

Abstract
The hepatitis C virus (HCV) infects approximately 3% of the world population or more than 185 million people worldwide. Each year, an estimated 350000-500000 deaths occur worldwide due to HCV-associated diseases including cirrhosis and hepatocellular carcinoma. HCV is the most common indication for liver transplantation in patients with cirrhosis worldwide. HCV is an enveloped RNA virus classified in the genus Hepacivirus in the Flaviviridae family. The HCV viral life cycle in a cell can be divided into six phases: (1) binding and internalization; (2) cytoplasmic release and uncoating; (3) viral polyprotein translation and processing; (4) RNA genome replication; (5) encapsidation (packaging) and assembly; and (6) virus morphogenesis (maturation) and secretion. Many host factors are involved in the HCV life cycle. Chaperones are an important group of host cytoprotective molecules that coordinate numerous cellular processes including protein folding, multimeric protein assembly, protein trafficking, and protein degradation. All phases of the viral life cycle require chaperone activity and the interaction of viral proteins with chaperones. This review will present our current knowledge and understanding of the role of chaperones in the HCV life cycle. Analysis of chaperones in HCV infection will provide further insights into viral/host interactions and potential therapeutic targets for both HCV and other viruses.
Keywords: Hepatitis C, Hepatitis C virus, Chaperones, Heat shock proteins, Viral life cycle
Core tip: Interaction of viral proteins with host chaperones is critical for the hepatitis C viral (HCV) life cycle. Some of these chaperones, such as cyclophilins have been studied in detail recently and have led to the advent of new therapies for HCV infection with high success rates. Further investigation of the role of chaperones in the viral life cycle may allow for development of novel therapies both for HCV and related viruses.
INTRODUCTION
The hepatitis C virus (HCV) infects approximately 3% of the world population or more than 185 million people worldwide[1,2]. While infection is less prevalent in developed countries including North America, other areas face prevalence rates as high as 3.5% or more[1]. Each year, an estimated 350000-500000 deaths occur worldwide due to HCV-associated diseases[1-3]. HCV is mainly responsible for liver transplantation in patients with cirrhosis worldwide[4-6]. Furthermore, HCV is the most common chronic bloodborne pathogen in the United States affecting 1.5% of the population and is the major etiologic factor responsible for the recent doubling of hepatocellular carcinoma[5,7-9].
HCV is an enveloped RNA virus classified in the genus Hepacivirus in the Flaviviridae family. It possesses an approximately 9.6 kb positive-sense RNA genome that is translated as a single polypeptide approximately 3000 amino acids in length[10,11]. It is subsequently proteolytically cleaved into 10 viral proteins including the structural proteins core, E1, and E2 as well as the non-structural (NS) proteins p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B[12]. Core is the viral nucleocapsid protein that encapsidates the viral genome in the virion. E1 and E2 are glycoproteins on the viral envelope that are involved in receptor-mediated viral entry. p7 is an integral membrane ion channel also called viroporin that functions to protect virions from acidification during maturation by allowing protons to flow[13]. NS2, NS3, and NS4A are the viral proteases, while NS4B is a helicase. NS5A, a 56-59 kDa multifunctional phosphoprotein, lacks any known enzymatic activity, is a component of the viral replicase complex, and has been implicated in regulation of HCV genome replication, internal ribosomal entry site (IRES)-mediated viral protein translation, and infectious virion assembly[14-18]. NS5B is the viral RNA-dependent RNA polymerase. In addition to these originally identified 10 proteins, another viral protein called the HCV F protein was observed[19,20] and later identified[21-23] to be expressed as a result of a ribosomal frameshift near the beginning of the core protein coding sequence. The F protein has been implicated in the regulation of protein degradation, inhibition of apoptosis, and immunoregulation[24-31].
The 5’ non-coding region (NCR) of the viral genome possesses an IRES, a cis-acting element found in some host RNA transcripts as well as in viruses that allows ribosomal translation initiation to occur internally within a transcript in lieu of 5’ 7-methylguanylate cap-dependent translation[12,32]. The HCV viral life cycle in a cell can be divided into six phases: (1) binding and internalization; (2) cytoplasmic release and uncoating; (3) viral polyprotein translation and processing; (4) RNA genome replication; (5) encapsidation (packaging) and assembly; and (6) virus morphogenesis (maturation) and secretion[33] (Figure 1).
Figure 1.
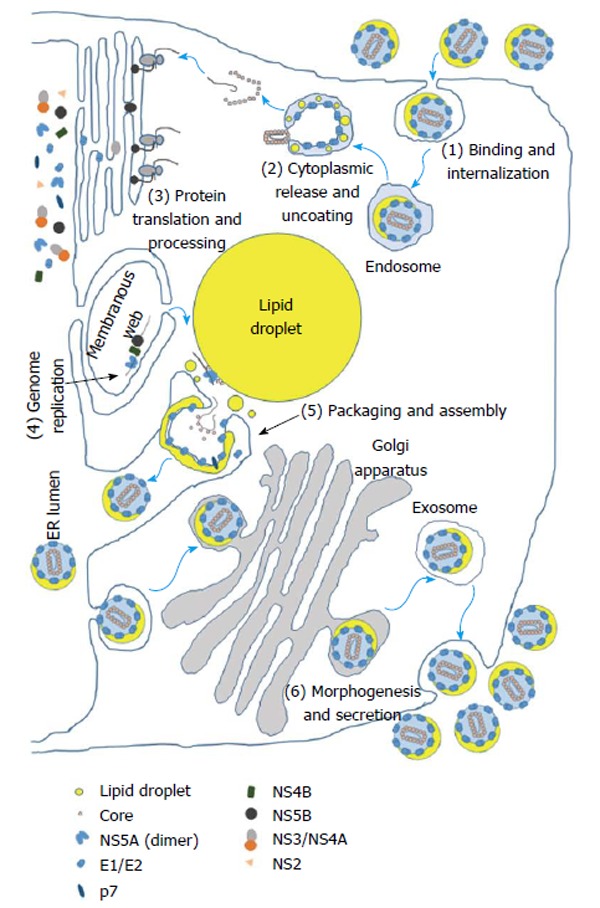
A schematic of the hepatitis C virus life cycle. The six steps of the viral life cycle are indicated in colored boxes with numbers. (1) Binding and internalization. HSC70 is part of the viral particle and may play a role in viral entry. Also HCV internalization occurs at least in part through clathrin-mediated endocytosis which involves HSC70; (2) Cytoplasmic release and uncoating. The chaperone activity of E1 and E2 may be involved in membrane fusion that releases the core-encapsidated viral genome into the cytosol; (3) Protein translation and processing. HSP70, together with the DNAJA2 member of HSP40 co-chaperones, is the main chaperone involved in IRES-mediated translation of the viral genome, while HSP90 may play some role as well. Calnexin, calreticulin, and CypA are also involved; (4) Genome replication. HSP90, some members of HSP40 co-chaperones, TRiC/CCT, FKBP38, SigR1, and some Cyps are involved in viral genome replication. Core and NS3 may play some roles in genome replication as well; (5) Packaging and assembly. HSC70, PDI, and MTTP are the principal chaperones involved in infectious virion assembly, and Cyps also play important roles; and (6) Morphogenesis and secretion. MTTP which is involved in the VLDL pathway also plays important roles in viral particle maturation and secretion. Cyps are also involved. Cyp: Cyclophilin; ER: Endoplasmic reticulum; FKBP: FK506-binding protein; HCV: Hepatitis C virus; HSC70: Heat shock cognate protein 70; HSP: Heat shock protein; MTTP: Microsomal triglyceride transfer protein; NS: Non-structural; PDI: Protein disulfide isomerase; SigR1: Sigma non-opioid intracellular receptor 1; TRiC/CCT: TCP-1 ring complex/chaperonin-containing TCP-1; VLDL: Very low-density lipoprotein.
The viral life cycle begins with the attachment of the enveloped virion to the cell followed by entry, which is mediated by interaction of the E1 and E2 glycoproteins in the viral membrane with a number of hepatocyte cell surface receptors and proteins which include the low-density lipoprotein receptor (LDLR), glycosaminoglycans (GAGs), CD81, scavenger receptor B1 (SR-B1), claudin 1, occludin, and the cholesterol absorption receptor Niemann-Pick C1-like 1[34]. Subsequently, the viral particle is internalized through clathrin-mediated endocytosis or an alternative clathrin-independent pathway after which, the viral and cellular membranes fuse through acidification of the endosomal compartment, and the core-encapsidated viral genome is released into the cytosol, uncoated, and subsequently translated[35,36]. The resulting polyprotein is cleaved with the help of the cellular proteases signalase and signal peptide peptidase and the viral proteases NS2-NS3 and NS3-NS4A[37]. Viral genome replication is carried out by NS5B utilizing a negative-sense viral genome intermediate[38]. New virions are assembled at the sites of cytosolic lipid droplets in the vicinity of endoplasmic reticulum (ER) membrane where core protein encapsidates the viral genome followed by budding of the nascent virion into the lumen of ER[39]. The virions follow the Golgi-dependent secretory pathway during which they undergo maturation by addition of lipid components significantly decreasing their buoyant density[40,41]. Finally the mature virions are secreted through exocytosis[42].
In order to establish successful infection, HCV depends on numerous host factors during its entire life cycle. In addition to performing virus-specific functions such as viral genome replication and virion assembly, HCV proteins alter cellular metabolism, critical signaling pathways, and organellar morphology and function to establish persistent infection and to escape the immune responses. Accumulation of misfolded viral proteins in the ER leads to ER stress and the unfolded protein response (UPR) which is a cellular program to help restore ER protein homeostasis by shutting down cellular protein synthesis, properly folding the misfolded proteins, targeting them to ER-associated degradation (ERAD) if folding is unsuccessful, and inducing apoptosis if the cell cannot cope with the ER stress[43]. HCV suppresses ERAD and apoptosis thereby maintaining cells under ER stress in order to persistently produce its own proteins. However, HCV maintains a balance between ER stress and the UPR and virus production through different mechanisms some of which are presented in this review[44-46]. Additionally, HCV replication in cells disrupts mitochondrial homeostasis leading to formation of irregular mitochondrial morphology, overproduction of reactive oxygen species (ROS), and oxidative stress[47]. Oxidative stress leads to activation of antioxidant programs to cope with the stress, and if unsuccessful, apoptosis is triggered. As is the case with ER stress, HCV not only induces oxidative stress, but also activates antioxidant programs and suppresses mitochondria-induced apoptosis[44,47,48]. Again, this leads to persistent infection and benefits virus production[44]. Thus, while HCV infection and some viral proteins may be capable of inducing apoptosis[49-51], it is generally agreed that apoptosis is effectively suppressed during infection. A few mechanisms that HCV utilizes to suppress apoptosis are also discussed in this review.
Virus infection of hepatocytes leads to rearrangements of ER membranes to generate double-membrane vesicles (DMVs) and to a lesser extent multi-membrane vesicles that are collectively referred to as the membranous web[52]. Viral genome replication occurs within the membranous web in replication complexes (RCs). Infection by all positive-strand RNA viruses results in the formation of membranous web. It is thought that the membranous web benefits viral replication by: (1) protecting viral RNA and proteins from degradation and intracellular antiviral defense; (2) increasing the local concentration of the factors involved in RNA replication; and (3) ensuring spatial proximity of viral RNA translation, viral genome replication, and virion assembly for efficient progression through the viral life cycle[39].
HCV also hijacks the hepatocyte very low-density lipoprotein (VLDL) pathway for the maturation and secretion of infectious viral particles[53]. Lipid secretion is reduced during infection, and maturing viral particles acquire VLDL characteristics, while secreted viral particles are bound to VLDL particles[40,54].
An important group of host factors intimately involved in essentially all steps of the HCV life cycle are molecular chaperones. The term chaperone reflects the significant role of these cytoprotective proteins in: (1) assisting client proteins to achieve native/functional conformation that is required for their function; (2) assembling/disassembling protein subunits; (3) preventing newly synthesized proteins or assembled protein subunits from forming nonfunctional aggregates and molecular crowding; (4) transporting proteins to particular subcellular compartments which is referred to as intracellular protein trafficking; and (5) targeting proteins to degradation if attempts to (re)fold or (re)assemble are not successful[55-57]. Newly synthesized proteins are assisted to fold properly by chaperones. Under stress conditions such as heat shock or viral infection, proteins can become misfolded, and chaperones attempt to refold such proteins. If folding is not successful, the protein gets targeted for proteasome-mediated degradation.
A large number of molecular chaperones belong to the family of heat shock proteins (HSPs) originally identified as proteins that helped refolding proteins that were denatured as a result of heat stress[58]. HSPs are a highly evolutionarily conserved family of proteins that are typically classified into four different systems based on their molecular weight: HSP70, HSP90, HSP60, and small HSPs[57]. The HSP70, HSP90, and HSP60 systems consist of the adenosine triphosphate (ATP)-dependent main chaperones that utilize their enzymatic activity to induce conformational changes in the client polypeptide by hydrolyzing ATP to adenosine diphosphate (ADP). In addition, a number of co-chaperones may assist and regulate the activity of the main chaperones. Small HSPs, on the other hand, do not possess enzymatic activity, and instead, perform their chaperone function by functioning as holdases, i.e., binding to client polyproteins, preventing their aggregation, and directing them to one of the ATP-dependent HSPs.
HCV has evolved a remarkable ability to interact with numerous chaperones to coordinate the diverse molecular systems and pathways that it requires for its propagation in hepatocytes (Table 1). This review presents our current knowledge and understanding of the chaperones that are involved in the HCV life cycle. First, HSPs are presented covering all four HSP systems HSP70, HSP90, HSP60, and small HSPs. Next, a diverse group of other molecular chaperones are discussed including BAG3, FK506-binding proteins (FKBPs), p23, prefoldin, apolipoprotein J [apoJ or clusterin (CLU)], protein disulfide isomerases (PDIs), microsomal triglyceride transfer protein (MTTP), calnexin (CANX), calreticulin (CALR), “endoplasmic reticulum degradation enhancer, mannosidase alpha-like 1” (EDEM1), EDEM3, sigma non-opioid intracellular receptor 1 (SigR1), prohibitin (PHB), and cyclophilins (Cyps). Finally the chaperone activity of the HCV proteins core, E1, E2, NS3, and NS4A are described. The gene names for the chaperones are also included in parentheses.
Table 1.
Chaperones and their roles in the hepatitis C virus viral life cycle
| Chaperone | Subcellular localization | Function in HCV infection/stage of viral life cycle |
| HSP70 family | ||
| GRP75 (HSPA9) | Mitochondrial | Varied expression/activity[66,72] |
| Interacts with NS5A[105] | ||
| GRP78 (HSPA5) | ER | Regulation of viral protein homeostasis and maintaining a balance between viral and cellular translation to prevent viral protein overload (involves induction of ER stress and the UPR)[43,85-96] |
| Increased expression and activity[72,85,88,93,95] | ||
| Associated with the viral genome[70,76] | ||
| HSC70 (HSPA8) | Cytosolic | Infectious virion assembly[18,74] |
| Potentially contributes to stability of virion structure and viral entry through clathrin-mediated endocytosis[35,74] | ||
| Associated with the viral genome[70,76] | ||
| Increased expression and activity[72,73] | ||
| Knockdown decreases lipid droplet size and virion assembly[18,74] | ||
| HSP70 (HSPA1A) | Cytosolic | IRES-mediated translation of viral genome[17,63,64,68,69] |
| Increased expression[65-67] | ||
| Knockdown decreases IRES activity and virus production[61,63] | ||
| HSP70B’ (HSPA6) | Cytosolic | Associated with 3’ NCR of HCV genome[70] |
| HSP40 family | ||
| DNAJA1 | Cytosolic | Co-immunoprecipitates with NS3-NS4A[105] |
| DNAJA2 | Cytosolic | IRES-mediated translation of viral genome[63] |
| DNAJA3 | Mitochondrial | Potentially HCV-induced mitochondrial dysfunction[61,127] |
| DNAJB1 | Cytosolic | Potentially regulates apoptosis[61,117] |
| Knockdown decreases virus production[61] | ||
| DNAJB6 | Cytosolic | Potentially viral RNA replication[105] |
| Interacts with NS5B[105] | ||
| Potentially overexpressed[108] | ||
| knockdown decreases viral RNA replication[105] | ||
| DNAJB9 | ER | Potentially regulates apoptosis[124] |
| Varied expression[108] | ||
| DNAJC1 | ER | Interacts with E1 and E2[107] |
| DNAJC7 | Cytosolic | Potentially regulates apoptosis[118] |
| Co-immunoprecipitates with NS3-NS4A[105] | ||
| DNAJC8 | Cytosolic | Upregulated[119] |
| DNAJC10 | ER | ER protein homeostasis likely benefiting virus production[126] |
| Proper folding of LDLR (viral entry)[126] | ||
| Likely overexpressed[125] | ||
| DNAJC14 | ER | Viral RNA replication[62,121,122] |
| HSP110 family | ||
| HSP105 (HSPH1) | Cytosolic | Overexpressed[129] |
| HSP70RY (HSPA4) | Cytosolic | Overexpressed[66,130] |
| Knockdown decreases viral RNA replication[130] | ||
| Hip (HSPBP1) | Cytosolic | Knockdown decreases virus production[62,134] |
| HSP90 family | ||
| GRP94 (HSP90B1) | ER | Regulation of viral protein homeostasis and maintaining a balance between viral and cellular translation to prevent viral protein overload (involves induction of ER stress and the UPR)[95,97,101] |
| Suppression of HCV-induced apoptosis[50] | ||
| Potentially HCV-induced liver fibrosis and autoimmune disease[155] | ||
| Overexpressed[95,101,130] | ||
| Knockdown decreases viral RNA replication[130] | ||
| HSP90 (HSP90AA1/HSP90AB1) | Cytosolic | HCV RNA replication[138,139,148,149] |
| Maturation and stability of HCV proteins[140-143] | ||
| IRES-mediated translation of viral genome[144] | ||
| Circumventing IFNβ response in peripheral B cells[151] | ||
| Potentially regulates miRNA levels in conjunction with GW182[145] | ||
| Interacts with NS5A and NS5B[105,107,143] | ||
| Overexpressed[130,152] | ||
| Knockdown decreases RNA replication[138] | ||
| HSP60 family (chaperonins) | ||
| HSP60 (HSPD1/HSPE1) | Mitochondrial | Regulates ROS production and apoptosis[159] |
| Interacts with core, NS3-NS4A, and viral genome[76,105,107,159] | ||
| Varied expression[66,130] | ||
| TRiC/CCT (TCP1/CCT2-8) | Cytosolic | Viral RNA replication by assisting in RC assembly[73] |
| Increased activity[129,130] | ||
| Increased TCP1, CCT2, and CCT5 expression[130] | ||
| Decreased CCT4 expression[129] | ||
| CCT4 co-immunoprecipitates with NS3-NS4A[105] | ||
| Knockdown of CCT5 decreases viral RNA replication[73] | ||
| Small HSPs | ||
| HSP22 (HSPB8) | Cytosolic | Potentially blocks apoptosis[166] |
| Overexpressed[119] | ||
| HSP27 (HSPB1) | Cytosolic | Potentially decreases apoptosis[164] |
| Binds NS5A[164] | ||
| Overexpressed[66] | ||
| Other chaperones | ||
| ApoJ (clusterin) (CLU) | Cytosolic | Binds to and stabilizes core and NS5A[190] |
| Overexpressed[190] | ||
| BAG3 (BAG3) | Cytosolic | Co-chaperone of HSP90 family |
| Likely blocks ER-stress-induced apoptosis[104] | ||
| Calnexin (CANX) | ER | E1/E2 folding and glycosylation[98,107,219,220,223-225] |
| HCV-induced ER stress and viral protein homeostasis[98] | ||
| Knockdown decreases virus production[62] | ||
| Calreticulin (CALR) | ER | E1/E2 glycosylation[98,107] |
| HCV-induced ER stress and viral protein homeostasis[98,101] | ||
| Overexpressed[101,130,226] | ||
| Knockdown decreases virus production[62] | ||
| Cyp40 (PPID) | Cytosolic | Lipid trafficking and virion secretion[303] |
| CypA (PPIA) | Cytosolic | RC formation and viral RNA replication[263,270] |
| NS5A and NS5B activation[276,280] | ||
| Viral polyprotein cleavage[283,301] | ||
| Regulates IFN response[304] | ||
| Lipid trafficking and virion assembly and secretion[291,303] | ||
| CypB (PPIB) | Cytosolic | RC formation and viral RNA replication[271,272] |
| NS5A and NS5B activation[271,272,274,276] | ||
| CypD (PPIF) | Mitochondrial | Inhibits mitochondrial function leading to ROS production[308] |
| EDEM1 (EDEM1) | ER | Downregulated[103,231] |
| Binds E1 and E2[230] | ||
| HCV-induced ER stress[230] | ||
| Targets misfolded glycoproteins to ERAD (viral protein homeostasis)[227,228] | ||
| EDEM3 (EDEM3) | ER | Binds E1 and E2[230] |
| HCV-induced ER stress[230] | ||
| Targets misfolded glycoproteins to ERAD (viral protein homeostasis)[227,228] | ||
| Erp72 (PDIA4) | Cytosolic | Increased activity[72] |
| FKBP38 (FKBP8) | Cytosolic | Co-chaperone of HSP90 family[137] |
| HCV RNA replication[137] | ||
| Blocks apoptosis[177] | ||
| Potentially regulates Ca2+ homeostasis by interacting with S100 proteins[175] | ||
| Interacts with NS5A[105,169] | ||
| Knockdown decreases HCV RNA replication[137] | ||
| FKBP54 (FKBP5) | Cytosolic | Interacts with NS5B[105] |
| GRP58 (PDIA3) | Cytosolic | Overexpressed[125,130] |
| Knockdown decreases viral RNA replication[130] | ||
| MTTP (MTTP) | Cytosolic | Part of the PDI/MTTP heterodimer involved in VLDL biogenesis[193] |
| Potentially causes HCV-induced liver steatosis[193,198] | ||
| Viral maturation and secretion[210,211] | ||
| Decreased expression and activity[193,198-200] | ||
| p23 (PTGES3) | Cytosolic | Co-chaperone of HSP90 family[179] |
| Potentially regulates telomerase activity[180,181] | ||
| PDI (P4HB) | ER | Folding and transfer of MTTP to ER as a PDI/MTTP heterodimer involved in VLDL biogenesis[193] |
| Increased activity[129] | ||
| PDIR (PDIA5) | Cytosolic | Increased activity[72] |
| Prefoldin | Cytosolic | Co-chaperone of TRiC/CCT[182] |
| (PFDN1-2/VBP1/PFDN4-6) | Binds F protein[183] | |
| Regulates cytoskeleton likely to balance virus production in hepatocytes[183] | ||
| Prohibitin (PHB/PHB2) | Mitochondrial | Inhibits mitochondrial respiratory function leading to ROS production[237-240] |
| Binds core[238] | ||
| Overexpressed[236,237] | ||
| SigR1 (SIGMAR1) | Cytosolic | Viral RNA replication immediately after entry[44,234] |
| Interorganellar communication between ER and mitochondria[44] | ||
| HCV chaperones | ||
| Core | Viral RNA stabilization, dimerization, and structural rearrangement[311-315] | |
| Folding of E1[316] | ||
| E1 | Proper folding of E2[224,318-320] | |
| E2 | Proper folding of E1[317] | |
| NS3 | Interconversion of viral RNA species[322] | |
| NS4A | Directs NS3 to ER[323] | |
| Increases NS3 stability[323] |
Apo: Apolipoprotein; BAG: BCL2-associated athanogene; Cyp: Cyclophilin; EDEM: Endoplasmic reticulum degradation enhancer, mannosidase alpha-like; ER: Endoplasmic reticulum; ERAD: ER-associated degradation; FKBP: FK506-binding protein; GRP: Glucose-regulated protein; GW: Glycine-tryptophan; HCV: Hepatitis C virus; Hip: HSP70-interacting protein; HSC70: Heat shock cognate protein 70; HSP: Heat shock protein; IFNβ: Interferon beta; IRES: Internal ribosomal entry site; LDLR: Low-density lipoprotein receptor; MTTP: Microsomal triglyceride transfer protein; NCR: Non-coding region; NS: Non-structural; ROS: Reactive oxygen species; PDI: Protein disulfide isomerase; RC: Replication complex; SigR1: Sigma non-opioid intracellular receptor 1; TRiC/CCT: TCP-1 ring complex/chaperonin-containing TCP-1; UPR: Unfolded protein response; VLDL: Very low-density lipoprotein.
HSP70/HSP40 SYSTEM
The HSP70 family of chaperones consists of a large number of proteins that are ubiquitously expressed throughout the cell. They play important roles in proper protein folding, protection of proteins from stress-induced damage, recovery/renaturing of damaged/aggregated proteins, protein degradation, protein translocation, and disassembly of protein complexes such as the DNA replication machinery[59,60]. This family of HSPs typically functions as a group of three proteins where the main HSP70 chaperone interacts with the client polypeptide through its substrate-binding domain (SBD), while the nucleotide-binding domain (NBD) binds to an ATP hydrolyzing it to ADP to induce conformational changes in SBD for its chaperone function. The hydrolysis is stimulated by substrate binding the chaperone resulting in a closed state where it tightly binds the substrate and helps with (re)folding it. Cofactor HSPs also known as co-chaperones, such as HSP40, typically interact with the NBD to modulate chaperone activity and to determine the clients of HSP70s via their specificity in binding particular target proteins. A nucleotide exchange factor (NEF) assists with the removal of hydrolyzed ADP which causes the chaperone to revert to its open conformation releasing the substrate.
HSP70 (HSPA1A)
HSP70 is an inducible chaperone that is expressed in conditions of stress such as heat shock and viral infection. HSP70 has been identified as one of the numerous host factors important for HCV production[61-64]. Knockdown of HSP70 led to decreased virus production[61,63] or replication in subgenomic replicon (SGR) systems[62,63]. Both HSP70 overexpression and autoantibodies against HSP70 in the sera of HCV-infected patients have also been reported[65]. Huh7 cells harboring an HCV SGR demonstrated upregulation of HSP70[66]. It was also found that expression of NS5A alone in huh7 cells was sufficient for upregulation of HSP70[67]. This upregulation was the result of NS5A-induced increased levels of nuclear factor of activated T cells 5 (NFAT5), one of the transcription factors responsible for HSP70 expression. The increased NFAT5 levels itself is mediated by NS5A-driven ROS production.
Our laboratory has shown NS5A to colocalize with HSP70 and HSP40 as well[63]. We further showed that knockdown of HSP70 inhibited NS5A-augmented IRES-mediated translation. The HSP synthesis inhibitor quercetin, a bioflavonoid, also suppressed the NS5A-augmented IRES-mediated translation[63,68]. In addition, we demonstrated that the NS5A/HSP70 interaction is direct and identified the site of NS5A/HSP70 interaction on NS5A to be a hairpin moiety at the C terminus of NS5A domain I[17]. Treatment of cells with a synthetic peptide corresponding to this hairpin moiety, which we termed the HSP-binding domain[69], disrupted the NS5A/HSP70 interaction and suppressed NS5A-augmented IRES-mediated translation and virus production[17]. Others have shown that overexpression of HSP70 leads to increased viral RNA and protein levels, while knockdown of HSP70 has the opposite effect[64]. HSP70 was found to interact with NS3-NS4A protein and NS5B as well. HSP70 increases RC formation by interacting with viral proteins in RCs, increasing the stability of viral proteins, and enhancing NS5A-driven viral IRES-mediated translation. Further, HSP70 was found to interact with the 3’ NCR of the viral genome[70].
Heat shock cognate protein 70 (HSPA8)
Heat shock cognate protein 70 (HSC70) is a constitutively-expressed housekeeping gene with diverse cellular functions including protein folding, signal transduction, apoptosis, autophagy, and many others[71]. Viral entry occurs at least in part through the HSC70-dependent clathrin-mediated endocytosis[35]. HSC70 activity was found to be significantly increased in an HCV SGR system[72], and HSC70 levels were increased in a proteomic analysis of RCs[73]. HSC70 was also identified to be part of the HCV viral particles, and the viral E2 protein was found to contain the HSC70-interacting histidine-proline-aspartic acid (HPD) motif[74] which is required for the interaction of the HSP40 co-chaperones with HSP70 family of chaperones[75]. Pretreatment of the virus with HSC70 antibody significantly diminished infectivity suggesting that HSC70 is a part of the viral particle[74]. In addition, HSC70, core, and E2 were found to colocalize around lipid droplets, the site of virion assembly. RNAi-mediated knockdown of HSC70 significantly decreased the volume of lipid droplets and viral secretion, but not viral RNA replication levels. These results suggest that HSC70 plays an important role during virion assembly and may play a structural role for the virion as well. It has been observed that HSC70 associates with positive-strand subgenomic viral RNA (corresponding to domains III and IV of the 5’ NCR and 36 nucleotides of core) and the 3’ NCR of the viral genome as well[70,76].
A number of compounds including IMB-DM122, N-substituted benzyl matrinic acid derivatives, and (+)-lycoricidine were shown to downregulate HSC70 mRNA expression leading to decreased virus production[77-79]. Our lab demonstrated that HSC70 directly binds to NS5A in vitro and colocalizes with NS5A in infected cells[18]. We further showed that knockdown of HSC70 significantly impacted intracellular infectious virion assembly thereby establishing distinct functions of HSC70 and HSP70 in the HCV life cycle. This is further supported by the fact that HSC70 and HSP70 do not interact with each other. Based on the available evidence, therefore, it seems that HSC70 is important for virion assembly.
HSP70B’ (HSPA6)
HSP70B’ is another member of the HSP70 family which is highly similar to HSPA1A in terms of sequence homology (82%) and function[80]. Both chaperones are stress inducible and work in conjunction to protect cells from stress. However, HSP70B’ is the secondary responder to stress after HSPA1A, and proteasome inhibition is a potent inducer of HSP70B’ expression[81]. HSP70B’ was found to be associated with the 3’ NCR of the HCV genome[70].
Glucose-regulated protein 78 (HSPA5)
Glucose-regulated protein 78 (GRP78), also known as the binding immunoglobulin protein (BiP), is another member of the HSP70 family and is the major molecular chaperone in the ER[82]. The ER is involved in vital cellular processes including protein folding, protein transport, the UPR, and calcium homeostasis. The UPR is an adaptive signaling program that is activated in response to accumulation of unfolded or misfolded proteins in the ER, referred to as ER stress. Proteins that are not successfully folded are either sent for refolding or tagged for degradation through the ERAD pathway[83]. If the UPR program is unable to successfully relieve cells from ER stress, it initiates mitochondria-mediated apoptosis[84]. Under certain conditions such as heat stress and pathogen infection, unfolded or misfolded proteins can accumulate in the ER leading to ER stress and activation of UPR. Stimulation of GRP78 transcription is an indication of ER stress and induction of UPR, which occurs in HCV infection likely to repress cellular protein translation in order to utilize cellular resources for the IRES-mediated translation of viral proteins and to suppress innate immunity in order to establish persistent infection[43,85-96]. GRP78 activity was also found to be significantly increased in an HCV SGR system[72].
UPR signaling can be initiated by three factors: Activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and double-stranded RNA-activated protein kinase R-like ER kinase (PERK)[43,92]. These three factors act as ER stress sensors and lead to induction of expression of GRP78, which is itself a negative regulator of the three ER stress sensors. ER stress may lead to the proteolytic cleavage of ATF6, an ER membrane-associated transmembrane protein. The 90 kDa ATF6 precursor, also known as pATF6α(P), is cleaved to form an approximately 50 kDa N-terminal fragment pATF6α(N) which translocates to the nucleus and activates transcription of ER chaperone genes such as GRP78 involved in the UPR. ER stress also leads to phosphorylation of IRE1 which results in the splicing of unspliced X-box-binding protein 1 to spliced XPB1 (sXBP1), a transcription factor that can induce expression of GRP78 and other genes involved in the UPR. Upon initiation of ER stress, PERK can also get activated and phosphorylate the eukaryotic initiation factor 2 alpha (eIF2α). Phosphorylated eIF2α (peIF2α) results in global inhibition of cellular protein synthesis and enhanced ATF4 expression which leads to induction of UPR genes. HCV can activate all three ER stress sensors.
It was found that the viral glycoprotein E2, and not E1, can induce transcription of GRP78 and that only E2 bound to GRP78[97]. Another group reported that both E1 and E2 bind GRP78[98]. However, it seems that GRP78 tends to bind to E1/E2 aggregates rather than monomeric glycoproteins. Expression of both E1 and E2 was also shown to lead to the UPR[99,100]. The HCV core protein has also been reported to induce expression of GRP78[101]. Induction of core, E1, E2, and p7 in mice liver led to ER stress and overexpression of GRP78[95]. Expression of HCV NS genes led to upregulation of GRP78[102]. The NS2 alone also induces ER stress and leads to upregulation of GRP78 protein levels[46]. NS4B alone can also induce ER stress and the UPR and upregulate GRP78 expression[87,103]. NS5A weakly binds GRP78, enhances GRP78 expression, and protects hepatocytes from ER stress-induced apoptosis leading to persistent infection[104,105]. It was also shown that HCV bearing certain mutations in NS5A and NS5B proteins (C2441S, P2938S or R2985P) displayed higher levels of GRP78 expression[94]. However, it was not clear whether NS5A alone can induce ER stress in these studies. Another group reported that NS5A does not lead to ER stress and the UPR[89,106]. An SGR system expressing all the NS proteins led to the UPR as well[106]. Thus, it is not clear whether the NS5 proteins alone can cause ER stress and the UPR. GRP78 was also shown to benefit virus production in a genome-wide expression analysis of multiple huh7-derived cell lines where interactions with core, E1, E2, p7, NS3, NS4B and NS5A were implicated[107]. Furthermore, GRP78 is a target of miR-30a, miR-30c, and miR-30e that were found to be downregulated in acute HCV infection potentially leading to GRP78 overexpression[108].
In addition to the ER-targeted E1 and E2 proteins, cytosol-targeted E1 and E2 proteins have also been described with opposing functions in the context of ER stress[109-112]. In the cytosol, E1 binds to the cytoplasmic domain of PERK. Furthermore, cytosolic E1 leads to downregulation of GRP78. Similarly, E2 binds to PERK as well, inhibits its kinase activity, reverses PERK-mediated global translation repression, and confers resistance to ER stress. In addition, NS2 leads to phosphorylation of eIF2α and decreased protein synthesis as well as reduction of IRES-mediated translation suggesting that NS2 can also provide a negative feedback regulation of ER stress by decreasing viral protein translation that is responsible for inducing ER stress[46].
Thus, it seems that GRP78, as well as other ER-resident chaperones, play an important role in regulating and maintaining viral protein homeostasis to ensure the availability of sufficient viral proteins to establish a persistent infection while minimizing cellular protein expression and preventing viral protein overload. GRP78 was also found to be associated with positive-strand subgenomic viral RNA (corresponding to domains III and IV of the 5’ NCR and 36 nucleotides of core) and the 3’ NCR of the viral genome[70,76].
A recent study reported that there was no significant difference in the mRNA levels of GRP74 and a number of other genes involved in ER stress and UPR between infected patients and healthy controls[113]. No difference in GRP78 protein levels were observed either. This may be attributed to the fact that typically HCV infects a small percentage of hepatocytes, and therefore, changes may not be detected.
GRP75 (HSPA9)
GRP75 also known as mtHSP70 or mortalin is the mitochondria-resident HSP70 family member. It plays a number of critical roles in the cells including anti-apoptosis, protein transport into mitochondria which may involve HSP60 as well, protection of cells from ROS, and mitochondrial biogenesis[114]. It has also been implicated in membrane trafficking and human immunodeficiency virus (HIV) virion release[115]. In the context of HCV, it has been reported that GRP75 activity was significantly increased in one HCV SGR system[72], while GRP75 protein was significantly downregulated in another SGR system[66]. These different results may reflect the HCV-mediated modulation of GRP75 activity/expression to accommodate its needs during the viral life cycle. Furthermore, NS5A was shown to co-immunoprecipitate with GRP75[105].
HSP40 family
The HSP40 family are co-chaperones of HSP70 proteins that regulate the activity of HSP70s and determine their client range by binding specific target proteins[60,116]. This large family of proteins are homologous with the bacterial DnaJ chaperone, and the term DNAJ is utilized in the gene nomenclature of the isoforms of this family. DNAJA1 and DNAJA2 are the most abundant cytosolic HSP40 co-chaperones[116]. DNAJA1 was reported to co-immunoprecipitate with the NS3-NS4A protein[105]. We have shown that DNAJA2 participates together with HSP70 in regulating the NS5A-augmented IRES-mediated translation of the viral genome[63]. The interaction of viral proteins with these co-chaperones may, therefore, modulate chaperone activity to benefit the viral life cycle. A genome-wide siRNA screening identified DNAJB1 to be important for HCV production[61]. DNAJB1 plays important roles in regulating apoptosis and cell proliferation[117]. DNAJC7 co-immunoprecipitates with NS3-NS4A protein[105]. DNAJC7 also regulates apoptosis by binding to the pro-apoptotic p53 protein and increasing its activity and stability[118]. Thus, it can be speculated that binding of NS3-NS4A may prevent the pro-apoptotic function of DNAJC7/p53 thereby suppressing apoptosis and contributing to persistent HCV infection. DNAJC8 was reported to be upregulated in quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) as well as microarray analyses of host gene expression in infected huh7 cells[119]. DNAJC8 has been shown to play an important role in regulating pre-mRNA splicing by the spliceosome[120]. This is achieved by the binding of DNAJC8 with “serine/arginine-rich splicing factor protein kinase 1”. DNAJB6 interacts with NS5B, and shRNA-mediated knockdown of DNAJB6 led to a significant decrease in viral RNA replication[105]. DNAJB6 may, therefore, be required for the stability or activity of NS5B for viral RNA replication. In addition, miR-17, miR-106a, and miR-106b with DNAJB6 as their target were found to be downregulated in acute HCV infection[108].
There are seven ER-resident HSP40 co-chaperones: DNAJB9, DNAJB11, DNAJC1, DNAJC3, DNAJC10, DNAJC23 and DNAJC25. DNAJC14 was found as a host factor involved in HCV replication in an siRNA screen where knockdown of DNAJC14 led to increased viral replication[62]. Further, DNAJC14 has been reported to be involved in RNA replication of yellow fever virus (YFV) and other flaviviruses including HCV[121] and has been shown to be important for RC assembly in YFV[122]. Overexpression of DNAJC14 blocked viral RNA replication in all flaviviruses tested including HCV, while NS2/3 cleavage was not inhibited. siRNA-mediated knockdown of DNAJC14 also demonstrated similar results indicating that both elevated and reduced levels of DNAJC14 interferes with viral RNA replication. Also DNAJC14 is recruited to YFV RCs consistent with the normal cellular function of DNAJC14 as an ER-localized co-chaperone involved in protein transport[121,123]. DNAJB9 was identified in a microarray analysis as one of the host genes with most consistently modified expression as a result of acute HCV infection[108]. Further, miR-17, miR-106a, and miR-106b that target DNAJB9 were found to be downregulated. DNAJB9 has been shown to be involved in regulation of apoptosis[124]. DNAJC10 expression was found to be increased in HeLa cells expressing HCV polyprotein[125]. DNAJC10 is also a member of the PDI family of chaperones (discussed below) which is responsible for removing non-native disulfide bonds in conjunction with BiP and targeting misfolded proteins for degradation[126]. Interaction of DNAJC10 with EDEM1, an ER chaperone (discussed below), is required for disulfide bond reduction. Interestingly, DNAJC10 is also required for the correct folding of LDLR, one of the cell surface receptors utilized by HCV for entry. DNAJC1 was identified as an antiviral protein in a genome-wide expression analysis of multiple huh7-derived cell lines where interactions with E1 and E2 were implicated[107].
Five members of the HSP40 family have been identified in mitochondria: DNAJA3, DNAJC11, DNAJC15, DNAJC19, and DANJC20. DNAJA3 was identified as an HCV-interacting protein[61]. DNAJA3 is normally involved in maintaining mitochondrial morphology, and altering DNAJA3 levels leads to mitochondrial fragmentation and reduced cell viability[127]. HCV infection leads to mitochondrial dysfunction, and DNAJA3 may play a role in this process.
NEFs
NEFs play an important role in normal chaperone functioning by facilitating replacement of the hydrolyzed ADP with an ATP[128]. Three families of NEFs have been identified for the HSP70 chaperones: (1) HSP110/GRP170; (2) HSP70-interacting protein (Hip) (HSPBP1)/BiP-associated protein (SIL1); and (3) the BCL2-associated athanogene (BAG) family of proteins. The HSP110/GRP170 family consists of three cytosolic members HSP105 (HSPH1), HSP70RY (Apg-2) (HSPA4), and OSP94 (Apg-1) (HSPA4L), and one mitochondrial member GRP170 (HYOU1).
It was found that HSP105 and HSP70RY expression levels increase in HCV SGR systems[66,129,130]. Also knockdown of HSP70RY in an SGR system decreased viral RNA replication levels[130]. This is expected as the levels and activity of HSP70 family members increase during HCV infection which may require more NEFs for their function. Furthermore, HSP110 levels increase in stressed cells likely to assist in coping with stress, and in the context of HCV infection, increased HSP110 levels may help cells with HCV-induced ER stress. Similar effects of overexpression of HSP110 has been reported in cancer and gastric ulcer where targeting HSP110 had beneficial effects[131-133]. In siRNA screens, it was found that knockdown of Hip led to a significant decrease in virus production levels[62,134]. The role of BAG3 in HCV infection is discussed below.
HSP90 SYSTEM
The HSP90 proteins are highly conserved evolutionarily and are involved in the folding of proteins especially those involved in signal transduction[135]. Thus, HSP90 possesses a more discrete range of clients compared with the HSP70 system. Like HSP70, HSP90 also undergoes conformational changes to assist with the folding of client proteins, a process which is driven by ATP hydrolysis, and co-chaperones also assist in regulating HSP90 function. HSP90 has been shown to be important for a large group of viruses including HCV[136]. The HSP90 family consists of the inducible cytosolic isoform HSP90α (HSP90AA1), the constitutively expressed cytosolic isoform HSP90β (HSP90AB1), the inducible ER isoform GRP94 (HSP90B1), and the mitochondrial isoform “tumor necrosis factor (TNF) receptor-associated protein 1” (TRAP1) (HSP90L).
HSP90 (HSP90AA1 and/or HSP90AB1)
HSP90 has been shown to be important for virus production[137]. siRNA-mediated knockdown of HSP90 as well as HSP90 inhibitors geldanamycin, “17-dimethylaminoethylamino-17-demethoxygeldanamycin” (17-DMAG), herbimycin A, and radicicol resulted in dose-dependent suppression of HCV in a replicon system[138]. Further, viral levels in chimeric mice with a humanized liver treated with 17-DMAG were significantly reduced. Other derivatives of geldanamycin as HSP90 inhibitors have also been reported to block HCV RNA replication[139].
HSP90 is required for the maturation of the viral polyprotein complex specially to generate functional NS2/3 protease[140]. HSP90 inhibitors were shown to block NS2/3 cleavage. Expression of HCV core in Saccharomyces cerevisiae impaired the growth of yeast cells, and it was found that HSC82, the yeast homolog of HSP90, is required for the stability of core protein[141]. Treatment of yeast cells with the HSP90 inhibitors geldanamycin, radicicol, herbimycin A, and herbimycin C suppressed core-induced growth impairment. HSP90 directly binds to NS3 through the NS3 helicase region and is required for NS3 stabilization[142,143]. In an SGR system, the HSP90 inhibitor “17-N-allylamino-17-demethoxygeldanamycin” (17-AAG) resulted in NS3 degradation specifically[142]. In the same SGR system, 17-AAG also suppressed HCV RNA replication in a dose-dependent manner. However, it was not clear if replication was affected directly or through decreased IRES translation. A subsequent study demonstrated the indirect interaction of HSP90 with the subunit C of eIF3c which involves and is dependent on the viral IRES RNA[144]. This interaction prevents the ubiquitination and the subsequent proteasome-dependent degradation of eIF3c which is required for IRES-mediated translation of the viral genome. Therefore, treatment with HSP90 inhibitors may prevent the chaperoning of eIF3c by HSP90 which leads to its degradation. Knockdown of eIF3c inhibited IRES-mediated translation, but not cellular 5’ 7-methylguanylate cap-dependent translation.
HSP90 was found to colocalize and co-immunoprecipitate with glycine-tryptophan (GW) 182, an important component of GW bodies which are involved in mRNA degradation and translational repression via miRNAs[145]. Both HSP90 and GW182 also colocalized with NS3, core, and NS5A. Knockdown of GW182 significantly decreased HCV RNA levels in infected cells, while overexpression of GW182 resulted in a significant increase in viral RNA levels. The HSP90 inhibitor 17-DMAG and knockdown of HSP90 significantly decreased GW182 and miR-122 levels leading to decreased HCV RNA levels. Ethanol was shown to upregulate both GW182 and HSP90 thereby facilitating HCV RNA replication. Interestingly, the same group discovered infectious exosomes from sera of HCV-infected patients or supernatants of infected huh7.5 cells that contained negative-strand viral RNA in association with Argonaute 2 [a component of the RNA-induced silencing complex (RISC)], HSP90, and miR-122[146]. These exosomes are capable of transmitting HCV infection in a CD81, SR-B1, and apolipoprotein E (apoE) receptor-independent manner, which was blocked by miR-122 and HSP90 inhibitors. An interaction between NS5A and HSP90 was also implicated in a genome-wide expression analysis of multiple huh7-derived cell lines[107]. Thus, viral proteins may modulate GW182 activity in an HSP90-dependent manner in order to regulate viral RNA replication and miRNA levels. A number of miRNAs have been shown to be modulated by HCV infection[108].
Treatment with the HSP90 inhibitor 17-DMAG was shown to destabilize phosphoinositide-dependent kinase 1 (PDK1), an upstream kinase of protein kinase C-related kinase 2 (PRK2)[147]. The PDK1-PRK2 signaling pathway leads to phosphorylation of NS5B, which is required for HCV RNA replication[148,149]. 17-DMAG-driven destabilization and degradation of PDK1 diminished NS5B phosphorylation levels leading to suppression of viral RNA replication[147]. An interaction between NS5B and HSP90 has also been reported in a yeast two-hybrid system[143]. NS5B co-immunoprecipitates with both isoforms of HSP90 as well[105].
Peripheral B cells have been proposed to serve as reservoirs for persistent HCV infection[150,151]. It was found that peripheral B cells in patients with chronic HCV infection circumvent the interferon beta (IFNβ)-mediated antiviral response in part by downregulating HSP90 which acts as a stabilizer of TANK-binding kinase 1 involved in phosphorylation of the interferon-regulatory factor 3 (IRF3) transcription factor that induces IFN expression[151]. Thus, by using this HSP90-mediated strategy, HCV in B cells evades detection by the immune system contributing to recurring infection even after liver transplant.
The constitutively expressed isoform of HSP90, HSP90AB1, was found to be significantly overexpressed in the mononuclear cells of HCV-infected patients[152]. Co-infection with HIV decreased the overexpression of HSP90AB1 in the same study. HSP90AB1 was also reproducibly enriched in the detergent-resistant membrane fraction of an SGR system[130].
HSP90 also plays an important role in HCV RNA replication in conjunction with FKBP38, a co-chaperone of HSP90 family, which is a member of the immunophilin family of proteins. The role of FKBP38 and its interaction with HSP90 is discussed in detail in the FKBP38 section below. Another HSP90 co-chaperone p23 is also involved in the HCV life cycle and is discussed below as well.
GRP94 (HSP90B1)
GRP94 is the ER-resident HSP90 isoform which is involved in folding of secreted proteins, ER stress, and the UPR[153]. It was found that the viral glycoprotein E2, and not E1, can lead to the ER stress response and induce transcription of GRP94[97]. This leads to activation of nuclear factor kappa B and induction of anti-apoptotic proteins[50]. In addition, knockdown of GRP94 abolished the anti-apoptotic activity of E2 suggesting that E2 inhibits apoptosis induced by HCV infection and leads to persistent viral infection in hepatocytes. The HCV core protein also contributes to ER stress by inducing the expression of GRP94[101]. Increased expression of GRP94 was also observed in the liver of mice conditionally expressing HCV structural proteins core, E1, E2 and p7[95]. No binding of GRP94 to either E1 or E2 glycoproteins was observed[98]. GRP94 was reproducibly enriched in the detergent-resistant membrane fraction of SGR cells[130]. HCV utilizes GRP94 as well as other ER-resident chaperones especially GRP78 to maintain viral protein homeostasis in the ER in order to establish persistent infection and suppress cellular protein translation. GRP94 was also shown to be beneficial for virus production in a genome-wide expression analysis of multiple huh7-derived cell lines where interactions with core, E2, NS3, and NS4B were implicated[107]. Knockdown of GRP94 in an SGR system led to a significant decrease in viral RNA replication levels as well[130].
GRP94 is prevented from translocating to the cell surface by “aminoacyl tRNA synthetase complex-interacting multifunctional protein 1” (AIMp1)/p43[154], which is a cofactor of aminoacyl tRNA synthetase complex and is involved in regulating transforming growth factor beta (TGF-β) signaling. Translocation of GRP94 to the cell surface leads to activation of dendritic cells and leads to autoimmune diseases. The HCV E2 protein has been reported to directly bind AIMp1/p43 and lead to its degradation through ubiquitination and the proteasome pathway[155]. In addition, E2 interferes with the AIMp1/p43-GRP78 interaction leading to lower cellular AIMp1/p43 levels. Decreased AIMp1/p43 levels in cells leads to elevated TGF-β signaling and cell surface expression of GRP94. Therefore, these mechanisms may be responsible for HCV-induced liver fibrosis and autoimmune diseases.
HSP60 SYSTEM
HSP60 chaperones also known as chaperonins are an important family of HSPs involved in protein folding and macromolecular assembly[156]. The HSP60 family consists of mitochondrial and cytosolic proteins. The mitochondrial HSP60 (encoded by HSPD1 and HSPE1 genes), also known as mtHSP60, is thought to have originated in the bacterial ancestors that were engulfed by early eukaryotic cells giving rise to the mitochondrial organelle. HSPD1 (the homolog of bacterial GroEL) forms tetradecamers, composed of two stacked heptameric rings with a central cavity that accommodates the target protein. HSPE1 (the homolog of bacterial GroES) forms one heptameric ring that serves as a cap for the HSPD1 structure. The HSPD1/HSPE1 complex functions in protein folding in an ATP-dependent manner. The eukaryotic/cytosolic chaperonin, also known as “TCP-1 ring complex/chaperonin-containing TCP-1” (TRiC/CCT), is homologous to the Archean thermosome complexes forming hexadecamers consisting of two octameric rings to assist in oligomeric protein assembly[157] and folding of approximately 10% of the proteome[158]. TRiC/CCT is composed of eight paralogous subunits encoded by TCP1 and CCT2-8 genes. The TRiC/CCT complex lacks a GroES-like homolog and instead uses a built-in cap system. Typically, the term HSP60 is used to refer to the mitochondrial chaperonin, whereas the eukaryotic cytosolic homolog is referred to as TRiC/CCT.
HSP60 (HSPD1/HSPE1)
Proteomic analyses of huh7 cells harboring an HCV SGR demonstrated downregulation of HSP60[66], while it was shown to be reproducibly enriched in the detergent-resistant membrane fraction of another SGR system[130]. However, these studies did not validate HSP60 levels by Western analysis or in the context of viral infection. HSP60 has been shown to interact with core[107,159]. This interaction led to production of ROS and sensitization of cells to TNFα-induced apoptosis[159]. Further, overexpression of HSP60 decreased ROS production and prevented apoptosis in core-expressing cells. Thus, binding of core to HSP60 seems to impair the function of HSP60 in regulating ROS production and apoptosis as a possible pro-oncogenic process. However, significant research is still required to elucidate the function of the HSP60 system in the context of HCV infection. Nevertheless, HSP60 has been shown to be important for Dengue virus production (also a positive-stranded RNA virus) although the exact function has not been elucidated[160]. Further, HSP60 is overexpressed in HBV and HIV infection[156,161]. Autoantibodies against HSP60 have been detected in sera of chronic HCV infected patients[162]. HSP60 has also been shown to co-immunoprecipitate with the NS3-NS4A protein[105] and associate with positive-strand subgenomic viral RNA (corresponding to domains III and IV of the 5’ NCR and 36 nucleotides of core)[76].
TRiC/CCT (TCP1/CCT2-8)
The activity of TRiC/CCT, the cytosolic chaperonin, was reported to be increased in an SGR system[129]. Also TCP1, CCT2, and CCT5 were reproducibly enriched in the detergent-resistant membrane fraction of an SGR system[130]. TRiC/CCT also plays an important role in the assembly of RCs which mediate HCV RNA replication[73]. This may be facilitated by an interaction between the subunit CCT5 of TRiC/CCT and NS5B. siRNA-mediated knockdown of CCT5 suppressed viral RNA replication. Treatment with an antibody against CCT5 also suppressed HCV RNA synthesis in an in vitro cell-free assay. These observations suggest that NS5B may recruit TRiC/CCT to the RCs to assemble components of RCs in order to facilitate HCV RNA replication. It was also reported that that CCT4 can co-immunoprecipitate with the NS3-NS4A protein[105]. CCT4 activity was decreased in an SGR system[129].
TRiC/CCT is regulated by a number of co-chaperones including prefoldin. The role of prefoldin in the HCV life cycle is discussed below.
SMALL HSPS
Small HSPs constitute a family of ten proteins with molecular mass in the range of 12-43 kDa with diverse functions including protein folding, development, and eye lens tissue formation to name a few[163]. They lack enzymatic activity and work as holdases in conjunction with the ATP-dependent chaperones to carry out their functions[57].
HSP27 (HSPB1)
Proteomic analyses of huh7 cells harboring an HCV SGR have demonstrated upregulation of HSP27[66]. HSP27 was found to bind NS5A (and not NS5B) in co-immunoprecipitation studies and colocalize by immunofluorescence under heat shock conditions[164]. The N-terminal regions of both proteins were found to be involved in the interaction (amino acids 1-122 of HSP27 and 1-181 of NS5A). While the function of this interaction is not known, it has been speculated that it may decrease infection-induced apoptosis. This is likely as HCV is known to modulate apoptosis in order to establish persistent infection. In fact, HSP27 is overexpressed and has anti-apoptotic roles in several cancers as well[165].
HSP22 (HSPB8)
HSP22 is a multifunctional chaperone involved in regulation of protein folding, macroautophagy, carcinogenesis, and apoptosis[166]. HSP22 was reported to be significantly overexpressed in infected huh7 cells as determined by qRT-PCR as well as microarray analyses of host gene expression[119]. HSP22 is an anti-apoptotic protein, and its upregulation by HCV may be one of the mechanisms that HCV utilizes to block apoptosis in hepatocytes.
OTHER CHAPERONES
In addition to HSPs, cells possess a number of other molecular chaperones and co-chaperones that play critical roles in numerous cellular functions by assisting with protein folding and stability in their respective pathways.
BAG3 (BAG3)
BAG3 is one of the BAG family of proteins and serves as a NEF for the HSP70 family of chaperones. BAG3 is the only heat stress-inducible BAG isoform and plays important roles in cell proliferation, apoptosis, adhesion, and migration[167]. It acts as an anti-apoptotic protein in different cancers. In the context of HCV infection, it was found that overexpression of NS5A in HepG2 cells upregulated a number of anti-apoptotic genes including BAG3 when the cells were treated with thapsigargin, an inducer of ER stress[104]. GRP78 was also overexpressed.
FKBP38 (FKBP8) and FKBP54 (FKBP5)
FKBP38 is a co-chaperone of the HSP90 family and a member of the immunophilin family of chaperone proteins which possess peptidylprolyl isomerase (PPIase) activity and also serve as receptors for the immunosuppressive drug FK506[168]. FKBP38 was identified as an NS5A interacting protein in a fetal liver cDNA library screen, and both NS5A and FKBP38 colocalize to mitochondria and the ER[169]. NS5A and FKBP38 were also shown to co-immunoprecipitate[105]. FKBP38 interacts with HSP90 and plays an important role in HCV RNA replication. FKBP38 forms a complex with HSP90 and NS5A where FKBP38 binds to both HSP90 and NS5A through different sites in its tetratricopeptide repeat domain[137]. Both knockdown of FKBP38 and treatment with geldanamycin suppresses HCV RNA replication in a replicon system indicating that the HSP90/NS5A/FKBP38 complex is important for the regulation of HCV RNA replication. In fact, the FKBP38/NS5A interaction is so critical for the virus that a single amino acid mutation in NS5A that disrupts its binding with FKBP38 impairs virus production[170]. The same group found that HSP90 binds to human butyrate-induced transcript 1 (hB-ind1)[171], which is a member of the Rho family of GTPases and a component of the Ras-related C3 botulinum toxin substrate 1 (Rac1) signaling pathway[172,173]. hB-ind1 was found to bind to NS5A and is involved in viral RNA replication through its interaction with HSP90. Thus, by interacting with NS5A, hB-ind1 recruits HSP90 and FKBP38 to the RCs. In addition, through immunofluorescence analyses, it was found that hB-ind1 colocalizes with NS5A, FKBP38, and double-stranded viral RNA at the site of the membranous web[174]. These results further support the role of HSP90 in viral RNA replication. Moreover, treatment with an HSP90 inhibitor decreased the HCV-induced UPR which points to a potential involvement of HSP90 in an hB-ind1-mediated protein folding mechanism in the membranous web in order to circumvent the virus-induced UPR.
It was also found that a few members of the S100 family of proteins, S100A1, S100A2, S100A6, S100B and S100P directly bind FKBP38 in cell-free in vitro assays in a Ca2+-dependent manner[175]. The S100 proteins are a family of 24 Ca2+ binding proteins which are involved in regulating inflammation, cell proliferation and differentiation, apoptosis, cell migration and invasion, and Ca2+ homeostasis[176]. The S100/FKBP38 interactions blocked both NS5A/FKBP38 and HSP90/FKBP38 interactions[175]. Furthermore, overexpression of S100A1, S100A2 and S100A6 suppressed HCV RNA replication. S100P was identified as one of the proteins with most consistently modified expression in acute HCV infection[108].
FKBP38 has also been reported to be involved in HCV suppression of apoptosis[177]. NS5A plays an important role in HCV pathogenesis by activating the mammalian target of rapamycin (mTOR) pathway. This leads to suppression of apoptosis and hepatocyte cell survival which is required for persistent infection. NS5A exerts its anti-apoptotic activity by blocking the interaction between FKBP38 and mTOR.
FKBP54 (p54), another FKBP family member, was reported to co-immunoprecipitate with NS5B[105]. FKBP54 is an important co-chaperone involved in regulating a number of signaling pathways, steroid hormone receptors, and autophagy[178].
p23 (PTGES3)
p23 (prostaglandin E synthase 3) is another HSP90 co-chaperone and an inhibitor of HSP90 ATP turnover[179]. In addition, p23 together with HSP90 are essential telomerase components, and telomerase activity as well as expression of multiple telomerase components were reported to be significantly induced in HCV infection of huh7.5 cell[180]. The same group also showed that expression of the La protein (Sjogren syndrome antigen B), a regulator of HCV IRES-mediated translation[181], significantly correlated with the expression of telomerase components including telomerase RNA, p23 and HSP90 in HCV-infected patient tissues. Thus, HCV may regulate telomerase activity in an HSP90-dependent manner which may potentially be linked to HCV-induced hepatocarcinogenesis.
Prefoldin (PFDN1-2/VBP1/PFDN4-6)
Prefoldin is the co-chaperone of the cytosolic chaperonin TRiC/CCT. It is a hexameric protein complex consisting of the six subunits encoded by the PFDN1-2, VBP1 (PFDN3), and PFDN4-6 genes[182]. Newly synthesized proteins at ribosomes bind to prefoldin which in cooperation with HSP70/HSP40 transports them to TRiC/CCT for proper folding and preventing protein aggregation. Prefoldin also plays an important role in clearing aggregated proteins as a result of ER stress or proteasome inhibitor treatment.
The HCV F protein, a 17 kDa product of ribosomal frameshift at the beginning of the core protein coding sequence, was found to bind prefoldin 2[183]. Prefoldin is involved in the proper folding of actin and tubulin subunits and plays an important role in the formation of the cytoskeleton. It was found that overexpression of the HCV F protein interfered with the prefoldin 1 and 2 interaction and resulted in an aberrant tubulin cytoskeleton. It was speculated that since an intact cytoskeleton is needed for HCV production in infected cells[184-187], the HCV F protein may modulate and decrease virus production in order to establish a persistent chronic infection[183].
ApoJ/clusterin (CLU)
ApoJ, also known as clusterin, is another chaperone with both intracellular and extracellular functions including protein folding and extracellular protein degradation and is involved in a number of age-related diseases including cardiovascular and neurodegenerative diseases and cancer likely by interacting with HSP60[188,189]. HCV infection led to increased clusterin expression both in cell culture and serum of infected patients[190]. siRNA-mediated silencing of clusterin led to decreased virus production without affecting viral RNA replication levels suggesting a subsequent step such as translation, assembly, or secretion is affected. It was found that clusterin binds to and stabilizes core and NS5A.
PDI (PDI family) and MTTP (MTTP)
The PDI family of proteins are ER chaperones that are responsible for disulfide bond formation[191]. The term PDI typically refers to the beta subunit of the prolyl 4-hydroxylase (P4H) enzyme, PDIA1 (P4HB), which is the first characterized member of the PDI family[192]. P4HB is involved in the folding and transfer of MTTP, a chaperone itself, from the cytosol into the lumen of ER[193,194]. P4HB and MTTP subsequently form a heterodimer, and MTTP then lipidates and stabilizes apolipoprotein B (apoB), a component of the VLDL produced by hepatocytes. ApoB associates with triglyceride containing particles generating VLDLs, and MTTP is involved in VLDL secretion as well[194,195].
It has been shown that core expression leads to decreased MTTP activity, in an HCV genotype 3-dependent manner[196] thereby reducing VLDL formation and secretion, which leads to accumulation of lipids in HCV-infected hepatocytes and subsequently liver steatosis[193,197,198]. Viral NS proteins have also been shown to decrease MTTP expression and activity and implicated in inhibition of VLDL secretion likely due to interaction of NS5A and apoB[199]. NS5A overexpression was also shown to decrease the expression of MTTP and increase lipid droplet size[200]. Furthermore, MTTP gene polymorphisms contribute to the accumulation of lipids in hepatocytes and may predict sustained virological response (SVR) to antiviral therapy in patients infected with genotype 4[201-204]. Thus, HCV infection is highly dependent on modulation of lipid metabolism, possibly in a genotype-specific manner[205-207], through interactions with MTTP[208]. During maturation, the newly assembled virions acquire low-density configuration prior to being secreted, a process that requires MTTP, and the secreted viral particles are bound to VLDL[54,209,210]. Secretion of viral particles depends on the apoB-positive lipoprotein particles in an MTTP-dependent manner, while virion assembly (and infectivity through LDLR and GAGs) requires apoE and is not MTTP and VLDL dependent[34,211-216].
P4HB activity was found to be increased in an HCV SGR system[129]. GRP58 (PDIA3), an important ER chaperone[191,217], was found to be overexpressed in HeLa cells expressing HCV polyprotein[125]. Further, GRP58 was reproducibly enriched in the detergent-resistant membrane fraction of an SGR system, and knockdown of GRP58 led to a significant decrease in viral RNA replication[130]. The activity of two other PDI family members ERp72 (PDIA4) and PDIR (PDIA5) were also significantly increased in an HCV SGR system[72,191]. ERp5 (PDIA6) activity was reduced in an SGR system. It should be noted that SGR systems do not produce infectious virus, and the activity/expression of PDIs may, therefore, not correspond with the context of viral infection.
The PDI family also includes DNAJC10, an HSP40 family member, which is discussed in the HSP40 section above.
Calnexin (CANX) and calreticulin (CALR)
Protein glycosylation among other post-translational modifications is carried out in the ER/Golgi apparatus. Calnexin and calreticulin are ER-resident chaperones that play a crucial role in the proper folding and glycosylation of glycoproteins. Both chaperones are part of a quality control mechanism in the ER that occurs in a cyclical manner[218]. Both HCV E1 and E2 being glycoproteins undergo the same cycles of quality control until they achieve the proper folding conformations required for the assembly of virions[98]. siRNA-mediated knockdown of calnexin and calreticulin decreased virus production[62].
Both E1 and E2 rapidly associate with calnexin immediately after synthesis in the ER, but dissociate slowly[61,98,107,219]. While E2 folding occurs rapidly and is complete upon cleavage of the E2-NS2 precursor polyprotein, folding of E1 is slow. Their association with calnexin parallels this timing suggesting that calnexin plays a role in proper folding of the E1/E2 glycoprotein complexes[220]. Calreticulin binds to E1 and E2 glycoproteins as well[98,107]. Whereas calnexin preferentially binds to monomeric glycoproteins, calreticulin seems to bind to E1/E2 aggregates. The N-linked oligosaccharides on these glycoproteins are important for the formation of E1/E2 complexes and for their interactions with some chaperones as treatment with tunicamycin, a glycosylation inhibitor, blocked the interaction of E1/E2 complexes with calnexin and calreticulin preventing their maturation and suppressing virus production[98,221,222]. Virus infectivity may also be impaired due to incorporation of immature glycoproteins in some virions[222]. Rather than being secreted, the E1/E2 complexes seem to remain in the ER and do not migrate past the cis-Golgi apparatus and are subsequently utilized in assembly of virions after undergoing proper folding and complex formation. Properly folded E1/E2 heterodimers no longer interact with calnexin[223-225].
NS2 was reported to co-immunoprecipitate with calnexin in infected cells[105]. All viral NS proteins were found to colocalize with the newly synthesized HCV RNA and calnexin at RCs which are ER-derived perinuclear structures[52]. In agreement with this observation, calnexin was reported to be associated with positive-strand subgenomic viral RNA (corresponding to domains III and IV of the 5’ NCR and 36 nucleotides of core)[76]. Calnexin is also a target of miR-130a, miR-130b and miR-310 that were shown to be downregulated in acute HCV infection[108]. HCV core protein causes ER stress thereby inducing the expression of calreticulin[101]. Calreticulin was reproducibly enriched in the detergent-resistant membrane fraction of an SGR system[130]. HCV infection was also found to increase calreticulin expression[226].
EDEM1 (EDEM1) and EDEM3 (EDEM3)
EDEMs that consist of three proteins EDEM1, EDEM2, and EDEM3 are lectin chaperones and regulators of ERAD that are involved in targeting misfolded glycoproteins to the ERAD pathway[227,228]. EDEMs binds to the target glycoproteins that are destined for degradation[229]. EDEMs also bind GRP78 and appear to provide the signal for degradation of the target glycoprotein[227]. EDEM1 and EDEM3, but not EDEM2, directly bind HCV glycoproteins and increase their ubiquitination[230]. Knockdown of EDEM1 and EDEM3 as well as treatment with kifunensine, an ERAD inhibitor, increased the half-life of E1 and E2 and virus production, and overexpression of the two EDEMs decreased virus production.
As mentioned above, misfolded proteins in the ER are targeted to the ERAD pathway if attempts to properly fold these proteins are unsuccessful[83]. While HCV production in cells leads to ER stress and the UPR, the virus has evolved strategies to prevent its proteins from being degraded through the ERAD pathway[44]. The ERAD pathway is activated downstream of the IRE1 pathway, and the IRE1 pathway is activated in response to HCV-induced ER stress and activation of the UPR[92]. However, despite activation of the IRE1 pathway, activation of the ERAD pathway is inhibited in HCV infection[231]. Thus, although sXBP1 is produced indicating activation of the IRE1 pathway, expression of EDEM1 is suppressed. This seems to be unique for HCV as other flaviviruses do not suppress EDEM expression in presence of sXBP1 production[83,231]. HCV NS4B similarly leads to production of sXBP1, but suppresses EDEM expression[103]. The lack of EDEM induction may also lead to increased IRES-mediated translation of viral proteins[231]. These results suggest that EDEMs may play a crucial role in regulating viral protein homeostasis and maintaining a balance in viral protein production to establish persistent infection.
SigR1 (SIGMAR1)
SigR1 is a cholesterol-binding chaperone in lipid-rich areas of ER and mitochondrion-associated ER membranes (MAMs)[232]. MAMs play an important role in pathogenesis of HCV by serving as interorganellar communication sites between ER and mitochondria both of which are crucial for HCV production[44]. SigR1 is normally involved in crucial processes including cellular response to stress, lipid and protein trafficking, cell survival, and neuroprotection[232,233]. SigR1 has been reported to play an important role for viral RNA replication immediately after virion entry, but not afterwards during persistent infection[44,234]. siRNA-mediated knockdown of SigR1 reduced viral RNA replication only in early stages of infection.
Prohibitin (PHB) and prohibitin 2 (PHB2)
The mitochondrial chaperone prohibitin is involved in a variety of processes including mitochondrial protein folding and membrane potential, cell cycle, and apoptosis[235]. It forms a ring structure composed of two subunits encoded by the PHB and PHB2 genes. The HCV core protein as well as viral infection lead to overexpression of prohibitin[236,237], which is a target of the HCV core protein[238]. Core binds to prohibitin and impairs its chaperone function thereby preventing the proper function of mitochondrial respiratory chain leading to overproduction of ROS which may result in hepatocarcinogenesis[237,238]. This is likely caused by the core-mediated suppression of the interaction between prohibitin and subunit I and IV of cytochrome C oxidase[239,240].
Cyps (PPI family)
Cyps are an important family of molecular chaperones most of which possess PPIase activity and are involved in diverse cellular processes including protein folding, scaffolding, protein trafficking, and apoptosis[241]. The genes that encode Cyps are referred to as PPIs. Cyps have been reported to be important for replication of HCV as well as other flaviviruses[242], and Cyp inhibitors such as cyclosporine A (CsA) have been shown to effectively block virus production when used alone or in combination with other antiviral agents such as IFN[243-262]. Cyps have been suggested to play important roles in the HCV life cycle including viral RNA replication, membranous web formation, viral polyprotein cleavage, lipid trafficking, virion assembly, suppression of IFN-based antiviral response, and induction of mitochondrial dysfunction.
It has been suggested that NS5B is recruited to the RCs in the membranous web by cyclophilin A (CypA) (PPIA) likely to ensure NS5B retains its proper conformation for viral RNA replication[263]. In fact, both NS5B and CypA share a common binding site on NS5A[264] suggesting that CypA delivers NS5B to the RCs at which point NS5B binds NS5A. This function of CypA is supported by the finding that treatment of cells with CsA reduces the levels of NS5B in RCs, but not NS5A or NS3[263]. In addition, mutant NS5B from CsA-resistant replicons retained their RC incorporation in presence of CsA. Other published Cyp inhibitor-selected mutations in NS5B have been reported to increase its RNA binding capacity[265-267]. Also the observed CsA resistance of the JFH1 strain (genotype 2a) is NS5B dependent[268]. PPIase mutant CypA maintained its NS5B binding[263]. However, the mutant CypA was unable to rescue HCV replication in CypA knockdown cells implicating its PPIase activity is important for HCV replication. Another study reported that CypA does not recruit NS5B or NS5A to RCs as CsA treatment did not affect the RC association of NS5B and NS5A, concluding the possibility of a CypA-independent recruitment of NS5B and NS5A to RCs[269]. A recent report seems to resolve this discrepancy[270]. It was found that Cyp inhibitor treatment did not affect the replicase activity of RCs after active RCs are established. This suggests that Cyp inhibitors exert their antiviral activity prior to formation of active RCs supporting the originally proposed CypA-mediated NS5B recruitment model.
In addition, NS5B binds to CypB (PPIB) which is required to stimulate the RNA-binding activity of NS5B and RNA synthesis[271-273]. Both CypA and CypB activate NS5B replicase function, particularly RNA binding, in vitro where CypB demonstrates viral genotype 1b specificity[274]. It was shown that the lack of PPIase activity in mutant CypA and CypB had some effect on NS5B activation, but the PPIase mutant CypA and CypB were still capable of activating NS5B to a significant extent suggesting that the PPIase activity is dispensable for NS5B activation. However, these experiments were performed in a cell free system, whereas the previous experiments showing the importance of PPIase activity in HCV replication were performed in a replicon system. Others have shown NS5B/CypB interaction to be mediated by CsA-associated helicase-like protein in GST pulldown assays[275].
Significant evidence also points to a role of Cyps in viral RNA replication through their PPIase activity likely inducing conformational changes in viral and/or host proteins for optimal functioning. NS5A is a substrate for the PPIase activity of CypA and CypB through many proline residues in NS5A domain II and the linker region between NS5A domains II and III (known as the low-complexity sequence II or LCS-II)[276-278]. A three amino acid structural motif, a proline-tryptophan turn, is essential for HCV RNA replication and proper interaction with CypA and influences the PPIase activity of CypA on NS5A domain II[279]. CypA also binds NS5A domain III and has PPIase activity towards some peptidylprolyl bonds in NS5A domain III[280]. The NS5A/CypA interaction and the PPIase activity of CypA, which are both disrupted by Cyp inhibitors, have been shown to be critical for HCV production[280-289], and the PPIase activity of CypA is required for the NS5A/CypA interaction[281]. Further, wild-type CypA rescued viral RNA replication under CypA knockdown, but a PPIase mutant did not[284]. Indeed, it was found that CypA interacts with NS5A and stimulates RNA binding of NS5A domain II in a PPIase-dependent manner[290,291]. Furthermore, some SNP mutations in the PPIase domain of CypA render hepatocytes resistant to HCV replication likely by decreasing the intracellular stability of CypA[292]. Mutant NS5A from Cyp inhibitor resistant virus still binds to CypA as wild-type NS5A in vitro[281,282,286], whereas in cell culture the interaction appears much stronger than with wild-type NS5A implying other cellular proteins are important for this interaction[170]. NS5B was found to further strengthen this interaction as well. Others have provided an alternative mechanism for resistance through NMR analyses showing that the resistant NS5A exhibited a trans to cis conformational shift possibly rendering NS5A less dependent on the PPIase activity of CypA for isomerization[285]. Importantly, the Cyp inhibitor-induced NS5A mutation can rescue viral replication under CypA knockdown conditions[282] although it still requires CypA at lower levels[293]. Thus, most of the evidence to date suggests that CypA is the most important Cyp in the context of HCV replication and that CypA and NS5A are the main targets of Cyp inhibitor-mediated antiviral activity as knockdown of CypB, CypC (PPIC), and CypD (PPIF) failed to suppress viral replication, and NS5A mutations have the major role in Cyp inhibitor resistance compared with NS5B and other viral proteins[263,265,283,284,293-298].
Cyp inhibitor treatment also prevents formation of DMVs that are required for RNA replication at RCs suggesting that Cyps are involved in formation of RCs as well[270]. While the NS3-NS5B polyprotein and even NS5A alone suffices for formation of DMVs, knockdown of CypA prevents DMV formation suggesting that Cyps and, in particular, CypA is required for DMV formation. In addition, the PPIase activity of CypA was found to be required for DMV formation indicating that both NS5A and CypA are crucial for formation of DMVs.
The JFH1 SGR (lacking NS2) is not very sensitive to CsA or NIM811 (another Cyp inhibitor)[299], and it was shown that full-length JFH1 was inhibited much more efficiently by CsA implicating NS2 to be important for CsA-mediated viral inhibition in a CypA-dependent manner[283,300,301]. Subsequently, it was found that NS2 itself is not a target of CsA, but that the rate-limiting NS2-NS3 cleavage determines sensitivity to CsA[301]. It has been suggested that NS3 also binds Cyps and that mutations in NS3 may also lead to CsA resistance[297,302]. Also it was found that the CypA dependence of HCV replication correlates with the NS5A-NS5B cleavage kinetics as demonstrated by substitution mutants at this cleavage site[283]. These findings indicate that viral polyprotein cleavage may at least in part be dependent on Cyps especially CypA.
CsA has also been shown to affect hepatocyte lipids pointing to an additional role of Cyps in lipid trafficking and in HCV pathogenesis[303]. Cyp inhibitor treatment disrupts the VLDL pathway of virus maturation described above resulting in increased lipid droplet size, accumulation of apoB on lipid droplets, removal of NS5A from lipid droplets, and inhibition of infectious virion assembly[291,303]. The Cyps involved were found to be CypA and Cyp40 (PPID).
Yet another role of CypA in viral infection has been suggested in the context of the IFN pathway[304]. It was found that CypA and IRF9, a component of the JAK/STAT pathway, directly bind each other via the PPIase domain of CypA and the newly-identified CypA binding site in the IRF-association domain of IRF9. Cyp inhibitors prevent this complex formation. Interestingly, NS5A and IRF9 compete for binding to CypA, and CypA inhibition led to increased IFN-induced transcriptional activity through interferon-sensitive response elements (ISREs). Thus, it seems that HCV utilizes NS5A to dampen the IFN response by replacing IRF9 in the CypA/IRF9 complex, in order to establish persistent infection in hepatocytes. Furthermore, it was observed that Cyp inhibitor treatment blocks phosphorylation of protein kinase R (PKR) and its target eIF2α which inhibits translation of interferon-stimulated genes[305,306]. Cyp inhibitors also blocked stress granule formation. CypA binds PKR, and this interaction was disrupted by Cyp inhibitor treatment as well[305]. However, it was reported that Cyp inhibitor-mediated inhibition of PKR phosphorylation is due to suppression/clearing of viral infection rather than being a direct effect[306]. Thus, the significance of the CypA/PKR interaction and its disruption by Cyp inhibitors is not clear.
It is also reported that CsA treatment of uninfected huh7 cells induces the UPR and upregulation of GRP78[307]. Further, treatment of cells with UPR-inducing agents suppressed HCV replication. This may suggest that CsA may also exert its antiviral activity by inducing UPR which likely leads to improper viral glycoprotein/protein folding, their aggregation, and subsequent degradation.
The Cyp inhibitor alisporivir has also been found to prevent and to some extent reverse the negative impacts of HCV infection on mitochondrial function revealing another potential role for Cyps in the context of viral infection[308]. In particular, alisporivir prevents HCV-mediated collapse of the mitochondrial membrane potential, overproduction of ROS, and mitochondrial calcium overload through inhibition of CypD-mediated opening of the mitochondrial permeability transition pore[308-310].
HCV PROTEINS AS CHAPERONES
Remarkably, some HCV proteins possess chaperone functions that are critical for virus production. For example, core, in particular the N-terminal domain I, has been shown to play important chaperone roles for viral RNA stabilization, dimerization, and structural rearrangements[311-315]. Also core appears to be involved in folding of the E1 glycoprotein[316]. Both viral glycoproteins E1 and E2 have been reported to possess chaperone functions. E2 has been reported to be required for proper E1 folding[317]. The disulfide bonds in E1 have been shown to be required for the proper function of E2 during viral assembly and entry[318], and E2 does not seem to be able to reach a native structure in the absence of E1[319]. Further, a monoclonal antibody was reported to recognize properly folded E2 only when complexed with E1[224]. Also the ectodomain of E2 was shown to fold only in presence of E1[320]. CANX may be important for the chaperone activities of HCV glycoproteins[220]. This is in agreement with the observation that E2, unlike E1, did not associate with cellular chaperones such as CANX in an infection-free system[319]. In many class II enveloped viruses, of which HCV is a member, one viral glycoprotein acts as a chaperone for the folding of the other one which carries out the membrane fusion after viral entry in order to release viral genome in the cytosol[321]. However, for HCV, the mechanism of membrane fusion and the role of glycoproteins is not fully understood. The NS3 protein which possesses a helicase domain has been reported to mediate functions beyond the known helicase activity as it is involved in “intermolecular annealing, resolves three-stranded RNA duplexes, and assists dsRNA and ssRNA inter-conversions to establish a steady state among RNA structures”[322]. NS4A directs NS3 to ER and increases the intracellular stability of NS3[323].
CONCLUSION
Chaperones play crucial roles in HCV infection, and essentially all phases of the viral life cycle depend on chaperone functions and the interaction of viral proteins with chaperones (Table 1). The critical roles of Cyps and HSP90 in HCV RNA replication among others, HSP70 in viral protein translation, HSC70 in virion assembly, and the ER chaperones GRP78 and GRP94 in viral protein stability and persistent infection are important examples. Better understanding of the role of chaperones in the viral life cycle will provide further insights into the mechanism of virus production and suppression of immune response. Recently, significant advancements have been achieved in HCV therapy, and IFN-free therapies utilizing combinations of direct-acting antivirals (DAAs) with or without ribavirin (RBV) are being used successfully to achieve SVR in the majority of cases. Besides very high costs associated with some therapies, other issues include variability in activity across different genotypes, such as genotype 3 that can result in failure to achieve SVR[324]. If RBV is required, significant side effects can occur such as hemolytic anemia[325]. Treatment with DAAs can also result in resistant virus as targeting viral proteins puts direct selective pressure for resistant mutants. Furthermore, a small percentage of patients are infected with intergenotypic recombinant strains of HCV which may not respond optimally to the current standard treatments[326,327]. Analysis of the role of chaperones in the viral life cycle may allow for development of novel strategies to target HCV infection. Targeting host factors may reduce selective pressure on the virus to generate resistant mutants. Furthermore, insights obtained by studying chaperones in HCV infection may allow for development of therapies for other viruses especially flaviviruses.
Footnotes
P- Reviewer: Bolhassani A, Tetsuya T S- Editor: Gong ZM L- Editor: A E- Editor: Liu SQ
Supported by NIH R01DK090794, SWF.
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 7, 2015
First decision: September 8, 2015
Article in press: December 18, 2015
References
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Hepatitis C, Fact Sheet N°164. 2014. [accessed 2015 Sept 30] Available from: http://www.who.int/mediacentre/factsheets/fs164/en/ [Google Scholar]
- 3.Gravitz L. Introduction: a smouldering public-health crisis. Nature. 2011;474:S2–S4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 4.Freeman RB, Steffick DE, Guidinger MK, Farmer DG, Berg CL, Merion RM. Liver and intestine transplantation in the United States, 1997-2006. Am J Transplant. 2008;8:958–976. doi: 10.1111/j.1600-6143.2008.02174.x. [DOI] [PubMed] [Google Scholar]
- 5.Biggins SW, Bambha KM, Terrault NA, Inadomi J, Shiboski S, Dodge JL, Gralla J, Rosen HR, Roberts JP. Projected future increase in aging hepatitis C virus-infected liver transplant candidates: a potential effect of hepatocellular carcinoma. Liver Transpl. 2012;18:1471–1478. doi: 10.1002/lt.23551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Younossi ZM, Kanwal F, Saab S, Brown KA, El-Serag HB, Kim WR, Ahmed A, Kugelmas M, Gordon SC. The impact of hepatitis C burden: an evidence-based approach. Aliment Pharmacol Ther. 2014;39:518–531. doi: 10.1111/apt.12625. [DOI] [PubMed] [Google Scholar]
- 7.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 8.Rich JD, Taylor LE. The beginning of a new era in understanding hepatitis C virus prevention. J Infect Dis. 2010;202:981–983. doi: 10.1086/656213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127:S27–S34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Baron S. Medical microbiology. 4th ed. Galveston, Tex. University of Texas Medical Branch at Galveston; 1996. [PubMed] [Google Scholar]
- 11.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J Virol. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010;6:e1001087. doi: 10.1371/journal.ppat.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Yan W, Coito C, Li Y, Gale M, Katze MG. The regulation of hepatitis C virus (HCV) internal ribosome-entry site-mediated translation by HCV replicons and nonstructural proteins. J Gen Virol. 2003;84:535–543. doi: 10.1099/vir.0.18658-0. [DOI] [PubMed] [Google Scholar]
- 15.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol. 2008;82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes M, Griffin S, Harris M. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J Gen Virol. 2009;90:1329–1334. doi: 10.1099/vir.0.009332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khachatoorian R, Arumugaswami V, Ruchala P, Raychaudhuri S, Maloney EM, Miao E, Dasgupta A, French SW. A cell-permeable hairpin peptide inhibits hepatitis C viral nonstructural protein 5A-mediated translation and virus production. Hepatology. 2012;55:1662–1672. doi: 10.1002/hep.25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khachatoorian R, Ganapathy E, Ahmadieh Y, Wheatley N, Sundberg C, Jung CL, Arumugaswami V, Raychaudhuri S, Dasgupta A, French SW. The NS5A-binding heat shock proteins HSC70 and HSP70 play distinct roles in the hepatitis C viral life cycle. Virology. 2014;454-455:118–127. doi: 10.1016/j.virol.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo SY, Selby M, Tong M, Ou JH. Comparative studies of the core gene products of two different hepatitis C virus isolates: two alternative forms determined by a single amino acid substitution. Virology. 1994;199:124–131. doi: 10.1006/viro.1994.1104. [DOI] [PubMed] [Google Scholar]
- 20.Ray RB, Lagging LM, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. 1996;70:4438–4443. doi: 10.1128/jvi.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou J. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001;20:3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7:710–721. doi: 10.1017/s1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varaklioti A, Vassilaki N, Georgopoulou U, Mavromara P. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J Biol Chem. 2002;277:17713–17721. doi: 10.1074/jbc.M201722200. [DOI] [PubMed] [Google Scholar]
- 24.Fiorucci M, Boulant S, Fournillier A, Abraham JD, Lavergne JP, Paranhos-Baccala G, Inchauspé G, Bain C. Expression of the alternative reading frame protein of Hepatitis C virus induces cytokines involved in hepatic injuries. J Gen Virol. 2007;88:1149–1162. doi: 10.1099/vir.0.82575-0. [DOI] [PubMed] [Google Scholar]
- 25.Shao SW, Wu WB, Bian ZQ, Yu JG, Zhao P, Zhao LJ, Zhu SY, Qi ZT. Hepatitis C virus F protein inhibits cell apoptosis by activation of intracellular NF-kappaB pathway. Hepatol Res. 2009;39:282–289. doi: 10.1111/j.1872-034X.2008.00452.x. [DOI] [PubMed] [Google Scholar]
- 26.Yuksek K, Chen WL, Chien D, Ou JH. Ubiquitin-independent degradation of hepatitis C virus F protein. J Virol. 2009;83:612–621. doi: 10.1128/JVI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu WT, Li HC, Lee SK, Ma HC, Yang CH, Chen HL, Lo SY. Both core and F proteins of hepatitis C virus could enhance cell proliferation in transgenic mice. Biochem Biophys Res Commun. 2013;435:147–152. doi: 10.1016/j.bbrc.2013.04.059. [DOI] [PubMed] [Google Scholar]
- 28.Yue M, Deng X, Zhai X, Xu K, Kong J, Zhang J, Zhou Z, Yu X, Xu X, Liu Y, et al. Th1 and Th2 cytokine profiles induced by hepatitis C virus F protein in peripheral blood mononuclear cells from chronic hepatitis C patients. Immunol Lett. 2013;152:89–95. doi: 10.1016/j.imlet.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, Yu X, Deng X, Yue M, Zhang J, Zhu D, Zhou Z, Zhai X, Xu K, Zhang Y. Hepatitis C virus alternate reading frame protein decreases interferon-α secretion in peripheral blood mononuclear cells. Mol Med Rep. 2014;9:730–736. doi: 10.3892/mmr.2013.1816. [DOI] [PubMed] [Google Scholar]
- 30.Xiao W, Jiang LF, Deng XZ, Zhu DY, Pei JP, Xu ML, Li BJ, Wang CJ, Zhang JH, Zhang Q, et al. PD-1/PD-L1 signal pathway participates in HCV F protein-induced T cell dysfunction in chronic HCV infection. Immunol Res. 2015:Epub ahead of print. doi: 10.1007/s12026-015-8680-y. [DOI] [PubMed] [Google Scholar]
- 31.Zhu DY, Deng XZ, Jiang LF, Xiao W, Pei JP, Li BJ, Wang CJ, Zhang JH, Zhang Q, Zhou ZX, et al. Potential Role of Hepatitis C Virus Alternate Reading Frame Protein in Negative Regulation of T-Bet Gene Expression. Inflammation. 2015;38:1823–1834. doi: 10.1007/s10753-015-0160-y. [DOI] [PubMed] [Google Scholar]
- 32.Pacheco A, Martinez-Salas E. Insights into the biology of IRES elements through riboproteomic approaches. J Biomed Biotechnol. 2010;2010:458927. doi: 10.1155/2010/458927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 34.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouillé Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda M, Suzuki R, Kataoka C, Watashi K, Aizaki H, Kato N, Matsuura Y, Suzuki T, Wakita T. Alternative endocytosis pathway for productive entry of hepatitis C virus. J Gen Virol. 2014;95:2658–2667. doi: 10.1099/vir.0.068528-0. [DOI] [PubMed] [Google Scholar]
- 37.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 39.Paul D, Madan V, Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe. 2014;16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki T. Morphogenesis of infectious hepatitis C virus particles. Front Microbiol. 2012;3:38. doi: 10.3389/fmicb.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Popescu CI, Riva L, Vlaicu O, Farhat R, Rouillé Y, Dubuisson J. Hepatitis C virus life cycle and lipid metabolism. Biology (Basel) 2014;3:892–921. doi: 10.3390/biology3040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J. Hepatitis C virus: a new class of virus associated with particles derived from very low-density lipoproteins. Arterioscler Thromb Vasc Biol. 2012;32:1099–1103. doi: 10.1161/ATVBAHA.111.241448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merquiol E, Uzi D, Mueller T, Goldenberg D, Nahmias Y, Xavier RJ, Tirosh B, Shibolet O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One. 2011;6:e24660. doi: 10.1371/journal.pone.0024660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vasallo C, Gastaminza P. Cellular stress responses in hepatitis C virus infection: Mastering a two-edged sword. Virus Res. 2015;209:100–117. doi: 10.1016/j.virusres.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 45.Shinohara Y, Imajo K, Yoneda M, Tomeno W, Ogawa Y, Kirikoshi H, Funakoshi K, Ikeda M, Kato N, Nakajima A, et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem Biophys Res Commun. 2013;432:326–332. doi: 10.1016/j.bbrc.2013.01.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von dem Bussche A, Machida R, Li K, Loevinsohn G, Khander A, Wang J, Wakita T, Wands JR, Li J. Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J Hepatol. 2010;53:797–804. doi: 10.1016/j.jhep.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brault C, Levy PL, Bartosch B. Hepatitis C virus-induced mitochondrial dysfunctions. Viruses. 2013;5:954–980. doi: 10.3390/v5030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver. Viruses. 2013;5:439–469. doi: 10.3390/v5020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng L, Adachi T, Kitayama K, Bungyoku Y, Kitazawa S, Ishido S, Shoji I, Hotta H. Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J Virol. 2008;82:10375–10385. doi: 10.1128/JVI.00395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SH, Song R, Lee MN, Kim CS, Lee H, Kong YY, Kim H, Jang SK. A molecular chaperone glucose-regulated protein 94 blocks apoptosis induced by virus infection. Hepatology. 2008;47:854–866. doi: 10.1002/hep.22107. [DOI] [PubMed] [Google Scholar]
- 51.Zhao P, Han T, Guo JJ, Zhu SL, Wang J, Ao F, Jing MZ, She YL, Wu ZH, Ye LB. HCV NS4B induces apoptosis through the mitochondrial death pathway. Virus Res. 2012;169:1–7. doi: 10.1016/j.virusres.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 52.El-Hage N, Luo G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J Gen Virol. 2003;84:2761–2769. doi: 10.1099/vir.0.19305-0. [DOI] [PubMed] [Google Scholar]
- 53.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ellis RJ. Molecular chaperones: assisting assembly in addition to folding. Trends Biochem Sci. 2006;31:395–401. doi: 10.1016/j.tibs.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 56.Höhfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 58.Kim YK, Jang SK. Continuous heat shock enhances translational initiation directed by internal ribosomal entry site. Biochem Biophys Res Commun. 2002;297:224–231. doi: 10.1016/s0006-291x(02)02154-x. [DOI] [PubMed] [Google Scholar]
- 59.Clerico EM, Tilitsky JM, Meng W, Gierasch LM. How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol. 2015;427:1575–1588. doi: 10.1016/j.jmb.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cyr DM, Ramos CH. Specification of Hsp70 function by Type I and Type II Hsp40. Subcell Biochem. 2015;78:91–102. doi: 10.1007/978-3-319-11731-7_4. [DOI] [PubMed] [Google Scholar]
- 61.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci USA. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez O, Fontanes V, Raychaudhuri S, Loo R, Loo J, Arumugaswami V, Sun R, Dasgupta A, French SW. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology. 2009;50:1756–1764. doi: 10.1002/hep.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen YJ, Chen YH, Chow LP, Tsai YH, Chen PH, Huang CY, Chen WT, Hwang LH. Heat shock protein 72 is associated with the hepatitis C virus replicase complex and enhances viral RNA replication. J Biol Chem. 2010;285:28183–28190. doi: 10.1074/jbc.M110.118323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chumpitazi BF, Bouillet L, Drouet MT, Kuhn L, Garin J, Zarski JP, Drouet C. Biological autoimmunity screening in hepatitis C patients by anti-HepG2 lysate and anti-heat shock protein 70.1 autoantibodies. Eur J Clin Microbiol Infect Dis. 2009;28:137–146. doi: 10.1007/s10096-008-0599-y. [DOI] [PubMed] [Google Scholar]
- 66.Fang C, Yi Z, Liu F, Lan S, Wang J, Lu H, Yang P, Yuan Z. Proteome analysis of human liver carcinoma Huh7 cells harboring hepatitis C virus subgenomic replicon. Proteomics. 2006;6:519–527. doi: 10.1002/pmic.200500233. [DOI] [PubMed] [Google Scholar]
- 67.Lim YS, Shin KS, Oh SH, Kang SM, Won SJ, Hwang SB. Nonstructural 5A protein of hepatitis C virus regulates heat shock protein 72 for its own propagation. J Viral Hepat. 2012;19:353–363. doi: 10.1111/j.1365-2893.2011.01556.x. [DOI] [PubMed] [Google Scholar]
- 68.Khachatoorian R, Arumugaswami V, Raychaudhuri S, Yeh GK, Maloney EM, Wang J, Dasgupta A, French SW. Divergent antiviral effects of bioflavonoids on the hepatitis C virus life cycle. Virology. 2012;433:346–355. doi: 10.1016/j.virol.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khachatoorian R, Ruchala P, Waring A, Jung CL, Ganapathy E, Wheatley N, Sundberg C, Arumugaswami V, Dasgupta A, French SW. Structural characterization of the HSP70 interaction domain of the hepatitis C viral protein NS5A. Virology. 2015;475:46–55. doi: 10.1016/j.virol.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris D, Zhang Z, Chaubey B, Pandey VN. Identification of cellular factors associated with the 3’-nontranslated region of the hepatitis C virus genome. Mol Cell Proteomics. 2006;5:1006–1018. doi: 10.1074/mcp.M500429-MCP200. [DOI] [PubMed] [Google Scholar]
- 71.Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther. 2012;136:354–374. doi: 10.1016/j.pharmthera.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 72.Singaravelu R, Blais DR, McKay CS, Pezacki JP. Activity-based protein profiling of the hepatitis C virus replication in Huh-7 hepatoma cells using a non-directed active site probe. Proteome Sci. 2010;8:5. doi: 10.1186/1477-5956-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Inoue Y, Aizaki H, Hara H, Matsuda M, Ando T, Shimoji T, Murakami K, Masaki T, Shoji I, Homma S, et al. Chaperonin TRiC/CCT participates in replication of hepatitis C virus genome via interaction with the viral NS5B protein. Virology. 2011;410:38–47. doi: 10.1016/j.virol.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 74.Parent R, Qu X, Petit MA, Beretta L. The heat shock cognate protein 70 is associated with hepatitis C virus particles and modulates virus infectivity. Hepatology. 2009;49:1798–1809. doi: 10.1002/hep.22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walsh P, Bursać D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Upadhyay A, Dixit U, Manvar D, Chaturvedi N, Pandey VN. Affinity capture and identification of host cell factors associated with hepatitis C virus (+) strand subgenomic RNA. Mol Cell Proteomics. 2013;12:1539–1552. doi: 10.1074/mcp.M112.017020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peng ZG, Fan B, Du NN, Wang YP, Gao LM, Li YH, Li YH, Liu F, You XF, Han YX, et al. Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA. Hepatology. 2010;52:845–853. doi: 10.1002/hep.23766. [DOI] [PubMed] [Google Scholar]
- 78.Chen DZ, Jiang JD, Zhang KQ, He HP, Di YT, Zhang Y, Cai JY, Wang L, Li SL, Yi P, et al. Evaluation of anti-HCV activity and SAR study of (+)-lycoricidine through targeting of host heat-stress cognate 70 (Hsc70) Bioorg Med Chem Lett. 2013;23:2679–2682. doi: 10.1016/j.bmcl.2013.02.089. [DOI] [PubMed] [Google Scholar]
- 79.Du NN, Peng ZG, Bi CW, Tang S, Li YH, Li JR, Zhu YP, Zhang JP, Wang YX, Jiang JD, et al. N-substituted benzyl matrinic acid derivatives inhibit hepatitis C virus (HCV) replication through down-regulating host heat-stress cognate 70 (Hsc70) expression. PLoS One. 2013;8:e58675. doi: 10.1371/journal.pone.0058675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9:1937–1954. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- 81.Noonan EJ, Place RF, Giardina C, Hightower LE. Hsp70B’ regulation and function. Cell Stress Chaperones. 2007;12:393–402. doi: 10.1379/CSC-278e.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Müller L, Zimmermann R. Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci. 2009;66:1556–1569. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol. 2006;80:11868–11880. doi: 10.1128/JVI.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777–2793. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ciccaglione AR, Marcantonio C, Tritarelli E, Equestre M, Vendittelli F, Costantino A, Geraci A, Rapicetta M. Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 2007;126:128–138. doi: 10.1016/j.virusres.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 86.Sekine-Osajima Y, Sakamoto N, Mishima K, Nakagawa M, Itsui Y, Tasaka M, Nishimura-Sakurai Y, Chen CH, Kanai T, Tsuchiya K, et al. Development of plaque assays for hepatitis C virus-JFH1 strain and isolation of mutants with enhanced cytopathogenicity and replication capacity. Virology. 2008;371:71–85. doi: 10.1016/j.virol.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 87.Li S, Ye L, Yu X, Xu B, Li K, Zhu X, Liu H, Wu X, Kong L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology. 2009;391:257–264. doi: 10.1016/j.virol.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 88.Funaoka Y, Sakamoto N, Suda G, Itsui Y, Nakagawa M, Kakinuma S, Watanabe T, Mishima K, Ueyama M, Onozuka I, et al. Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J Virol. 2011;85:5986–5994. doi: 10.1128/JVI.02583-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol. 2002;64:1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- 90.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohl BP, Tedbury PR, Griffin S, Harris M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J Virol. 2012;86:10724–10732. doi: 10.1128/JVI.01667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan SW. Unfolded protein response in hepatitis C virus infection. Front Microbiol. 2014;5:233. doi: 10.3389/fmicb.2014.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009;5:e1000291. doi: 10.1371/journal.ppat.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mishima K, Sakamoto N, Sekine-Osajima Y, Nakagawa M, Itsui Y, Azuma S, Kakinuma S, Kiyohashi K, Kitazume A, Tsuchiya K, et al. Cell culture and in vivo analyses of cytopathic hepatitis C virus mutants. Virology. 2010;405:361–369. doi: 10.1016/j.virol.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 95.Tumurbaatar B, Sun Y, Chan T, Sun J. Cre-estrogen receptor-mediated hepatitis C virus structural protein expression in mice. J Virol Methods. 2007;146:5–13. doi: 10.1016/j.jviromet.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandra PK, Gunduz F, Hazari S, Kurt R, Panigrahi R, Poat B, Bruce D, Cohen AJ, Bohorquez HE, Carmody I, et al. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS One. 2014;9:e108616. doi: 10.1371/journal.pone.0108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liberman E, Fong YL, Selby MJ, Choo QL, Cousens L, Houghton M, Yen TS. Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein. J Virol. 1999;73:3718–3722. doi: 10.1128/jvi.73.5.3718-3722.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choukhi A, Ung S, Wychowski C, Dubuisson J. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J Virol. 1998;72:3851–3858. doi: 10.1128/jvi.72.5.3851-3858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan SW, Egan PA. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. doi: 10.1096/fj.04-3455fje. [DOI] [PubMed] [Google Scholar]
- 100.Chan SW, Egan PA. Effects of hepatitis C virus envelope glycoprotein unfolded protein response activation on translation and transcription. Arch Virol. 2009;154:1631–1640. doi: 10.1007/s00705-009-0495-5. [DOI] [PubMed] [Google Scholar]
- 101.Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- 102.Ciccaglione AR, Costantino A, Tritarelli E, Marcantonio C, Equestre M, Marziliano N, Rapicetta M. Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch Virol. 2005;150:1339–1356. doi: 10.1007/s00705-004-0487-4. [DOI] [PubMed] [Google Scholar]
- 103.Zheng Y, Gao B, Ye L, Kong L, Jing W, Yang X, Wu Z, Ye L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J Microbiol. 2005;43:529–536. [PubMed] [Google Scholar]
- 104.Jiang X, Kanda T, Wu S, Nakamoto S, Wakita T, Shirasawa H, Yokosuka O. Hepatitis C virus nonstructural protein 5A inhibits thapsigargin-induced apoptosis. PLoS One. 2014;9:e113499. doi: 10.1371/journal.pone.0113499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Germain MA, Chatel-Chaix L, Gagné B, Bonneil É, Thibault P, Pradezynski F, de Chassey B, Meyniel-Schicklin L, Lotteau V, Baril M, et al. Elucidating novel hepatitis C virus-host interactions using combined mass spectrometry and functional genomics approaches. Mol Cell Proteomics. 2014;13:184–203. doi: 10.1074/mcp.M113.030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J Virol. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.MacPherson JI, Sidders B, Wieland S, Zhong J, Targett-Adams P, Lohmann V, Backes P, Delpuech-Adams O, Chisari F, Lewis M, et al. An integrated transcriptomic and meta-analysis of hepatoma cells reveals factors that influence susceptibility to HCV infection. PLoS One. 2011;6:e25584. doi: 10.1371/journal.pone.0025584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X, Wang T, Wakita T, Yang W. Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology. 2010;398:57–67. doi: 10.1016/j.virol.2009.11.036. [DOI] [PubMed] [Google Scholar]
- 109.Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J Virol. 2003;77:3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Egan PA, Sobkowiak M, Chan SW. Hepatitis C Virus Envelope Protein E1 Binds PERK and Represses the Unfolded Protein Response. Open Virol J. 2013;7:37–40. doi: 10.2174/1874357901307010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Selby M, Erickson A, Dong C, Cooper S, Parham P, Houghton M, Walker CM. Hepatitis C virus envelope glycoprotein E1 originates in the endoplasmic reticulum and requires cytoplasmic processing for presentation by class I MHC molecules. J Immunol. 1999;162:669–676. [PubMed] [Google Scholar]
- 112.Pavio N, Taylor DR, Lai MM. Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J Virol. 2002;76:1265–1272. doi: 10.1128/JVI.76.3.1265-1272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McPherson S, Powell EE, Barrie HD, Clouston AD, McGuckin M, Jonsson JR. No evidence of the unfolded protein response in patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2011;26:319–327. doi: 10.1111/j.1440-1746.2010.06368.x. [DOI] [PubMed] [Google Scholar]
- 114.Dores-Silva PR, Barbosa LR, Ramos CH, Borges JC. Human mitochondrial Hsp70 (mortalin): shedding light on ATPase activity, interaction with adenosine nucleotides, solution structure and domain organization. PLoS One. 2015;10:e0117170. doi: 10.1371/journal.pone.0117170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Flachbartová Z, Kovacech B. Mortalin - a multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol. 2013;57:3–15. doi: 10.4149/av_2013_01_3. [DOI] [PubMed] [Google Scholar]
- 116.Baaklini I, Wong MJ, Hantouche C, Patel Y, Shrier A, Young JC. The DNAJA2 substrate release mechanism is essential for chaperone-mediated folding. J Biol Chem. 2012;287:41939–41954. doi: 10.1074/jbc.M112.413278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Park SY, Choi HK, Seo JS, Yoo JY, Jeong JW, Choi Y, Choi KC, Yoon HG. DNAJB1 negatively regulates MIG6 to promote epidermal growth factor receptor signaling. Biochim Biophys Acta. 2015;1853:2722–2730. doi: 10.1016/j.bbamcr.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 118.Kubo N, Wu D, Yoshihara Y, Sang M, Nakagawara A, Ozaki T. Co-chaperon DnaJC7/TPR2 enhances p53 stability and activity through blocking the complex formation between p53 and MDM2. Biochem Biophys Res Commun. 2013;430:1034–1039. doi: 10.1016/j.bbrc.2012.11.121. [DOI] [PubMed] [Google Scholar]
- 119.Blackham S, Baillie A, Al-Hababi F, Remlinger K, You S, Hamatake R, McGarvey MJ. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J Virol. 2010;84:5404–5414. doi: 10.1128/JVI.02529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482–495. doi: 10.1101/gad.1752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yi Z, Sperzel L, Nürnberger C, Bredenbeek PJ, Lubick KJ, Best SM, Stoyanov CT, Law LM, Yuan Z, Rice CM, et al. Identification and characterization of the host protein DNAJC14 as a broadly active flavivirus replication modulator. PLoS Pathog. 2011;7:e1001255. doi: 10.1371/journal.ppat.1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yi Z, Yuan Z, Rice CM, MacDonald MR. Flavivirus replication complex assembly revealed by DNAJC14 functional mapping. J Virol. 2012;86:11815–11832. doi: 10.1128/JVI.01022-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee HJ, Kim JM, Kim KH, Heo JI, Kwak SJ, Han JA. Genotoxic stress/p53-induced DNAJB9 inhibits the pro-apoptotic function of p53. Cell Death Differ. 2015;22:86–95. doi: 10.1038/cdd.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vandermeeren AM, Gómez CE, Patiño C, Domingo-Gil E, Guerra S, González JM, Esteban M. Subcellular forms and biochemical events triggered in human cells by HCV polyprotein expression from a viral vector. Virol J. 2008;5:102. doi: 10.1186/1743-422X-5-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oka OB, Pringle MA, Schopp IM, Braakman I, Bulleid NJ. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol Cell. 2013;50:793–804. doi: 10.1016/j.molcel.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elwi AN, Lee B, Meijndert HC, Braun JE, Kim SW. Mitochondrial chaperone DnaJA3 induces Drp1-dependent mitochondrial fragmentation. Int J Biochem Cell Biol. 2012;44:1366–1376. doi: 10.1016/j.biocel.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 128.Bracher A, Verghese J. GrpE, Hsp110/Grp170, HspBP1/Sil1 and BAG domain proteins: nucleotide exchange factors for Hsp70 molecular chaperones. Subcell Biochem. 2015;78:1–33. doi: 10.1007/978-3-319-11731-7_1. [DOI] [PubMed] [Google Scholar]
- 129.Blais DR, Brûlotte M, Qian Y, Bélanger S, Yao SQ, Pezacki JP. Activity-based proteome profiling of hepatoma cells during hepatitis C virus replication using protease substrate probes. J Proteome Res. 2010;9:912–923. doi: 10.1021/pr900788a. [DOI] [PubMed] [Google Scholar]
- 130.Hara H, Aizaki H, Matsuda M, Shinkai-Ouchi F, Inoue Y, Murakami K, Shoji I, Kawakami H, Matsuura Y, Lai MM, et al. Involvement of creatine kinase B in hepatitis C virus genome replication through interaction with the viral NS4A protein. J Virol. 2009;83:5137–5147. doi: 10.1128/JVI.02179-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Park JM, Kim JW, Hahm KB. HSPA4, the „Evil Chaperone” of the HSP Family, Delays Gastric Ulcer Healing. Dig Dis Sci. 2015;60:824–826. doi: 10.1007/s10620-015-3597-9. [DOI] [PubMed] [Google Scholar]
- 132.Yang Z, Zhuang L, Szatmary P, Wen L, Sun H, Lu Y, Xu Q, Chen X. Upregulation of heat shock proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in tumour tissues is associated with poor outcomes from HBV-related early-stage hepatocellular carcinoma. Int J Med Sci. 2015;12:256–263. doi: 10.7150/ijms.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saito Y, Yamagishi N, Hatayama T. Nuclear localization mechanism of Hsp105beta and its possible function in mammalian cells. J Biochem. 2009;145:185–191. doi: 10.1093/jb/mvn155. [DOI] [PubMed] [Google Scholar]
- 134.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eckl JM, Richter K. Functions of the Hsp90 chaperone system: lifting client proteins to new heights. Int J Biochem Mol Biol. 2013;4:157–165. [PMC free article] [PubMed] [Google Scholar]
- 136.Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta. 2012;1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nakagawa S, Umehara T, Matsuda C, Kuge S, Sudoh M, Kohara M. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun. 2007;353:882–888. doi: 10.1016/j.bbrc.2006.12.117. [DOI] [PubMed] [Google Scholar]
- 139.Shan GZ, Peng ZG, Li YH, Li D, Li YP, Meng S, Gao LY, Jiang JD, Li ZR. A novel class of geldanamycin derivatives as HCV replication inhibitors targeting on Hsp90: synthesis, structure-activity relationships and anti-HCV activity in GS4.3 replicon cells. J Antibiot (Tokyo) 2011;64:177–182. doi: 10.1038/ja.2010.161. [DOI] [PubMed] [Google Scholar]
- 140.Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci USA. 2001;98:13931–13935. doi: 10.1073/pnas.241510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kubota N, Inayoshi Y, Satoh N, Fukuda T, Iwai K, Tomoda H, Kohara M, Kataoka K, Shimamoto A, Furuichi Y, et al. HSC90 is required for nascent hepatitis C virus core protein stability in yeast cells. FEBS Lett. 2012;586:2318–2325. doi: 10.1016/j.febslet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 142.Ujino S, Yamaguchi S, Shimotohno K, Takaku H. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus nonstructural protein NS3. J Biol Chem. 2009;284:6841–6846. doi: 10.1074/jbc.M806452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dolan PT, Zhang C, Khadka S, Arumugaswami V, Vangeloff AD, Heaton NS, Sahasrabudhe S, Randall G, Sun R, LaCount DJ. Identification and comparative analysis of hepatitis C virus-host cell protein interactions. Mol Biosyst. 2013;9:3199–3209. doi: 10.1039/c3mb70343f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ujino S, Nishitsuji H, Sugiyama R, Suzuki H, Hishiki T, Sugiyama K, Shimotohno K, Takaku H. The interaction between human initiation factor eIF3 subunit c and heat-shock protein 90: a necessary factor for translation mediated by the hepatitis C virus internal ribosome entry site. Virus Res. 2012;163:390–395. doi: 10.1016/j.virusres.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 145.Bukong TN, Hou W, Kodys K, Szabo G. Ethanol facilitates hepatitis C virus replication via up-regulation of GW182 and heat shock protein 90 in human hepatoma cells. Hepatology. 2013;57:70–80. doi: 10.1002/hep.26010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014;10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim MG, Moon JS, Kim EJ, Lee SH, Oh JW. Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochem Biophys Res Commun. 2012;421:112–118. doi: 10.1016/j.bbrc.2012.03.126. [DOI] [PubMed] [Google Scholar]
- 148.Kim SJ, Kim JH, Sun JM, Kim MG, Oh JW. Suppression of hepatitis C virus replication by protein kinase C-related kinase 2 inhibitors that block phosphorylation of viral RNA polymerase. J Viral Hepat. 2009;16:697–704. doi: 10.1111/j.1365-2893.2009.01108.x. [DOI] [PubMed] [Google Scholar]
- 149.Kim SJ, Kim JH, Kim YG, Lim HS, Oh JW. Protein kinase C-related kinase 2 regulates hepatitis C virus RNA polymerase function by phosphorylation. J Biol Chem. 2004;279:50031–50041. doi: 10.1074/jbc.M408617200. [DOI] [PubMed] [Google Scholar]
- 150.Ito M, Murakami K, Suzuki T, Mochida K, Suzuki M, Ikebuchi K, Yamaguchi K, Mizuochi T. Enhanced expression of lymphomagenesis-related genes in peripheral blood B cells of chronic hepatitis C patients. Clin Immunol. 2010;135:459–465. doi: 10.1016/j.clim.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 151.Ito M, Masumi A, Mochida K, Kukihara H, Moriishi K, Matsuura Y, Yamaguchi K, Mizuochi T. Peripheral B cells may serve as a reservoir for persistent hepatitis C virus infection. J Innate Immun. 2010;2:607–617. doi: 10.1159/000317690. [DOI] [PubMed] [Google Scholar]
- 152.Boukli NM, Shetty V, Cubano L, Ricaurte M, Coelho-Dos-Reis J, Nickens Z, Shah P, Talal AH, Philip R, Jain P. Unique and differential protein signatures within the mononuclear cells of HIV-1 and HCV mono-infected and co-infected patients. Clin Proteomics. 2012;9:11. doi: 10.1186/1559-0275-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marzec M, Eletto D, Argon Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012;1823:774–787. doi: 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Han JM, Park SG, Liu B, Park BJ, Kim JY, Jin CH, Song YW, Li Z, Kim S. Aminoacyl-tRNA synthetase-interacting multifunctional protein 1/p43 controls endoplasmic reticulum retention of heat shock protein gp96: its pathological implications in lupus-like autoimmune diseases. Am J Pathol. 2007;170:2042–2054. doi: 10.2353/ajpath.2007.061266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim MS, Kim S, Myung H. Degradation of AIMP1/p43 induced by hepatitis C virus E2 leads to upregulation of TGF-β signaling and increase in surface expression of gp96. PLoS One. 2014;9:e96302. doi: 10.1371/journal.pone.0096302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nakamura H, Minegishi H. HSP60 as a drug target. Curr Pharm Des. 2013;19:441–451. [PubMed] [Google Scholar]
- 157.Hemmingsen SM, Woolford C, van der Vies SM, Tilly K, Dennis DT, Georgopoulos CP, Hendrix RW, Ellis RJ. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature. 1988;333:330–334. doi: 10.1038/333330a0. [DOI] [PubMed] [Google Scholar]
- 158.Leitner A, Joachimiak LA, Bracher A, Mönkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814–825. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kang SM, Kim SJ, Kim JH, Lee W, Kim GW, Lee KH, Choi KY, Oh JW. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009;279:230–237. doi: 10.1016/j.canlet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 160.Padwad YS, Mishra KP, Jain M, Chanda S, Karan D, Ganju L. RNA interference mediated silencing of Hsp60 gene in human monocytic myeloma cell line U937 revealed decreased dengue virus multiplication. Immunobiology. 2009;214:422–429. doi: 10.1016/j.imbio.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 161.Cappello F, Marino Gammazza A, Palumbo Piccionello A, Campanella C, Pace A, Conway de Macario E, Macario AJ. Hsp60 chaperonopathies and chaperonotherapy: targets and agents. Expert Opin Ther Targets. 2014;18:185–208. doi: 10.1517/14728222.2014.856417. [DOI] [PubMed] [Google Scholar]
- 162.Fukuda Y, Yotsuyanagi H, Ooka S, Sekine T, Koike J, Takano T, Suzuki M, Itoh F, Nishioka K, Kato T. Identification of a new autoantibody in patients with chronic hepatitis. Hum Immunol. 2004;65:1530–1538. doi: 10.1016/j.humimm.2004.08.186. [DOI] [PubMed] [Google Scholar]
- 163.Bakthisaran R, Tangirala R, Rao ChM. Small heat shock proteins: Role in cellular functions and pathology. Biochim Biophys Acta. 2015;1854:291–319. doi: 10.1016/j.bbapap.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 164.Choi YW, Tan YJ, Lim SG, Hong W, Goh PY. Proteomic approach identifies HSP27 as an interacting partner of the hepatitis C virus NS5A protein. Biochem Biophys Res Commun. 2004;318:514–519. doi: 10.1016/j.bbrc.2004.04.052. [DOI] [PubMed] [Google Scholar]
- 165.Wang X, Chen M, Zhou J, Zhang X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review) Int J Oncol. 2014;45:18–30. doi: 10.3892/ijo.2014.2399. [DOI] [PubMed] [Google Scholar]
- 166.Acunzo J, Katsogiannou M, Rocchi P. Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol. 2012;44:1622–1631. doi: 10.1016/j.biocel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 167.Zhu H, Liu P, Li J. BAG3: a new therapeutic target of human cancers? Histol Histopathol. 2012;27:257–261. doi: 10.14670/HH-27.257. [DOI] [PubMed] [Google Scholar]
- 168.Edlich F, Lücke C. From cell death to viral replication: the diverse functions of the membrane-associated FKBP38. Curr Opin Pharmacol. 2011;11:348–353. doi: 10.1016/j.coph.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 169.Wang J, Tong W, Zhang X, Chen L, Yi Z, Pan T, Hu Y, Xiang L, Yuan Z. Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 2006;580:4392–4400. doi: 10.1016/j.febslet.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 170.Okamoto T, Omori H, Kaname Y, Abe T, Nishimura Y, Suzuki T, Miyamura T, Yoshimori T, Moriishi K, Matsuura Y. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J Virol. 2008;82:3480–3489. doi: 10.1128/JVI.02253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Taguwa S, Okamoto T, Abe T, Mori Y, Suzuki T, Moriishi K, Matsuura Y. Human butyrate-induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J Virol. 2008;82:2631–2641. doi: 10.1128/JVI.02153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Courilleau D, Chastre E, Sabbah M, Redeuilh G, Atfi A, Mester J. B-ind1, a novel mediator of Rac1 signaling cloned from sodium butyrate-treated fibroblasts. J Biol Chem. 2000;275:17344–17348. doi: 10.1074/jbc.M000887200. [DOI] [PubMed] [Google Scholar]
- 173.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–374. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Taguwa S, Kambara H, Omori H, Tani H, Abe T, Mori Y, Suzuki T, Yoshimori T, Moriishi K, Matsuura Y. Cochaperone activity of human butyrate-induced transcript 1 facilitates hepatitis C virus replication through an Hsp90-dependent pathway. J Virol. 2009;83:10427–10436. doi: 10.1128/JVI.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tani J, Shimamoto S, Mori K, Kato N, Moriishi K, Matsuura Y, Tokumitsu H, Tsuchiya M, Fujimoto T, Kato K, et al. Ca(2+) /S100 proteins regulate HCV virus NS5A-FKBP8/FKBP38 interaction and HCV virus RNA replication. Liver Int. 2013;33:1008–1018. doi: 10.1111/liv.12151. [DOI] [PubMed] [Google Scholar]
- 176.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med. 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 177.Peng L, Liang D, Tong W, Li J, Yuan Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J Biol Chem. 2010;285:20870–20881. doi: 10.1074/jbc.M110.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zannas AS, Wiechmann T, Gassen NC, Binder EB. Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology. 2016;41:261–274. doi: 10.1038/npp.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rehn AB, Buchner J. p23 and Aha1. Subcell Biochem. 2015;78:113–131. doi: 10.1007/978-3-319-11731-7_6. [DOI] [PubMed] [Google Scholar]
- 180.Shirasaki T, Honda M, Mizuno H, Shimakami T, Okada H, Sakai Y, Murakami S, Wakita T, Kaneko S. La protein required for internal ribosome entry site-directed translation is a potential therapeutic target for hepatitis C virus replication. J Infect Dis. 2010;202:75–85. doi: 10.1086/653081. [DOI] [PubMed] [Google Scholar]
- 181.Ali N, Siddiqui A. The La antigen binds 5’ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc Natl Acad Sci USA. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Abe A, Takahashi-Niki K, Takekoshi Y, Shimizu T, Kitaura H, Maita H, Iguchi-Ariga SM, Ariga H. Prefoldin plays a role as a clearance factor in preventing proteasome inhibitor-induced protein aggregation. J Biol Chem. 2013;288:27764–27776. doi: 10.1074/jbc.M113.476358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tsao ML, Chao CH, Yeh CT. Interaction of hepatitis C virus F protein with prefoldin 2 perturbs tubulin cytoskeleton organization. Biochem Biophys Res Commun. 2006;348:271–277. doi: 10.1016/j.bbrc.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 184.Lai CK, Jeng KS, Machida K, Lai MM. Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J Virol. 2008;82:8838–8848. doi: 10.1128/JVI.00398-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roohvand F, Maillard P, Lavergne JP, Boulant S, Walic M, Andréo U, Goueslain L, Helle F, Mallet A, McLauchlan J, et al. Initiation of hepatitis C virus infection requires the dynamic microtubule network: role of the viral nucleocapsid protein. J Biol Chem. 2009;284:13778–13791. doi: 10.1074/jbc.M807873200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Counihan NA, Rawlinson SM, Lindenbach BD. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 2011;7:e1002302. doi: 10.1371/journal.ppat.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bost AG, Venable D, Liu L, Heinz BA. Cytoskeletal requirements for hepatitis C virus (HCV) RNA synthesis in the HCV replicon cell culture system. J Virol. 2003;77:4401–4408. doi: 10.1128/JVI.77.7.4401-4408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Trougakos IP. The molecular chaperone apolipoprotein J/clusterin as a sensor of oxidative stress: implications in therapeutic approaches - a mini-review. Gerontology. 2013;59:514–523. doi: 10.1159/000351207. [DOI] [PubMed] [Google Scholar]
- 189.Chaiwatanasirikul KA, Sala A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011;2:e219. doi: 10.1038/cddis.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lin CC, Tsai P, Sun HY, Hsu MC, Lee JC, Wu IC, Tsao CW, Chang TT, Young KC. Apolipoprotein J, a glucose-upregulated molecular chaperone, stabilizes core and NS5A to promote infectious hepatitis C virus virion production. J Hepatol. 2014;61:984–993. doi: 10.1016/j.jhep.2014.06.026. [DOI] [PubMed] [Google Scholar]
- 191.Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Hum Genomics. 2012;6:6. doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci. 1994;19:331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 193.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 194.Gordon DA. Recent advances in elucidating the role of the microsomal triglyceride transfer protein in apolipoprotein B lipoprotein assembly. Curr Opin Lipidol. 1997;8:131–137. doi: 10.1097/00041433-199706000-00002. [DOI] [PubMed] [Google Scholar]
- 195.Burnett JR, Barrett PH. Apolipoprotein B metabolism: tracer kinetics, models, and metabolic studies. Crit Rev Clin Lab Sci. 2002;39:89–137. doi: 10.1080/10408360208951113. [DOI] [PubMed] [Google Scholar]
- 196.Mirandola S, Realdon S, Iqbal J, Gerotto M, Dal Pero F, Bortoletto G, Marcolongo M, Vario A, Datz C, Hussain MM, et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology. 2006;130:1661–1669. doi: 10.1053/j.gastro.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 197.André P, Perlemuter G, Budkowska A, Bréchot C, Lotteau V. Hepatitis C virus particles and lipoprotein metabolism. Semin Liver Dis. 2005;25:93–104. doi: 10.1055/s-2005-864785. [DOI] [PubMed] [Google Scholar]
- 198.Yamaguchi A, Tazuma S, Nishioka T, Ohishi W, Hyogo H, Nomura S, Chayama K. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig Dis Sci. 2005;50:1361–1371. doi: 10.1007/s10620-005-2788-1. [DOI] [PubMed] [Google Scholar]
- 199.Domitrovich AM, Felmlee DJ, Siddiqui A. Hepatitis C virus nonstructural proteins inhibit apolipoprotein B100 secretion. J Biol Chem. 2005;280:39802–39808. doi: 10.1074/jbc.M510391200. [DOI] [PubMed] [Google Scholar]
- 200.Parvaiz F, Manzoor S, Iqbal J, McRae S, Javed F, Ahmed QL, Waris G. Hepatitis C virus nonstructural protein 5A favors upregulation of gluconeogenic and lipogenic gene expression leading towards insulin resistance: a metabolic syndrome. Arch Virol. 2014;159:1017–1025. doi: 10.1007/s00705-013-1892-3. [DOI] [PubMed] [Google Scholar]
- 201.Zampino R, Ingrosso D, Durante-Mangoni E, Capasso R, Tripodi MF, Restivo L, Zappia V, Ruggiero G, Adinolfi LE. Microsomal triglyceride transfer protein (MTP) -493G/T gene polymorphism contributes to fat liver accumulation in HCV genotype 3 infected patients. J Viral Hepat. 2008;15:740–746. doi: 10.1111/j.1365-2893.2008.00994.x. [DOI] [PubMed] [Google Scholar]
- 202.Mirandola S, Osterreicher CH, Marcolongo M, Datz C, Aigner E, Schlabrakowski A, Realdon S, Gerotto M, Alberti A, Stickel F. Microsomal triglyceride transfer protein polymorphism (-493G/T) is associated with hepatic steatosis in patients with chronic hepatitis C. Liver Int. 2009;29:557–565. doi: 10.1111/j.1478-3231.2008.01892.x. [DOI] [PubMed] [Google Scholar]
- 203.Siqueira ER, Oliveira CP, Correa-Giannella ML, Stefano JT, Cavaleiro AM, Fortes MA, Muniz MT, Silva FS, Pereira LM, Carrilho FJ. MTP -493G/T gene polymorphism is associated with steatosis in hepatitis C-infected patients. Braz J Med Biol Res. 2012;45:72–77. doi: 10.1590/S0100-879X2011007500160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Saad Y, Shaker O, Nassar Y, Ahmad L, Said M, Esmat G. A polymorphism in the microsomal triglyceride transfer protein can predict the response to antiviral therapy in Egyptian patients with chronic hepatitis C virus genotype 4 infection. Gut Liver. 2014;8:655–661. doi: 10.5009/gnl13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ryan MC, Desmond PV, Slavin JL, Congiu M. Expression of genes involved in lipogenesis is not increased in patients with HCV genotype 3 in human liver. J Viral Hepat. 2011;18:53–60. doi: 10.1111/j.1365-2893.2010.01283.x. [DOI] [PubMed] [Google Scholar]
- 206.Rojas Á, del Campo JA, Maraver M, Aparcero R, García-Valdecasas M, Diago M, Carmona I, Andrade RJ, Solà R, Romero-Gómez M. Hepatitis C virus infection alters lipid metabolism depending on IL28B polymorphism and viral genotype and modulates gene expression in vivo and in vitro. J Viral Hepat. 2014;21:19–24. doi: 10.1111/jvh.12209. [DOI] [PubMed] [Google Scholar]
- 207.Bridge SH, Sheridan DA, Felmlee DJ, Crossey MM, Fenwick FI, Lanyon CV, Dubuc G, Seidah NG, Davignon J, Thomas HC, et al. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: evidence for genotype-specific regulation of lipoprotein metabolism. J Hepatol. 2015;62:763–770. doi: 10.1016/j.jhep.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 208.Mirandola S, Bowman D, Hussain MM, Alberti A. Hepatic steatosis in hepatitis C is a storage disease due to HCV interaction with microsomal triglyceride transfer protein (MTP) Nutr Metab (Lond) 2010;7:13. doi: 10.1186/1743-7075-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gastaminza P, Kapadia SB, Chisari FV. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol. 2006;80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Icard V, Diaz O, Scholtes C, Perrin-Cocon L, Ramière C, Bartenschlager R, Penin F, Lotteau V, André P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS One. 2009;4:e4233. doi: 10.1371/journal.pone.0004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Owen DM, Huang H, Ye J, Gale M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology. 2009;394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, et al. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology. 2010;51:43–53. doi: 10.1002/hep.23278. [DOI] [PubMed] [Google Scholar]
- 215.Cun W, Jiang J, Luo G. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol. 2010;84:11532–11541. doi: 10.1128/JVI.01021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol. 2009;83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Castillo V, Oñate M, Woehlbier U, Rozas P, Andreu C, Medinas D, Valdés P, Osorio F, Mercado G, Vidal RL, et al. Functional Role of the Disulfide Isomerase ERp57 in Axonal Regeneration. PLoS One. 2015;10:e0136620. doi: 10.1371/journal.pone.0136620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Dubuisson J, Rice CM. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J Virol. 1996;70:778–786. doi: 10.1128/jvi.70.2.778-786.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dubuisson J. Folding, assembly and subcellular localization of hepatitis C virus glycoproteins. Curr Top Microbiol Immunol. 2000;242:135–148. doi: 10.1007/978-3-642-59605-6_7. [DOI] [PubMed] [Google Scholar]
- 221.Chapel C, Garcia C, Roingeard P, Zitzmann N, Dubuisson J, Dwek RA, Trépo C, Zoulim F, Durantel D. Antiviral effect of alpha-glucosidase inhibitors on viral morphogenesis and binding properties of hepatitis C virus-like particles. J Gen Virol. 2006;87:861–871. doi: 10.1099/vir.0.81503-0. [DOI] [PubMed] [Google Scholar]
- 222.Wohlfarth C, Efferth T. Natural products as promising drug candidates for the treatment of hepatitis B and C. Acta Pharmacol Sin. 2009;30:25–30. doi: 10.1038/aps.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Deleersnyder V, Pillez A, Wychowski C, Blight K, Xu J, Hahn YS, Rice CM, Dubuisson J. Formation of native hepatitis C virus glycoprotein complexes. J Virol. 1997;71:697–704. doi: 10.1128/jvi.71.1.697-704.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cocquerel L, Quinn ER, Flint M, Hadlock KG, Foung SK, Levy S. Recognition of native hepatitis C virus E1E2 heterodimers by a human monoclonal antibody. J Virol. 2003;77:1604–1609. doi: 10.1128/JVI.77.2.1604-1609.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Meunier JC, Fournillier A, Choukhi A, Cahour A, Cocquerel L, Dubuisson J, Wychowski C. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J Gen Virol. 1999;80(Pt 4):887–896. doi: 10.1099/0022-1317-80-4-887. [DOI] [PubMed] [Google Scholar]
- 226.Ahmed QL, Manzoor S, Tariq M, Khalid M, Ashraf W, Parvaiz F, Imran M. Hepatitis C virus infection in vitro triggers endoplasmic reticulum stress and downregulates insulin receptor substrates 1 and 2 through upregulation of cytokine signaling suppressor 3. Acta Virol. 2014;58:238–244. doi: 10.4149/av_2014_03_238. [DOI] [PubMed] [Google Scholar]
- 227.Määttänen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 228.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Saeed M, Suzuki R, Watanabe N, Masaki T, Tomonaga M, Muhammad A, Kato T, Matsuura Y, Watanabe H, Wakita T, et al. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem. 2011;286:37264–37273. doi: 10.1074/jbc.M111.259085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem. 2004;279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 232.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 233.Ruscher K, Wieloch T. The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J Pharmacol Sci. 2015;127:30–35. doi: 10.1016/j.jphs.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 234.Friesland M, Mingorance L, Chung J, Chisari FV, Gastaminza P. Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection. J Virol. 2013;87:6377–6390. doi: 10.1128/JVI.03557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhou TB, Qin YH. Signaling pathways of prohibitin and its role in diseases. J Recept Signal Transduct Res. 2013;33:28–36. doi: 10.3109/10799893.2012.752006. [DOI] [PubMed] [Google Scholar]
- 236.Dang SS, Sun MZ, Yang E, Xun M, Ma L, Jia ZS, Wang WJ, Jia XL. Prohibitin is overexpressed in Huh-7-HCV and Huh-7.5-HCV cells harboring in vitro transcribed full-length hepatitis C virus RNA. Virol J. 2011;8:424. doi: 10.1186/1743-422X-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Tsutsumi T, Matsuda M, Aizaki H, Moriya K, Miyoshi H, Fujie H, Shintani Y, Yotsuyanagi H, Miyamura T, Suzuki T, et al. Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology. 2009;50:378–386. doi: 10.1002/hep.22998. [DOI] [PubMed] [Google Scholar]
- 238.Fujinaga H, Tsutsumi T, Yotsuyanagi H, Moriya K, Koike K. Hepatocarcinogenesis in hepatitis C: HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology. 2011;81 Suppl 1:11–17. doi: 10.1159/000333253. [DOI] [PubMed] [Google Scholar]
- 239.Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Koike K. The oncogenic role of hepatitis C virus. Recent Results Cancer Res. 2014;193:97–111. doi: 10.1007/978-3-642-38965-8_6. [DOI] [PubMed] [Google Scholar]
- 241.Kumari S, Roy S, Singh P, Singla-Pareek SL, Pareek A. Cyclophilins: proteins in search of function. Plant Signal Behav. 2013;8:e22734. doi: 10.4161/psb.22734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Qing M, Yang F, Zhang B, Zou G, Robida JM, Yuan Z, Tang H, Shi PY. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob Agents Chemother. 2009;53:3226–3235. doi: 10.1128/AAC.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Watashi K, Shimotohno K. Cyclophilin and viruses: cyclophilin as a cofactor for viral infection and possible anti-viral target. Drug Target Insights. 2007;2:9–18. [PMC free article] [PubMed] [Google Scholar]
- 245.Gaither LA, Borawski J, Anderson LJ, Balabanis KA, Devay P, Joberty G, Rau C, Schirle M, Bouwmeester T, Mickanin C, et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology. 2010;397:43–55. doi: 10.1016/j.virol.2009.10.043. [DOI] [PubMed] [Google Scholar]
- 246.Inoue K, Sekiyama K, Yamada M, Watanabe T, Yasuda H, Yoshiba M. Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. J Gastroenterol. 2003;38:567–572. doi: 10.1007/s00535-002-1104-5. [DOI] [PubMed] [Google Scholar]
- 247.Inoue K, Yoshiba M. Interferon combined with cyclosporine treatment as an effective countermeasure against hepatitis C virus recurrence in liver transplant patients with end-stage hepatitis C virus related disease. Transplant Proc. 2005;37:1233–1234. doi: 10.1016/j.transproceed.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 248.Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 249.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 250.Ma S, Boerner JE, TiongYip C, Weidmann B, Ryder NS, Cooreman MP, Lin K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50:2976–2982. doi: 10.1128/AAC.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Paeshuyse J, Kaul A, De Clercq E, Rosenwirth B, Dumont JM, Scalfaro P, Bartenschlager R, Neyts J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 252.Mathy JE, Ma S, Compton T, Lin K. Combinations of cyclophilin inhibitor NIM811 with hepatitis C Virus NS3-4A Protease or NS5B polymerase inhibitors enhance antiviral activity and suppress the emergence of resistance. Antimicrob Agents Chemother. 2008;52:3267–3275. doi: 10.1128/AAC.00498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nakagawa M, Sakamoto N, Enomoto N, Tanabe Y, Kanazawa N, Koyama T, Kurosaki M, Maekawa S, Yamashiro T, Chen CH, et al. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem Biophys Res Commun. 2004;313:42–47. doi: 10.1016/j.bbrc.2003.11.080. [DOI] [PubMed] [Google Scholar]
- 254.Teraoka S, Mishiro S, Ebihara K, Sanaka T, Yamaguchi Y, Nakajima I, Kawai T, Yagisawa T, Honda H, Fuchinoue S. Effect of cyclosporine on proliferation of non-A, non-B hepatitis virus. Transplant Proc. 1988;20:868–876. [PubMed] [Google Scholar]
- 255.Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, Siwak E, Cielniak I, Higersberger J, Kierkus J, et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- 256.Coelmont L, Kaptein S, Paeshuyse J, Vliegen I, Dumont JM, Vuagniaux G, Neyts J. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob Agents Chemother. 2009;53:967–976. doi: 10.1128/AAC.00939-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Flisiak R, Feinman SV, Jablkowski M, Horban A, Kryczka W, Pawlowska M, Heathcote JE, Mazzella G, Vandelli C, Nicolas-Métral V, et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naïve hepatitis C patients. Hepatology. 2009;49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 258.Inoue K, Watanabe T, Yamada M, Yoshikumi H, Ogawa O, Yoshiba M. Efficacy of interferon Beta combined with cyclosporine induction and intensified therapy for retreatment of chronic hepatitis C. Transplant Proc. 2009;41:246–249. doi: 10.1016/j.transproceed.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 259.Hopkins S, Scorneaux B, Huang Z, Murray MG, Wring S, Smitley C, Harris R, Erdmann F, Fischer G, Ribeill Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob Agents Chemother. 2010;54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lawitz E, Godofsky E, Rouzier R, Marbury T, Nguyen T, Ke J, Huang M, Praestgaard J, Serra D, Evans TG. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res. 2011;89:238–245. doi: 10.1016/j.antiviral.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 261.Hopkins S, DiMassimo B, Rusnak P, Heuman D, Lalezari J, Sluder A, Scorneaux B, Mosier S, Kowalczyk P, Ribeill Y, et al. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J Hepatol. 2012;57:47–54. doi: 10.1016/j.jhep.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 262.Chatterji U, Garcia-Rivera JA, Baugh J, Gawlik K, Wong KA, Zhong W, Brass CA, Naoumov NV, Gallay PA. The combination of alisporivir plus an NS5A inhibitor provides additive to synergistic anti-hepatitis C virus activity without detectable cross-resistance. Antimicrob Agents Chemother. 2014;58:3327–3334. doi: 10.1128/AAC.00016-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol. 2009;83:6554–6565. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Rosnoblet C, Fritzinger B, Legrand D, Launay H, Wieruszeski JM, Lippens G, Hanoulle X. Hepatitis C virus NS5B and host cyclophilin A share a common binding site on NS5A. J Biol Chem. 2012;287:44249–44260. doi: 10.1074/jbc.M112.392209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fernandes F, Poole DS, Hoover S, Middleton R, Andrei AC, Gerstner J, Striker R. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology. 2007;46:1026–1033. doi: 10.1002/hep.21809. [DOI] [PubMed] [Google Scholar]
- 266.Robida JM, Nelson HB, Liu Z, Tang H. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J Virol. 2007;81:5829–5840. doi: 10.1128/JVI.02524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Liu Z, Robida JM, Chinnaswamy S, Yi G, Robotham JM, Nelson HB, Irsigler A, Kao CC, Tang H. Mutations in the hepatitis C virus polymerase that increase RNA binding can confer resistance to cyclosporine A. Hepatology. 2009;50:25–33. doi: 10.1002/hep.22987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Abe K, Ikeda M, Ariumi Y, Dansako H, Wakita T, Kato N. HCV genotype 1b chimeric replicon with NS5B of JFH-1 exhibited resistance to cyclosporine A. Arch Virol. 2009;154:1671–1677. doi: 10.1007/s00705-009-0502-x. [DOI] [PubMed] [Google Scholar]
- 269.Chatterji U, Bobardt MD, Lim P, Gallay PA. Cyclophilin A-independent recruitment of NS5A and NS5B into hepatitis C virus replication complexes. J Gen Virol. 2010;91:1189–1193. doi: 10.1099/vir.0.018531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chatterji U, Bobardt M, Tai A, Wood M, Gallay PA. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob Agents Chemother. 2015;59:2496–2507. doi: 10.1128/AAC.04958-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 272.Heck JA, Meng X, Frick DN. Cyclophilin B stimulates RNA synthesis by the HCV RNA dependent RNA polymerase. Biochem Pharmacol. 2009;77:1173–1180. doi: 10.1016/j.bcp.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Watashi K, Shimotohno K. Chemical genetics approach to hepatitis C virus replication: cyclophilin as a target for anti-hepatitis C virus strategy. Rev Med Virol. 2007;17:245–252. doi: 10.1002/rmv.534. [DOI] [PubMed] [Google Scholar]
- 274.Weng L, Tian X, Gao Y, Watashi K, Shimotohno K, Wakita T, Kohara M, Toyoda T. Different mechanisms of hepatitis C virus RNA polymerase activation by cyclophilin A and B in vitro. Biochim Biophys Acta. 2012;1820:1886–1892. doi: 10.1016/j.bbagen.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 275.Morohashi K, Sahara H, Watashi K, Iwabata K, Sunoki T, Kuramochi K, Takakusagi K, Miyashita H, Sato N, Tanabe A, et al. Cyclosporin A associated helicase-like protein facilitates the association of hepatitis C virus RNA polymerase with its cellular cyclophilin B. PLoS One. 2011;6:e18285. doi: 10.1371/journal.pone.0018285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem. 2009;284:13589–13601. doi: 10.1074/jbc.M809244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Grisé H, Frausto S, Logan T, Tang H. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates Alisporivir susceptibility. J Virol. 2012;86:4811–4822. doi: 10.1128/JVI.06641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yang F, Robotham JM, Grise H, Frausto S, Madan V, Zayas M, Bartenschlager R, Robinson M, Greenstein AE, Nag A, et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 2010;6:e1001118. doi: 10.1371/journal.ppat.1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Dujardin M, Madan V, Montserret R, Ahuja P, Huvent I, Launay H, Leroy A, Bartenschlager R, Penin F, Lippens G, et al. A Proline-Tryptophan Turn in the Intrinsically Disordered Domain 2 of NS5A Protein Is Essential for Hepatitis C Virus RNA Replication. J Biol Chem. 2015;290:19104–19120. doi: 10.1074/jbc.M115.644419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, Bartenschlager R, Penin F, Lippens G, Hanoulle X. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem. 2011;286:20441–20454. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Chatterji U, Lim P, Bobardt MD, Wieland S, Cordek DG, Vuagniaux G, Chisari F, Cameron CE, Targett-Adams P, Parkinson T, et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol. 2010;53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hopkins S, Bobardt M, Chatterji U, Garcia-Rivera JA, Lim P, Gallay PA. The cyclophilin inhibitor SCY-635 disrupts hepatitis C virus NS5A-cyclophilin A complexes. Antimicrob Agents Chemother. 2012;56:3888–3897. doi: 10.1128/AAC.00693-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009;5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, Sakamoto N, Vuagniaux G, Parkinson T, Gallay P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem. 2009;284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One. 2010;5:e13687. doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Fernandes F, Ansari IU, Striker R. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One. 2010;5:e9815. doi: 10.1371/journal.pone.0009815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Waller H, Chatterji U, Gallay P, Parkinson T, Targett-Adams P. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J Virol Methods. 2010;165:202–210. doi: 10.1016/j.jviromet.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Gregory MA, Bobardt M, Obeid S, Chatterji U, Coates NJ, Foster T, Gallay P, Leyssen P, Moss SJ, Neyts J, et al. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob Agents Chemother. 2011;55:1975–1981. doi: 10.1128/AAC.01627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Gallay PA, Bobardt MD, Chatterji U, Trepanier DJ, Ure D, Ordonez C, Foster R. The Novel Cyclophilin Inhibitor CPI-431-32 Concurrently Blocks HCV and HIV-1 Infections via a Similar Mechanism of Action. PLoS One. 2015;10:e0134707. doi: 10.1371/journal.pone.0134707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Foster TL, Gallay P, Stonehouse NJ, Harris M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol. 2011;85:7460–7464. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Nag A, Robotham JM, Tang H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J Virol. 2012;86:12616–12624. doi: 10.1128/JVI.01351-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.von Hahn T, Schiene-Fischer C, Van ND, Pfaender S, Karavul B, Steinmann E, Potthoff A, Strassburg C, Hamdi N, Abdelaziz AI, et al. Hepatocytes that express variants of cyclophilin A are resistant to HCV infection and replication. Gastroenterology. 2012;143:439–447.e1. doi: 10.1053/j.gastro.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 293.Yang F, Robotham JM, Nelson HB, Irsigler A, Kenworthy R, Tang H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J Virol. 2008;82:5269–5278. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Goto K, Watashi K, Inoue D, Hijikata M, Shimotohno K. Identification of cellular and viral factors related to anti-hepatitis C virus activity of cyclophilin inhibitor. Cancer Sci. 2009;100:1943–1950. doi: 10.1111/j.1349-7006.2009.01263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ansari IU, Allen T, Berical A, Stock PG, Barin B, Striker R. Phenotypic analysis of NS5A variant from liver transplant patient with increased cyclosporine susceptibility. Virology. 2013;436:268–273. doi: 10.1016/j.virol.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Garcia-Rivera JA, Bobardt M, Chatterji U, Hopkins S, Gregory MA, Wilkinson B, Lin K, Gallay PA. Multiple mutations in hepatitis C virus NS5A domain II are required to confer a significant level of resistance to alisporivir. Antimicrob Agents Chemother. 2012;56:5113–5121. doi: 10.1128/AAC.00919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Arai M, Tsukiyama-Kohara K, Takagi A, Tobita Y, Inoue K, Kohara M. Resistance to cyclosporin A derives from mutations in hepatitis C virus nonstructural proteins. Biochem Biophys Res Commun. 2014;448:56–62. doi: 10.1016/j.bbrc.2014.04.053. [DOI] [PubMed] [Google Scholar]
- 298.Ansari IU, Striker R. Subtype specific differences in NS5A domain II reveals involvement of proline at position 310 in cyclosporine susceptibility of hepatitis C virus. Viruses. 2012;4:3303–3315. doi: 10.3390/v4123303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ishii N, Watashi K, Hishiki T, Goto K, Inoue D, Hijikata M, Wakita T, Kato N, Shimotohno K. Diverse effects of cyclosporine on hepatitis C virus strain replication. J Virol. 2006;80:4510–4520. doi: 10.1128/JVI.80.9.4510-4520.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ciesek S, Steinmann E, Wedemeyer H, Manns MP, Neyts J, Tautz N, Madan V, Bartenschlager R, von Hahn T, Pietschmann T. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology. 2009;50:1638–1645. doi: 10.1002/hep.23281. [DOI] [PubMed] [Google Scholar]
- 301.Madan V, Paul D, Lohmann V, Bartenschlager R. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology. 2014;146:1361–1372.e1-9. doi: 10.1053/j.gastro.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 302.Puyang X, Poulin DL, Mathy JE, Anderson LJ, Ma S, Fang Z, Zhu S, Lin K, Fujimoto R, Compton T, et al. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob Agents Chemother. 2010;54:1981–1987. doi: 10.1128/AAC.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Anderson LJ, Lin K, Compton T, Wiedmann B. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol J. 2011;8:329. doi: 10.1186/1743-422X-8-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Bobardt M, Hopkins S, Baugh J, Chatterji U, Hernandez F, Hiscott J, Sluder A, Lin K, Gallay PA. HCV NS5A and IRF9 compete for CypA binding. J Hepatol. 2013;58:16–23. doi: 10.1016/j.jhep.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Daito T, Watashi K, Sluder A, Ohashi H, Nakajima S, Borroto-Esoda K, Fujita T, Wakita T. Cyclophilin inhibitors reduce phosphorylation of RNA-dependent protein kinase to restore expression of IFN-stimulated genes in HCV-infected cells. Gastroenterology. 2014;147:463–472. doi: 10.1053/j.gastro.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 306.Bobardt M, Chatterji U, Lim P, Gawlik K, Gallay P. Both Cyclophilin Inhibitors and Direct-Acting Antivirals Prevent PKR Activation in HCV-Infected Cells. Open Virol J. 2014;8:1–8. doi: 10.2174/1874357901408010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Nakagawa M, Sakamoto N, Tanabe Y, Koyama T, Itsui Y, Takeda Y, Chen CH, Kakinuma S, Oooka S, Maekawa S, et al. Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology. 2005;129:1031–1041. doi: 10.1053/j.gastro.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 308.Quarato G, D’Aprile A, Gavillet B, Vuagniaux G, Moradpour D, Capitanio N, Piccoli C. The cyclophilin inhibitor alisporivir prevents hepatitis C virus-mediated mitochondrial dysfunction. Hepatology. 2012;55:1333–1343. doi: 10.1002/hep.25514. [DOI] [PubMed] [Google Scholar]
- 309.Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Benedicto I, Rosado JA, Salido GM, Gonzalez-Gallego J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol. 2009;50:872–882. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 310.Piccoli C, Scrima R, Quarato G, D’Aprile A, Ripoli M, Lecce L, Boffoli D, Moradpour D, Capitanio N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology. 2007;46:58–65. doi: 10.1002/hep.21679. [DOI] [PubMed] [Google Scholar]
- 311.Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004;32:2623–2631. doi: 10.1093/nar/gkh579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fossé P, Darlix JL. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006;34:2618–2633. doi: 10.1093/nar/gkl240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ivanyi-Nagy R, Lavergne JP, Gabus C, Ficheux D, Darlix JL. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 2008;36:712–725. doi: 10.1093/nar/gkm1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Sharma Kk, Didier P, Darlix JL, de Rocquigny H, Bensikaddour H, Lavergne JP, Pénin F, Lessinger JM, Mély Y. Kinetic analysis of the nucleic acid chaperone activity of the hepatitis C virus core protein. Nucleic Acids Res. 2010;38:3632–3642. doi: 10.1093/nar/gkq094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Sharma KK, de Rocquigny H, Darlix JL, Lavergne JP, Pénin F, Lessinger JM, Mély Y. Analysis of the RNA chaperoning activity of the hepatitis C virus core protein on the conserved 3’X region of the viral genome. Nucleic Acids Res. 2012;40:2540–2553. doi: 10.1093/nar/gkr1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Merola M, Brazzoli M, Cocchiarella F, Heile JM, Helenius A, Weiner AJ, Houghton M, Abrignani S. Folding of hepatitis C virus E1 glycoprotein in a cell-free system. J Virol. 2001;75:11205–11217. doi: 10.1128/JVI.75.22.11205-11217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Michalak JP, Wychowski C, Choukhi A, Meunier JC, Ung S, Rice CM, Dubuisson J. Characterization of truncated forms of hepatitis C virus glycoproteins. J Gen Virol. 1997;78(Pt 9):2299–2306. doi: 10.1099/0022-1317-78-9-2299. [DOI] [PubMed] [Google Scholar]
- 318.Wahid A, Helle F, Descamps V, Duverlie G, Penin F, Dubuisson J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J Virol. 2013;87:1605–1617. doi: 10.1128/JVI.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Brazzoli M, Helenius A, Foung SK, Houghton M, Abrignani S, Merola M. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology. 2005;332:438–453. doi: 10.1016/j.virol.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 320.Ortega-Atienza S, Lombana L, Gómez-Gutiérrez J, Yélamos B, Peterson DL, Gavilanes F. Production and characterization of the ectodomain of E2 envelope glycoprotein of hepatitis C virus folded in the presence of full-length E1 glycoprotein. Protein Expr Purif. 2014;104C:20–25. doi: 10.1016/j.pep.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 321.Vaney MC, Rey FA. Class II enveloped viruses. Cell Microbiol. 2011;13:1451–1459. doi: 10.1111/j.1462-5822.2011.01653.x. [DOI] [PubMed] [Google Scholar]
- 322.Huang ZS, Wang CC, Wu HN. HCV NS3 protein helicase domain assists RNA structure conversion. FEBS Lett. 2010;584:2356–2362. doi: 10.1016/j.febslet.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 323.Wölk B, Sansonno D, Kräusslich HG, Dammacco F, Rice CM, Blum HE, Moradpour D. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J Virol. 2000;74:2293–2304. doi: 10.1128/jvi.74.5.2293-2304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Donaldson EF, Harrington PR, O’Rear JJ, Naeger LK. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology. 2015;61:56–65. doi: 10.1002/hep.27375. [DOI] [PubMed] [Google Scholar]
- 325.Shiffman ML. What future for ribavirin? Liver Int. 2009;29 Suppl 1:68–73. doi: 10.1111/j.1478-3231.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- 326.Foster GR. Mutant Ninja viruses. Hepatology. 2015;61:421–423. doi: 10.1002/hep.27540. [DOI] [PubMed] [Google Scholar]
- 327.Hedskog C, Doehle B, Chodavarapu K, Gontcharova V, Crespo Garcia J, De Knegt R, Drenth JP, McHutchison JG, Brainard D, Stamm LM, et al. Characterization of hepatitis C virus intergenotypic recombinant strains and associated virological response to sofosbuvir/ribavirin. Hepatology. 2015;61:471–480. doi: 10.1002/hep.27361. [DOI] [PubMed] [Google Scholar]