


Abstract
The potential roles of the numerous repetitive elements found in the genomes of multi-cellular organisms remain speculative. Several studies have suggested a role in stabilizing specific 3D genomic contacts. To test this hypothesis, we exploited inter-chromosomal contacts frequencies obtained from Hi-C experiments and show that the folding of the human, mouse and Drosophila genomes is associated with a significant co-localization of several specific repetitive elements, notably many elements of the SINE family. These repeats tend to be the oldest ones and are enriched in transcription factor binding sites. We propose that the co-localization of these repetitive elements may explain the global conservation of genome folding observed between homologous regions of the human and mouse genome. Taken together, these results support a contribution of specific repetitive elements in maintaining and/or reshaping genome architecture over evolutionary times.
INTRODUCTION
A significant fraction of the human genome is highly repetitive, with over two-thirds of the sequence consisting of families of repeats, also termed repetitive elements (1). Debates regarding potential in vivo functions of these elements have been long standing. Controversial references to ‘junk’ or ‘selfish’ DNA were put forward early on, implying that repetitive DNA segments are remainders from past evolution or autonomous self-replicating sequences hacking the cell machinery to proliferate, respectively (2–4). References to genomic ‘dark matter’ have also been used, by analogy to physics where this term designates an element of major importance in shaping our world but not yet fully understood. Concomitantly to the advent of the genomic era, the influence of repetitive elements on DNA-related metabolic processes as well as on genome evolution has been increasingly investigated (5–7), although the debate remains open regarding the overall functional relevance of these elements (8,9).
Repetitive elements found in eukaryotic genomes fall into different classes, depending on their mode of multiplication and/or structure. The disposition of repetitive elements consists either in arrays of tandemly repeated sequences, or in repeats dispersed throughout the genome (for review see 10). Tandem repeats consist mostly of satellite DNA, a structure made of multiple adjacent occurrences of a DNA motif. Satellite repeats are localized preferentially at centromeres, telomeres and within heterochromatic regions. Dispersed repeats consist principally in transposable elements (TEs), ranging in sizes from a hundred to a few thousands base pairs. TEs have the ability to move and eventually multiply within genomes, and two main classes can be distinguished. On one hand, retrotransposons comprise the Long and Short Interspersed Nuclear Element (LINEs and SINEs, respectively) and Long-Terminal Repeats (LTR) families. Retrotransposons of all three families can be transcribed, and the resulting RNA molecule subsequently reverse transcribed into DNA by a reverse transcriptase often encoded by the TE itself. The DNA form of the retrotransposon can then be re-integrated into the genome, resulting in a ‘copy and paste’-like dissemination mechanism. On the other hand, DNA transposons move principally through ‘cut and paste’-like mechanisms catalyzed by transposase enzymes and do not involve an RNA intermediate.
Originally discovered by Barbara McClintock (11), TEs displacements have been increasingly recognized as a potential source of genetic variation and regulation. Models aiming at bridging gene co-regulation with the scattering of putative regulatory sequences within the genome were developed early on (12). Today, comparative genomics studies have reinforced the idea that TEs have, or had, the ability to reshape gene regulatory networks of vertebrate's genomes over evolutionary times (6,9,13,14). Interestingly, it was recently suggested that Alu and MIR elements, both members of the SINE family, could act as regulatory sequences such as enhancers (15,16).
Together with these regulatory roles, a structural role of repeated DNA in shaping the 3D folding of genomes has also been proposed (17,18). This hypothesis is only supported by a limited set of experimental evidence, such as the formation of TE mediated chromatin loops in mammals (19), Drosophila (20) and fission yeast (21,22).
In recent years, the development of genomic derivatives of chromosome conformation capture experiments (Hi-C; 23,24) has led to the generation of contact maps describing the average contact frequencies between all DNA regions of a genome from a cell population. It is generally admitted that the contact frequencies of non-adjacent chromatin regions, as quantified through a genomic 3C experiment, reflect their relative average proximity within the nuclear volume, hence the population average genomic organization.
In this analysis, we took advantage of recently published contact maps of three different metazoans genomes to investigate further the potential influence of repetitive elements on the 3D folding of genomes (25–28). We show that in the human, mouse and fly genomes, several classes of repetitive elements present a high tendency for co-localization within the nuclear space. It appears that the identified repeats tend to be evolutionary old and enriched with transcription factor binding sites (TFBS). We also show that the 3D organization of syntenic blocks in mammalian genome is conserved between human and mouse. Higher contact frequencies between distant regions correlate with enrichment at these positions for retro-transposons families that are evolutionary close. This effect seems globally conserved through cell differentiation, with only local 3D reorganization.
MATERIALS AND METHODS
The pipeline we used is represented on Figure 1. Hi-C reads from different raw datasets (Supplementary Table S1 sheet H), of various sizes, experimental approaches and laboratories were aligned using Bowtie 2 in very sensitive mode (29). We used an iterative alignment procedure similar to the one presented in (30). To avoid misalignments over repetitive regions, two different strategies were designed (Figure 1A). The first and simpler approach consisted in applying a stringent mapping quality filtering while discarding ambiguous mapping results. We used a threshold of 40 for the mapping quality, which for instance retained ∼72% of the reads of the human genome datasets. It should be noted that even if a read is overlapping partially with a repeat element referenced in UCSC server it remains possible to map it unambiguously thanks to the differences between the regions neighboring the repeat. The second strategy was much more stringent, since only the reads that did not overlap with any of the repeat elements, were retained for further analysis (31). This procedure only kept ∼ 23% of the reads for the human genome datasets. Only inter-chromosomal contacts, defined as a pair of uniquely mapped reads on two different chromosomes, were kept for the analysis in order to avoid artifacts that could arise from the correlated distributions of repeated elements along the chromosomes.
Figure 1.

(Color online) Most important steps of the pipeline to detect repetitive elements presenting a significant 3D co-localization score (CS). (A) Illustration of the two different strategies of alignment used for the present analysis. The first strategy consists in keeping all mappings with a Mapping Quality above a certain threshold. The second strategy is much more stringent and keeps only reads that do not overlap any sequences referenced in the RepeatMasker track of UCSC. (B) Flow-chart describing the main steps of our analysis. (C) Illustration of the CS computation: alongside the matrix of Hi-C contacts between human chromosomes 1 and 2 is aligned the repeat density profile. The black lines on the repeats density profiles represent the threshold above which the bin is considered as enriched with the repeat. The CS is then the average of all matrix elements Mij for which bins i and j are enriched with the repeat. These elements are highlighted in purple on the contacts map.
Normalization of chromosomal contact maps
The human, mouse and Drosophila genomes were binned into sections of 10 or 100 consecutive restriction fragments (resulting in to bins with average sizes of 30 or 300 kb; see Supplementary Figure S1). Matrices were processed using the sequential component normalization procedure (SCN) described in (32), a balancing procedure similar to the iterative normalization described in (30). Correlation matrices were computed so that each element Cij is the Pearson correlation coefficient between the lines i and j of the original contact matrix M.
Genomic annotation of repetitive elements and transcription factors binding sites
The positions of the repeated sequences for each subfamily were retrieved from the UCSC server (RepeatMasker, hg19 assembly). Some repeats have many occurrences along the genome (e.g. AluJb), resulting in most of the bins defined above harboring at least one occurrence. In such a case, only bins in which the repeat is over-represented were considered. More precisely, a uniform law along the genome was considered as a null model, bins with P-value < 0.05 were kept (we used the binomial law of libRmath). For instance, the AluJb subfamily has 144 945 occurrences in the human genome, distributed over 46 786 bins, some being highly enriched in repeats while others containing only a couple. Only the bins containing five or more occurrences were conserved for the computation of CS, representing 9204 bins in total (see Supplementary Figure S1C). This filtering was applied to 196 subfamilies of repeats for which the number of occurrences within the genome was >5000. On the opposite, the 388 bins containing an instance of the LTR13 subfamily were kept, since this repeat is only present as 480 occurrences within the genome (Supplementary Figure S1C). Moreover, most of the bins in the genome are enriched with several different repeat subfamilies (average: 13.6, see Supplementary Figure S1B).
A similar analysis was performed for transcription factors and histone modifications (using Peaks files—that correspond to regions of enriched signal in the Chip-seq experiments—from UCSC server see Supplementary Table S1). For the set of tRNA genes, we used the track tRNA genes of UCSC. For the nucleolus-associated chromatin domains (NADs), we used the positions identified in (33) (adapted to the hg19 assembly, liftover tool from UCSC).
Computation of a co-localization score (CS)
We computed the co-localization score (CS) for each feature A (A being a DNA repeat or a TFBS) as the average inter-chromosomal contact frequency between all bins that contain A.
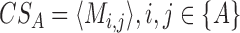 |
Statistical significance of co-localization scores
The statistical significance of the CS of each feature of interest was assessed using a random sampling method (34). Since genome folding as determined with Hi-C is known to be influenced by specific 1D annotations (either for technical or biological reasons), the pools of bins from which these random sets were generated were carefully chosen according to features that were already known to correlate with chromosome organization. More precisely, the analysis was done using three different null models. The first null model takes into account the GC content of the bins, the second one the sequencing coverage in the Hi-C experiment, and the third one the distribution of the bins on different chromosomes. The GC content affects the density in restriction fragments and is probably the most obvious source of biases in Hi-C experiments. The GC content is also highly correlated with the two-compartment (active and inactive chromatin) organization of the human genome (30). For the coverage null model, we choose a random set of bins conserving the Hi-C coverage distribution of the group of interest (which alleviate potential biases due to the Hi-C experiment itself). For the chromosomes distribution null model, the number of bins belonging to each chromosome is conserved.
That way, we generated 1000 random sets of bins under each different null model to which the actual CS could be compared.
We then fitted a log-normal law to the distribution of CS of the random sets obtained. The parameters extracted from the fit allow us to assign a P-value for the CS of each group (see Supplementary Figure S2 for two representative examples).
Starting with a desired significance level of 0.01 and taking into account the fact that we do multiple testing (corresponding to the 1395 repetitive elements tested), with the Bonferroni correction, we put the cutoff of P-value at 10e-05 (0.01/1395 ∼ 10e-05) in our detection of significance of CSs.
A negative control was used to check that this threshold was relevant (see Supplementary Figure S3, left) which corresponds to the positions of repeated elements shifted with a minimal distance (150 Mbp).
Receiver operating characteristic (ROC) curves
To measure the enrichment in old repetitive elements among the repetitive elements that present a significant co-localization, we used a ROC analysis. We took the chronological ordering of the 360 human TE from Giordano et al. (Supplementary Table S2 of (35)) and labeled each TE that presented a significant co-localization according to our analysis. We took the repeats with a significant CS in human embryonic stem cells i.e. Supplementary Table S1, sheet A. We then applied the receiver operating characteristic of the R package ROCR with default parameters. The area under curve (AUC) and the P-value were calculated with the R function roc.area. The P-value produced is related to the Mann-Whitney U statistics. True positives correspond to repeats having a significant CS and the false positives correspond to repeats not having a significant CS.
We used a similar approach to show enrichment within the pool of repeats presenting highly significant CS with repeats that are enriched with TFBS. We ordered all the repeat elements according to the P-value of their CS and identified each repeat enriched with TFBS. True positives correspond to repeats enriched with TFBS and false positives to repeats not enriched with TFBS. Enrichment for TFBS were computed exploiting the following datasets:
human
- Supplementary Table S1 of (36) that provides Human repeat-associated binding sites (RABS) for OCT4, NANOG and CTCF for hESC (human embryonic stem cell).
- Supplementary Table S3 of (37) that provides DHS-associated repeats (DARs) enriched for TFs ChIP-Seq for hESC.
- Supplementary Table S4 of (37) that provides DARs enriched for TFs motifs for hESC.
mouse
- Supplementary Table S2 of (36) that provides Mouse RABS for OCT4, NANOG and CTCF for mESC.
Comparison of repetitive elements co-localization in IMR90 and hESC
To compare the CS of repeats in different cell lines, we retained repeats present in at least 500 bins. The log ratio between the CS in the two cell types was then computed. The distribution of this log ratio can be considered as normal. We then computed the same distribution for repetitive elements families that have been identified as frequently carrying binding sites for three different transcription factors: CTCF, NANOG and OCT4 (Supplementary Table S1 of (36)). To statistically show the difference in the mean between different groups, we used the Mann–Whitney test of R (with the option alternative = ‘less’).
Synteny analysis
Using the synteny blocks coordinates given by OrthoClusterDB (38), we converted the coordinates of the human and mouse assemblies using the ‘liftover’ tool from UCSC. When necessary, we duplicated the bins in one organism that spanned over two or more bins in the other organism in order to keep the same number of bins for each block in the human and in the mouse genomes.
RESULTS
Several subfamilies of repetitive elements co-localize in hESC nuclei
The potential co-localization of specific repetitive elements in hESC was investigated using inter-chromosomal contacts obtained from several Hi-C datasets generated through different protocols and laboratories (25).
The 1395 different repeats subfamilies from the RepeatMasker table of UCSC were pooled into six families: satellites/low complexity sequences, SINE, LINE, LTR, DNA-transposons and RNA repeats (31). For each subfamily a CS was computed (‘Materials and Methods’ section), to assess the tendency for regions of the genome carrying these repeats to make physical contacts in the nuclear space. Importantly, and to avoid potential false positive contacts resulting from misalignment of the reads along the genome, CSs were computed through two independent approaches (‘Materials and Methods’ section). The first and simplest approach was to retain all pairs of reads that unambiguously aligned against the genome when using stringent and restrictive mapping parameters (Figure 1A). The second approach was more stringent, since only unambiguous pairs of reads that did not overlap with any repeat were considered. The significance of each CS was then estimated using null models designed to account for three known potential biases: the sequencing coverage of the Hi-C experiment, the GC content and the distribution of repeats along chromosomes (see Figure 1B and Supplementary Figure S2, ‘Materials and Methods’ section). For each dataset, a subset of repeat subfamilies exhibited CS significantly higher than expected by chance, with the three null models giving very similar results (Supplementary Table S1, sheet B, Spearman correlation > 0.7 between different null models). On Figure 2 are presented the results obtained with the constant Hi-C coverage null model, which takes into account a potential inherent bias to the Hi-C experiment itself (see ‘Materials and Methods’ section).
Figure 2.

(Color online) CS of repetitive elements in human Embryonic Stem Cells. CS and P-values for all repetitive elements for the human embryonic stem cell. For each class of repeat (i–vi), the CSs and the P-value of all repeat sub-families are plotted. (i) Red dots: low complexity sequences (LC). Purple dots: satellite repeats. (ii) Green dots: SINEs (SINE older than 25MY are colored in dark green). (iii) Cyan dots: LINEs. (iv) Blue dots: LTR. (v) Yellow dots: DNA transposons (vi) Orange dots: RNA repeats.
The null model constraining the GC distribution was the most stringent one, i.e. yielding to the smallest number of significant repeats (see Supplementary Figure S4). This is expected, since the GC content strongly correlates with the binary compartmentalization of the genome (30). Significant co-localization under this null model therefore excludes the possibility to be simply explained by the spatial segregation of open and closed chromatin (39). In addition, other chromosomal features such as PolII occupancy, DNAse accessibility and replication timing profiles were also used as null models and gave similar results (data not shown).
All families contain subfamilies with significantly high CSs (Figure 2; see Supplementary Table S1, sheet A for the complete list). First, 36 GC-rich micro- and mini-satellites as well as one satellite (ALR/Alpha) (Figure 2i, purple dots) were found to significantly co-localize. Low complexity sequences found to have a significant CS (red dots). Interestingly, most (36 out of 50) SINEs present a significant CS (Figure 2ii), with old and young Alu exhibiting high and low CSs, respectively (dark green dots in Figure 2ii represent SINEs older than 25 MY). Regarding other families, several repeats exhibit slight increases though nevertheless significant CSs, including L2 LINEs (Figure 2iii), the LTR repeat LTR13 (Figure 2iv) and DNA transposons of the MER family (Figure 2v). Finally, a few RNA repeats were found to have a significant CS (Figure 2vi), including the ancestral Alu sequence 7SR RNA.
In order to better contextualize the CSs obtained for the different repeats, we computed the CS of genomic elements known to co-localize such as nucleolar associated domains (NAD; Supplementary Figure S3) and of a large set of transcription factors (TF; e.g. NANOG). A total of 99 out of the 102 elements tested presented significant CS when confronted to the coverage null model (see Supplementary Table S1, sheet G), supporting the validity of the approach. The most significant elements correspond to sets of bins enriched in DNAse hyper-sensitive sites and RNA polymerase II (Pol II).
Importantly, most of the CS remained significant with a more stringent mapping strategy (Figure 1A) i.e. when all the reads overlapping even partially with the elements annotated in the RepeatMasker track of UCSC were discarded (80/133). This important control alleviates all possible concerns about mapping biases (Supplementary Figure S5 and Supplementary Table S1, sheet A). The results are also the same when considering bins of equal sized (see Supplementary Figure S6).
Repetitive elements with significant co-localization score are evolutionary older and enriched in transcription factor binding sites
We noticed that older Alu subfamilies such as AluJb or AluJo tend to exhibit high CSs. To test whether this was a general trend affecting the majority of repetitive elements presenting a high CS, we performed a Receiver Operating Characteristic (ROC) analysis. The 360 human TE were ordered according to their estimated divergence over evolutionary times (35), revealing that significant CSs were essentially found within the oldest repetitive elements subfamilies of the human genome (see Figure 3A). This observation raises the interesting hypothesis that these repeats may have been fixed in the host genome at these specific positions because of a functional role related to the increased contact frequencies.
Figure 3.

(Color online) Evolutionary age and enrichment for TFBS of the co-localizating repetitive elements. (A) Receiver operating characteristic curve showing that the age of repetitive elements associated with significant CS is higher than that of repetitive elements not associated with significant CS. Black line represents the null condition that the age of the two sets of repetitive elements were distributed similarly. The bar plots represent the proportions of repeat elements older than 25 MYA for all repeats and for a subset of repeats with significant CS. The proportion is higher for the group of repeats with significant CS (Fisher test, P-value = 0.00043). (B) Receiver operating characteristic curve showing that the CS associated with repetitive elements enriched with TFBS is higher than that of repetitive elements not associated with enrichment for TFBS. Black line represents the null condition that the CS of the two sets of repetitive elements were distributed similarly. Red line corresponds to hESC cells and green line to mESC. The bar plots represent the proportions of repeat elements enriched with TFBS for all repeats and for repeats with significant CS. The proportion is higher for the group of repeats with significant CS (Fisher test, P-value = 0.012). (C) Distribution of the log ratios between the CS for all the repeats between two cell types (hESC and IMR90). We considered either all the repeats or only a subset of repeats bound by three TFs (CTCF, NANOG, OCT4 or both NANOG and OCT4).
We then analyzed whether the repetitive elements presenting significant CS were enriched with TFBS (‘Materials and Methods’ section). Such repeat-associated binding sites (RABS) have previously been identified in several cell types (36) and confirmed by the ENCODE consortium in other cell types (40). We used ROC analysis to show that repeats with significant CS tend to be enriched with RABS (hESC, Figure 3B). These enrichments provide hypothesis regarding a mechanistic and functional role for the observed association of repetitive elements (see ‘Discussion’ section).
The spatial association of some repetitive elements is cell type dependent and relies on cell type specific binding factors
CSs were computed for two other cell types (human embryonic lung fibroblasts cells (IMR90; 25) and in lymphoblastoid cells (GM12878; 27), giving overall similar results to those obtained with hESC and suggesting that the average large-scale folding of the genome could be influenced by the genome sequence itself rather than by cell-type specific reorganization (see Supplementary Figure S7 and Supplementary Table S1, sheet C and D).
Using different datasets on the same cell type, we were also able to show that our results do not depend on read length (see Supplementary Figure S7C).
Interestingly, despite an overall CSs conservation, some local changes in the inter-chromosomal contact maps generated using different cells types could nevertheless be observed. In order to investigate whether or not these changes could be associated with specific repetitive elements, we computed the log ratio between the CS of each repeat in hESC and IMR90 (two datasets generated through the same protocol). The distribution of this log ratio is symmetric and centered around zero. We then computed the same distribution for repetitive elements that have been identified as bound by three different transcription factors: CTCF, NANOG and OCT4 (12). We found that the CS of the repeats containing binding sites for both OCT4 and NANOG simultaneously, which are expressed only in hESC, are significantly higher in this cell line compare to IMR90 (Mann–Whitney test, P-value of 0.015) whereas there was no significant change for CTCF, which is expressed in both cell types (Figure 3C).
Repetitive elements co-localize in the mouse and Drosophila genomes
To determine whether co-localizations of similar repetitive elements in human were also present in other metazoans such as mouse and Drosophila (25,26), CSs and P-values (under constant Hi-C coverage null hypothesis) in these species were computed. For mouse embryonic stem cells (mESC), a trend for co-localization of repeats was observed, very similar to the one found in human (Figure 4A, Supplementary Figure S8 and Table S1, sheet E). A surprising difference was that numerous AT-rich simple repeats were significantly found to co-localize in mice (10 AT-rich satellite and 3 Low complexity elements). The mouse genome is actually enriched with such AT-rich sequences when compared to the human genome (41). Similar significant repeats elements were recovered from mouse cortex cells (25; not shown), confirming the conservation of DNA repeat co-localization in pluripotent and differentiated cells. In order to see whether co-localization of repetitive elements occurs outside the mammalian phylum, CSs were also computed for the repeat subfamilies of the Drosophila genome. Flies, contrary to vertebrates, do not have SINEs elements. Still, significant enrichments in contacts between several classes of repetitive elements were observed (Figure 4B; Supplementary Table S1, sheet F), including Gypsy and Invaders subfamilies from the LTR family and the ProtoP elements from the DNA transposons family (18). Therefore, despite important divergences in the evolutionary history of repeats in these three genomes, a subset of repetitive elements always exhibits high enrichments in inter-chromosomal contacts suggesting mobile elements may be involved in the regulation of the folding of many metazoan genomes.
Figure 4.

(Color online) CS of all the different repetitive elements in mouse and Drosophila. (A) CS and corresponding P-values (constant Hi-C coverage null model) of repetitive elements in mouse ESC (25). (B) in Drosophila embryo (26).
genome organization of human and mouse syntenic blocks is conserved and correlates with SINEs positions
To analyze the potential interplay between the CS of repetitive elements, the genome organization and the evolutionary history of the mouse and human genomes, we reorganized the contact map of the mouse genome according to its synteny conservation with the human genome. Contacts between the homologous regions conserved in the two genomes can then be readily compared in light of repeat positions (Figure 5). The reordered contact map of the mouse genome appears strikingly similar to the human one (see also Supplementary Figure S9A). We quantified this similarity by computing the Pearson correlation coefficient between each line of the two matrices (a line corresponding to a region of ∼300 kb), which can rise as high as 0.80 (color scale between the two matrices on Figure 5). The density of MIR (mammalian interspersed repeat, a SINE repeat), a TE whose expansion within these genomes predates the divergence between human and rodents, was calculated for both genomes (bar plots above the two matrices; 42,43). As expected from their common origin, the density of MIR repeats is conserved between the two genomes (0.59, Pearson correlation). Interestingly, high MIR densities correlate with strong inter-chromosomal contacts in both genomes which form a typical banding pattern common to both maps (blue bar plot on Figure 5). The genomic densities of two other SINEs with high CSs, either primate (Alu) or rodent specific (B1) exhibit strikingly similar distributions (0.60 Pearson correlation), while also correlating with regions co-localizing in space (red and green bar plots on Figure 5, respectively). Contrary to MIRs, these two repeats, which are both derivatives of the 7SL RNA element (44), have expanded after the rodent/primate divergence. These results taken together raise the interesting hypothesis that repetitive, independent fixation of SINEs in homologous regions exhibiting high contact frequencies may be favored over evolutionary times.
Figure 5.

(Color online) Correlation matrices of the inter-chromosomal contacts in human and mouse re-ordered according to syntenic regions. Correlation matrices obtained from whole inter-chromosomal contact of human (Left) and mouse autosome chromosomes (Right). For the human genome, the 22 chromosomes are shown using the color code below the matrix. For the mouse genome, the chromosomes are reordered as a function of their synteny conservation with human as illustrated by the resulting mosaic color code. The two color maps show the correlation in contact frequencies strength between distant parts of the genome, with high and low level of contacts in red and yellow, respectively. The color scale in the middle corresponds to the Pearson coefficient between two lines of each matrix. MIR densities in each species are indicated by the blue bar plot atop of the matrices. Genomic densities of two other SINEs with high CSs, either primate (Alu) or rodent specific (B1) were also plotted (red and green bar plots, on the left and on the right, respectively).
DISCUSSION
A potential role for repeated sequences in regulating the 3D folding of the genome
The analysis above suggest that a subset of repetitive elements (including both TEs and satellites sequences) are genomic imprints of the 3D folding of metazoan genomes. An ensuing question is whether they play an active or passive role in this folding. In the first case, the observed preferential contact between two distant parts of the genome is driven by the presence of a similar repeat element. In the second case, the presence of the repeats in the vicinity of contact point is a consequence of something else. An ideal way to answer this question would be to remove the cause (the DNA repetitive elements) and to ask whether or not the consequence (the co-localization of distant regions in the genome) is still observed. This is obviously out of reach as of today.
However, arguments supporting an active role for repeat element in the 3D folding of genomes can be sustained based on the results presented here. A prediction regarding the existence of such a role would be that syntenic regions that exhibit similar content in repetitive elements in mouse and human are more likely to exhibit similar 3D contacts. We tested this prediction using the similarity between contact maps obtained for syntenic regions in mouse and human. The results show that higher correlations in the 3D neighbors are reached between bins harboring related repetitive elements both in human and mouse (see Supplementary Figure S7B for Alu and B1 SINEs analysis). On the other hand, homologous bins that contain many repetitive elements in only one of the two species have anti-correlated contact profiles (see Supplementary Figure S7B).
These findings do not rule out the possibility that regions exhibiting preferential contacts share a similar content of retro-transposons because retro-transposons are more likely to spread into genomic regions that are already in close spatial proximity. In order to get an idea of the retro-transposition dynamics along the genome, one possibility is to look at the genome wide positions of newly expanded TEs populations. For instance, newly transposed Alus are found uniformly throughout the genome, with a slight preference for the AT-rich, inactive regions (45). In this context, it appears difficult to envision that the observed enrichment of TEs at contact points results primarily from their transposition dynamics.
Taken altogether, these pieces of evidence suggest that, besides being passive imprints of the genome folding, retro-transposons may also influence the self-organized folding of the genome (46,47).
Further experimental support to the co-localization of repetitive elements
Although the quantitative analysis of genome-wide contact maps remains a delicate exercise, a converging body of evidences from imaging and molecular experiments in a variety of organisms supports the potential role of repetitive elements in organizing genome folding. Co-localization of DNA sequences within nuclei has notably been investigated using fluorescence in situ hybridization (FISH) in several species. For instance, fluorescent probes targeting centromeric and telomeric repetitive elements in human and mouse lymphocytes have revealed their co-localization (48). Retrotransposons and satellite sequences were shown to form foci in mouse retina cells and other cell types using probes targeting B1-SINE, L1 and major satellites (49). Interestingly, the latter report revealed that the co-localization of these repeats was maintained in different cells types despite important differences in the global arrangements of chromosomes in nuclei. Co-localization of Alu repeats in the center of the nucleus has been shown in human fibroblast (50) as well as co-localization and insulating role of Gypsy retrotransposon in Drosophila cells (20,51). Finally, Tf retrotransposons positioned throughout the genome form clusters at centromeres in fission yeast Schizosaccharomyces pombe (21), an enrichment also identifiable from genomic 3C data analysis (52). Importantly, the repeats shown to our knowledge to co-localize experimentally in human, mouse and Drosophila all presented a high CS in the assay described above. Experimental validations of the co-localization of other repeats exhibiting significant CS is still missing, but may unveil folding rules driven in part by repetitive elements.
Potential molecular mechanisms accounting for the co-localization of repetitive elements
An immediate question arising from the results described above concerns the potential molecular mechanisms involved. As of today, three different pathways, not mutually exclusive, may influence or drive the organization of repetitive elements.
First, specific proteins are known to mediate long-range contacts. For instance, the clustering of fission yeast retrotransposon is mediated by the positioning of the Ku heterodimer complex also involved in non-homologous end-joining, and regulated by epigenetic marks, at a discrete position on the LTR that covers a few hundred base pairs (22). Our results suggest that similar mechanisms may be involved in metazoan as well. One of the main result supporting this protein driven mechanism is the observation that in hESC repetitive elements harboring NANOG and OCT4 binding sites where found to co-localize whereas this co-localization disappear in IMR90 in which these factors are repressed. Another partner could be CTCF, which was the first factor shown to correlate with the Hi-C contacts (53). This complex physically bridges regions of chromatin otherwise linearly distant or on separate chromosomes (54). CTCF is found associated with B2 SINEs in mouse (42,55) and its presence can explain the high CSs of these elements in mouse but also for other repeats. Interestingly, LTR13 which was also found to exhibit a high CS in human is enriched for such CTCF binding (36). In addition, several SINEs sequences were known to harbor binding sites for many other transcription factors and/or act as insulators e.g. MIR, B2 (42,56) and Alu (57). Preferential contacts of specific repetitive elements with the nuclear lamina, notably AT-rich low complexity and micro-satellite, are also a potential factor of chromosomal organization (58).
Besides this mechanism, an intriguing possibility could be the formation of inter-strand DNA structures leading to the co-localization of mini-satellites and low complexity sequences (59). Notably, G-quadruplex (G4) can form between two DNA strands containing at least two stretches of three guanine residues separated by 3–8 nucleotides. Although this structure is stable in vitro, the functional relevance of G4 in vivo in mammalian cells remains controversial. Nevertheless, the formation of such inter-strand G4 might explains why mini-satellites, which contain stretches of three or more guanine residues exhibit high CSs (see Supplementary Table S1, sheet A). An important enrichment of these sequences at genes promoters and replication origins (60) combined with the formation of G4 structures could to participate in the co-localization of early replicating chromatin within the nucleus. Inter-strand interactions of similar DNA structures can be extended into a more general, and exciting, mechanism, i.e. direct recognition of homologous regions. This recognition has been proposed on the basis of theoretical arguments involving the sequence dependent disposition of charges along the DNA molecule (61). In vitro work carried using magnetic tweezers and naked DNA showed that this recognition is efficient between regions of homology of 5 kb or more (62). In a recent in vivo work in Neurospora crassa, two DNA segments were shown to align with each other's under the condition that they present the same triplets of nucleotides positioned with a ∼11 bp periodicity, i.e. the double helix coil. This alignment was present even in the absence of known pairing complexes involved in homology recognition (63). This mechanism could therefore play a role in bringing together long, similar satellite repeats which can extend over millions of bp.
The third mechanism that could promote 3D contacts could be transcription itself. This hypothesis relies on evidences regarding the existence of transcription factories, i.e. sites of active transcription containing up to a hundred RNA polymerases at a time (64). Many retrotransposons carry PolII and PolIII promoters (65). Evidences for transcription of these elements at the genome scale suggest that these promoters may drive the co-localization of these elements in such transcription foci (66).
A role of retrotransposons in the evolution of genomes 3D folding
In this paper we showed that many repetitive elements could act as specific anchor points that spatially organize chromosomes. This organization is conserved between related species since syntenic blocks in human and mouse exhibit strikingly similar 3D contacts, the distribution of MIR, Alu and B SINEs repeats along the genome being strongly correlated with these contacts (Figure 5). MIR amplified before the primate/rodent split and could be responsible for the maintenance of the ancestral genome fold in the two organisms. An intriguing fact is that the correlation between the contacts and the repetitive content of these genomes is also observed for Alu and B SINE although they spread after the primate/rodent split. This naturally leads to the suggestion that these homologous repeats in contemporary genomes were selectively retained at similar positions as the result of independent selection processes. A possibility is that these elements play a role in the maintenance and/or fine tuning of 3D contacts between these regions. This hypothesis is compatible with a role for retro-elements independent amplification waves in sequentially reshaping spatial contacts. Since MIRs and Alus are significantly enriched upstream orthologous genes in both genomes (57–67) consecutive waves of TE insertion could result in fine remodeling and evolution of both regulatory networks (see (6)) and genome architecture. Overall, the observations described here pave the way for future investigations aiming at deciphering experimentally the precise influence of these repeats in shaping genome architecture.
AVAILABILITY
Programs and commands developed for the analysis (C and R language) are available here:
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are thankful to Jean-Baptiste Boulé, Christophe Escudé, Annick Lesne, Thierry Forné, Marie-Claude Marsolier-Kergoat and Jean-Marc Victor and his team for helpful discussions and advices. We thank Matthew Campbell for comments on the manuscript and ‘Spatial Regulation of Genomes’ team for their support and discussions.
FUNDING
ANR PIRIBIO [ANR-09-PIRI-0024]; European Research Council under the 7th Framework Program (FP7/2007-2013)/ERC grant agreement [260822 to R.K.]; UPMC Convergence program CVG_1110. Funding for open access charge: European Research Council under the 7th Framework Program (FP7/2007-2013)/ERC grant agreement [260822 to R.K.]
Conflict of interest statement. None declared.
REFERENCES
- 1.De Koning A.P.J., Gu W., Castoe T.A., Batzer M.A., Pollock D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- 3.Ohno S. So much ‘junk’ DNA in our genome. Brookhaven Symp. Biol. 1972;23:366–370. [PubMed] [Google Scholar]
- 4.Orgel L.E., Crick F.H., Sapienza C. Selfish DNA. Nature. 1980;288:645–646. doi: 10.1038/288645a0. [DOI] [PubMed] [Google Scholar]
- 5.Arkhipova I.R., Batzer M.A., Brosius J., Feschotte CÈ., Moran J.V., Schmitz J., Jurka J. Genomic impact of eukaryotic transposable elements. Mob. DNA. 2012;3:19. doi: 10.1186/1759-8753-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourque G. Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr. Opin. Genet. Dev. 2009;19:607–612. doi: 10.1016/j.gde.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Huang C.R.L., Burns K.H., Boeke J.D. Active transposition in genomes. Annu. Rev. Genet. 2012;46:651–675. doi: 10.1146/annurev-genet-110711-155616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eddy S.R. The ENCODE project: missteps overshadowing a success. Curr. Biol. 2013;23:R259–R261. doi: 10.1016/j.cub.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Souza F.S.J., de Franchini L.F., Rubinstein M. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong. Mol. Biol. Evol. 2013;30:1239–1251. doi: 10.1093/molbev/mst045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard G.-F., Kerrest A., Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 2008;72:686–727. doi: 10.1128/MMBR.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClintock B. Controlling element and the gene. Cold Spring Harb. Symp. Quant. Biol. 1956;21:197–216. doi: 10.1101/sqb.1956.021.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Britten R.J., Davidson E.H. Gene regulation for higher cells: a theory. Science. 1969;165:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 13.Brosius J. The contribution of RNAs and retroposition to evolutionary novelties. Genetica. 2003;118:99–115. [PubMed] [Google Scholar]
- 14.Lynch V.J., Leclerc R.D., May G., Wagner G.P. Transposon regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 2011;43:1154–1159. doi: 10.1038/ng.917. [DOI] [PubMed] [Google Scholar]
- 15.Jjingo D., Conley A.B., Wang J., Mariño-Ramírez L., Lunyak V.V., Jordan I.K. Mammalian-wide interspersed repeat (MIR)-derived enhancers and the regulation of human gene expression. Mob. DNA. 2014;5:14. doi: 10.1186/1759-8753-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su M., Han D., Boyd-Kirkup J., Yu X., Han J.-D.J. Evolution of Alu Elements toward Enhancers. Cell Rep. 2014;7:376–385. doi: 10.1016/j.celrep.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Kumar R.P., Senthilkumar R., Singh V., Mishra R.K. Repeat performance: how do genome packaging and regulation depend on simple sequence repeats. BioEssays. 2010;32:165–174. doi: 10.1002/bies.200900111. [DOI] [PubMed] [Google Scholar]
- 18.Shapiro J.A., von Sternberg R. Why repetitive DNA is essential to genome function. Biol. Rev. 2005;80:227–250. doi: 10.1017/s1464793104006657. [DOI] [PubMed] [Google Scholar]
- 19.Apostolou E., Thanos D. Virus infection induces NF-κB-dependent interchromosomal associations mediating monoallelic IFN-β gene expression. Cell. 2008;134:85–96. doi: 10.1016/j.cell.2008.05.052. [DOI] [PubMed] [Google Scholar]
- 20.Byrd K., Corces V.G. Visualization of chromatin domains created by the gypsy insulator of Drosophila. J. Cell Biol. 2003;162:565–574. doi: 10.1083/jcb.200305013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cam H.P., Noma K., Ebina H., Levin H.L., Grewal S.I.S. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature. 2008;451:431–436. doi: 10.1038/nature06499. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka A., Tanizawa H., Sriswasdi S., Iwasaki O., Chatterjee A., Speicher D., Levin H., Noguchi E., Noma K. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Mol. Cell. 2012;48:532–546. doi: 10.1016/j.molcel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 24.De Laat W., Dekker J. 3C-based technologies to study the shape of the genome. Methods. 2012;58:189–191. doi: 10.1016/j.ymeth.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou C., Li L., Qin Z., Corces V. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell. 2012;48:471–484. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalhor R., Tjong H., Jayathilaka N., Alber F., Chen L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotech. 2012;30:90–98. doi: 10.1038/nbt.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Meth. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 32.Cournac A., Marie-Nelly H., Marbouty M., Koszul R., Mozziconacci J. Normalization of a chromosomal contact map. BMC Genomics. 2012;13:436. doi: 10.1186/1471-2164-13-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Németh A., Conesa A., Santoyo-Lopez J., Medina I., Montaner D., Péterfia B., Solovei I., Cremer T., Dopazo J., Längst G. Initial genomics of the human nucleolus. PLoS Genet. 2010;6:e1000889. doi: 10.1371/journal.pgen.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witten D.M., Noble W.S. On the assessment of statistical significance of three-dimensional colocalization of sets of genomic elements. Nucleic Acids Res. 2012;40:3849–3855. doi: 10.1093/nar/gks012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giordano J., Ge Y., Gelfand Y., Abrusán G., Benson G., Warburton P.E. Evolutionary history of mammalian transposons determined by genome-wide defragmentation. PLoS Comput. Biol. 2007;3:e137. doi: 10.1371/journal.pcbi.0030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunarso G., Chia N.-Y., Jeyakani J., Hwang C., Lu X., Chan Y.-S., Ng H.-H., Bourque G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010;42:631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 37.Jacques P.-É., Jeyakani J., Bourque G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013;9:e1003504. doi: 10.1371/journal.pgen.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng M.-P., Vergara I.A., Frech C., Chen Q., Zeng X., Pei J., Chen N. OrthoClusterDB: an online platform for synteny blocks. BMC Bioinformatics. 2009;10:192. doi: 10.1186/1471-2105-10-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundaram V., Cheng Y., Ma Z., Li D., Xing X., Edge P., Snyder M.P., Wang T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014;24:1963–1976. doi: 10.1101/gr.168872.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chinwalla A.T., Cook L.L., Delehaunty K.D., Fewell G.A., Fulton L.A., Fulton R.S., Graves T.A., Hillier L.W., Mardis E.R., McPherson J.D., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 42.Bourque G., Leong B., Vega V.B., Chen X., Lee Y.L., Srinivasan K.G., Chew J.-L., Ruan Y., Wei C.-L., Ng H.H., et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18:1752–1762. doi: 10.1101/gr.080663.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smit A.F., Riggs A.D. MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation. Nucleic Acids Res. 1995;23:98–102. doi: 10.1093/nar/23.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kriegs J.O., Churakov G., Jurka J., Brosius J., Schmitz J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 2007;23:158–161. doi: 10.1016/j.tig.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Medstrand P., van de Lagemaat L.N., Mager D.L. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 2002;12:1483–1495. doi: 10.1101/gr.388902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 47.Misteli T. Self-organization in the genome. PNAS. 2009;106:6885–6886. doi: 10.1073/pnas.0902010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weierich C., Brero A., Stein S., von Hase J., Cremer C., Cremer T., Solovei I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 2003;11:485–502. doi: 10.1023/a:1025016828544. [DOI] [PubMed] [Google Scholar]
- 49.Solovei I., Kreysing M., Lanctôt C., Kösem S., Peichl L., Cremer T., Guck J., Joffe B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell. 2009;137:356–368. doi: 10.1016/j.cell.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 50.Bolzer A., Kreth G., Solovei I., Koehler D., Saracoglu K., Fauth C., Müller S., Eils R., Cremer C., Speicher M.R., et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerasimova T.I., Byrd K., Corces V.G. A chromatin insulator determines the nuclear localization of DNA. Mol. Cell. 2000;6:1025–1035. doi: 10.1016/s1097-2765(00)00101-5. [DOI] [PubMed] [Google Scholar]
- 52.Tanizawa H., Iwasaki O., Tanaka A., Capizzi J.R., Wickramasinghe P., Lee M., Fu Z., Noma K. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010;38:8164–8177. doi: 10.1093/nar/gkq955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Botta M., Haider S., Leung I.X.Y., Lio P., Mozziconacci J. Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol. Syst. Biol. 2010;6:426. doi: 10.1038/msb.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips J.E., Corces V.G. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt D., Schwalie P., Wilson M., Ballester B., Gonçalves A., Kutter C., Brown G., Marshall A., Flicek P., Odom D. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lunyak V.V., Prefontaine G.G., Núñez E., Cramer T., Ju B.-G., Ohgi K.A., Hutt K., Roy R., García-Díaz A., Zhu X., et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science. 2007;317:248–251. doi: 10.1126/science.1140871. [DOI] [PubMed] [Google Scholar]
- 57.Polak P., Domany E. Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. 2006;7:133. doi: 10.1186/1471-2164-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.-B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–280. doi: 10.1101/gr.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox R., Mirkin S.M. Characteristic enrichment of DNA repeats in different genomes. PNAS. 1997;94:5237–5242. doi: 10.1073/pnas.94.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Méchali M., Yoshida K., Coulombe P., Pasero P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013;23:124–131. doi: 10.1016/j.gde.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 61.Kornyshev A.A., Leikin S. Sequence recognition in the pairing of DNA duplexes. Phys. Rev. Lett. 2001;86:3666–3669. doi: 10.1103/PhysRevLett.86.3666. [DOI] [PubMed] [Google Scholar]
- 62.Danilowicz C., Lee C.H., Kim K., Hatch K., Coljee V.W., Kleckner N., Prentiss M. Single molecule detection of direct, homologous, DNA/DNA pairing. PNAS. 2009;106:19824–19829. doi: 10.1073/pnas.0911214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gladyshev E., Kleckner N. Direct recognition of homology between double helices of DNA in Neurospora crassa. Nat. Commun. 2014;5:3509. doi: 10.1038/ncomms4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li G., Ruan X., Auerbach R., Sandhu K., Zheng M., Wang P., Poh H., Goh Y., Lim J., Zhang J., et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lunyak V.V., Atallah M. Genomic relationship between SINE retrotransposons, Pol III-Pol II transcription, and chromatin organization: the journey from junk to jewel. Biochem. Cell Biol. 2011;89:495–504. doi: 10.1139/o11-046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faulkner G.J., Kimura Y., Daub C.O., Wani S., Plessy C., Irvine K.M., Schroder K., Cloonan N., Steptoe A.L., Lassmann T., et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009;41:563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 67.Tsirigos A., Rigoutsos I. Alu and B1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol. 2009;5:e1000610. doi: 10.1371/journal.pcbi.1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.