

Abstract
Intracellular trafficking of viruses can be influenced by a variety of inter-connected cellular sorting and degradation pathways involving endo-lysosomal vesicles, the ubiquitin-proteasome system, and autophagy-based or endoplasmic reticulum-associated machinery. In the case of recombinant adeno-associated viruses (AAV), proteasome inhibitors are known to prevent degradation of ubiquitinated AAV capsids, thereby leading to increased nuclear accumulation and transduction. However, the impact of other cellular degradation pathways on AAV trafficking is not well understood. In the current study, we screened a panel of small molecules focused on modulating different cellular degradation pathways and identified eeyarestatin I (EerI) as a novel reagent that enhances AAV transduction. EerI improved AAV transduction by an order of magnitude regardless of vector dose, genome architecture, cell type, or serotype. This effect was preceded by sequestration of AAV within enlarged vesicles that were dispersed throughout the cytoplasm. Specifically, EerI treatment redirected AAV particles toward large vesicles positive for late endosomal (Rab7) and lysosomal (LAMP1) markers. Notably, MG132 and EerI (proteasomal and endoplasmic reticulum-associated degradation inhibitors, respectively) appear to enhance AAV transduction by increasing the intracellular accumulation of viral particles in a mutually exclusive fashion. Taken together, our results expand on potential strategies to redirect recombinant AAV vectors toward more productive trafficking pathways by deregulating cellular degradation mechanisms.
Keywords: confocal microscopy, gene therapy, intracellular trafficking, proteasome, virus, adeno-associated virus, transduction, vesicle
Introduction
Eukaryotic cells utilize tightly regulated pathways to sort and degrade internalized cargo by exploiting lysosomal proteases, the ubiquitin-proteasome system, and endoplasmic reticulum (ER)3-associated or autophagy-based machinery. ER-associated degradation, or ERAD, is a critical eukaryotic process that involves extraction and ubiquitination of misfolded proteins followed by their degradation by the proteasomal machinery (1). Several viral pathogens exploit this process to infect and replicate within host cells (2, 3). For instance, polyomaviruses appear to interact with ER lumen components to rearrange capsid proteins and subsequently retrotranslocate into the cytosol (4). Another constitutive degradation pathway essential for maintaining cellular homeostasis is autophagy (5). As with ERAD, several viruses have now been shown to subvert or mimic autophagy to facilitate replication and/or dissemination (6–8). The specific degradation pathway and the subsequent intracellular fate of viral proteins as well as their genomic cargo within the host cell are often dictated by a variety of preceding vesicular sorting events.
Recently, vesicular transport of non-enveloped parvoviruses such as the minute virus of mice (MVM) through the ER and Golgi has been shown to accelerate progeny virus release (9). A particularly interesting member of the same parvovirus family is the helper-dependent adeno-associated virus (AAV), which replicates upon co-infection with adenoviruses or other viruses such as herpes simplex virus or papillomavirus. This small, non-pathogenic Dependoparvovirus contains a 4.7-kb ssDNA genome packaged within an icosahedral capsid ∼25 nm in diameter (10). Different AAV serotypes recognize various cell surface glycans such as heparan sulfate, sialic acid, or galactose as primary receptors for attachment (11). Subsequent internalization of AAV particles into endocytic vesicles is thought to be mediated by integrins and/or specific transmembrane receptors. In addition, several diverse and cell-specific mechanisms of endocytic uptake ranging from macropinocytosis to the CLIC/GEEC (CLathrin-Independent Carriers, GPI-Enriched Endocytic Compartment) pathway have been described (12, 13). Despite these differences, perinuclear accumulation within the Golgi apparatus (14–18) and exploitation of the nuclear import machinery for nuclear entry appear to be broadly conserved, downstream trafficking events (19).
Although these studies provide a detailed map of AAV transport within the host cell, it remains unclear whether the modulation of cellular degradation pathways such as ERAD or autophagy outlined earlier can influence AAV trafficking. Most studies to date have focused on proteasome inhibitors such as MG132 (20), Llnl (21), and bortezomib or carfilzomib (22, 23), which have been shown to increase AAV transduction through increased nuclear/nucleolar accumulation of viral particles. In the current study, we tested the effect of several small molecules that modulate the ubiquitin-proteasome system, autophagy, and/or ERAD on AAV transduction. The overall goal of the study was to understand the interplay (or lack thereof) between these different cellular degradation pathways in facilitating or restricting AAV trafficking within host cells. In doing so, we identified an ERAD inhibitor (eeyarestatin I/EerI) that deregulates endocytic sorting of AAV particles and redirects viral transport toward Rab7/Lamp1+ vesicles prior to nuclear entry. More importantly, we established an approach to facilitate improved trafficking of AAV capsids to the nucleus through mutually exclusive, yet synergistic approaches.
Materials and Methods
Cell Culture
HeLa, HepG2, and Huh7 cells were maintained in Dulbecco's modified Eagle's medium with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin B (Sigma-Aldrich). Human fibroblasts (AG05244)were obtained from Coriell Cell Repositories (Camden, NJ) and were maintained in Dulbecco's modified Eagle's medium with 15% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained at 37 °C and 5% CO2.
Antibodies, Chemicals, and Cell Labeling Reagents
Mouse anti-VCP (ab11433), rabbit anti-VCP (ab109240), and mouse anti-actin (ab3280) antibodies were obtained from Abcam (Cambridge, MA). Rabbit anti-EEA1 (C45B10) and rabbit anti-Golgin97 (D8P2K) were obtained from Cell Signaling (Danvers, MA). Rabbit anti-STX5 (110053) was obtained from Synaptic Systems (Goettingen, Germany). Goat anti-mouse-HRP antibody (32430) was obtained from Thermo Fisher. Anti-capsid protein antibody B1 (24) was used to blot for capsid protein, whereas anti-capsid antibody A20 (25) was used for immunoprecipitation and immunostaining. EerI (E1286), PR-619 (SML0430), PYR-41 (N2915), 3-methyladenine (M9281), nicardipine (N7510), and spautin-1 (SML0440) were obtained from Sigma-Aldrich. MG132 (10012628) was obtained from Cayman Chemical (Ann Arbor, MI). Bortezomib (S1013) was obtained from Selleck Chemicals (Houston, TX). BacMam 2.0 baculovirus delivering Emerald Green GFP (emGFP)-tagged Rab7a (late endosomal marker, C10588) and LAMP1 (lysosomal marker, C10596), were obtained from Life Technologies.
Recombinant AAV Production
Recombinant AAV packaging chicken β-actin (CBA) promoter-driven firefly luciferase (fLuc) as well as single-stranded and self-complementary vectors packaging a truncated CBA promoter driving GFP reporters were produced in HEK293 cells using the triple plasmid transfection protocol and purified, and the titers determined as described earlier (26, 27).
Transduction and Cell Viability Assays
Cells were plated at a density of 5 × 104 cells/well in 24-well plates and allowed to adhere overnight. Unless otherwise indicated, cells were treated with DMSO vehicle control or EerI for 4 h before transduction with AAV2-CBA-fLuc at 1,000 (vector genomes) vg/cell. Cells were lysed 24 h after using the luciferase assay system from Promega (Madison, WI) according to the manufacturer's instructions and read on a Wallac® 1420 Victor3 automated plate reader. Cell viability was assayed using the CellTiter Glo® luminescent cell viability assay from Promega according to the manufacturer's instructions on the same instrument. Experiments were all performed in quadruplicate. For time course studies, EerI was provided at the designated time points by adding concentrated EerI stock solution in medium to a final concentration of 10 μm.
Cell Surface Binding and Uptake Assays
HeLa cells were plated at a density of 1 × 105 cells/well in 24-well plates and allowed to adhere overnight. Cells were prechilled at 4 °C for 30 min, and then incubated with AAV2 particles at 10,000 vg/cell for 1 h at 4 °C. Cells were then washed with ice-cold PBS to remove unbound virions. Cells were then scraped off the plate, and whole genomic DNA was extracted using a DNeasy kit (Qiagen). In addition, cells being utilized to monitor viral uptake were transferred to 37 °C for 1 h, following which cells were trypsinized and washed three times with PBS to remove all un-internalized virions. Viral DNA was then extracted using a DNeasy kit (Qiagen).
Confocal Fluorescence Microscopy
Cells were plated on slide covers in 24-well plates at a density of 5 × 104 cells/well and allowed to adhere overnight. Cells were plated at a density of 2.5 × 104 cells/well and infected concurrently with the BacMam baculovirus encoding for GFP-tagged endosomal markers (20 copies/cell). Baculovirus was removed from cells after 24 h, and cells were allowed to recover for an additional 24 h prior to further studies. Cells were then treated with EerI 4 h before incubation with AAV2 at 50,000 vg/cell. After 8 h, cells were fixed with 2% paraformaldehyde for 15 min and then permeabilized with 0.2% Triton X-100 for 5 min. Cells were stained with primary antibody overnight at 4 °C, and then stained with secondary antibody for 1 h at 37 °C. Cells were then mounted with Prolong Gold antifade with DAPI (Life Technologies) and imaged using a Zeiss 710 scanning confocal microscope.
Image Analysis
Quantification of AAV particle colocalization with subcellular markers was carried out using ImageJ software with the Colocalization Analysis tools from the Wright Cell Imaging Facility website. In the case of BacMam 2.0 incubated cells, care was taken to include only cells expressing the GFP-tagged vesicle markers in the colocalization analysis. Data are represented as Pearson's correlation coefficients. Briefly, a Pearson's correlation coefficient of 0 represents random localization, whereas a value of 1 represents perfect colocalization. Therefore, a higher coefficient represents greater colocalization of AAV particles with the corresponding subcellular marker.
Statistical Analysis
All data are expressed as mean with error bars representing S.E. A two-tailed unpaired student t test was used for all statistical analysis. p values less than 0.05 were considered significant. Asterisks are used to indicate p values as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.005.
Results
Eeyarestatin I Increases AAV Transduction
Small molecule inhibitors that augment cellular degradation mechanisms have been used extensively to dissect virus-host interactions (4, 28, 29). We first treated HeLa cells with various small molecules that modulate the cellular ubiquitin proteasome system and then incubated the cells with AAV2-CBA-fLuc. Inhibitors of the proteasome, MG132 and bortezomib, increased transduction by approximately a log order (Fig. 1A), as reported previously (20, 30). Interestingly, PR-619, a pan-deubiquitinase inhibitor, did not alter transduction, whereas PYR-41, an inhibitor of the ubiquitin-activating enzyme UBA1, increased transduction as effectively as proteasome inhibitors (Fig. 1A). We then investigated a potential role for autophagy in AAV transduction, which has recently been shown to be critical in the infection pathway of numerous viruses (6–8). However, neither the autophagy inducer, nicardipine nor the autophagy inhibitor, spautin-1 altered AAV transduction (Fig. 1B). We then tested ERAD using two inhibitors, EerI and kifunensine (Kif). Interestingly, although Kif treatment did not significantly alter transduction, EerI treatment led to a transduction increase of approximately a log order (Fig. 1C).
FIGURE 1.
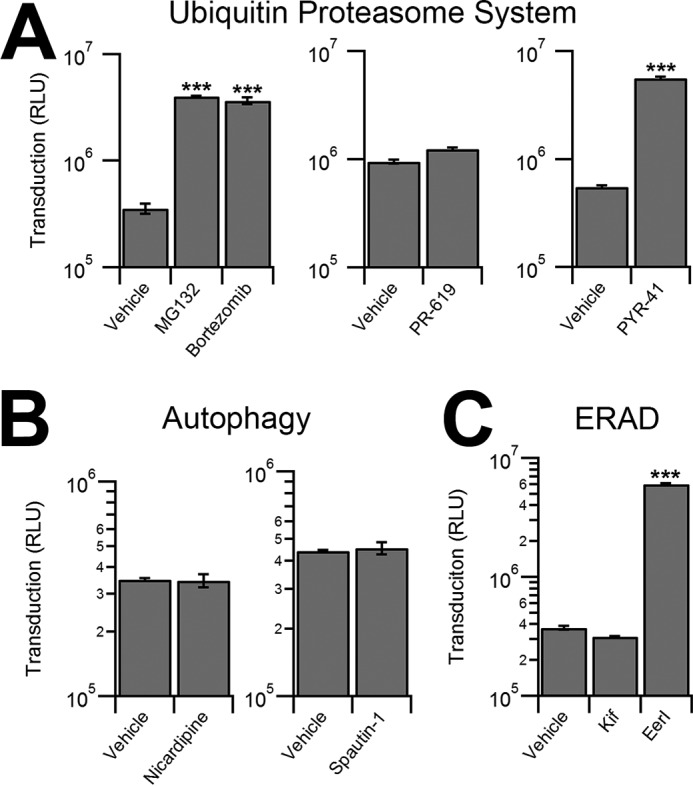
Effect of different chemical inhibitors of cellular degradation pathways on AAV transduction. A, luciferase reporter expression in HeLa cells pretreated with vehicle controls or inhibitors of the ubiquitin proteasome system and transduced with AAV2-Luc. Effect of proteasome inhibitors MG132 (10 μm), bortezomib (500 nm), pan-deubiquitinase inhibitor PR-619 (5 μm), or UBE1 inhibitor, PYR-41 (50 μm) is shown. RLU, relative light units. B, HeLa cells pretreated with vehicle control or autophagy modulators Nicardipine (5 μm) or Spautin-1 (10 μm) transduced with AAV2-Luc. C, HeLa cells pretreated with vehicle control or ERAD inhibitors EerI (10 μm) or Kif (1 μm) transduced with AAV2-Luc vectors.***, p < 0.005.
Because EerI has been shown to directly interact with the AAA+ ATPase valosin-containing protein, also known as VCP/p97 (31), we utilized three additional VCP inhibitors, ML240, NMS-873, or DBeQ (N2N4-dibezylquinazoline-2,4-diamine) to recapitulate these results. However, as shown in Fig. 2A, none of the VCP inhibitors increased AAV transduction with the exception of EerI. It should be noted that siRNA-mediated knockdown of VCP/p97 was accompanied by significant cytotoxicity (>50%) precluding efforts to directly address the potential (indirect) role of VCP/p97 in AAV transduction (data not shown). To confirm whether the increase in transduction efficiency was due to pleiotropic effects of EerI, we transfected HeLa cells with the pTR-CBA-fLuc packaging plasmid as control, allowed 48 h for transgene expression, and then treated the cells with EerI for 24 h. We observed no change in fLuc expression as a result of EerI treatment (Fig. 2B), confirming that the effect seen exclusively affects AAV transduction and does not enhance transcription or protein stability in general.
FIGURE 2.

EerI increases AAV transduction in a serotype-, cell type-, and vector genome-independent manner. A, luciferase reporter expression in HeLa cells pretreated with vehicle control, 1 μm ML240, 1 μm NMS-873, 10 μm DBeQ, or 10 μm EerI was transduced with AAV2-Luc. RLU, relative light units. B, luciferase expression in HeLa cells transfected with the pTR-CBA-Luc plasmid and treated with various concentrations of EerI 48 h post-transfection. C, HeLa cells pretreated with various concentrations of EerI and transduced with AAV2-Luc (gray bars) or subjected to a CellTiter-Glo assay to determine cell viability (circles). D, HeLa cells pretreated with either DMSO control (squares) or EerI (triangles) and transduced with AAV2-Luc at increasing vector genome copies (vg) per cell. E, HeLa cells were pretreated with DMSO control (gray bars) or EerI (white bars) and transduced with various AAV serotypes packaging the Luc transgene cassette. F, HepG2, Huh7, and human fibroblasts (hFibroblast) were pretreated with EerI at concentrations individually optimized to limit cytotoxicity (HepG2, 8 μm; Huh7, 4 μm; human fibroblasts, 4 μm) and transduced with AAV2-CBA-Luc. G, HeLa cells were pretreated with DMSO control or EerI and then transduced with either single-stranded ssAAV2-GFP vectors or self-complementary scAAV2-GFP vectors. Cells were fixed 24 h and imaged using confocal microscopy. *, p < 0.05; **, p < 0.01; ***, p < 0.005.
Next, we investigated the effect of altering EerI concentration or virus dose on transduction. We observed a dose-dependent increase of AAV transduction at EerI concentrations up to 15 μm, after which a decrease in transduction efficiency was observed due to cytotoxicity (Fig. 2C). Further, at an optimal EerI concentration of 10 μm, we observed a uniformly beneficial effect on transduction efficiency in HeLa cells over a broad range of viral doses ranging from 10 to 10,000 vg/cell (Fig. 2D).
EerI Enhances AAV Transduction Independent of Capsid, Vector Genome, or Cell Type
We next sought to investigate whether varying AAV capsid types or cell types affected the ability of EerI to enhance transduction. We investigated this possibility by incubating HeLa cells with numerous AAV serotypes at 1,000 vg/cell. As seen in Fig. 2E, EerI uniformly increases the transduction of all serotypes tested, although at varying levels. In addition, transduction was increased in other human cell types tested, albeit at different optimal EerI concentrations based on the toxicity profile (Fig. 2F). Further, we determined that EerI enhances the intensity of GFP reporter gene expression mediated by AAV vectors packaging single-stranded (ss) or self-complementary (sc) GFP reporter cassettes in a similar fashion (Fig. 2G). Taken together, these data demonstrate that EerI increases transduction regardless of capsid, second strand synthesis, or cell type.
EerI Redistributes AAV from a Perinuclear Pattern to Large, Dispersed Vesicles in the Cytoplasm
Given the broad impact of EerI on AAV transduction, we assessed whether these effects were preceded by notable changes in the intracellular trafficking of AAV particles through confocal fluorescence microscopy studies. Specifically, we pretreated HeLa cells with EerI and incubated the cells with AAV2 particles, followed by immunofluorescent labeling of AAV capsids and VCP at 2, 4, and 8 h post-incubation. We observed no significant correlation between the intracellular patterns of VCP (the apparent target of EerI discussed later) and AAV capsid immunostaining at 2, 4 or 8 h post-incubation with or without EerI treatment (Fig. 3, A–C). As seen in higher magnification images (bottom panels), a steady increase in the perinuclear accumulation of AAV particles was observed over time. At 8 h post-incubation (Fig. 3C), we observed that although some AAV particles remained in the perinuclear region, a majority of AAV particles redistributed from the perinuclear space to a large and dispersed punctate pattern throughout the cytoplasm upon VCP inhibition. This observation strikingly contrasts with DMSO-treated cells, in which AAV virions remain concentrated in the perinuclear region (Fig. 3C, bottom panel).
FIGURE 3.

EerI redirects AAV particles from a perinuclear pattern to a dispersed cytosolic punctate pattern. A–C, confocal micrographs of HeLa cells pretreated with EerI and transduced with AAV2-Luc at 50,000 vg/cell. Cells were fixed at 2 h (A), 4 h (B), and 8 h (C), immunostained for VCP (green) and AAV2 capsid (magenta) as outlined under “Materials and Methods,” and counterstained with DAPI (blue). White inset boxes indicate areas imaged at higher magnification. Scale bar = 20 μm. p.i., post incubation.
EerI Redirects AAV Particles to Late Endosomes and Lysosomes
In an effort to identify the nature of the punctate structures formed in the cytoplasm upon EerI treatment, we utilized the BacMam 2.0 system from Life Technologies, which allowed the delivery of GFP-tagged versions of different endosomal markers, specifically Rab7 (late endosome) and LAMP1 (lysosome) using baculoviral expression vectors. In addition, we utilized an EEA1 antibody to stain for early endosomes. At 8 h post-incubation, AAV particles did not appear to colocalize with EEA1+ vesicles (data not shown), indicating that AAV is not associated with early endosomes at this time interval. However, AAV particles colocalized prominently with Rab7+ (Fig. 4A) and more extensively with LAMP1+ vesicles in EerI-treated cells (Fig. 4B). This increased colocalization was further confirmed by quantitation of fluorescent signal using the ImageJ software as outlined under “Materials and Methods.” In particular, we determined the Pearson's coefficients (Table 1) for AAV particle colocalization with different subcellular markers including EEA1 (early endosomes), Rab7a (late endosomes), LAMP1 (lysosomes), Golgin-97 (Golgi), and STX5 (syntaxin 5). A statistically significant increase in Pearson's coefficients was noted for redistribution of AAV particles to Rab7 and LAMP1+ vesicles.
FIGURE 4.

AAV particles accumulate within enlarged Rab7+ and Lamp1+ vesicles upon treatment with EerI. A and B, confocal micrographs of HeLa cells infected with BacMam baculovirus encoding for GFP-Rab7a (A) or GFP-LAMP1 (B). Cells were pretreated with DMSO or EerI at 72 h post-plating and incubated with AAV2-Luc at 50,000 vg/cell. Cells were then fixed at 8 h, immunostained for AAV capsid (magenta), and counterstained with DAPI (blue). White inset boxes indicate areas imaged at higher magnification. Scale bar = 20 μm.
TABLE 1.
Pearson coefficient values for colocalization of subcellular markers with AAV capsid immunofluorescence (A20 monoclonal antibody)
Values are depicted as means ± S.E. (*, p < 0.05: significant; n.s.: not significant).
| Subcellular marker | DMSO | EerI | p value |
|---|---|---|---|
| EEA1 | 0.198 ± 0.018 | 0.252 ± 0.040 | n.s |
| Rab7a | 0.216 ± 0.019 | 0.342 ± 0.024 | *, p < 0.05 |
| LAMP1 | 0.269 ± 0.036 | 0.439 ± 0.025 | *, p < 0.05 |
| Golgin97 | 0.379 ± 0.025 | 0.312 ± 0.032 | n.s |
| STX5 | 0.453 ± 0.009 | 0.334 ± 0.042 | n.s |
EerI Influences an Early, Post-entry Trafficking Event during AAV Transduction
To further understand the effect of EerI on AAV transduction, we systematically investigated the impact of EerI treatment on each step of the AAV intracellular trafficking pathway. First, we assessed whether EerI affects early steps during transduction, i.e. AAV capsid binding to the cell surface and endocytic uptake. As seen in Fig. 5, A and B, EerI treatment does not alter the number of bound virions attached to the cell surface nor internalized virions at 1 h post-incubation. Further evidence supporting the notion that EerI influences a downstream step in the AAV trafficking pathway was obtained by treating cells at different time intervals before or after viral incubation. As shown in Fig. 5C, the beneficial effect of EerI on AAV transduction gradually weakened when added at later time intervals. Specifically, EerI addition as late as 4 h after incubation with viral particles increased AAV transduction, albeit to a lesser degree when compared with drug pretreatment. By 8 h post-AAV incubation, the addition of EerI increased transduction efficiency by less than 1.5-fold. When considered together with confocal fluorescence data, these results suggest that EerI treatment alters the endocytic sorting of AAV particles after cellular uptake, but prior to nuclear entry.
FIGURE 5.

EerI and MG132 increase AAV transduction through distinct, yet cumulative mechanisms. A, cell surface binding of AAV particles incubated on HeLa cells that were pretreated with DMSO or EerI. Viral particles were incubated on cells at 4 °C for 1 h, following which cells were washed to remove unbound virus and cell surface-associated vector genome DNA was quantified by quantitative PCR. RLU, relative light units. B, HeLa cells incubated with AAV particles at 4 °C for 1 h were washed to remove unbound virions and then shifted to 37 °C for 1 h to allow cellular uptake. Cells were then treated with trypsin to remove surface-bound virions, and internalized virions were quantitated by quantitative PCR. C, luciferase expression in HeLa cells treated with DMSO control or EerI at various time intervals prior to transduction with AAV2-Luc vectors. D, confocal micrographs of HeLa cells pretreated with EerI or MG132 and incubated with AAV particles at 50,000 vg/cell. Cells were fixed at 8 h, immunostained for AAV capsids (magenta), and counterstained with DAPI (blue). Scale bar = 20 μm. E, recovery of internalized vector genome-containing particles at different time intervals from HeLa cells pretreated with DMSO, EerI, or MG132 and transduced with AAV2-Luc as described above. F, luciferase expression in HeLa cells pretreated with DMSO control, EerI, MG132, or EerI and MG132 in combination and transduced with AAV2-Luc. At 4 h post-incubation, the drugs and virus were removed and replaced with fresh prewarmed medium. *, p < 0.05; **, p < 0.01; ***, p < 0.005.
EerI and MG132 Enhance AAV Transduction in a Mutually Exclusive Manner
We next sought to characterize any similarities and/or differences in the effect(s) of proteasome inhibitors and EerI on AAV transduction. Confocal microscopy experiments revealed a stark difference in capsid accumulation upon treatment with either EerI or MG132. As seen in Fig. 5D, where EerI treatment results in accumulation in enlarged vesicles, MG132 treatment leads to a more disseminated capsid accumulation in the perinuclear region. Further, proteasomal inhibition is thought to directly enhance AAV transduction by preventing capsid degradation in the cytosol. As a consequence, enhanced recovery of intact AAV capsids and genomes from the nuclear fraction has been reported by several groups (20–23, 32). In the current study, upon EerI treatment, we observed a striking increase in the number of AAV genomes recovered after synchronized cellular uptake at later time intervals (Fig. 5E). These results were essentially identical to those obtained upon proteasomal inhibition with MG132. Specifically, no differences in recovered vg were observed at early time intervals up to 4 h. However, as much as 80–90% of AAV genomes was recovered at 16–24 h upon treatment with EerI in contrast to 25–35% of AAV genomes obtained from untreated cells. These data might indicate that EerI treatment indirectly helps protect AAV genomes from rapid degradation.
The above described results prompted us to further consider the possibility that EerI might not only affect AAV trafficking, but also influence AAV capsid degradation in a pleiotropic manner. To assess this possibility, cells were co-treated with EerI and MG132, alone or in combination. It is noteworthy to mention that these studies were optimized to minimize the combined cytotoxicity arising from dual drug treatment. As seen in Fig. 5F, EerI enhanced AAV transduction by 4-fold, whereas MG132 was twice as effective, resulting in an ∼8-fold increase in transduction. Importantly, combined drug treatment was synergistic and enhanced AAV transduction by nearly 15-fold. These results demonstrate that cellular degradation pathways can influence AAV infection in distinct ways, which, when modulated in conjunction, can cumulatively enhance AAV transduction.
Discussion
EerI is widely viewed as an ERAD inhibitor that is also capable of augmenting the unfolded protein response (33). In addition, EerI has been shown to block Sec61-mediated protein translocation from the cytosol into the ER (34) and was recently shown to affect productive ER exit of BK polyomavirus (4). Intriguingly, EerI has also been reported to interfere with both retrograde as well as anterograde trafficking of endocytic cargo (35). Specifically, treatment with this drug not only delayed the trafficking of Shiga toxin to the perinuclear region and the Golgi, but also delayed the escape of diphtheria toxin into the cytosol from acidified vesicles. In the current study, we observed that EerI treatment redirects the intracellular trafficking of AAV particles from a conventional, Golgi-directed retrograde pathway toward late endosomes and/or lysosomes. Specifically, a large number of AAV particles were segregated in large Rab7/LAMP1+ vesicles. These observations are supported in part by earlier studies that reported a dramatic increase in the size of endosomes upon EerI treatment (36). Further, these observations are consistent with earlier studies demonstrating that AAV transport through Rab7+ late endosomes/lysosomes is highly productive due to their high motility and faster retrograde velocities when compared with early/recycling endosomes in neurons (37). Such a scenario is possible due to the low pH and high protease activity within such vesicles that could more effectively prime the AAV capsid (38) by exposing the VP1 phospholipase A2 (PLA2) domain (39) and nuclear localization signals (40).
Another important observation is that EerI treatment affected AAV trafficking to the microtubule-organizing center in the perinuclear region. Previous studies have indicated that AAV transduction efficiency is affected by knockdown of proteins involved in sorting retrograde cargo, such as syntaxin 5, calnexin, KDEL receptor (KDELR), and other Golgi/ER-associated proteins (16, 18). In the current study, EerI treatment appears to decrease the colocalization of AAV particles with markers such as STX5 and Golgin-97 (Table 1), although these results were not statistically significant. Nevertheless, our results suggest that efficient vesicular trafficking can enhance AAV transduction. This overall approach toward improved AAV trafficking (depicted in Fig. 6) is further supported by (i) the increased recovery of vector genome copy numbers from within cells at time intervals as late as 16–24 h following EerI treatment, similar to that observed with the proteasomal inhibitor, MG132 and (ii) the cumulative effects of EerI and MG132 on AAV transduction.
FIGURE 6.
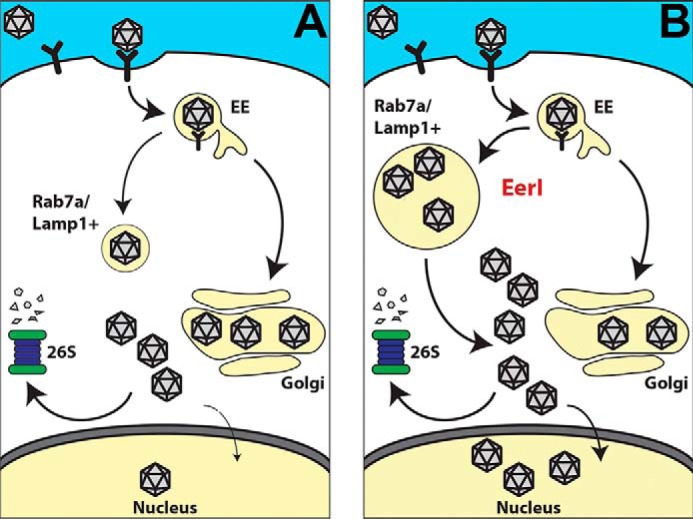
Schematic outlining a potential approach to enhance transduction by redirecting the vesicular trafficking of AAV particles. A, following cellular uptake, AAV virions are subjected to endosome-to-lysosome transport as well as retrograde transport from early endosomes (EE) to the Golgi leading to perinuclear accumulation. Cytosolic escape of AAV particles results in processing by the ubiquitin-proteasome system, which in turn limits nuclear entry and transduction efficiency. B, in cells treated with EerI, virions appear to accumulate within Rab7a/Lamp1+ vesicles instead of a perinuclear pattern due to an unknown mechanism. Sequestration from proteasomal processing and efficient “priming” of AAV capsids could result in increased nuclear entry and subsequent transduction.
At the mechanistic level, EerI has been shown to directly bind VCP, an essential component of the ERAD machinery (31, 41). However, we note that attempts to modulate AAV transduction by using other small molecule VCP inhibitors (Fig. 2A), using siRNA-mediated knockdown of VCP/p97, or colocalizing AAV capsids with VCP/p97 through confocal microscopy or immunoprecipitation studies were unsuccessful (data not shown). These studies are also particularly challenging due to overt toxicity displayed during RNAi-mediated knockdown of VCP as well as overexpression of WT/dominant negative forms of VCP. Thus, it is important to consider the pleotropic effects of chemical inhibitors such as EerI in such studies. This aspect is also consistent with earlier studies, where EerI has been shown to augment cellular processes separate from ERAD (34–36, 42). Thus, the exact molecular mechanism(s) that are implicated in the current study remain to be determined.
A key question is whether such studies have the potential for translational impact in recombinant AAV vector-mediated gene therapy. In this regard, numerous chemical inhibitors of VCP, including EerI, are currently being evaluated for their potential as chemotherapeutics (43, 44). However, similar to proteasome inhibitors such as bortezomib, the effective dose and toxicity of such agents will need to be determined in preclinical animal models as well as the clinic prior to potential applications in the gene therapy setting. Nevertheless, our study constitutes an important step forward in understanding the interplay between different cellular degradation pathways that influence AAV trafficking and highlight potential strategies to modulate the same.
Author Contributions
G. E. B. and A. A. designed the overall study. G. E. B. carried out all experiments and data analysis. G. E. B. and A. A. wrote the manuscript.
This study was supported by National Institutes of Health Grants R01HL089221 and P01HL112761) (to A. A.) through the NHLBI. This work was also supported by National Institutes of Health Training Grant 5T32 GM007092 (to G. E. B.) through the NIGMS. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- AAV
- adeno-associated virus
- EerI
- eeyarestatin I
- Kif
- kifunensine
- VCP
- valosin-containing protein
- CBA
- chicken β-actin
- fLuc
- firefly luciferase
- DMSO
- dimethyl sulfoxide
- vg
- vector genomes.
References
- 1.Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byun H., Gou Y., Zook A., Lozano M. M., and Dudley J. P. (2014) ERAD and how viruses exploit it. Front. Microbiol. 5, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noack J., Bernasconi R., and Molinari M. (2014) How viruses hijack the ERAD tuning machinery. J. Virol. 88, 10272–10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett S. M., Jiang M., and Imperiale M. J. (2013) Role of cell-type-specific endoplasmic reticulum-associated degradation in polyomavirus trafficking. J. Virol. 87, 8843–8852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boya P., Reggiori F., and Codogno P. (2013) Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkegaard K. (2009) Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 335, 323–333 [DOI] [PubMed] [Google Scholar]
- 7.Jackson W. T. (2015) Viruses and the autophagy pathway. Virology. 479–480, 450–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lennemann N. J., and Coyne C. B. (2015) Catch me if you can: the link between autophagy and viruses. PLoS Pathog. 11, e1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bär S., Rommelaere J., and Nüesch J. P. (2013) Vesicular transport of progeny parvovirus particles through ER and Golgi regulates maturation and cytolysis. PLoS Pathog. 9, e1003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowles D. E., Rabinowitz J. E., and Samulski R. J. (2006) Parvoviruses, pp. 15–23, Edward Arnold Ltd., New York [Google Scholar]
- 11.Huang L. Y., Halder S., and Agbandje-McKenna M. (2014) Parvovirus glycan interactions. Curr. Opin. Virol. 7, 108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nonnenmacher M., and Weber T. (2011) Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell. Host Microbe 10, 563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg M. S., Nicolson S., Bhatt A. P., McLendon M., Li C., and Samulski R. J. (2014) Recombinant adeno-associated virus utilizes cell-specific infectious entry mechanisms. J. Virol. 88, 12472–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bantel-Schaal U., Hub B., and Kartenbeck J. (2002) Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the Golgi compartment. J. Virol. 76, 2340–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson J. S., Li C., DiPrimio N., Weinberg M. S., McCown T. J., and Samulski R. J. (2010) Mutagenesis of adeno-associated virus type 2 capsid protein VP1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction. J. Virol. 84, 8888–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson J. S., Gentzsch M., Zhang L., Ribeiro C. M., Kantor B., Kafri T., Pickles R. J., and Samulski R. J. (2011) AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis. PLoS Pathog. 7, e1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao P. J., and Samulski R. J. (2012) Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J. Virol. 86, 10462–10473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nonnenmacher M. E., Cintrat J. C., Gillet D., and Weber T. (2015) Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction. J. Virol. 89, 1673–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicolson S. C., and Samulski R. J. (2014) Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J. Virol. 88, 4132–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douar A. M., Poulard K., Stockholm D., and Danos O. (2001) Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation. J. Virol. 75, 1824–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan D., Yue Y., Yan Z., Yang J., and Engelhardt J. F. (2000) Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest. 105, 1573–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson J. S., and Samulski R. J. (2009) Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J. Virol. 83, 2632–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell A. M., and Samulski R. J. (2013) Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J. Virol. 87, 13035–13041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wobus C. E., Hügle-Dörr B., Girod A., Petersen G., Hallek M., and Kleinschmidt J. A. (2000) Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74, 9281–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wistuba A., Weger S., Kern A., and Kleinschmidt J. A. (1995) Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing Rep and Cap proteins. J. Virol. 69, 5311–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen S., Horowitz E. D., Troupes A. N., Brown S. M., Pulicherla N., Samulski R. J., Agbandje-McKenna M., and Asokan A. (2013) Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J. Biol. Chem. 288, 28814–28823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen S., Troupes A. N., Pulicherla N., and Asokan A. (2013) Multiple roles for sialylated glycans in determining the cardiopulmonary tropism of adeno-associated virus 4. J. Virol. 87, 13206–13213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mateo R., Nagamine C. M., Spagnolo J., Méndez E., Rahe M., Gale M. Jr, Yuan J., and Kirkegaard K. (2013) Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 87, 1312–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raaben M., Posthuma C. C., Verheije M. H., te Lintelo E. G., Kikkert M., Drijfhout J. W., Snijder E. J., Rottier P. J., and de Haan C. A. (2010) The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84, 7869–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathwani A. C., Cochrane M., McIntosh J., Ng C. Y., Zhou J., Gray J. T., and Davidoff A. M. (2009) Enhancing transduction of the liver by adeno-associated viral vectors. Gene Ther. 16, 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q., Shinkre B. A., Lee J. G., Weniger M. A., Liu Y., Chen W., Wiestner A., Trenkle W. C., and Ye Y. (2010) The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS ONE 5, e15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan Z., Zak R., Luxton G. W., Ritchie T. C., Bantel-Schaal U., and Engelhardt J. F. (2002) Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J. Virol. 76, 2043–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKibbin C., Mares A., Piacenti M., Williams H., Roboti P., Puumalainen M., Callan A. C., Lesiak-Mieczkowska K., Linder S., Harant H., High S., Flitsch S. L., Whitehead R. C., and Swanton E. (2012) Inhibition of protein translocation at the endoplasmic reticulum promotes activation of the unfolded protein response. Biochem. J. 442, 639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cross B. C., McKibbin C., Callan A. C., Roboti P., Piacenti M., Rabu C., Wilson C. M., Whitehead R., Flitsch S. L., Pool M. R., High S., and Swanton E. (2009) Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 122, 4393–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aletrari M. O., McKibbin C., Williams H., Pawar V., Pietroni P., Lord J. M., Flitsch S. L., Whitehead R., Swanton E., High S., and Spooner R. A. (2011) Eeyarestatin 1 interferes with both retrograde and anterograde intracellular trafficking pathways. PLoS ONE 6, e22713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramanathan H. N., and Ye Y. (2012) The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res. 22, 346–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castle M. J., Perlson E., Holzbaur E. L., and Wolfe J. H. (2014) Long-distance axonal transport of AAV9 is driven by dynein and kinesin-2 and is trafficked in a highly motile Rab7-positive compartment. Mol. Ther. 22, 554–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venkatakrishnan B., Yarbrough J., Domsic J., Bennett A., Bothner B., Kozyreva O. G., Samulski R. J., Muzyczka N., McKenna R., and Agbandje-McKenna M. (2013) Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 87, 4974–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girod A., Wobus C. E., Zádori Z., Ried M., Leike K., Tijssen P., Kleinschmidt J. A., and Hallek M. (2002) The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 83, 973–978 [DOI] [PubMed] [Google Scholar]
- 40.Grieger J. C., Snowdy S., and Samulski R. J. (2006) Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J. Virol. 80, 5199–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson P. I., and Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 42.Ramadan K., Bruderer R., Spiga F. M., Popp O., Baur T., Gotta M., and Meyer H. H. (2007) Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature. 450, 1258–1262 [DOI] [PubMed] [Google Scholar]
- 43.Magnaghi P., D'Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C., Polucci P., Ballinari D., Perrera C., Leone A., Cervi G., Casale E., Xiao Y., Wong C., Anderson D. J., Galvani A., Donati D., O'Brien T., Jackson P. K., and Isacchi A. (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556 [DOI] [PubMed] [Google Scholar]
- 44.Yi P., Higa A., Taouji S., Bexiga M. G., Marza E., Arma D., Castain C., Le Bail B., Simpson J. C., Rosenbaum J., Balabaud C., Bioulac-Sage P., Blanc J. F., and Chevet E. (2012) Sorafenib-mediated targeting of the AAA+ ATPase p97/VCP leads to disruption of the secretory pathway, endoplasmic reticulum stress, and hepatocellular cancer cell death. Mol. Cancer. Ther. 11, 2610–2620 [DOI] [PubMed] [Google Scholar]