

Abstract
Chromodomain Helicase DNA-binding protein 4 (CHD4) is a chromatin-remodeling enzyme that has been reported to regulate DNA-damage responses through its N-terminal region in a poly(ADP-ribose) polymerase-dependent manner. We have identified and determined the structure of a stable domain (CHD4-N) in this N-terminal region. The-fold consists of a four-α-helix bundle with structural similarity to the high mobility group box, a domain that is well known as a DNA binding module. We show that the CHD4-N domain binds with higher affinity to poly(ADP-ribose) than to DNA. We also show that the N-terminal region of CHD4, although not CHD4-N alone, is essential for full nucleosome remodeling activity and is important for localizing CHD4 to sites of DNA damage. Overall, these data build on our understanding of how CHD4-NuRD acts to regulate gene expression and participates in the DNA-damage response.
Keywords: chromatin remodeling, DNA binding protein, DNA damage, nucleosome, nucleosome remodeling deacetylase (NuRD), CHD4, HMG-box, poly(ADP-ribose)
Introduction
The nucleosome remodeling and deacetylase (NuRD)3 complex is a conserved transcription co-regulatory complex that is found in all complex animals. It is unique in that it contains both nucleosome remodeling and deacetylase activity (1–6); other co-regulator complexes such as Sin3 (7), Rpd3S (8), and PRC2 (9) only carry a single enzymatic activity. The NuRD complex has been shown to have essential roles in both activation and repression of gene transcription and has links to stem cell renewal and differentiation (10), cell cycle control (11), cancer (for review see Ref. 12), and the DNA-damage response, including DNA double strand break (DSB) repair (13). DSBs are highly cytotoxic; if repaired erroneously or left unrepaired, they can lead to cancer, neurodegeneration, and immunodeficiency (14).
One of the defining components of the NuRD complex is chromodomain helicase DNA-binding protein 4 (CHD4). CHD4 mediates the chromatin remodeling activity of the NuRD complex via a SNF2-type DNA translocase domain (15), and evidence has accumulated that CHD4 is important for an effective DNA-damage response and for the maintenance of genomic integrity. For example, the expression level of CHD4 has been shown to increase upon UV irradiation (16). CHD4 has been also reported to be a target of the DNA-damage response apical kinases ATM and ATR (17–19) and to be recruited to DSB sites as part of the NuRD complex (20). This recruitment occurs in a poly(ADP-ribose) polymerase (PARP)-dependent manner. It has been shown that a 750-residue N-terminal fragment of CHD4 (residues 1–758) was sufficient for its recruitment and that this region binds poly(ADP-ribose) (PAR) polymers with the same apparent affinity as full-length CHD4 (20), suggesting that the N-terminal portion of CHD4 contains one or more PAR binding motifs. Similarly, the Drosophila homologue of CHD4 (dMi-2) has been shown to be recruited to active heat shock genes in a PARP-dependent manner (21). Proteomic studies have also indicated that CHD4 and other proteins of the CHD family can undergo poly-ADP-ribosylation; however, the biological effects of this modification are still unknown (22, 23).
Poly(ADP-ribose) molecules are linear or multibranched chains of ADP-ribose. These polymers are synthesized by PARPs using NAD+ as a substrate and are attached covalently to proteins as a post-translational modification (for review see Ref. 24). Although still somewhat mysterious, PARylation has been shown to inhibit protein-protein and protein-DNA interactions, promote or alter protein localization, and regulate protein modifications such as ubiquitylation (for review see Ref. 25). As signaling molecules, PARylated proteins have roles in a number of cellular processes, including DNA repair and programmed cell death. In recent years, several PAR binding domains have been identified and their structures determined. For example, the DNA repair factor aprataxin polynucleotide kinase-like (APLF) has been shown to interact with PAR through its two PAR-binding zinc finger (PBZ) domains (26). The structure of the PAR binding macrodomain, a module present in histone macroH2A1.1, has been determined in complex with ADP-ribose (27). The apoptosis-inducing factor (AIF), which has a critical role in PARP1-mediated cell death (28), has also been shown to bind PAR with high affinity; however, there is no structural information on this interaction. These data suggest that several distinct modes of PAR recognition exist and that interactions occur either through stacking between tyrosine/phenylalanine residues and the adenine rings or through electrostatic interactions between basic residues and the phosphate moiety of ADP-ribose.
In addition to its SNF2 DNA translocase domain, CHD4 possesses two conserved chromodomains that are a feature of all CHD proteins and that are known to regulate the activity of the SNF2 domain (29) (Fig. 1a). Two additional plant homeodomains (PHDs) are present upstream of the chromodomains and are also found in the paralogues CHD3 and CHD5. The CHD4 PHDs bind the N-terminal tail of histone H3, exhibiting a preference for trimethylated or acetylated Lys-9 (30, 31) and potentially conferring some substrate specificity to the CHD4-NuRD complex. Unlike CHD1 (32–34), which carries a DNA binding SANT-SLIDE domains in the C-terminal region, CHD4 has not yet been shown to contain any C-terminal DNA binding domains. The N-terminal region of CHD4, which we define here as the ∼360 residues upstream of the PHD domains, is conserved in CHD3/4 orthologues but absent in other members of the CHD family. This sequence lacks appreciable similarity to known domains and is rich in stretches of basic and acidic residues.
FIGURE 1.

CHD4 contains a conserved and uncharacterized N-terminal region. a, domain organization of human CHD4 and the related yeast proteins ISW1 and CHD1. PHD, chromo (CHR), and SNF2-type helicase domains are shown as well as the HAND, SANT, and SLIDE DNA binding domains. b, sequence alignment of the N-terminal region of human CHD4-(1–364) and several eukaryotic orthologues. The secondary structure of CHD4-N is shown above the alignment. The sequence alignment was generated by ALINE (72).
The focus of this study was to probe the role of the N-terminal region in the function of human CHD4. We identified a conserved domain in the N-terminal region of CHD4 (CHD4-N) and determined its three-dimensional structure by solution NMR spectroscopy. CHD4-N displays structural similarity to the high mobility group (HMG)-box fold, a known DNA binding module that has roles in a wide variety of nuclear processes (for review see Ref. 35) but no known affinity for PAR. We performed microscale thermophoresis (MST) assays and NMR experiments to assess the nucleic acid binding properties of CHD4-N and carried out chromatin remodeling assays and transient transfections coupled with laser micro-irradiation to assess whether the N-terminal region of CHD4 plays a role in localizing CHD4 to DSB sites. Our data show that the CHD4-N domain can recognize PAR and that the remainder of the N-terminal region of CHD4 enhances the chromatin remodeling activity of CHD4 and is important for directing CHD4 to DSB sites.
Experimental Procedures
Sequence Analysis
Analysis of the N-terminal sequence of CHD4-(1–364) was performed using a combination of multiple sequence alignments, secondary structure prediction in Jpred (36), and disorder predictions (37, 38).
Cloning, Expression, and Purification of CHD4-N in Bacteria
Residues 145–225 (CHD4-N) of human CHD4 were cloned into a pGEX-6P vector to produce an N-terminal GST fusion protein. CHD4-N was expressed in Escherichia coli Rosetta 2 cells grown for 20 h at 25 °C in rich medium or, for isotope labeling, in minimal medium prepared as described previously (39). Cells were harvested and lysed and CHD4-N purified as described previously (39). CHD4-N has the additional sequence GPLGS on the N-terminal end, a remnant from the protease cleavage site. Purified CHD4-N was assessed by SDS-PAGE and size-exclusion chromatography coupled to a multiangle laser light scattering detector. For NMR experiments, the protein was concentrated to 0.3–1 mm in a buffer composed of 20 mm sodium phosphate, pH 7.5, and 150 mm NaCl. D2O (5% v/v) and 2,2-dimethyl-2-silapentane-5-sulfonic acid (170 μm, as a chemical shift reference) were added to the solution.
NMR Assignments and Structure Calculations
NMR spectra were recorded at 25 °C on 600 or 800 MHz Bruker Avance spectrometers equipped with TCI cryoprobes, processed using TopSpinTM (Bruker, Karlsruhe), and analyzed with SPARKY (T. D. Goddard and D. G. Kneller, University of California at San Francisco). 1H, 15N, and 13C assignments were made using standard triple resonance spectra, and backbone dihedral angles were calculated with TALOS+ (40), as described previously (39). Two-dimensional, 15N-separated and 13C-separated NOESY spectra were analyzed to obtain distance constraints, and initial structure calculations were performed using CYANA (41). From a family of 500 structures, the 100 lowest energy structures were subjected to water refinement using the RECOORD protocol (42). The 20 lowest energy water-refined structures were analyzed by PROCHECK NMR (43). Protein coordinates have been deposited in the Protein Data Bank under the accession code 2n5n.
15N HSQC Titrations
All 15N HSQC spectra were recorded at 25 °C. 15N-Labeled CHD4-N was used at 50 μm (170 μm in the case of adenosine 5′-diphosphate (ADP, Sigma) titration) in buffer containing 20 mm sodium phosphate, pH 7.5, 50 mm NaCl, 5% D2O, and 0.17 mm 2,2-dimethyl-2-silapentane-5-sulfonic as a chemical shift reference. Titrations were performed with ADP, an ADP-ribose dinucleotide (Sigma), or poly(A) (single-stranded DNA ssA15, Integrated DNA Technologies) dissolved in the same buffer as CHD4-N. Spectra were recorded for CHD4-N alone and after each addition of ADP, ADP-ribose dinucleotide, or poly(A). Molar ratios of CHD4-N:ligand for which spectra were collected were 1:0, 1:0.5, 1:1, 1:2, 1:5, 1:10. 15N HSQC spectra were also recorded for 20 μm 15N-labeled CHD4-N in a buffer composed of 10 mm Tris, pH 8, and 1 mm EDTA in the presence or absence of poly(ADP-ribose) (Trevigen®); the poly(ADP-ribose) was added to a concentration of 6 μm in ADP-ribose subunits. All spectra were processed using TopSpinTM (Bruker).
Microscale Thermophoresis
MST experiments were performed on a Monolith NT.115 (Nanotemper Technologies, Munich, Germany). Each titration curve consisted of 12 points prepared from a serial dilution of CHD4-N and a constant concentration of the fluorescein-labeled ligand. Experiments were carried out using 50 nm fluorescein-labeled DNA (Integrated DNA Technologies); sequences tested were: (i) 15 nucleotides of single stranded poly(A) (ssA15), (ii) 15-nucleotide poly(T) (ssT15), and (iii) 15 base pairs of double-stranded poly(AT) (prepared from ssT15 and fluorescein-labeled ssA15 hybridized in-house and purified by size exclusion chromatography). Titrations with poly(ADP-ribose) were performed using a mixture of 200 nm fluorescein-labeled streptavidin (Vector Laboratories) and 1 nm biotinylated PAR (Trevigen®). Commercially available PAR consists of a mixture of polymers that range from 2 to 300 subunits; the average of subunits per polymer was considered to be 150 for the purpose of concentration determination. MST assays were performed with 20, 50, or 100% LED power using a blue filter and a 20 or 50% MST power. The normalized fluorescence readings were plotted as a function of CHD4-N concentration and fitted to a standard Langmuir binding isotherm, as implemented in the Nanotemper software.
Cloning of FLAG-CHD4 and FLAG-GFP-CHD4 Constructs
A pcDNA3 plasmid containing human chd4 and an N-terminal FLAG sequence was used to clone several mutant constructs. Using site-directed ligase-independent mutagenesis, described in Ref. 44, the truncated constructs CHD4-ΔN and CHD4-Δ364, which lack residues 150–216 and 1–364, respectively, were created. The mutants CHD4-K757A and CHD4-α4A (which alters the sequence 202MMVLGAKWREFSTNN216 to AMVLGAKAAEFSTAA) were produced using standard PCR mutagenesis. For live imaging experiments, we used the previously described human gfp-HA-chd4 construct in pCMV-Sport6 as the wild-type construct (20). To clone the flag-gfp-tagged versions of CHD4-ΔN, CHD4-Δ364, and CHD4-α4A, we used the method described by Gibson et al. (45).
Laser Micro-irradiation and Live Imaging
U2OS cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS) from BioSera. Plasmid transfections were performed using TransIT-LT1 (Mirus Bio) following the manufacturer's guidelines. Localized lines of DNA damage were induced by laser micro-irradiation essentially as described previously (46, 47). Briefly, U2OS cells were plated on glass-bottomed dishes (Willco-Wells) pre-sensitized with 10 μm BrdU (Sigma) in phenol red-free medium (Invitrogen) for ∼48 h at 37 °C. Subsequent exposure to a laser beam was performed using a FluoView 1000 confocal microscope (Olympus) equipped with a 37 °C heating stage (Ibidi) and a 405-nm laser diode (6 milliwatt) focused through a 60× UPlanSApo/1.35 oil objective and resulting in a spot size of 0.5–1 μm. Laser beam exposure times of 250 ms (fast scanning mode) were used at a setting of 0.4-milliwatt output (50 scans) to yield pre-sensitization-dependent DNA damage, restricted to laser tracks without detectable cytotoxicity. Images of live cells were acquired using the same microscope, objective, and software as described above. For live-intensity quantifications of GFP-CHD4 laser lines, average GFP-CHD4 intensities along laser tracks were measured, and ratios of GFP-CHD4 average line intensities over average nucleoplasm intensities were calculated and compared using FV-10 software (Olympus). Approximately 10 cells were quantified per GFP-CHD4 variant per time point. Normalized average intensities (1 for the GFP-CHD4-WT) are presented.
Expression and Purification of FLAG-CHD4 in Mammalian Cells
HEK293 cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 in DMEM supplemented with 10% (v/v) FBS, 2 mm l-glutamine, 1× minimum essential media non-essential amino acids, 1 mm sodium pyruvate, 50 units/ml penicillin, and 50 μg/ml streptomycin (all ingredients from GibcoTM). pcDNA3 plasmids encoding for FLAG-CHD4 constructs were transfected into HEK293 cells at 70% confluence, and CHD4 proteins were expressed for 48–72 h at 37 °C and 5% CO2. Cells were harvested and resuspended in 50 mm HEPES, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, 1 mm DTT, 1 mm PMSF, and 1× cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science). Cells were incubated on ice for 30 min, vortexed, and spun down for 5 min at 3300 × g. The supernatant, which corresponds to the cytoplasmic fraction, was removed. The pellet was resuspended in 50 mm Tris, pH 8, 0.5 m NaCl, 1 mm EDTA, 1% Triton-X, 1.5 mm MgCl2, 1 mm DTT, 1 mm PMSF, and 1× EDTA-free protease inhibitors. The nuclear fraction was then lysed by sonication on ice, incubated on ice for 30 min to allow chromatin to precipitate, and cleared by centrifugation for 20 min at 16,000 × g at 4 °C. The resulting nuclear extract was then incubated with α-FLAG M2 Affinity Gel beads (Sigma) overnight on a rocker at 4 °C. The next day the protein was eluted with 300 μg/ml 3X-FLAG-peptide (ApexBio) in 20 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm DTT, and 10% v/v glycerol. Protein samples were analyzed by SDS-PAGE and visualized using SYPRO-Ruby and Coomassie stains.
Nucleosome Reconstitutions
Nucleosomes were assembled on DNA fragments derived from the 601 nucleosome positioning sequence (48) and purified recombinant Xenopus laevis histone octamers to give a typical final nucleosome concentration of 1–2 μm. Assembly was performed by salt gradient dialysis using a double-dialysis method (48) as follows. Reactions were placed in microdialysis buttons, which were placed inside a dialysis bag containing 30 ml of 1× Tris-EDTA (TE) and 2 m NaCl; the dialysis bag was then dialyzed overnight against 2 liters of 1× TE at room temperature and then against a further 1 liter of 1× TE for 3–6 h.
The histone octamers used in these reactions were assembled using standard protocols from purified recombinant histones (49), either all as unlabeled proteins or containing AlexaFluor488-labeled H2A. Labeling of H2A was achieved via the incorporation of a single cysteine residue at position 120. A synthetic gene encoding H2A-T120C was purchased from GeneArt® and cloned into a rhamnose-inducible pRham vector (Lucigen). H2AT120C was expressed at 37 °C overnight in Rosetta2 (DE3) pLysS E. coli cells in ZYP-5052 auto-induction media (50) containing an additional 0.2% rhamnose and then purified from inclusion bodies using standard protocols (49). Labeling of purified H2A-T120C was performed under denaturing conditions in 20 mm Tris, pH 7.0, 7 m guanidine-HCl, 5 mm EDTA, and 5 mm Tris(2-carboxyethyl)phosphine with a ∼5-fold molar excess of AlexaFluor488 C5 maleimide overnight at 4 °C. Reactions were quenched via the addition of 30 mm β-mercaptoethanol and then purified via gel filtration on a Superdex 200 10/300 column in 20 mm Tris, pH 7.0, 7 m guanidine-HCl, 0.1% β-mercaptoethanol. Purified labeled H2A was dialyzed against deionized water with 0.05% (v:v) β-mercaptoethanol overnight at 4 °C, aliquoted, and lyophilized for long term storage. Labeling efficiency was ∼65–70%.
DNA fragments were produced by PCR using MyTaq DNA polymerase (Bioline) and fluorophore/quencher-labeled primers purchased from Sigma or ATDBio. The PCR products were purified via 0.5× Tris borate-EDTA 5% polyacrylamide gel electrophoresis and electroelution. The notation xWy denotes the 147-bp 601 sequence with flanking DNA of x and y bp on the upstream and downstream side, respectively.
ATP-driven Nucleosome Remodeling Reactions
Nucleosomes were assembled on Cy3 or BHQ1-labeled 0W47 DNA to generate asymmetric end-positioned nucleosomes for gel-based and real-time remodeling reactions, respectively. All remodeling reactions were performed at 30 °C. Gel-based remodeling reactions were carried out essentially as described in Ryan et al. (32). Real-time quenched FRET remodeling reactions were performed under similar conditions but monitored in a FluoStar Optima plate reader using Corning black non-binding surface half-area 96-well plates and 485P and 520P excitation and emission filters, respectively. The reactions contained 50 nm BHQ1–0W47 AlexaFluor488-labeled nucleosomes, 50 mm Tris, pH 7.5, 50 mm NaCl, 3 mm MgCl2, and the enzyme concentrations indicated in the figures. Reactions were monitored for 1 min before the addition of 1 mm ATP to ensure fluorescence changes were ATP-dependent and then monitored for a further 5–10 min. The following single exponential function was fit to each individual remodeling data set.
where a is the asymptote, b is the total response (i.e. a minus the starting value), and c is a constant. The initial reaction rates were then calculated using the solution of the first derivative of f(x) when x = 0, as follows.
Relative rates were calculated against the nominated control sample (e.g. wild-type CHD4 or control treatment nucleosomes).
Nucleosome/DNA-stimulated PARylation Reactions
PARylation and control reactions were performed at room temperature for 35 min. BHQ1–0W47 AlexaFluor488-labeled nucleosomes (0.5 μm) or an equimolar amount of free BHQ1–0W47 DNA were incubated in 50 mm Tris, pH 7.5, 50 mm NaCl, 3 mm MgCl2 in the presence of 1 unit/μl of high-specific activity PARP1 (Trevigen®) and 250 μm NAD+. Reactions were stopped via the addition of the PARP inhibitor 3-aminobenzamide (3-AB) to 1 mm either before or after the addition of PARP1. Control treatment reactions contained NAD+ and 3-AB but not PARP1.
Results
CHD4 Contains a Conserved Domain at the N Terminus
Sequence analysis of CHD4 (residues 1–364) revealed the presence of an 80-residue region (145–225) that is highly conserved across complex animals (Fig. 1b). For example, sequence identity between human and zebrafish CHD4 is ∼90% in this region, whereas orthologues of CHD4 in plants lack this N-terminal region. The conserved sequence is predicted to encode a stable structure according to secondary structure and disorder prediction software, suggesting that it might constitute a functional domain. We expressed and purified a polypeptide comprising human CHD4(145–225), which we termed CHD4-N, and used standard solution NMR approaches to determine its three-dimensional structure. The solution structure obtained is of high precision, with a root mean square deviation over the backbone atoms of ordered residues (residues 150–217) of 0.40 Å and, for all heavy atoms, 0.9 Å when the 20 lowest energy structures are superposed (Fig. 2a). Experimental restraints and structural statistics are reported in Table 1. The CHD4-N geometry has no outliers according to the Ramachandran plot generated by PROCHECK NMR (43), suggesting that the CHD4-N structure is of high quality.
FIGURE 2.

Structure of CHD4-N. a, superposition of the 20 lowest-energy structures (Cα in red and side chains in green, left) and ribbon diagram (right) of CHD4-N. All four helices and the long linker connecting helices 1 and 2 are labeled. b, residues involved in hydrophobic packing in the buried surface formed by helices 2, 3, and 4. CHD4-N is viewed as from the top of panel a after a 45° clockwise rotation. c, electrostatic surface charge of CHD4-N. The left panel is in the same orientation as a. d, size-exclusion chromatography-multiangle laser light scattering detector data for CHD4-N. The expected molecular mass for the domain is 9290 Da. The average molecular mass of the eluted peak is 9800 ± 150 Da.
TABLE 1.
Experimental constraints and structural statistics for CHD4-N
| NOE-based distance constraints | |
| Total | 1291 |
| Intraresidue (i, i) | 344 |
| Sequential (i, i +1) | 307 |
| Medium range (2 ≤ |i − j| ≤ 4) | 298 |
| Long range (|i − j| >4) | 342 |
| Total dihedral angle constraints | 133 |
| Root mean square deviation | |
| All backbone atoms (N, Cα, C′) (Å) | 3.2 |
| All heavy atoms (N, C, O, S) (Å) | 3.1 |
| PROCHECK Ramachandran statistics | |
| Residues in most favored region (%) | 92.9 |
| Residues in additional allowed regions (%) | 7.1 |
| Residues in disallowed regions (%) | 0 |
| Deviation from idealized geometry | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.1 |
CHD4-N Forms a Helical Bundle
The structure of CHD4-N consists of a four-helix bundle (Fig. 2a). Helices 1 and 2 are on opposite sides of the domain and are separated by a long linker, whereas helices 2 and 3 and helices 3 and 4 are linked by short turns. Helix 1 packs against helix 4, which in turn packs against helix 3. At the helix 2 end, CHD4-N presents a hydrophobic core comprising His-165 and Phe-167 of the long linker, Tyr-172 of helix 2, Tyr-178, Phe-181, and Phe-184 of helix 3, and Trp-209 and Phe-212 of helix 4 (Fig. 2b). Electrostatic surface charge analysis reveals the presence of two charged regions on opposite faces of the protein: a basic region along helices 3 and 4 and an acidic region along the long linker (Fig. 2c). Although in principle this observation could indicate dimerization or oligomerization of CHD4-N, size-exclusion chromatography-multiangle laser light scattering detector data show that CHD4-N is monomeric in solution (Fig. 2d). On the other hand, the combination of basic and acidic surfaces could suggest a possible interaction with chromatin in which the basic surface contacts DNA, whereas the acidic region simultaneously makes interactions with the highly basic histone proteins.
CHD4-N Is an HMG Box-like Domain
Structure matching of CHD4-N, performed by the structure search engine PDBeFold, identifies the fold as similar to that of HMG-box domains. The HMG-box is a versatile protein module that in general binds preferentially to distorted DNA structures (for review see Ref. 35). This domain is found in both HMG-only-containing proteins or as part of multi-domain proteins, such as transcription factors or chromatin remodeling factors.
The three most similar HMG-box structures displayed 26% sequence similarity with CHD4-N, and structural comparisons give root mean square deviation values of between 1.2–1.5 Å over 38–39 Cα atoms (Fig. 3a). All structural superpositions and sequence similarity occurred between the DNA binding region of the HMG-box and helices 3 and 4 of CHD4-N (Fig. 3, b–d), the helices that form a positively charged surface. These observations are consistent with the idea that the helix 3/4 region might mediate binding to DNA. At the same time our structural data demonstrate that there is no homology between CHD4-N and structurally characterized PAR binding domains.
FIGURE 3.

CHD4-N has an HMG-box-like fold. a, superposition of CHD4-N (red) with the HMG-box domain from several proteins: yeast NHP6A (PDB code 1j5n, light gray), fly HMG-D (PDB code 3nm9, dark gray), and mouse HMGX2 (PDB code 2crj, black). b, structure and electrostatic surface charge of the complex of NHP6A bound to SRY DNA (PDB code 1j5n). The protein is shown in the same orientation as a. c, alignment of CHD4-N (red) and NHP6A (gray), showing conserved residues across helices 3 and 4 of CHD4-N as highlighted in the alignment d. d, sequence alignment of CHD4-N and various eukaryotic HMG-box domains (UniProt entry numbers in brackets). CHD4-N secondary structure is shown in red above the alignment, and residues involved in DNA-binding for NHP6A (b) and for CHD4-N (Fig. 4e) are indicated with green and yellow diamonds, respectively. The sequence alignment was generated by ALINE (72).
CHD4-N Can Bind DNA
In light of the structural similarity with the HMG-box domain and the reported PAR binding activity of the N-terminal portion of CHD4, 15N HSQC titrations were performed to assess the binding of CHD4-N to nucleic acids. 15N HSQC titrations using a single-stranded DNA oligonucleotide revealed that CHD4-N can bind to a 15-nucleotide poly(A) homopolymer (ssA15, Fig. 4a). We then used MST to measure the affinity of CHD4-N for ssA15. In MST experiments the diffusion of a fluorescently labeled macromolecule in a temperature gradient is measured in the presence of increasing concentrations of a binding partner (51). Changes in diffusion are plotted to create a binding isotherm. Titration of fluorescein-labeled ssAA15 with CHD4-N confirmed that the two species form a 1:1 complex with a dissociation constant, KD, of 32 ± 3 μm (Fig. 4b). Similarly, MST experiments performed using single-stranded poly(T) (ssT15) showed 1:1 binding that fitted with a KD of 11 ± 1 μm (Fig. 4c). These data suggest that CHD4-N recognizes the DNA backbone structure rather than making specific interactions with the bases. To further assess DNA binding specificity, MST assays were performed with the corresponding double-stranded DNA (d(A·T)15). The calculated KD in these assays was 13 ± 1 μm (Fig. 4d), indicating that CHD4-N does not distinguish single-stranded from double-stranded DNA.
FIGURE 4.

CHD4-N binds DNA. a, 15N HSQC titration of 15N-CHD4-N with ssA15. Spectra corresponding to protein:DNA molar ratios of 1:0 to 1:10 are shown. b–d, MST titration experiments of CHD4-N with ssA15 (b), ssT15 (c) and (d) d(A.T)15. MST graphs show representative experiments. KD values are given with the S.E. from at least three independent experiments. e, chemical shift (CS) changes (calculated as in Ayed et al. (73)) for CHD4-N residues upon the addition of 10 mol eq of ssA15. Residues that underwent chemical shift changes (CSC) of more than |CSCmean + 1σ| are highlighted in yellow. f, CHD4-N colored according to the same color scheme as in e. The structure is in the same orientation as in Fig. 2a.
To explore whether sequences N- or C-terminal to the core CHD4-N domain influenced DNA binding affinity, we expressed and purified eight polypeptides spanning different fractions of residues 1–355 of CHD4 (Fig. 5). Two could not be expressed (1–310 and 1–355). Affinities for the interaction of each of the others with ssA15 (measured by MST) are shown in Fig. 5. Overall, proteins with C-terminal extensions displayed ∼10-fold higher affinity for ssA15 than CHD4-N. We selected the polypeptide with the highest affinity, CHD4-(145–310) and also measured its binding to ssT15. Binding of CHD4-(145–310) to ssT15 was also higher than for CHD4-N (2.7 μm compared with 11 μm, respectively). This indicates that the additional C-terminal sequence that is rich in basic amino acids improved binding to single-stranded DNA in a nonspecific manner.
FIGURE 5.
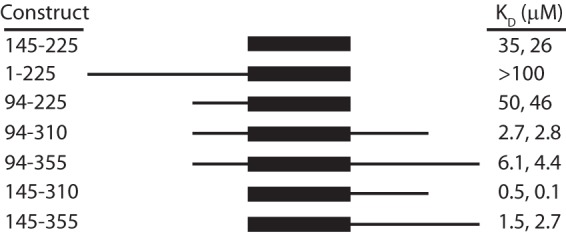
Longer CHD4 polypeptides bind DNA. Constructs that were designed to encode longer segments of the N-terminal part of CHD4 as shown were designed, expressed, and purified. Their affinity for ssA15 was measured by MST. KD values from two independent measurements (one carried out with the corresponding cleaved protein or with the GST-fusion protein, respectively) are shown.
CHD4-N and HMG-box Domains Have Similar Binding Surfaces
The DNA binding properties of CHD4-N were further examined in a 15N HSQC titration with ssA15; up to 10 mol eq of ssA15 was added. The interaction was in fast exchange on the chemical shift timescale, and residues that had the largest chemical shift changes after the addition of DNA were Lys-200, Met-201, Met-202, Val-204, Leu-205, Gly-206, Ala-207, Trp-209, and Arg-210 (Fig. 4, e and f). This set of residues lies essentially on one surface of helix 4 of CHD4-N, the same surface that was predicted to interact with nucleic acids due to its basic surface charge and structural homology with other HMG-box domains.
CHD4-N Binds PAR with Higher Affinity Than DNA
Given the published observation that the N-terminal portion of CHD4 has PAR binding activity, we performed binding experiments to determine if CHD4-N would bind to substrates that could mimic poly(ADP-ribose) molecules. Previously, PBZ (PAR-binding zinc finger) domains of the APLF (polynucleotide kinase-like) protein have been shown to bind similarly to ADP and ADP-ribose but not to adenine alone (26), suggesting that the substrate recognition occurs at the ribose-phosphate moiety. However, 15N HSQC titrations of 15N-CHD4-N with either ADP alone or a dinucleotide of ADP-ribose (up to 10 mol eq) did not give rise to any chemical shift changes. It is possible that ADP-ribose dinucleotide is too short to effectively mimic PAR, so we, therefore, tested the binding of CHD4-N to polymers of ADP-ribose. Commercially available PAR consists of a heterogeneous mixture of oligomers and polymers of different lengths and branched architectures, ranging from an estimated 2 to 300 subunits. This heterogeneity prevents accurate determination of a molar concentration. We estimated the average size of PAR molecules to be 150 subunits and calculated concentrations on that basis. Biotinylated PAR was incubated with fluorescein-tagged streptavidin, and this complex was used as the titrand in MST experiments with CHD4-N. MST data showed that CHD4-N binds PAR with an apparent KD of ∼0.27 μm (Fig. 6a), ∼50 times tighter than the affinity for DNA. Note that because PAR was the titrand, the calculated KD does not differ significantly for different estimated polymer lengths between 2 and 300 subunits. Furthermore, the shape of the binding curve did not indicate the presence of cooperativity even though multiple CHD4-N monomers could likely bind to a single PAR polymer.
FIGURE 6.

CHD4-N binds PAR with higher affinity than DNA. a, a representative MST titration of poly(ADP-ribose) with CHD4-N. The KD is given with the S.E. from five independent experiments. The no PAR titration refers to a control experiment where the streptavidin-biotin-PAR was substituted by streptavidin alone. b, 15N HSQC of 15N-CHD4-N in the absence (red) and in the presence (blue) of PAR. c, chemical shift (CS) changes (CSC) (calculated as in Ayed et al. (73)) for CHD4-N residues upon the addition of 0.3 mol eq of PAR. Residues that underwent chemical shift changes of more than |CSCmean + 1σ| are highlighted in yellow. Residues for which peaks disappeared or became significantly more intense are marked with cyan crosses or circles, respectively. d, ribbon diagram of CHD4-N showing residues for which signals in the 15N HSQC are perturbed after the addition of PAR. The same color scheme as in c is used. The top panel structure is in the same orientation as in Fig. 2a.
In contrast to the situation for DNA binding, the affinity of the longer CHD4-(145–310) polypeptide for PAR was ∼7-fold lower (2.0 μm) than was the affinity of CHD4-N. This effect could be due to the additional C-terminal residues preventing access to the PAR-binding site. Thus, these residues did not make a contribution to the interaction with PAR, suggesting that the minimal PAR binding region is the CHD4-N domain.
To confirm the binding of CHD4-N to PAR, we ran 15N HSQC spectra of CHD4-N in the absence and presence of PAR (Fig. 6b). The addition of PAR (the molar ratio of protein:PAR was 1:0.3; only this quantity was available) gave rise to a range of selective changes in the CHD4-N spectrum. In this case the timescale for binding to PAR appeared to be slower than for binding to DNA, consistent with the higher affinity. Additional line broadening might also arise from the high molecular weight of the PAR ligand, although carbohydrates typically display very fast internal dynamics compared with proteins, which would serve to mitigate this effect. The majority of the peaks that suffered chemical shift changes upon binding lie on the linker between helices 1 and 2 and the surrounding surface (Fig. 6, c and d), suggesting that the surface in CHD4-N involved in PAR binding is not the same as the surface that binds DNA.
The CHD4-N Domain Is Not Essential to Recruit CHD4 to DSB Sites
To evaluate the effect of CHD4-N on CHD4 recruitment to DNA damage sites, we performed laser micro-irradiation experiments in a similar manner to those described previously (20). It has been shown that, when laser micro-irradiation is used to generate localized DNA damage in human U2OS cells, CHD4 co-localizes with γH2AX, a well established marker of DNA DSBs in a PARP-dependent manner. We tested several GFP-tagged mutants of CHD4 in these experiments. CHD4-WT corresponds to the full-length wild-type protein, CHD4-ΔN lacks the CHD4-N domain, CHD4-α4A is a full-length protein with five mutations to alanine on the DNA-binding surface of helix 4 of CHD4-N, and CHD4-Δ364 lacks the whole N-terminal region upstream of the PHD domains (Fig. 7a). As shown in Fig. 7b, GFP-tagged-CHD4-WT, -CHD4-ΔN, and -CHD4-α4A are effectively localized to the nucleus and accumulate at DSB sites with similar kinetics (Fig. 7c). In contrast, GFP-CHD4-Δ364, which lacks both CHD4-N and the surrounding sequence that is predicted to be disordered, was present in both the nucleus and cytoplasm and was unable to accumulate at DSB sites.
FIGURE 7.
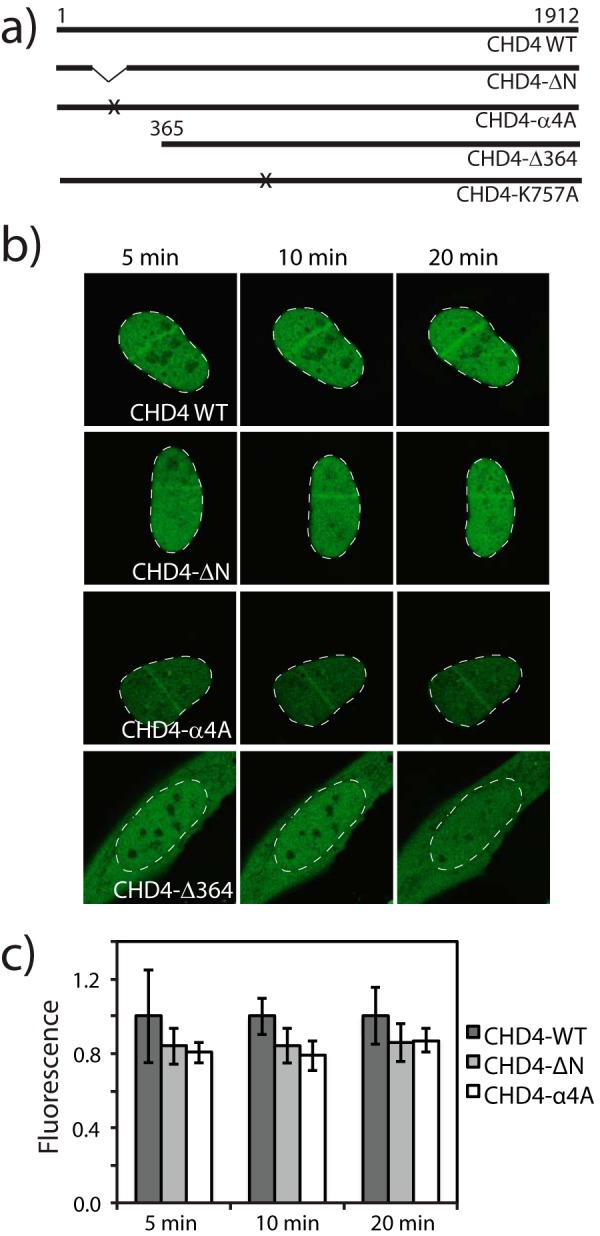
The CHD4-N domain is not essential for the recruitment of CHD4 to DNA damage sites, but the N-terminal region of CHD4 has a nuclear localization role. a, diagram of the CHD4 constructs tested. The crosses indicate approximately the sites of mutagenesis: five alanine mutations for CHD4-α4A and one Ala mutation for CHD4-K757A. b, live imaging of GFP-CHD4 variants at the indicated time points after laser micro-irradiation in U2OS cells. The nucleus of each cell is outlined with a dashed white line. c, quantification of GFP-CHD4 recruitment at the DSB site. Average fluorescence intensities were normalized relatively to the wild-type protein. Error bars are from the S.D. calculated for a population of n = 10.
Deletion of the N-terminal of CHD4 Reduces Nucleosome Remodeling Activity
We next assessed the role of the N-terminal region of CHD4 in the nucleosome remodeling activity of CHD4. First, we purified full-length FLAG-tagged recombinant CHD4 from HEK293 cells using anti-FLAG-Sepharose beads. Then we established that the purified protein was able to reposition nucleosomes in an ATP-dependent manner in a standard gel-based nucleosome repositioning assay. The addition of CHD4 and ATP to an asymmetric end-positioned nucleosome results in the appearance of a second band on a native DNA gel (Fig. 8a), consistent with CHD4 centering the nucleosome through its intrinsic remodeling activity. To obtain a more quantitative estimate of CHD4 activity, we established a real-time FRET-based nucleosome repositioning. This assay is similar to that described by Yang et al. (52) except that the FRET dye pair has been modified to simplify measurements, increase throughput, and improve sensitivity.4 Briefly, asymmetric end-positioned nucleosomes with 0- and 47-bp linker DNA on each side were assembled in which the DNA was labeled on the 0-bp linker with the dark quencher BHQ1. AlexaFluor488 was conjugated to a cysteine mutant (T120C) of histone H2A; this residue is close to the exit point of the DNA in the nucleosome (Fig. 8, b and c). The proximity of the BHQ1 moiety to the AlexaFluor488 in these nucleosomes results in strong quenching of AlexaFluor488 fluorescence. Any movement of the BHQ1 away from the AlexaFluor488 dye, such as during ATP-dependent repositioning by a remodeling enzyme, results in a robust increase in fluorescence that can be monitored in real time (Fig. 8d).
FIGURE 8.

Deletion of the CHD4 N-terminal region reduces nucleosome repositioning activity. a, native PAGE (0.5× Tris borate-EDTA, 5% acrylamide) analysis of nucleosome repositioning assays using recombinant human CHD4. End-positioned Cy3-labeled 0W47 nucleosomes (50 nm) were incubated for 30 min with different amounts of enzyme (1 or 10 nm) with or without ATP. The gel was scanned for Cy3 fluorescence. b, structure of the nucleosome core particle (PDB code 1kx5) showing dye attachment sites used in real-time repositioning experiments (H2A is shown in blue; H2B, H3, and H4 in gray; DNA in orange). The dark quencher BHQ1 (purple) is attached to the 5′-end of the DNA, and AlexaFluor488 (green) is attached via maleimide linkage to Cys120 in H2AT120C. c, schematic representation of the ATP- and remodeler-dependent movement of the BHQ1 dye relative to AlexaFluor488. In the starting asymmetric (0W47) nucleosomes, the proximity of BHQ1 to the AlexaFluor488 moiety resulted in strong quenching of AlexaFluor488 fluorescence. The movement of BHQ1 away from the AlexaFluo488 during remodeling released quenching, and AlexaFluor488 fluorescence emission consequently increased. Coloring is the same as in b. d, nucleosome remodeling activity is dependent on the concentration of CHD4. Real-time fluorescence repositioning traces (520 nm) for 50 nm BHQ1–0W47 AlexaFluor488-H2A nucleosomes were recorded in the presence of increasing CHD4 concentrations. e, real-time repositioning traces of BHQ1–0W47 AlexaFluor488-H2A nucleosomes (50 nm) recorded in the presence of 10 nm wild-type CHD4 or the indicated CHD4 mutants. f, relative rates of repositioning of 50 nm BHQ1–0W47 AlexaFluor488-H2A nucleosomes by CHD4 mutant proteins in comparison to wild-type CHD4. Data are the average of four experiments using 10 nm enzyme. Error bars represent the S.E. Large errors for K757A are due to the model inadequately fitting the essentially null data and not large fluctuations in the data itself.
Using these real-time remodeling assays we compared the repositioning activity of wild-type CHD4, an inactive ATPase mutant (K757A), and the N-terminal mutants used in the laser microradiation experiments (proteins were expressed in HEK293 cells as FLAG fusion proteins and purified by FLAG affinity chromatography). As expected, the inactive ATPase mutant has no measurable repositioning activity, whereas wild-type CHD4, CHD4-ΔN, and CHD4-α4A all robustly reposition nucleosomes at a similar rate. Interestingly, CHD4-Δ364 is a much less efficient enzyme and repositions nucleosomes at ∼20% that of the rate of the wild-type protein (Fig. 8, e and f).
PARylation of Nucleosomes/PARP1 Inhibits the Nucleosome Remodeling Activity of CHD4
Given the interaction between CHD4 and PAR and that PARylation is a common nucleosomal modification (53–56), we next tested if PARylation has any effect on CHD4 remodeling activity. Real-time nucleosome repositioning assays were performed as above, except that the nucleosomes were treated with PARP1, NAD+, and the PARP1 inhibitor 3-AB as outlined in Fig. 9a. Repositioning assays were performed with wild-type CHD4 using either standard untreated nucleosomes or nucleosomes treated with NAD+ and 3-AB alone (control treatment) or with NAD+ and PARP1 that was inhibited by 3-AB at the start of the treatment (inhibited PARP). Fig. 9b shows that neither NAD+/3-AB nor inhibited PARP1 have a discernible effect on the rate of nucleosome repositioning by CHD4. In comparison, nucleosome repositioning by CHD4 is greatly reduced when nucleosomes have been pretreated with active PARP1. These results indicate that PARylation, either of nucleosomes or PARP1 itself, inhibits the remodeling activity of CHD4. PARylation of CHD4 cannot account for the observed effect as PARP1 is inhibited by 3-AB before encountering CHD4 in the repositioning assay. Furthermore, these effects are not due to major disruption of the substrate nucleosomes, which look essentially identical in all cases by native PAGE.
FIGURE 9.
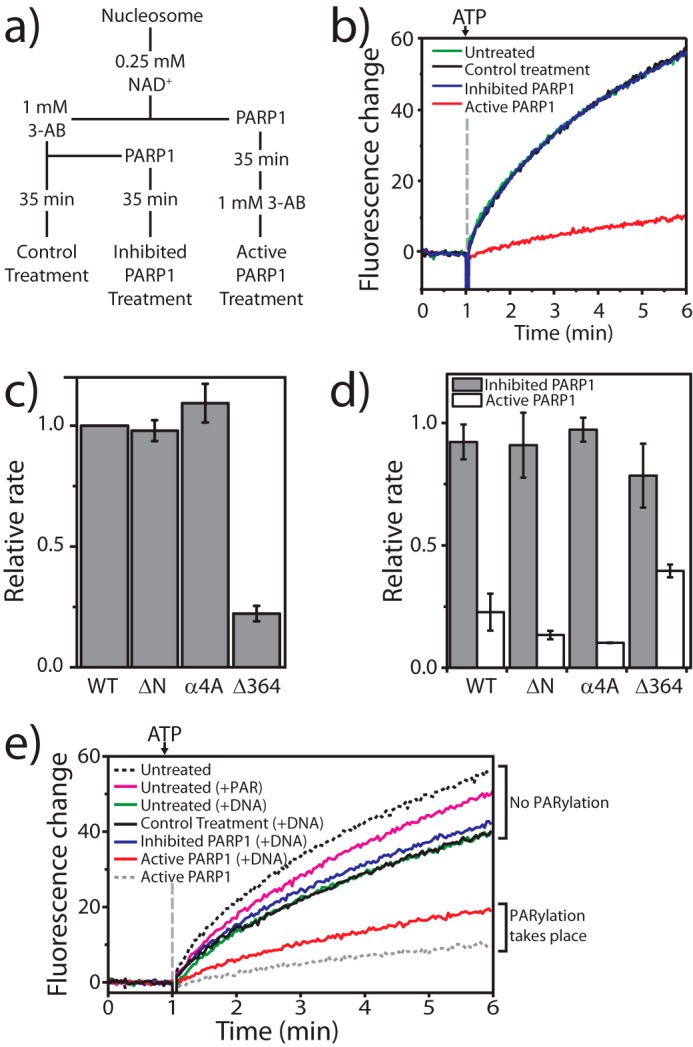
PARylation of PARP1/nucleosomes inhibits nucleosome repositioning by CHD4. a, scheme outlining the treatment of nucleosomes with “active” or “inhibited” PARP1 used to generate PARylated and control substrates for real-time nucleosome repositioning assays. b, real-time nucleosome repositioning data for 10 nm wild-type CHD4 and 50 nm BHQ1–0W47- and AlexaFluor488-H2A-labeled nucleosomes derived from untreated stock or pretreated as described in a. Only nucleosomes incubated with active PARP1 give rise to reduce nucleosome repositioning activity by CHD4. Note that data for the “untreated” and “control treatment” samples are directly underneath the inhibited PARP1′ curve and, therefore, not visible. c, rates of repositioning of control treatment nucleosomes by CHD4 N-terminal mutant proteins relative to that of wild-type CHD4. Data are the average of three experiments and are recorded using the same conditions as in b. d, relative rates of repositioning of nucleosomes treated with active or inhibited PARP1 expressed as a fraction of the rate of repositioning of control-treated nucleosomes (shown in c)) using the same mutant. Data are the average of three experiments. All error bars represent the S.E. e, PARylated PARP1 inhibits CHD4 remodeling activity. DNA was used to stimulate PARP1 activity under active or inhibited conditions, essentially following the scheme described for nucleosomes in a and added to real-time fluorescence repositioning traces for 50 nm BHQ1–0W47 AlexaFluor488-H2A nucleosomes recorded in the presence of 10 nm CHD4 and 1 μm PAR. The black and gray dashed curves correspond to the green and red curves in b, respectively. The magenta line shows that nucleosome repositioning is inhibited only to a small degree by free PAR (1 μm).
Next, we assessed the activity of our N-terminal mutants in these assays. Fig. 9c shows that the relative rates of repositioning by the mutant proteins on control-treated nucleosomes mirrors the pattern seen with untreated nucleosomes in Fig. 8d, indicating NAD+ and 3-AB do not affect the activity of the mutants. Treatment of nucleosomes with inhibited PARP1 also does not lead to any significant changes in the rates of repositioning (Fig. 9d, gray bars). In contrast, nucleosomes treated with active PARP1 are repositioned much more slowly by either wild-type CHD4 or by any of the mutants (Fig. 9d, white bars). CHD4-Δ364 appears to be slightly less affected than the other mutants. However, it should be noted that the activity of CHD4-Δ364 is already significantly reduced compared with wild type (Fig. 9c); the reduction shown in Fig. 9d is in addition to that change and equates to <10% of normal wild-type activity. Given that the trend is similar across all CHD4 mutants, these data suggest that the N-terminal region of CHD4 does not specifically drive the PARylation-dependent reduction of CHD4 activity.
The inhibition of remodeling we observe after treatment of nucleosomes with active PARP1 could result from PARylation of the nucleosomes, PARP1 itself, or both. To investigate this issue, we set up PARylation reactions targeting PARP1 only. It is known that PARP1 activity is stimulated by nucleic acids (54), and so PARylation reactions were performed essentially as described in Fig. 9a, except that the nucleosomes were substituted with free BHQ1–0W47 DNA. This treatment maintains stimulation of PARP1 activity but means that PARP1 itself is the only available substrate. Subsequently, PARP1 was inhibited with 3-AB and the material mixed with nucleosomes in preparation for real-time repositioning experiments. The final reaction mixture contained equimolar amounts of nucleosomes, free DNA, and PARP1 at an equivalent concentration to previous experiments. Fig. 9e shows that in the absence of PARylation all repositioning reactions proceed at essentially the same rate. Note that the presence of the free DNA itself partially inhibits CHD4 activity (compare the black dashed line with +DNA treatments), most likely by simple competition with nucleosomal DNA for CHD4 binding. In contrast, the presence of auto-PARylated PARP1 (red curve) results in a marked decrease in the rate of nucleosome repositioning by CHD4 (compare the red with blue curves). However, the level of inhibition achieved under these conditions is not as great as when both PARP1 and nucleosomes are available as PARP1 substrates (compare the red line and dashed gray line).
Discussion
CHD4 Contains an HMG-box-like Domain That Can Bind PAR
The CHD4-NuRD complex has been shown to be important in the DNA-damage response. In particular, the N-terminal ∼750-residue region of CHD4 has been reported to be sufficient to recruit the NuRD complex to sites of damage (20). This same region was also able to bind PAR in immunoprecipitation experiments, suggesting that it would contain a PAR binding motif. In addition, Drosophila melanogaster CHD4, which shares 30% similarity with human CHD4 in the corresponding region, was shown to interact in vitro with PAR through basic motifs in this region (21). We have identified a conserved and ordered domain at the N-terminal region of CHD4. This domain, CHD4-N, does not resemble other PAR binding domains for which structures are known but is instead related to HMG-box domains. In vitro binding assays show that CHD4-N binds single-stranded and double-stranded DNA with similar affinities, suggesting that interactions with such “standard” nucleic acids are probably not sequence-specific and most likely occur at the phosphate backbone. When compared with HMG-box-DNA complex structures, CHD4-N helices 3 and 4 superpose with the helices of HMG-box domains that are commonly involved in DNA binding. Our NMR data showed that this surface in CHD4-N was involved in DNA binding, suggesting that CHD4-N and HMG-box domains interact with nucleic acids in a similar way.
In contrast, even though CHD4-N bears no resemblance to known PAR binding domains, we have demonstrated that CHD4-N can bind PAR in vitro. Despite the substantial heterogeneity of the PAR preparation used in these experiments, we can infer unequivocally both that the binding is direct and that it is non-cooperative. We can also infer that CHD4-N binds PAR with a higher affinity than it binds DNA. Our NMR data indicate that residues from the acidic linker between helices 1 and 2 of CHD4-N are most involved in the interaction with PAR, suggesting that the PAR binding surface is distinct from the DNA binding surface discussed above.
The observation that CHD4-N did not bind to either monomeric or dimeric ADP-ribose suggests that recognition involves a structural element of PAR that is not present in these two moieties. The most likely candidate is the branch structure that is characteristic of PAR, in which the terminal ribose of an ADP-ribose unit connects to two additional units through both its C1′ and C2′ carbons. At this stage, however, the nature of the interaction cannot be deduced. Further analysis will require the chemical synthesis of specific PAR substructures and the purification of homogeneous preparations of PAR species, both processes that are technically challenging. Surprisingly, the corresponding HMG-box-like region of Drosophila CHD4 did not show any interaction with PAR (21), but instead the basic sequences upstream and downstream of the HMG-box-like domain were sufficient to mediate PAR binding, suggesting that PAR binding activity might be partitioned among several N-terminal regions in CHD family members.
HMG Boxes Are Known for Their Ability to Recognize Unusual Nucleic Acid Structures
The HMG-box is one of the most abundant chromatin binding domains in metazoans (57). Typically, these domains bind kinked or bent DNA structures such as those found at entry or exit points to the nucleosome (57, 58). HMG-box domains have not, however, previously been shown to bind poly(ADP-ribose).
Although we have shown that CHD4-N binds more tightly to PAR than to DNA, we cannot rule out that CHD4-N could be responsible for recognizing particular DNA structures in an analogous fashion to HMG1/2. In addition, the sequence around CHD4-N harbors stretches of basic and acidic residues that could improve binding to DNA. Basic extensions at either the N or C terminus of the HMG domain have been reported to increase the DNA binding affinity and stabilize the bent conformation of DNA, as seen in the transcription factor LEF-1, the yeast chromatin-associated NHP6A protein, and the D. melanogaster non-histone chromosomal HMG-D protein (59–61).
Furthermore, several other ATP-dependent chromatin-remodeling complexes either contain or associate with HMG-box proteins (62–66). In these cases, however, the nature of the nucleic acid targets of the HMG-box domains is not known. Sequence identity between these HMG domains and CHD4-N is ∼20–25% around the DNA binding helices 3 and 4, not sufficient to infer that these HMG domains could also bind PAR.
CHD4, PAR, and Chromatin Remodeling
In addition to its well established connection with DNA repair, PARylation has also been shown to have effects in nucleosome remodeling; indeed, it is likely that these represent two faces of the same biochemical function. When ISWI is a target of poly-ADP-ribosylation, both ISWI ATPase activity and binding to DNA and nucleosomes are impaired (67). In contrast, the ATPase and chromatin remodeling activities of ALC1 are activated upon PARylation of PARP1 (68, 69). This activation is a result of cooperative binding of PARylated PARP1 and ALC1 to nucleosomes. The interplay between PARylation and CHD4 appears to be different again. Our data show that the remodeling activity of CHD4 is strongly inhibited in the presence of PARylation. Yet, unlike ISWI, it is not the remodeler that is PARylated but rather nucleosomes and/or PARP1 itself. PARylated PARP1 is sufficient to elicit some inhibition, but the effect is enhanced when nucleosomes are also exposed to active PARP1, a situation that could result either from direct PARylation of nucleosomes or from nucleosomes acting as a greater stimulus of PARP1 activity (compared with free DNA). Further work is required to deconvolute these two possibilities.
Given our observation that the isolated CHD4-N domain binds PAR with submicromolar affinity, it was surprising that mutants targeting CHD4-N behaved largely like wild type in response to PARylation in nucleosome-repositioning assays. In addition, these mutants also had wild-type-like behavior in the laser-induced DNA damage experiments. Thus, CHD4-N appears not to be necessary for the recruitment of CHD4 to DSB sites and the PARylation-dependent inhibition of its nucleosome remodeling activity (at least on single nucleosomes). On the other hand, a larger region encompassing CHD4-N (CHD4-(1–364)) is required for the efficient accumulation of CHD4 at DSB sites and is also critical for the in vitro nucleosome repositioning activity of CHD4.
How does PARylated PARP1 inhibit CHD4 remodeling activity? One possibility is that binding of this species to CHD4 prevents it from binding nucleosomes, acting as a simple competitor in the same way free DNA also reduces CHD4 remodeling activity. Alternatively, binding to PARylated PARP1 might interfere directly with the catalytic cycle of CHD4, perhaps preventing a conformational change within the enzyme that is required for remodeling. Conversely, a direct PARP1-nucleosome interaction (PARylated PARP1 is known to bind nucleosomes; Ref. 70) might reduce the efficacy of CHD4 as a remodeler.
Finally, what is the biological context for this PARP1-mediated inhibition of CHD4 activity? We offer two possible models. First, in the context of DNA-damage repair, the presence of PARylated PARP1 at specific loci (e.g. broken DNA ends) might demarcate sites at which CHD4 should not act. In this scenario CHD4 would be recruited to damaged chromatin via PARylated PARP1 but be unable to directly remodel that chromatin. Instead, CHD4 action would be directed to areas up- and downstream of this site, either through interactions with other NuRD subunits or by subsequently being repositioned for action via the binding of its N-terminal region to other PARylated proteins. In addition, post-translational modification of CHD4 might also have a role to play in regulating its activity in this context. CHD4 has already been shown to be phosphorylated in a DNA-damage-dependent manner (20). Second, it might be that the CHD4-PARP1 interaction instead plays a role in the regulation of chromatin structure and gene transcription. PARP1 has been shown both to compact and to decondense chromatin under different circumstances and also to both stimulate and inhibit the transcription of specific genes (71). Thus, PARP1 might act to antagonize CHD4 activity in certain contexts. Choosing between these (or other) models will require a deeper understanding of the biochemical mechanisms through which both PARP1 and CHD4 act.
Author Contributions
A. P. G. S. and J. P. M. coordinated the study. A. P. G. S. performed all the experiments presented in Figs. 1–6 and produced clones/proteins for Figs. 7–9. D. P. R. designed, performed, and analyzed the experiments shown in Figs. 8 and 9. Experiments shown in Fig. 7 were designed by Y. G. and S. P. J. and performed and analyzed by Y. G. J. K. K. L. produced the CHD4 catalytic mutant used in experiments from Fig. 8. M. V. provided guidance in designing and analyzing the MST experiments. A. P. G. S., D. P. R., Y. G., and J. P. M. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Ann Kwan for maintaining the NMR facility at The University of Sydney.
This work was supported by Fellowships and Project Grants (to J. P. M. and D. P. R.) from the National Health and Medical Research Council of Australia. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 2n5n) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. P. G. Silva, D. P. Ryan, Y. Galanty, J. K. K. Low, M. Vandevenne, S. P. Jackson, and J. P. Mackay, manuscript in preparation.
- NuRD
- nucleosome remodeling and deacetylase
- DSB
- double strand break
- CHD4
- chromodomain helicase DNA-binding protein 4
- PAR
- poly(ADP-ribose)
- PARP
- poly(ADP-ribose) polymerase
- PHDs
- plant homeodomain
- CHD4-N
- N-terminal domain of CHD4
- MST
- microscale thermophoresis
- HSQC
- heteronuclear single quantum correlation
- 3-AB
- 3-aminobenzamide
- HMG
- high mobility group.
References
- 1.Tong J. K., Hassig C. A., Schnitzler G. R., Kingston R. E., and Schreiber S. L. (1998) Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395, 917–921 [DOI] [PubMed] [Google Scholar]
- 2.Wade P. A., Jones P. L., Vermaak D., and Wolffe A. P. (1998) A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr. Biol. 8, 843–846 [DOI] [PubMed] [Google Scholar]
- 3.Xue Y., Wong J., Moreno G. T., Young M. K., Côté J., and Wang W. (1998) NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 2, 851–861 [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., LeRoy G., Seelig H. P., Lane W. S., and Reinberg D. (1998) The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95, 279–289 [DOI] [PubMed] [Google Scholar]
- 5.Feng Q., and Zhang Y. (2003) The NuRD complex: linking histone modification to nucleosome remodeling. Curr. Top. Microbiol. Immunol. 274, 269–290 [DOI] [PubMed] [Google Scholar]
- 6.Denslow S. A., and Wade P. A. (2007) The human Mi-2/NuRD complex and gene regulation. Oncogene 26, 5433–5438 [DOI] [PubMed] [Google Scholar]
- 7.Pile L. A., Schlag E. M., and Wassarman D. A. (2002) The SIN3/RPD3 deacetylase complex is essential for G2 phase cell cycle progression and regulation of SMRTER corepressor levels. Mol. Cell Biol. 22, 4965–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B., Gogol M., Carey M., Pattenden S. G., Seidel C., and Workman J. L. (2007) Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 21, 1422–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., and Reinberg D. (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16, 2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng P. M., and Lufkin T. (2011) Embryonic stem cells: protein interaction networks. Biomol. Concepts 2, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sims J. K., and Wade P. A. (2011) Mi-2/NuRD complex function is required for normal S phase progression and assembly of pericentric heterochromatin. Mol. Biol. Cell 22, 3094–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai A. Y., and Wade P. A. (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11, 588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smeenk G., Wiegant W. W., Vrolijk H., Solari A. P., Pastink A., and van Attikum H. (2010) The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 190, 741–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown J. S., and Jackson S. P. (2015) Ubiquitylation, neddylation and the DNA damage response. Open. Biol. 5, 150018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopfner K. P., Gerhold C. B., Lakomek K., and Wollmann P. (2012) Swi2/Snf2 remodelers: hybrid views on hybrid molecular machines. Curr. Opin. Struct. Biol. 22, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burd C. J., Kinyamu H. K., Miller F. W., and Archer T. K. (2008) UV radiation regulates Mi-2 through protein translation and stability. J. Biol. Chem. 283, 34976–34982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R. 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., and Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 18.Mu J. J., Wang Y., Luo H., Leng M., Zhang J., Yang T., Besusso D., Jung S. Y., and Qin J. (2007) A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 282, 17330–17334 [DOI] [PubMed] [Google Scholar]
- 19.Stokes M. P., Rush J., Macneill J., Ren J. M., Sprott K., Nardone J., Yang V., Beausoleil S. A., Gygi S. P., Livingstone M., Zhang H., Polakiewicz R. D., and Comb M. J. (2007) Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 104, 19855–19860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polo S. E., Kaidi A., Baskcomb L., Galanty Y., and Jackson S. P. (2010) Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 29, 3130–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murawska M., Hassler M., Renkawitz-Pohl R., Ladurner A., and Brehm A. (2011) Stress-induced PARP activation mediates recruitment of Drosophila Mi-2 to promote heat shock gene expression. PLoS Genet. 7, e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M. O., and Nielsen M. L. (2013) Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285 [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y., Wang J., Ding M., and Yu Y. (2013) Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 10, 981–984 [DOI] [PubMed] [Google Scholar]
- 24.Schreiber V., Dantzer F., Ame J. C., and de Murcia G. (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 7, 517–528 [DOI] [PubMed] [Google Scholar]
- 25.Gibson B. A., and Kraus W. L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 [DOI] [PubMed] [Google Scholar]
- 26.Li G. Y., McCulloch R. D., Fenton A. L., Cheung M., Meng L., Ikura M., and Koch C. A. (2010) Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 107, 9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timinszky G., Till S., Hassa P. O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E. H., Scheffzek K., Hottiger M. O., and Ladurner A. G. (2009) A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929 [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Kim N. S., Haince J. F., Kang H. C., David K. K., Andrabi S. A., Poirier G. G., Dawson V. L., and Dawson T. M. (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 4, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hauk G., McKnight J. N., Nodelman I. M., and Bowman G. D. (2010) The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol. Cell 39, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mansfield R. E., Musselman C. A., Kwan A. H., Oliver S. S., Garske A. L., Davrazou F., Denu J. M., Kutateladze T. G., and Mackay J. P. (2011) Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J. Biol. Chem. 286, 11779–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musselman C. A., Mansfield R. E., Garske A. L., Davrazou F., Kwan A. H., Oliver S. S., O'Leary H., Denu J. M., Mackay J. P., and Kutateladze T. G. (2009) Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem. J. 423, 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryan D. P., Sundaramoorthy R., Martin D., Singh V., and Owen-Hughes T. (2011) The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 30, 2596–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma A., Jenkins K. R., Héroux A., and Bowman G. D. (2011) Crystal structure of the chromodomain helicase DNA-binding protein 1 (Chd1) DNA-binding domain in complex with DNA. J. Biol. Chem. 286, 42099–42104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamada K., Frouws T. D., Angst B., Fitzgerald D. J., DeLuca C., Schimmele K., Sargent D. F., and Richmond T. J. (2011) Structure and mechanism of the chromatin remodelling factor ISW1a. Nature 472, 448–453 [DOI] [PubMed] [Google Scholar]
- 35.Stros M., Launholt D., and Grasser K. D. (2007) The HMG-box: a versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell. Mol. Life Sci. 64, 2590–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cole C., Barber J. D., and Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linding R., Jensen L. J., Diella F., Bork P., Gibson T. J., and Russell R. B. (2003) Protein disorder prediction: implications for structural proteomics. Structure 11, 1453–1459 [DOI] [PubMed] [Google Scholar]
- 38.Yang Z. R., Thomson R., McNeil P., and Esnouf R. M. (2005) RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21, 3369–3376 [DOI] [PubMed] [Google Scholar]
- 39.Silva A. P., Kwan A. H., and Mackay J. P. (2014) 1H,13C and 15N resonance assignments of an N-terminal domain of CHD4. Biomol. NMR Assign. 8, 137–139 [DOI] [PubMed] [Google Scholar]
- 40.Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 42.Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., and Bonvin A. M. (2005) RECOORD: a recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672 [DOI] [PubMed] [Google Scholar]
- 43.Laskowski R. A., Moss D. S., and Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 44.Chiu J., March P. E., Lee R., and Tillett D. (2004) Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 46.Limoli C. L., and Ward J. F. (1993) A new method for introducing double-strand breaks into cellular DNA. Radiat. Res. 134, 160–169 [PubMed] [Google Scholar]
- 47.Lukas C., Falck J., Bartkova J., Bartek J., and Lukas J. (2003) Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 5, 255–260 [DOI] [PubMed] [Google Scholar]
- 48.Thåström A., Lowary P. T., Widlund H. R., Cao H., Kubista M., and Widom J. (1999) Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 288, 213–229 [DOI] [PubMed] [Google Scholar]
- 49.Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]
- 50.Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 51.Wienken C. J., Baaske P., Rothbauer U., Braun D., and Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 52.Yang J. G., Madrid T. S., Sevastopoulos E., and Narlikar G. J. (2006) The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat. Struct. Mol. Biol. 13, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 53.D'Amours D., Desnoyers S., D'Silva I., and Poirier G. G. (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 249–268 [PMC free article] [PubMed] [Google Scholar]
- 54.Kraus W. L., and Lis J. T. (2003) PARP goes transcription. Cell 113, 677–683 [DOI] [PubMed] [Google Scholar]
- 55.Pinnola A., Naumova N., Shah M., and Tulin A. V. (2007) Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. J. Biol. Chem. 282, 32511–32519 [DOI] [PubMed] [Google Scholar]
- 56.Rouleau M., Aubin R. A., and Poirier G. G. (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J. Cell Sci. 117, 815–825 [DOI] [PubMed] [Google Scholar]
- 57.Gerlitz G., Hock R., Ueda T., and Bustin M. (2009) The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 87, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malarkey C. S., and Churchill M. E. (2012) The high mobility group box: the ultimate utility player of a cell. Trends Biochem. Sci. 37, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love J. J., Li X., Case D. A., Giese K., Grosschedl R., and Wright P. E. (1995) Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature 376, 791–795 [DOI] [PubMed] [Google Scholar]
- 60.Allain F. H., Yen Y. M., Masse J. E., Schultze P., Dieckmann T., Johnson R. C., and Feigon J. (1999) Solution structure of the HMG protein NHP6A and its interaction with DNA reveals the structural determinants for non-sequence-specific binding. EMBO J. 18, 2563–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dow L. K., Jones D. N., Wolfe S. A., Verdine G. L., and Churchill M. E. (2000) Structural studies of the high mobility group globular domain and basic tail of HMG-D bound to disulfide cross-linked DNA. Biochemistry 39, 9725–9736 [DOI] [PubMed] [Google Scholar]
- 62.Formosa T., Eriksson P., Wittmeyer J., Ginn J., Yu Y., and Stillman D. J. (2001) Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 20, 3506–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orphanides G., Wu W. H., Lane W. S., Hampsey M., and Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 64.Papoulas O., Daubresse G., Armstrong J. A., Jin J., Scott M. P., and Tamkun J. W. (2001) The HMG-domain protein BAP111 is important for the function of the BRM chromatin-remodeling complex in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 5728–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ray S., and Grove A. (2009) The yeast high mobility group protein HMO2, a subunit of the chromatin-remodeling complex INO80, binds DNA ends. Nucleic Acids Res. 37, 6389–6399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang W., Chi T., Xue Y., Zhou S., Kuo A., and Crabtree G. R. (1998) Architectural DNA binding by a high-mobility-group/kinesin-like subunit in mammalian SWI/SNF-related complexes. Proc. Natl. Acad. Sci. U.S.A. 95, 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sala A., La Rocca G., Burgio G., Kotova E., Di Gesù D., Collesano M., Ingrassia A. M., Tulin A. V., and Corona D. F. (2008) The nucleosome-remodeling ATPase ISWI is regulated by poly-ADP-ribosylation. PLos Biol. 6, e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gottschalk A. J., Timinszky G., Kong S. E., Jin J., Cai Y., Swanson S. K., Washburn M. P., Florens L., Ladurner A. G., Conaway J. W., and Conaway R. C. (2009) Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 106, 13770–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gottschalk A. J., Trivedi R. D., Conaway J. W., and Conaway R. C. (2012) Activation of the SNF2 family ATPase ALC1 by poly(ADP-ribose) in a stable ALC1.PARP1.nucleosome intermediate. J. Biol. Chem. 287, 43527–43532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muthurajan U. M., Hepler M. R., Hieb A. R., Clark N. J., Kramer M., Yao T., and Luger K. (2014) Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc. Natl. Acad. Sci. U.S.A. 111, 12752–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim M. Y., Zhang T., and Kraus W. L. (2005) Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 19, 1951–1967 [DOI] [PubMed] [Google Scholar]
- 72.Bond C. S., and Schüttelkopf A. W. (2009) ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 65, 510–512 [DOI] [PubMed] [Google Scholar]
- 73.Ayed A., Mulder F. A., Yi G. S., Lu Y., Kay L. E., and Arrowsmith C. H. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Biol. 8, 756–760 [DOI] [PubMed] [Google Scholar]