

Abstract
Emerging integrative analysis of genomic and anatomical imaging data which has not been well developed, provides invaluable information for the holistic discovery of the genomic structure of disease and has the potential to open a new avenue for discovering novel disease susceptibility genes which cannot be identified if they are analyzed separately. A key issue to the success of imaging and genomic data analysis is how to reduce their dimensions. Most previous methods for imaging information extraction and RNA-seq data reduction do not explore imaging spatial information and often ignore gene expression variation at the genomic positional level. To overcome these limitations, we extend functional principle component analysis from one dimension to two dimensions (2DFPCA) for representing imaging data and develop a multiple functional linear model (MFLM) in which functional principal scores of images are taken as multiple quantitative traits and RNA-seq profile across a gene is taken as a function predictor for assessing the association of gene expression with images. The developed method has been applied to image and RNA-seq data of ovarian cancer and kidney renal clear cell carcinoma (KIRC) studies. We identified 24 and 84 genes whose expressions were associated with imaging variations in ovarian cancer and KIRC studies, respectively. Our results showed that many significantly associated genes with images were not differentially expressed, but revealed their morphological and metabolic functions. The results also demonstrated that the peaks of the estimated regression coefficient function in the MFLM often allowed the discovery of splicing sites and multiple isoforms of gene expressions.
Keywords: imaging, RNA-seq, imaging genomics, functional principal component analysis, functional linear model
INTRODUCTION
There is increasing consensus that imaging measures show closer associations with genomic variants and the penetrance of an individual genomic variant is expected to be higher at the imaging level than at the clinical diagnostic and outcome level. Imaging measures as an endophenotype have a higher power to identify genomic variants that significantly contribute to the development of diseases [1,2]. Integrated genomic and imaging data analysis is a new powerful approach used to uncover the individual variability and mechanism of disease development [3]. Both imaging and genomics generate a huge amount of data that present critical bottlenecks in their analysis. Despite its great success, integrative analysis of unprecedented high dimensional imaging and genomic data faces great conceptual and computational challenges [4].
A key issue to the success of imaging and genomic data analysis is how to reduce dimensions of both imaging and genomic data. Previously investigated methods for imaging information extraction include single region-of-interest (ROI) methods, voxelwise approaches, principal component analysis (PCA), singular value decomposition, self-organizing Map (SOM) and multidimensional scaling (MDS) [5]. However, these multivariate dimension reduction methods do not explore imaging spatial information. They take the set of spectral images as an unordered set of high dimensional pixels [6]. Spatial information is very important for image cluster and classification analysis. To overcome limitations of multivariate dimension reduction and to utilize spatial information of the image signal, we extend the widely used one dimensional functional principal component analysis (FPCA) [7] to high dimensional FPCA to extract imaging signals.
The traditional methods for assessing the relationship between gene expressions measured by microarray and phenotypes are linear regressions [8,9]. However, the rapidly developed next-generation sequencing (NGS) technologies have become the platform of choice for gene expression profiling. RNA-seq for expression profiling offers a comprehensive picture of the transcriptome, with less background noise and a wider dynamic range of expression [10]. Unlike microarrays for measuring gene expression, RNA-seq provides multiple layers of resolutions and transcriptome complexity: the expression at exon, SNP, and positional level, splicing, transcription start sites, polyadenylation sites, post-transcriptional RNA editing across the entire gene, and isoform and allele-specific expression [11]. The current linear regression for modeling association of gene expressions with phenotypes quantifies the expression level of a gene/transcript by a single number that summarizes all the reads mapped to that gene/transcript. A single number measuring gene expression level ignores gene expression variation at the genomic positions. Therefore, linear regression is appropriate for microarray expression data, but may not be good for RNA-seq data.
To overcome these limitations, we propose a multiple functional linear model (MFLM) in which functional principal component scores of images are taken as multiple quantitative traits and RNA-seq profile across a gene is taken as a function predictor for assessing the association of gene expression with imaging signals which can take gene splicing and expression variation at genomic positional levels into account.
RESULTS
To evaluate its performance, the proposed MFLM for integrative imaging and RNA-seq data analysis was applied to images and RNA-seq datasets of ovarian cancer (OV) and kidney renal clear cell carcinoma (KIRC) which were downloaded from the The Cancer Genome Atlas (TCGA) datasets. The ovarian cancer dataset consists of 231 tumor tissue samples with histology images and RNA-seq profiles of 16,598 genes (after quality control). The KIRC dataset consists of 188 (121 tumor and 67 normal tissue samples) with histology images and RNA-seq profiles of 16,775 genes (after quality control). RNA-seq data were created by Illumina HiSeq 2000 PE paired-end RNA sequencing. More detailed information can be downloaded from the TCGA website (http://cancergenome.nih.gov/).
The pathology images are used to study the manifestations of disease. Some tissue samples from the patients are obtained by either surgery, biopsy or autopsy. These tissues are either frozen or placed in formaldehyde for fixation which stabilizes the tissues to prevent decay. Then the fixed samples are sectioned into thin slices and stained with one or more dyes. Finally, the prepared pathology slides are placed under the optical microscope and captured by the charged-couple device (CCD) camera. The pathology images have the ability to identify the pathological change of the patient’s tissue at the cellular level such as the shape of the nucleus and the texture of the cell.
FPCA for imaging signal extraction
In our study, we compared our two dimensional FPCA with the traditional PCA by capturing space variation of image signals. To evaluate the performance of the two methods on image compression, we compared the original histology images and reconstructed images by two dimensional FPCA and PCA. The result clearly demonstrated that the reconstructed images by FPCA were much closer to the original images than that by PCA (Figure S1). In addition, 90.3% of the total imaging variation could be explained by the top 30 functional principal components, while only 63.6% of the total imaging variation was explained by the top 30 traditional principal components. Therefore, two dimensional FPCA is a better and more authentic image compression algorithm for image signal capturing, with minimal loss of information and fewer principal components usage than traditional PCA.
Behavior of the MFLM for integrative analysis of RNA-seq and imaging data
In the process of integrative analysis of RNA-seq and image data, histology image data were compressed with our proposed two dimensional FPCA, and the FPC scores were taken as phenotypes. We considered gene expression values at single-base resolution and represented the expression profile of a gene by a functional curve, called a “gene expression function”. The pipeline for RNA-seq data processing is given as follows. Bam files were obtained from the TCGA project and raw reads for each gene were extracted from Samtools [12] (revised version to recode the reads into binary data to decrease the storage memory). We used “easy RNASeq” to perform quantile normalization for normalizing the read counts and imputation [13]. We used the Karhunen-Loeve decomposition [7] to decompose the random gene expression function into orthogonal FPCs. The multiple FPC scores for imaging signal extraction were regressed on the FPC scores that were obtained from decomposition of the gene expression functions. In other words, we proposed to use MFLM for integrative analysis of RNA-seq and imaging data (Materials and Methods). Two FPCs that accounted for 81.7% and 88.6% variation of imaging signals for ovarian and KIRC, respectively, were selected as phenotypes. The number of selected FPCs for the RNA-seq which accounted for 95% of the variation of gene expression ranges from 2 to 60. P-values for declaring significant association after applying the Bonferroni correction for multiple tests in ovarian cancer and KIRC analysis were 3.012 × 10−6 and 2.98 × 10−6, respectively. To indirectly examine the validity of MFLM for assessing the association of gene expression with the histology images, we plotted a QQ plot of the test in the MFLM (Figure 1). The QQ plots clearly showed that the false positive rates of the MFLM for detection of the association of gene expression with histology images in both ovarian cancer and KIRC studies were controlled.
Figure 1. QQ plot for the KIRC dataset and Ovarian Cancer dataset.
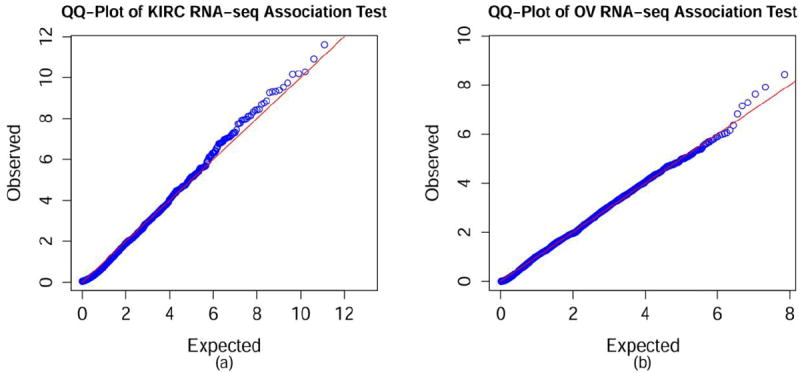
(A) QQ plot for the KIRC dataset. (B) QQ plot for the Ovarian Cancer dataset.
MFLM for integrative analysis of RNA-seq and histology images
Three statistical methods: MFLM with FPC scores as phenotypes, MFLM with image descriptors [14] as phenotypes and multivariate regression model with FPC score as phenotypes and a single gene expression value (level 3 in TCGA datasets) as a regressor were applied to the ovarian cancer and KIRC datasets. For the ovarian cancer dataset, MFLM with FPC scores as phenotypes, MFLM with image descriptors and multivariate regression identified 24, 2 and 0 genes whose expressions were associated with image signals, respectively. Similarly, for the KIRC dataset, MFLM with FPC scores as phenotypes, MFLM with image descriptors and multivariate linear model (MLM) identified 84, 6 and 1 genes whose expressions were associated with image signals, respectively. The results were summarized in Tables 1 and 2.
Table 1.
P-values of three statistics for testing association of expression with images in ovarian cancer study.
| Gene |
P-value
|
||
|---|---|---|---|
| MFLM_FPC | MFLM_Descriptor | MLM | |
| ZNF805 | 2.31E-10 | 0.434406 | 0.932869 |
| LOC653501 | 3.86E-09 | 0.024875 | 0.904242 |
| TMEM170B | 1.23E-08 | 0.006176 | 0.91104 |
| DRP2 | 2.38E-08 | 0.097565 | 0.589285 |
| OR6V1 | 5.27E-08 | 0.002066 | 0.176132 |
| GPR113 | 7.09E-08 | 3.67E-07 | 0.569642 |
| LOC389765 | 1.51E-07 | 0.076075 | NA* |
| ZNF484 | 4.47E-07 | 0.006771 | 0.934051 |
| DNAL1 | 7.00E-07 | 0.006636 | 0.859388 |
| ITGA10 | 8.72E-07 | 0.200773 | 0.6412 |
| NBEAL1 | 9.43E-07 | 0.007667 | 0.711715 |
| IBA57 | 1.03E-06 | 0.006602 | NA |
| C16orf52 | 1.13E-06 | 0.021002 | 0.905685 |
| PHKA1 | 1.31E-06 | 0.027968 | 0.715083 |
| PTPRG | 1.39E-06 | 0.949854 | 0.657859 |
| IFT88 | 1.64E-06 | 1.09E-05 | 0.810783 |
| PARD3B | 1.78E-06 | 0.889289 | 0.448784 |
| TRAPPC11 | 1.85E-06 | 0.367912 | NA |
| LIMD1 | 2.11E-06 | 0.468624 | 0.871478 |
| FAM73A | 2.13E-06 | 0.003969 | 0.927942 |
| CAPN14 | 2.45E-06 | 0.01782 | 0.479734 |
| CPEB3 | 2.55E-06 | 0.026235 | 0.987898 |
| CDCA2 | 2.80E-06 | 0.972605 | 0.374079 |
| PUS3 | 3.08E-06 | 0.78059 | 0.921797 |
NA: Expression (level 3) data were not available.
Table 2.
P-values of three statistics for testing association of expression with images in KIRC study.
| Gene |
P-value
|
Gene |
P-value
|
||||
|---|---|---|---|---|---|---|---|
| MFLM (FPC) | MFLM (Descriptor) | MLM | MFLM(FPC) | MFLM(Descriptor) | MLM | ||
| HELZ | 6.62E-16 | 6.08E-01 | 8.79E-01 | ZNF81 | 9.95E-08 | 2.06E-07 | 7.25E-01 |
| 9-Mar | 2.12E-15 | 1.02E-06 | 7.58E-01 | GAB2 | 1.04E-07 | 1.34E-02 | 6.38E-01 |
| MSH5-SAPCD1 | 8.98E-13 | 9.79E-03 | NA* | MMP24-AS1 | 1.29E-07 | 3.26E-06 | NA |
| SLC2A12 | 2.52E-12 | 9.94E-03 | 2.76E-08 | LOC647859 | 1.43E-07 | 8.56E-02 | 1.23E-03 |
| BRWD1 | 1.26E-11 | 5.61E-03 | 9.54E-01 | C2orf68 | 1.49E-07 | 4.83E-03 | 7.84E-01 |
| RFX7 | 5.29E-11 | 1.00E+00 | 9.58E-01 | SDR39U1 | 1.57E-07 | 8.83E-04 | 5.88E-01 |
| C22orf39 | 6.55E-11 | 1.29E-03 | 5.77E-01 | ZRANB3 | 1.66E-07 | 1.03E-03 | 9.59E-01 |
| NSD1 | 7.06E-11 | 1.67E-02 | 9.74E-01 | PSMC4 | 1.71E-07 | 1.39E-02 | 8.87E-01 |
| RTF1 | 1.82E-10 | 9.49E-01 | 8.58E-01 | FLJ12825 | 1.74E-07 | 1.39E-04 | 7.08E-01 |
| MBD5 | 3.00E-10 | 1.08E-04 | 9.33E-01 | ARHGEF11 | 2.26E-07 | 8.24E-03 | 8.55E-01 |
| ZSCAN16-AS1 | 4.16E-10 | 6.08E-02 | NA | LOC100289019 | 2.61E-07 | 8.50E-04 | NA |
| SESN1 | 4.84E-10 | 3.42E-01 | 6.71E-01 | SUFU | 2.79E-07 | 1.99E-01 | 5.84E-01 |
| ITGA9 | 5.12E-10 | 2.11E-02 | 9.52E-01 | ZNF555 | 3.75E-07 | 2.16E-02 | 3.75E-01 |
| PPM1K | 5.60E-10 | 1.48E-01 | 1.11E-04 | KHNYN | 3.85E-07 | 1.54E-01 | 4.62E-01 |
| USP42 | 1.39E-09 | 9.79E-01 | 9.06E-01 | ANKRD11 | 4.80E-07 | 1.00E+00 | 8.92E-01 |
| FAM47E-STBD1 | 1.77E-09 | 1.11E-02 | NA | BOLA2 | 4.82E-07 | 9.88E-02 | 8.33E-01 |
| ZNF710 | 2.05E-09 | 1.22E-01 | 9.82E-01 | BOLA2B | 4.82E-07 | 9.88E-02 | NA |
| TECPR2 | 3.59E-09 | 9.53E-04 | 5.63E-01 | SAPCD1 | 4.97E-07 | 4.24E-01 | NA |
| RASSF8-AS1 | 3.88E-09 | 3.08E-03 | NA | SLC9A4 | 6.26E-07 | 2.27E-02 | 1.87E-01 |
| CCDC93 | 4.04E-09 | 1.00E+00 | 9.45E-01 | CRYBG3 | 6.30E-07 | 5.18E-03 | 5.59E-02 |
| NAV2 | 4.90E-09 | 4.11E-02 | 1.17E-01 | SLC15A2 | 6.78E-07 | 1.18E-04 | 3.63E-05 |
| CYB5B | 6.75E-09 | 5.52E-04 | 7.11E-01 | BRD4 | 7.77E-07 | 5.46E-01 | 9.85E-01 |
| ANKRD17 | 7.55E-09 | 1.00E+00 | 4.47E-01 | ATP6V1C2 | 7.80E-07 | 2.86E-03 | 1.88E-01 |
| CCDC181 | 7.98E-09 | 5.24E-03 | NA | SMAD2 | 9.23E-07 | 9.38E-01 | 7.52E-01 |
| SPHK2 | 1.08E-08 | 3.26E-03 | 1.83E-02 | ST3GAL6 | 1.19E-06 | 5.01E-01 | 2.16E-01 |
| KCNN3 | 1.15E-08 | 9.47E-01 | 9.17E-01 | ZMIZ1 | 1.26E-06 | 3.75E-01 | 9.46E-01 |
| ZFYVE16 | 1.16E-08 | 1.98E-02 | 7.65E-01 | USP34 | 1.36E-06 | 1.74E-03 | 8.24E-01 |
| CMTM1 | 1.23E-08 | 9.99E-01 | 3.66E-01 | RALGAPA2 | 1.38E-06 | 3.48E-03 | 1.57E-01 |
| LINC00875 | 1.69E-08 | 1.00E+00 | NA | FRMD4A | 2.03E-06 | 5.14E-03 | 7.95E-01 |
| NOTCH1 | 1.81E-08 | 1.96E-02 | 1.80E-01 | PSMA5 | 2.12E-06 | 7.81E-02 | 9.35E-01 |
| BLZF1 | 1.87E-08 | 2.05E-03 | 8.94E-01 | RIPPLY1 | 2.18E-06 | 1.00E+00 | 3.89E-06 |
| CHD2 | 3.55E-08 | 1.28E-01 | 9.47E-01 | ERCC6 | 2.20E-06 | 1.21E-01 | 7.01E-01 |
| MTUS1 | 4.71E-08 | 8.63E-02 | 2.18E-01 | MINK1 | 2.29E-06 | 1.19E-02 | 9.14E-01 |
| REV3L | 4.96E-08 | 2.28E-02 | 9.59E-01 | DIP2C | 2.38E-06 | 2.51E-03 | 9.13E-01 |
| LRIG2 | 5.00E-08 | 6.78E-03 | 8.40E-01 | PHLDB2 | 2.51E-06 | 3.45E-03 | 6.34E-01 |
| DENND1C | 5.83E-08 | 9.41E-01 | 2.40E-01 | TBC1D24 | 2.54E-06 | 7.79E-01 | 2.13E-01 |
| TMEM50B | 6.77E-08 | 4.16E-03 | 8.31E-02 | APBA3 | 2.55E-06 | 7.20E-02 | 1.29E-01 |
| CELF1 | 7.92E-08 | 1.00E+00 | 9.13E-01 | TRAK1 | 2.60E-06 | 7.67E-01 | 1.53E-01 |
| ZSCAN20 | 8.41E-08 | 1.23E-06 | 9.37E-01 | DLC1 | 2.60E-06 | 7.23E-02 | 8.35E-01 |
| MINA | 8.76E-08 | 1.61E-03 | 4.43E-03 | NISCH | 2.62E-06 | 1.00E+00 | 2.28E-01 |
| C5AR2 | 9.01E-08 | 4.97E-02 | NA | CPEB3 | 2.64E-06 | 9.82E-01 | 4.91E-03 |
| SSH2 | 9.68E-08 | 1.00E-02 | 3.00E-01 | UFD1L | 2.94E-06 | 3.03E-03 | 7.78E-01 |
NA: Expression (level 3) data were not available.
Several remarkable features from these results were observed. First, the P-values calculated from the MFLM with FPC scores as phenotypes were much smaller than that calculated from the MFLM with image descriptors as phenotypes. Two methods assumed the same functional linear model (FLM) for RNA-seq data, but with a different approach to imaging signal reduction. The FPCA can reduce the dimensions of the imaging data more substantially than the traditional image descriptors. Therefore, the degrees of the test statistic in the MFLM with FPC score as phenotypes were much smaller than that in the MFLM with descriptors as phenotypes, which lead to the smaller P-values of the tests in the MFLM with the FPC scores as phenotypes. Second, we observed very few significant associations of the gene expression in the MLM. The MLM used the same FPCA for imaging data reduction, but model the gene expression level in a gene as a single value. The results demonstrated that the widely used single value representation of the expression level in the gene overlooked the expression variation across the gene, which led to large P-values of the tests. Third, expressions of genes which were associated with the imaging signal may or may not be differentially expressed (Table S1, Figure S2). In other words, significant association of gene expression with imaging signals can provide additional information which differential expressions cannot offer. For example, genes NOTCH1, ARHGEF11 and BRD4 that were associated with imaging signals, but not differentially expressed between tumor and normal tissues were reported to regulate interactions between physically adjacent cells and induce G2/M arrest and trigger apoptosis in renal cell carcinoma [15], associated with kidney injury in the Dahl salt-sensitive rat [16] and kidney disease [17]. Fourth, the MFLM with FPC scores as phenotypes could identify associated genes that showed alternative splicing expression patterns.
To illustrate this, we presented average expression of microtubule associated tumor suppressor 1 (MTUS1) in the KIRC study (Figure 2). So far, seven isoforms of MTUS1 have been discovered. We observed from Figure 2 that a higher expression level in exon 1, exon 2 and exon 15 in the normal samples than that in tumor samples, and alternatively spliced transcript variations encoding different isoforms between tumor and normal samples were substantial. This might indicate that splicing sites affect the tissue structure variation which was measured by imaging signals. MTUS1 is interacted with microtubules to control cellular architecture and organize microtubule arrays. Express variation of MTUS1 influences variation in microtubule structure, which in turn causes variation of histology images. Disruption of microtubule-dependent processes is involved in cancer development and metastasis [18]. We should point out that many methods and tools have already been proposed to analyze alternative splicing in RNA-seq between samples, such as DEXseq and DSGseq [19,20]. Fifth, imaging data convey relatively closer association with the disease than traditional phenotypes [21]. The genes significantly associated with imaging will have profound implication in cellular function and disease development.
Figure 2. Expression of gene MTUS1.
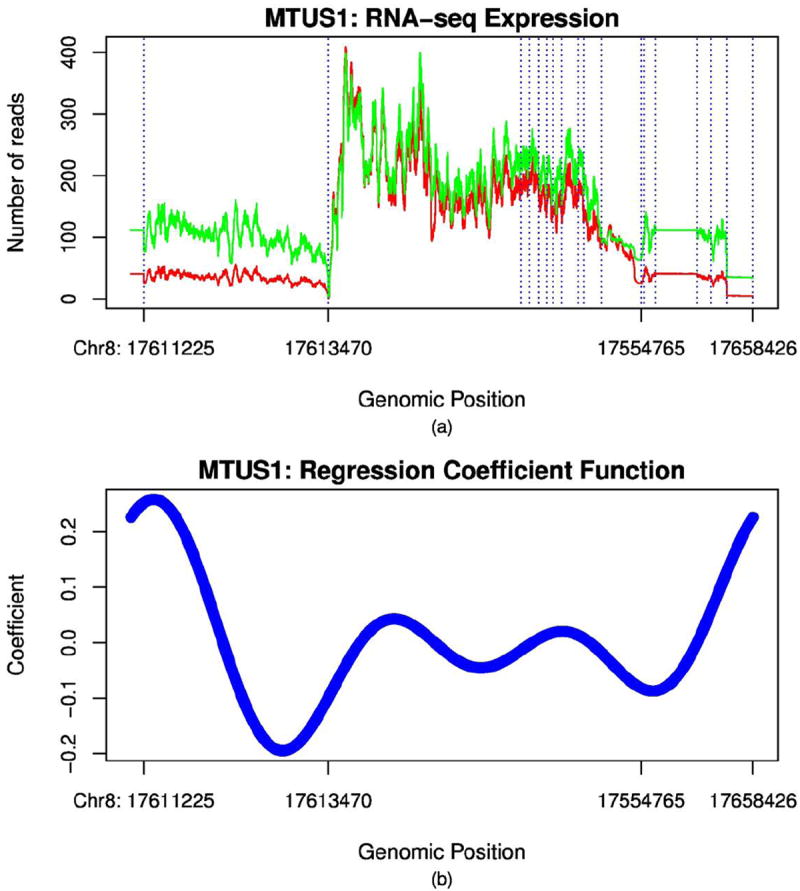
(A) RNA-seq curve of Gene MTUS1. Number of reads of gene MTUS1 as a function of the genomic position in the KIRC study, where the green line represents the gene expression profile of normal and the red line represents the gene expression profile of the cancer patient, dashed vertical lines represent exon recombination site where the splicing occurs. Introns were excluded in the plot. (B) Regression coefficient function of gene MTUS1 in the MFLM.
In the ovarian cancer study, among the 24 significantly associated genes with histology images, protein tyrosine phosphatase receptor type G (PTPRG) that regulate a variety of cellular processes including cell growth, differentiation, mitotic cycle, and oncogenic transformation, is a functional tumor suppressor gene and involved in ovarian tumorigenesis [22,23]. Cytoplasmic polyadenylation element binding protein 3 (CPEB3) that controls cell cycle progression, regulates senescence, establishes cell polarity, promotes tumorigenesis and metastasis [24], and plays a role in ovarian cancer development [25]. In the KIRC study, integrin, alpha 9 (ITGA9) that participates in regulation of myotube formation [26], is reported to be involved in renal carcinomas [27], NOTCH1 that regulates interactions between physically adjacent cells, is reported to trigger apoptosis in renal cell carcinoma [28], and Rho guanine nucleotide exchange factor (ARHGEF11) whose expression induces the reorganization of the actin cytoskeleton and the formation of membrane ruffling and filopodia, is associated with kidney injury [29] and key regulators of tumorigenesis [30].
Image associated gene form protein-protein interaction networks
A large proportion of genes whose expression variation was associated with imaging signal variation formed protein-protein interaction networks (Figure 3). In the ovarian cancer study, 30 out of 130 proteins significantly associated genes identified by false discovery rate 0.05 are interacted with each other to form a network. Hub gene SETDB1, encoding a histone methyltransferase in the network, is an oncogene and is involved in the development of several cancers [31]. Another hub gene Glul that catalyzes the synthesis of glutamine from glutamate and ammonia is involved in cell proliferation, inhibition of apoptosis, and cell signalling, and plays key roles in several cancers [32]. We also observed from the KIRC study that 28 out of the 84 proteins significantly associated genes with imaging signals are interacted to form a network, in which 10 genes are differentially expressed between tumor and normal tissues. A hub gene REV3L with 11 degrees in the network is the catalytic subunit of DNA translesion synthesis polymerase ζ. It involves a variety of DNA-damaging, genome stability, cytotoxicity, and resistance to chemotherapeutic agents. Surprisingly, although REV3L is not differentially expressed, it is reported to be associated with lung, breast, colon cancers and gliomas [33-35]. The interacted genes KCNN3, ANKRD17, BRD4, NOTCH1, SMAD2, ZMIZ1, UFD1L, and MINK1 are associated with various cancers [36-42]. Most of these genes are not differentially expressed, but are involved in the formation of cell and tissue structures. Their gene expression variations cause imaging signal variations and are thereby captured by integrative RNA-seq and imaging analysis.
Figure 3. Protein-protein interaction networks in the ovarian cancer study and KIRC study.
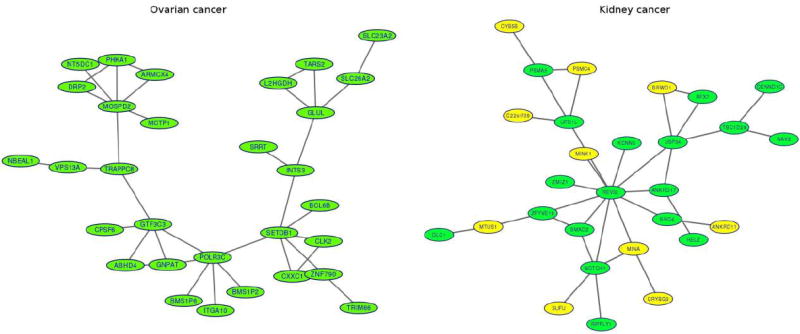
(A) Protein-protein interaction networks in the ovarian cancer study. Proteins of 30 out of 130 significantly associated genes identified by false discovery rate 0.05 are interacted with each other to form a network in the ovarian cancer study. (B) Protein-protein interaction networks in the KIRC study. Proteins of 28 out of 84 significantly associated genes with images are interacted each other in protein-protein database to form a network in the KIRC study where genes in yellow color were differentially expressed between tumor and normal tissues and dotted vertical lines denote location of splicing sites.
Image associated genes and alternative splicing
We performed FPCA on RNA-seq profiles of each image associated gene and obtained their FPC scores in the ovarian cancer and KIRC studies. Then, we used a hierarchical algorithm to cluster genes based on their FPC scores. The results were shown in Figure 4 and Figure S3. We used DAVID (the Database for Annotation, Visualization and Integrated Discovery) Bioinformatics Resources [43], to extract biological features/meaning of the genes including the role of the gene in metabolization and cell growth regulation as well the gene function location such as membrane, cytoplasm or nuclear. DAVID bioinformatics gene function annotation analysis showed that most image associated genes play important roles in alternative splicing (Figure 2 and Figure S2). We observed that the genes with the similar patterns of alternative splicing sites are grouped together (Figure S4). As Figure S4 showed, the four genes of TTC23, CPEB3, CAPN14 and PHKA1 were clustered together since all these genes have large numbers of splicing sites in the end of the genes (3′), and another four genes of CDCA2, TRAPPC11, PTPRG and ITGA10 were clustered together because all these genes have large number of splicing sites in the start of the genes (5′). There is increasing consensus that alternative splicing may affect large and conservative regions of the protein structures and often leads to changes in cell morphologies and phenotypes such as actin cytoskeleton remodeling, regulation of cell-cell junction formation and regulation of cell migrations [44,45]. Variations in alternative splicing of gene expression leads to variations in cell morphologies and phenotypes, thus influencing variations of imaging measures of the cells. This opens a new pathway to cancer development and progression.
Figure 4. Clusters of image associated genes in the ovarian cancer study by-means clustering algorithms. Dendrogram of the differential expressed or image-significant associated genes based on FPCA scores.
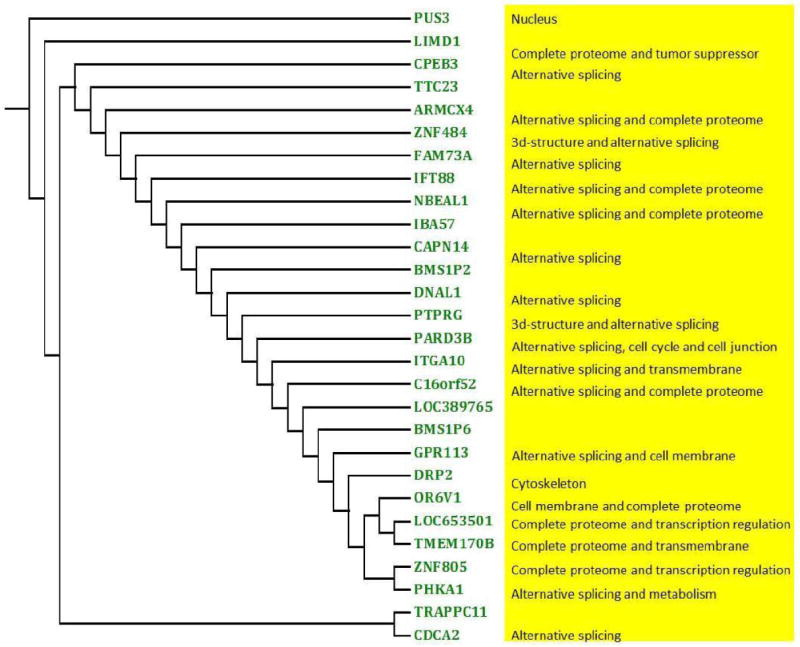
The result showed that the majority of these genes were related with alternative splicing.
Image associated genes and ingenuity pathway analysis
We used the Ingenuity Pathway Analysis (IPA) (Ingenuity® Systems, (http://www.ingenuity.com)) that is a web-based functional analysis tool for comprehensive omic data to study the function of genes significantly associated with image. In other words, the list of the identified genes which were significantly associated with imaging signals were input into the IPA. The IPA transforms a list of genes (with or without accompanying expression information) into a set of relevant networks based on extensive records maintained in the Ingenuity Pathways Knowledge Base (IPKB) [1,2]. This knowledge base has been abstracted into a large network, called the Global Molecular Network, composed of thousands of genes and gene products that interact with each other. In the ovarian cancer study, unloading 24 significantly associated genes with imaging into the IPA software, the identified network with the highest score 27 (P-value < 10−27) is the cancer network. We observed that 11 out of the 24 genes were included in the cancer network (Figure 5). Figure 5 showed that these genes are mostly regulated by miR-128, miR-92, miR-34 and miR-27a. These microRNAs play an important role in tumorigenesis and cancer development, especially in ovarian cancer [46-50]. Figure 5 also showed that the gene CPEB3 was a translation regulator and played an important role in the development of ovarian cancer [51]. In the KIRC study, IPA analysis of 84 genes that were significantly associated with imaging identified the network of cellular function and maintenance, hematological system development and function, and inflammatory response with the highest score 54 (Figure S5). We observed that 23 out of the 84 genes were included in the network. Genes SMAD2, NOTCH1, ERCC6 and NSD1 are transcription regulators that mediate multiple signaling pathways. It was reported that SMAD2 might serve as novel prognostic markers in clear cell renal cell carcinoma patients [52], NOTCH1 played an important role in oncogenesis of the KIRC [53]. Translation regulator CELF1 regulate pre-mRNA alternative splicing resulting in multiple transcript variants encoding different isoforms. CELF1 suppressed the proliferation of cancer [54]. Figure S5 also showed that most genes in this network interact with the Akt, p38MARK, PI3K, and NFkB complex. The PI3K/Akt, NF-κB, and MAPK pathways have been reported to be involved in nephrogenesis, and these pathways are activated in human renal cell carcinoma [55-58].
Figure 5. The top protein-protein physical/functional interaction network generated by Ingenuity Pathway Analysis for ovarian cancer.
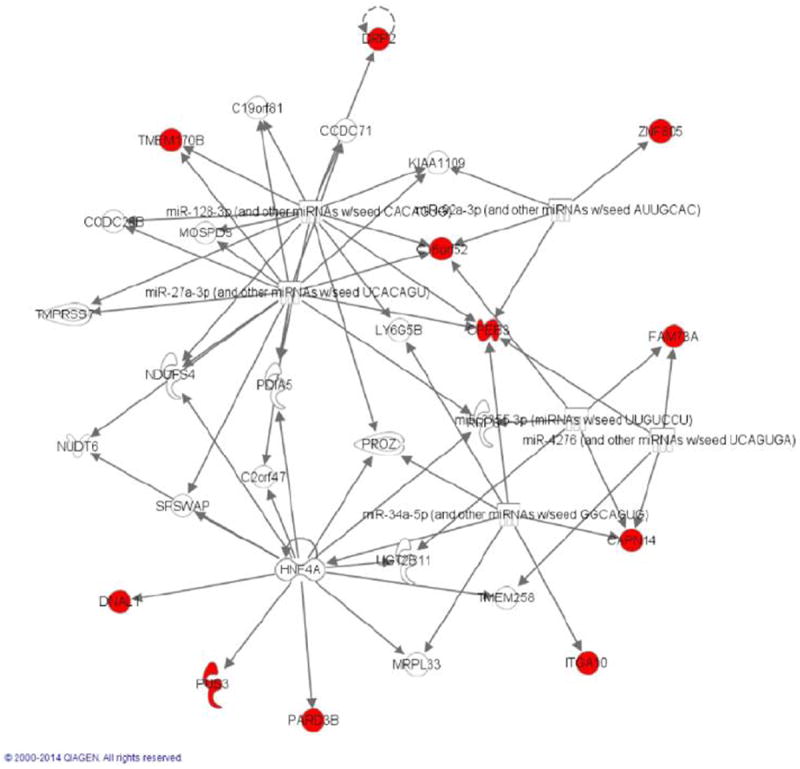
Genes in red node were identified as significantly associated genes with imaging signals in our study.
DISCUSSION
The current major focus of RNA-seq data analysis is to identify differentially expressed genes [59] and the major paradigm of RNA-seq data analysis is to test differences in gene expression level that is measured by a single value summarizing statistic. However, there is increasingly recognition that the differential expression feature of genes may not be a unique source to cause disease. Changes in cell morphologies and motility can also influence development and progression of diseases. In this paper we have presented a MFLM with FPC scores of imaging measures as phenotypes for the integrative analysis of imaging and RNA-seq data and offered a new alternative paradigm for RNA-seq data analysis. We have also shifted the paradigm of RNA-seq data analysis from the single value representation of gene expression to the random function representation of RNA-seq profiles which takes gene expression variation at the genomic positional level into account. Our study has made several remarkable findings.
The first finding is that imaging and RNA-seq analysis can detect cancer susceptibility genes that are not differentially expressed. Changes in cell morphologies, motility and phenotypes play important roles in the development and progression of the cancer. Genes causing these changes may not be differentially expressed between tumor and normal tissue samples and hence cannot be detected by gene differential expression analysis. The integrative analysis of imaging and RNA-seq data opens a new avenue for identifying cancer causing genes.
The second finding is that the function feature of image associated genes is alternative splicing. Surprisingly, we found that the peaks of regression coefficient functions in the MFLM of imaging and RNA-seq data analysis were located in the splicing sites. Alternative splicing often changes the protein structures, cell morphologies and phenotypes [44,45]. These changes generate variation of histology images of tumor tissues, which in turn provide information for discovery of image associated genes.
The third finding is that the widely used single value representation of the expression level in the gene overlooks the expression variation at the genomic positional level across the gene and hence has great limitations to identify image associated genes.
As demonstrated in the real data analysis, the MFLM showed great promise as a tool for integrative analysis of imaging and RNA-seq data. However, to date, very few integrative analyses of imaging and RNA-seq data have been performed. The results presented in this paper are among the first such studies and hence are considered preliminary. The number of selected orthogonal basis functions in the expansion of RNA-seq function will influence the performance of the integrative analysis of imaging and RNA-seq data. Genome-wide imaging and RNA-seq data analysis still pose great challenges. The main purpose of this paper is to stimulate discussion on the optimal strategies for genome-wide imaging and RNA-seq data analysis.
MATERIALS AND METHODS
Two dimensional functional principal component analysis
One dimensional functional principal component analysis (FPCA) has been well developed (7). Now we extend one dimensional FPCA to two dimensional FPCA. Consider a two dimensional region. Let s and t denote coordinates in the s axis and t axis, respectively. Let x(s, t) be a centered image signal located at s and t of the region. The signal x(s, t) is a function of locations s and t.
Consider a linear combination of functional values:
where β(s, t) is a weight function. To capture the variations in the random functions, we chose weight function β(s, t) to maximize the variance of f, which, after imposing a constraint to make the solution unique, leads to the following optimization problem:
| [1] |
where R(s1, t1, s2, t2,) = cov(x(s1, t1), x(s2, t2)) is the covariance function of the image signal function x(s, t). By variation calculus [60], we obtain the eigenequation as a solution to the optimization problem [1]:
| [2] |
for an appropriate eigenvalue λ, where βj (s, t) is an eigenfunction. The random functions xi (s, t) can be expanded in terms of eigenfunctions as
| [3] |
where , i = 1,…,N, j = 1,…,J are FPC scores (See Supplementary Materials 1).
Multivariate functional linear model for integrative analysis of imaging and RNA-seq data
We take K FPC scores as K quantitative traits. Assume that n individuals are sampled. Let yik, k = 1,2,…, K, be K trait values of the i -th individual. Consider a genomic region [a, b]. Let xi(t) be a RNA-seq profile, the number of reads as a function of the genomic position t of the i-th individual defined in the regions [a, b]. The multivariate functional linear model (MFLM) for integrative analysis of imaging and RNA-seq data can be defined as
| [4] |
where α0k is an overall mean, αk (t) is a regression coefficient function for the k -th trait, k = 1, …, K, εik are independent and identically distributed normal variables with mean of zero and covariance matrix Σ.
We assume that both trait values and RNA-seq profiles are centered. The RNA-seq profiles xi (t) are expanded in terms of the orthonormal basis function as:
| [5] |
where φj(t) are sequences of the orthonormal basis functions. Substituting equation [5] into equation [4], we obtain
| [6] |
where . The parameters αkj are referred to as genetic additive effect scores for the k -th trait.
Equation [6] can be rewritten in a matrix form:
The standard least square estimators of α and the variance covariance matrix Σ are given by
Denote the matrix (ξT ξ)−1 ξT by A. Then, the estimator of the parameter α is given by
The variance-covariance matrix of the estimator of the parameter α is given by
| [7] |
An essential problem in the QTL analysis or in the integrative analysis of imaging and RNA-seq data is to test the association of a gene with imaging phenotype. Formally, we investigate the problem of testing the following hypothesis:
which is equivalent to testing the hypothesis:
Define the test statistic for testing the association of a gene with K quantitative traits as
| [8] |
Then, under the null hypothesis H0 : α = 0, T is asymptotically distributed as a central or distribution if J components are taken in the expansion equation [5] (Supplementary Materials 2).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The project described was supported by grants 1R01AR057120–01, 1R01HL106034-01 and 1R01 MH101054 from the National Institutes of Health, NHLBI and NIMH, respectively. The authors wish to acknowledge the contributions of the research institutions, study investigators, field staff and study participants in creating the TCGA datasets for biomedical research.
Footnotes
SUPPLEMENTARY MATERIALS
The supplementary materials can be found online with this article at DOI 10.1007/s40484-015-0048-8.
Junhai Jiang, Nan Lin, Shicheng Guo, Jinyun Chen and Momiao Xiong declare that they have no conflict of interest.
COMPLIANCE WITH ETHICS GUIDELINES
Junhai Jiang, Nan Lin, Shicheng Guo, Jinyun Chen and Momiao Xiong conformed to the Helsinki Declaration of 1875, as revised in 2000(5) concerning Human and Animal Rights, and that they followed out policy concerning Informed Consent as shown on Springer.com.
References
- 1.Hibar DP, Kohannim O, Stein JL, Chiang MC, Thompson PM. Multilocus genetic analysis of brain images. Front Genet. 2011;2:73. doi: 10.3389/fgene.2011.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J, Calhoun VD. A review of multivariate analyses in imaging genetics. Front Neuroinform. 2014;8:29. doi: 10.3389/fninf.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stingo FC, Guindani M, Vannucci M, Calhoun VD. An integrative Bayesian modeling approach to imaging genetics. J Am Stat Assoc. 2013;108:876. doi: 10.1080/01621459.2013.804409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chi EC, Allen GI, Zhou H, Kohannim O, Lange K, Thompson PM. Imagine genetics via sparse canonical correlation analysis. Proceedings / IEEE International Symposium on Biomedical Imaging: from nano to macro IEEE International Symposium on Biomedical Imaging; 2013. pp. 740–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burges CJC. Dimension reduction: A guided tour. Found Trends Mach Learn. 2010;2:275–365. doi: 10.1561/2200000002. [DOI] [Google Scholar]
- 6.Gupta MR, Jacobson NNP. Wavelet principal component analysis and its application to hyperspectral images. In IEEE Int Conf Image Processing. 2006:1585–1588. [Google Scholar]
- 7.Ramsay JO, Silverman BW. Functional Data Analysis. 2. Heidelberg: Springer; 2005. pp. 147–172. [Google Scholar]
- 8.Ray M, Zhang W. Integrating gene expression and phenotypic information to analyze Alzheimer’s disease. J Alzheimers Dis. 2009;16:73–84. doi: 10.3233/JAD-2009-0917. [DOI] [PubMed] [Google Scholar]
- 9.Wu T, Sun W, Yuan S, Chen CH, Li KC. A method for analyzing censored survival phenotype with gene expression data. BMC Bioinformatics. 2008;9:417. doi: 10.1186/1471-2105-9-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Z, Zhu Y. Systematic comparison of RNA-Seq normalization methods using measurement error models. Bioinformatics. 2012;28:2584–2591. doi: 10.1093/bioinformatics/bts497. [DOI] [PubMed] [Google Scholar]
- 11.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delhomme N, Padioleau I, Furlong EE, Steinmetz LM. easyRNASeq: a bioconductor package for processing RNA-Seq data. Bioinformatics. 2012;28:2532–2533. doi: 10.1093/bioinformatics/bts477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowe DG. Object recognition from local scale-invariant features. The Proceedings of the Seventh IEEE International Conference on Computer Vision; 1999. pp. 1150–1157. [Google Scholar]
- 15.Wu K, Zhang L, Lin Y, Yang K, Cheng Y. Inhibition of γ-secretase induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Oncol Lett. 2014;8:55–61. doi: 10.3892/ol.2014.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams JM, Johnson AC, Stelloh C, Dreisbach AW, Franceschini N, Regner KR, Townsend RR, Roman RJ, Garrett MR. Genetic variants in Arhgef11 are associated with kidney injury in the Dahl salt-sensitive rat. Hypertension. 2012;60:1157–1168. doi: 10.1161/HYPERTENSIONAHA.112.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, Rusinova E, Gerona-Nevarro G, Moshkina N, Joshua J, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287:28840–28851. doi: 10.1074/jbc.M112.359505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez P, Tirnauer JS. Tumor suppressor interactions with microtubules: keeping cell polarity and cell division on track. Dis Model Mech. 2010;3:304–315. doi: 10.1242/dmm.004507. [DOI] [PubMed] [Google Scholar]
- 19.Liu R, Loraine AE, Dickerson JA. Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems. BMC Bioinformatics. 2014;15:364. doi: 10.1186/s12859-014-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang W, Qin Z, Feng Z, Wang X, Zhang X. Identifying differentially spliced genes from two groups of RNA-seq samples. Gene. 2013;518:164–170. doi: 10.1016/j.gene.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 21.Rasetti R, Weinberger DR. Intermediate phenotypes in psychiatric disorders. Curr Opin Genet Dev. 2011;21:340–348. doi: 10.1016/j.gde.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Della Peruta M, Martinelli G, Moratti E, Pintani D, Vezzalini M, Mafficini A, Grafone T, Iacobucci I, Soverini S, Murineddu M, et al. Protein tyrosine phosphatase receptor type γ is a functional tumor suppressor gene specifically downregulated in chronic myeloid leukemia. Cancer Res. 2010;70:8896–8906. doi: 10.1158/0008-5472.CAN-10-0258. [DOI] [PubMed] [Google Scholar]
- 23.van Niekerk CC, Poels LG. Reduced expression of protein tyrosine phosphatase gamma in lung and ovarian tumors. Cancer Lett. 1999;137:61–73. doi: 10.1016/S0304-3835(98)00344-9. [DOI] [PubMed] [Google Scholar]
- 24.D’Ambrogio A, Nagaoka K, Richter JD. Translational control of cell growth and malignancy by the CPEBs. Nat Rev Cancer. 2013;13:283–290. doi: 10.1038/nrc3485. [DOI] [PubMed] [Google Scholar]
- 25.Hansen CN, Ketabi Z, Rosenstierne MW, Palle C, Boesen HC, Norrild B. Expression of CPEB, GAPDH and U6snRNA in cervical and ovarian tissue during cancer development. APMIS. 2009;117:53–59. doi: 10.1111/j.1600-0463.2008.00015.x. [DOI] [PubMed] [Google Scholar]
- 26.Ooishi R, Shirai M, Funaba M, Murakami M. Microphthalmia-associated transcription factor is required for mature myotube formation. Biochim Biophys Acta. 2012;1820:76–83. doi: 10.1016/j.bbagen.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Senchenko VN, Liu J, Loginov W, Bazov I, Angeloni D, Seryogin Y, Ermilova V, Kazubskaya T, Garkavtseva R, Zabarovska VI, et al. Discovery of frequent homozygous deletions in chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast carcinomas. Oncogene. 2004;23:5719–5728. doi: 10.1038/sj.onc.1207760. [DOI] [PubMed] [Google Scholar]
- 28.Wu K, Zhang L, Lin Y, Yang K, Cheng Y. Inhibition of γ-secretase induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Oncol Lett. 2014;8:55–61. doi: 10.3892/ol.2014.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams JM, Johnson AC, Stelloh C, Dreisbach AW, Franceschini N, Regner KR, Townsend RR, Roman RJ, Garrett MR. Genetic variants in Arhgef11 are associated with kidney injury in the Dahl salt-sensitive rat. Hypertension. 2012;60:1157–1168. doi: 10.1161/HYPERTENSIONAHA.112.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu J, Wu X, Dong Q, Romeo MJ, Lin X, Gutkind JS, Berman DM. A nonsynonymous single-nucleotide polymorphism in the PDZ-Rho guanine nucleotide exchange factor (Ser1416Gly) modulates the risk of lung cancer in Mexican Americans. Cancer. 2006;106:2707–2715. doi: 10.1002/cncr.21944. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Paredes M, Martinez de Paz A, Simó-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vázquez-Cedeira M, Lazo PA, Carneiro F, et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014;33:2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou C, Chen H, Han L, Wang A, Chen LA. Identification of featured biomarkers in different types of lung cancer with DNA microarray. Mol Biol Rep. 2014;41:6357–6363. doi: 10.1007/s11033-014-3515-9. [DOI] [PubMed] [Google Scholar]
- 33.Knobel PA, Kotov IN, Felley-Bosco E, Stahel RA, Marti TM. Inhibition of REV3 expression induces persistent DNA damage and growth arrest in cancer cells. Neoplasia. 2011;13:961–970. doi: 10.1593/neo.11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci USA. 2010;107:20786–20791. doi: 10.1073/pnas.1011409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varadi V, Bevier M, Grzybowska E, Johansson R, Enquist K, Henriksson R, Butkiewicz D, Pamula-Pilat J, Tecza K, Hemminki K, et al. Genetic variation in genes encoding for polymerase ζ subunits associates with breast cancer risk, tumour characteristics and survival. Breast Cancer Res Treat. 2011;129:235–245. doi: 10.1007/s10549-011-1460-z. [DOI] [PubMed] [Google Scholar]
- 36.Chantôme A, Potier-Cartereau M, Clarysse L, Fromont G, Marionneau-Lambot S, Guéguinou M, Pagès JC, Collin C, Oullier T, Girault A, et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013;73:4852–4861. doi: 10.1158/0008-5472.CAN-12-4572. [DOI] [PubMed] [Google Scholar]
- 37.Ioana M, Angelescu C, Burada F, Mixich F, Riza A, Dumitrescu T, Alexandru D, Ciurea T, Cruce M, Saftoiu A. MMR gene expression pattern in sporadic colorectal cancer. J Gastrointestin Liver Dis. 2010;19:155–159. [PubMed] [Google Scholar]
- 38.Yan Y, Yang FQ, Zhang HM, Li J, Li W, Wang GC, Che JP, Zheng JH, Liu M. Bromodomain 4 protein is a predictor of survival for urothelial carcinoma of bladder. Int J Clin Exp Pathol. 2014;7:4231–4238. [PMC free article] [PubMed] [Google Scholar]
- 39.Bokhari AA, Lee LR, Raboteau D, Hamilton CA, Maxwell GL, Rodriguez GC, Syed V. Progesterone inhibits endometrial cancer invasiveness by inhibiting the TGFβ pathway. Cancer Prev Res (Phila) 2014;7:1045–1055. doi: 10.1158/1940-6207.CAPR-14-0054. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, Shin A, Jee SH, Kim DH, Cai Q, et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet. 2014;46:533–542. doi: 10.1038/ng.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Ran ZH, Tong JL, Nie F, Zhu MM, Xu XT, Xiao SD. RNA interference (RNAi) of Ufd1 protein can sensitize a hydroxycamptothecin-resistant colon cancer cell line SW1116/HCPT to hydroxycamptothecin. J Dig Dis. 2011;12:110–116. doi: 10.1111/j.1751-2980.2011.00478.x. [DOI] [PubMed] [Google Scholar]
- 42.Hwang J, Pallas DC. STRIPAK complexes: structure, biological function, and involvement in human diseases. Int J Biochem Cell Biol. 2014;47:118–148. doi: 10.1016/j.biocel.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landau WM, Liu P. Dispersion estimation and its effect on test performance in RNA-seq data analysis: a simulation-based comparison of methods. PLoS One. 2013;8:e81415. doi: 10.1371/journal.pone.0081415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birzele F, Csaba G, Zimmer R. Alternative splicing and protein structure evolution. Nucleic Acids Res. 2008;36:550–558. doi: 10.1093/nar/gkm1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 47.Li M, Fu W, Wo L, Shu X, Liu F, Li C. miR-128 and its target genes in tumorigenesis and metastasis. Exp Cell Res. 2013;319:3059–3064. doi: 10.1016/j.yexcr.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 48.Xu L, Xiang J, Shen J, Zou X, Zhai S, Yin Y, Li P, Wang X, Sun Q. Oncogenic MicroRNA-27a is a target for genistein in ovarian cancer cells. Anticancer Agents Med Chem. 2013;13:1126–1132. doi: 10.2174/18715206113139990006. [DOI] [PubMed] [Google Scholar]
- 49.Ohyagi-Hara C, Sawada K, Kamiura S, Tomita Y, Isobe A, Hashimoto K, Kinose Y, Mabuchi S, Hisamatsu T, Takahashi T, et al. miR-92a inhibits peritoneal dissemination of ovarian cancer cells by inhibiting integrin α5 expression. Am J Pathol. 2013;182:1876–1889. doi: 10.1016/j.ajpath.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 50.Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 2010;16:1119–1128. doi: 10.1158/1078-0432.CCR-09-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansen CN, Ketabi Z, Rosenstierne MW, Palle C, Boesen HC, Norrild B. Expression of CPEB, GAPDH and U6snRNA in cervical and ovarian tissue during cancer development. APMIS. 2009;117:53–59. doi: 10.1111/j.1600-0463.2008.00015.x. [DOI] [PubMed] [Google Scholar]
- 52.Park JH, Lee C, Suh JH, Chae JY, Moon KC. Nuclear expression of Smad proteins and its prognostic significance in clear cell renal cell carcinoma. Hum Pathol. 2013;44:2047–2054. doi: 10.1016/j.humpath.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 53.Wu K, Zhang L, Lin Y, Yang K, Cheng Y. Inhibition of γ-secretase induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Oncol Lett. 2014;8:55–61. doi: 10.3892/ol.2014.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu LN, Xue YJ, Zhang LJ, Ma XM, Chen JF. Si-RNA mediated knockdown of CELF1 gene suppressed the proliferation of human lung cancer cells. Cancer Cell Int. 2013;13:115. doi: 10.1186/1475-2867-13-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sourbier C, Lindner V, Lang H, Agouni A, Schordan E, Danilin S, Rothhut S, Jacqmin D, Helwig JJ, Massfelder T. The phosphoinositide 3-kinase/Akt pathway: a new target in human renal cell carcinoma therapy. Cancer Res. 2006;66:5130–5142. doi: 10.1158/0008-5472.CAN-05-1469. [DOI] [PubMed] [Google Scholar]
- 56.Sourbier C, Danilin S, Lindner V, Steger J, Rothhut S, Meyer N, Jacqmin D, Helwig JJ, Lang H, Massfelder T. Targeting the nuclear factor-kappaB rescue pathway has promising future in human renal cell carcinoma therapy. Cancer Res. 2007;67:11668–11676. doi: 10.1158/0008-5472.CAN-07-0632. [DOI] [PubMed] [Google Scholar]
- 57.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, Zhang ZF, VandenBeldt K, Duesbery NS, Resau JH, et al. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008;68:81–88. doi: 10.1158/0008-5472.CAN-07-5311. [DOI] [PubMed] [Google Scholar]
- 58.Dormoy V, Danilin S, Lindner V, Thomas L, Rothhut S, Coquard C, Helwig JJ, Jacqmin D, Lang H, Massfelder T. The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol Cancer. 2009;8 doi: 10.1186/1476-4598-8-123. 123.2803450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sagan H. Introduction to the Calculus of Variations. New York: Dover Publications, Inc; 1969. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.