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev. Cell 13, 391–404 [DOI] [PubMed] [Google Scholar]
- 48.Chen X. W., Leto D., Xiong T., Yu G., Cheng A., Decker S., and Saltiel A. R. (2011) A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol. Biol. Cell 22, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karunanithi S., Xiong T., Uhm M., Leto D., Sun J., Chen X. W., and Saltiel A. R. (2014) A Rab10:RalA G protein cascade regulates insulin-stimulated glucose uptake in adipocytes. Mol. Biol. Cell 25, 3059–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jewell J. L., Oh E., Ramalingam L., Kalwat M. A., Tagliabracci V. S., Tackett L., Elmendorf J. S., and Thurmond D. C. (2011) Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J. Cell Biol. 193, 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.