


Abstract
Mycobacterium tuberculosis (Mtb) Cmr (Rv1675c) is a CRP/FNR family transcription factor known to be responsive to cAMP levels and during macrophage infections. However, Cmr's DNA binding properties, cellular targets and overall role in tuberculosis (TB) complex bacteria have not been characterized. In this study, we used experimental and computational approaches to characterize Cmr's DNA binding properties and identify a putative regulon. Cmr binds a 16-bp palindromic site that includes four highly conserved nucleotides that are required for DNA binding. A total of 368 binding sites, distributed in clusters among ∼200 binding regions throughout the Mycobacterium bovis BCG genome, were identified using ChIP-seq. One of the most enriched Cmr binding sites was located upstream of the cmr promoter, and we demonstrated that expression of cmr is autoregulated. cAMP affected Cmr binding at a subset of DNA loci in vivo and in vitro, including multiple sites adjacent to members of the DosR (DevR) dormancy regulon. Our findings of cooperative binding of Cmr to these DNA regions and the regulation by Cmr of the DosR-regulated virulence gene Rv2623 demonstrate the complexity of Cmr-mediated gene regulation and suggest a role for Cmr in the biology of persistent TB infection.
INTRODUCTION
The World Health Organization (WHO) estimated 9 million new tuberculosis (TB) cases in 2013 and 1.5 million TB deaths, of which 0.36 million were Human Immunodeficiency Virus (HIV)-associated (1). Global impact of the TB epidemic is exacerbated by the synergy of Mycobacterium tuberculosis (Mtb), the causative agent of TB, with HIV, as well as the steadily increasing rates of drug resistance that are a byproduct of the lengthy treatment regimens (2,3). The bulk of TB disease occurs via reactivation of pre-existing dormant infections, resurfacing during favorable conditions for the bacterium, like compromised immune status of the host (4). It is estimated that one-third of the world's population is latently infected with Mtb, thus providing a huge reservoir for potential reactivation (5). Therefore, TB continues to be one of the deadliest communicable diseases.
Significant progress has been made in surveillance and control strategies since TB was declared a global emergency in 1993. However, wide gaps still exist in our understanding of Mtb biology, hindering the development of new therapeutics. The success of Mtb as a human pathogen depends on its ability to persist within host macrophages or granulomas for extended periods of time, and escape eradication by the host immune response (6). These processes require that Mtb regulate its gene expression in response to the complex environmental stresses within the host. The Mtb genome encodes at least 200 transcription factors, which underscores the importance of transcriptional regulation in Mtb (7,8). Extensive studies on individual transcription factors (9,10), and regulatory networks defined using large-scale studies on multiple transcription factors (8) have provided valuable insight into the physiology of Mtb. Sigma factors, two-component and kinase phosphorelay systems, and a vast array of small signaling molecules (7,11,12) also contribute to this regulatory response. Second messenger signaling molecules like cyclic nucleotides, (p)ppGpp and divalent calcium relay external signals to appropriate effector molecules within the cell, such as transcription factors, that control gene regulation (13,14). Cyclic nucleotide molecules like c-di-AMP and c-di-GMP also have been associated with Mtb virulence and dormancy (15,16).
cAMP (cyclic adenosine 3′,5′-monophosphate) is one of the most broadly used second messenger molecules, first discovered for its roles in hormone signal transduction in eukaryotes (reviewed in (13)). Subsequently, the role of cAMP in catabolite repression, virulence and signaling pathways have been studied in many bacterial and fungal pathogens, including Mtb (13). Mtb has at least 15 adenylyl cyclases, which convert adenosine triphosphate (ATP) to cAMP in response to diverse environmental cues, thus making the role of cAMP in Mtb more prominent and complex than in most other bacterial pathogens (17–19). Bacterial cAMP levels within viable Mycobacterium bovis BCG Pasteur and Mtb increase upon macrophage infection as do the cAMP levels within the host macrophages (20). This surge in macrophage cAMP was shown to be dependent on an Mtb adenylyl cyclase, Rv0386 (21).
Regulation of gene expression is a primary outcome of cAMP production, and in bacteria this is usually achieved via cAMP binding to effector proteins, such as transcription factors. cAMP binding often causes allosteric changes that alter the activation state of these effector proteins and affects gene expression. The transcription factors CRPMt and Cmr are two of the 10 cNMP (cyclic nucleotide monophosphate)-binding proteins in Mtb initially predicted by an in silico study (17). However, CRPMt and Rv0998, a cAMP-dependent lysine acetyltransferase (22), are the only downstream effectors of cAMP that have been well characterized in Mtb.
CRPMt (Rv3676) and Cmr (Rv1675c) belong to the CRP/FNR superfamily of transcription factors, and each contains a putative N-terminal nucleotide sensing domain and a C-terminal helix-turn-helix (HTH) binding domain (19,23). Deletion of the gene Rv3676 (crp), which encodes CRP in Mtb attenuates growth of the bacterium in laboratory culture medium, macrophages and a BALB/c mouse model (24), suggesting its role in virulence. A contributing factor to Mtb Δcrp's slow growth in vitro was attributed to CRPMt's role in amino acid biosynthesis, particularly serine production (25). CRPMt also regulates expression of rpfA, one of the five genes encoding resuscitation promoting factor proteins in Mtb (24), and whiB1, an essential NO-responsive transcription factor in Mtb (26,27). In Mtb, CRPMt recognizes a consensus sequence similar to that of Escherichia coli CRP (9,24,28), but CRPMt is less dependent on cAMP for DNA-binding (9,24). A binding site upstream of whiB1 is one of the few exceptions at which CRPMt requires cAMP for DNA binding (26,29). The genome-wide binding profile of CRPMt was recently investigated (30,31) but Cmr's DNA-binding and gene regulatory properties remain largely unexplored.
Cmr was identified initially as the regulator of a group of cAMP and macrophage-responsive genes (32). These genes were deregulated in a Δcmr mutant within macrophages or upon addition of exogenous cAMP to mycobacterial growth media (33). Cmr is non-essential for Mtb growth in vitro (32,34,35), but TraSH analysis showed a moderate effect of Cmr on in vivo growth in a murine model system (http://orca2.tamu.edu/U19/genes/detail/Rv1675c/). In the present study, we identified ∼200 genome-wide Cmr binding regions using ChIP-seq and show that Cmr is an autorepressible global regulator that contributes to the expression of the DosR regulon. Cmr binding sites were often clustered together within the chromosome, suggesting cooperative binding of Cmr, which was affected by cAMP levels. Cmr's association with DosR/DevR regulon members suggests a complex interplay between two global transcription factors involved in Mtb virulence and persistence.
MATERIALS AND METHODS
Bacterial strains and culture
Mtb H37Rv (ATCC 25618), M. bovis BCG (Pasteur strain, Trudeau Institute) and all recombinant strains in these parent backgrounds were grown in mycomedium (Middlebrook 7H9 medium (Difco) supplemented with 0.2% [vol/vol] glycerol, 10% [vol/vol] oleic acid-albumin-dextrose-catalase (OADC), 0.05% [vol/vol] Tween-80). Fresh cultures were inoculated from frozen stocks for every experiment. Bacteria were typically used after 7 days of growth (late log phase). Bacterial cultures were grown to late log phase at 37°C in 25-cm2 tissue culture flasks in mycomedium at a shallow depth of 2 mm under ambient conditions (20% O2, 0% CO2) in a controlled atmospheric incubator, and maintained either shaking or standing. E. coli strains were grown in Luria-Bertani broth or on Luria-Bertani agar plates. Kanamycin at 25 μg/ml or hygromycin at 50 μg/ml was added for recombinant strains. All cultures were grown at 37°C unless specified otherwise.
Cmr knockout strains of both M. bovis BCG and Mtb were generated using homologous recombination as described previously (32), to replace a portion of cmr open reading frame with a kanamycin cassette. The kanamycin cassette interrupts cmr open reading frame and replaces amino acids 64–165. Single copy complementation strain was constructed as described in (32). Briefly, the cmr open reading frame and its corresponding upstream intergenic promoter sequence was polymerase chain reaction (PCR)-amplified and cloned into the integrating expression vector pMBC409, which contains a hygromycin-resistance marker, to generate the single copy-complementation strain pMBC668. The plasmid was transformed into M. bovis BCG Δcmr, and Mtb H37Rv Δcmr strains. cmr open reading frame and its Shine–Delgarno sequence was PCR-amplified and cloned downstream of a tuf promoter into expression vector pMBC283-oriM, to generate a multi-copy tuf promoter driven complementation plasmid pMBC357. The plasmid was transformed into M. bovis BCG wild-type (WT) and M. bovis BCG Δcmr strains. M. bovis BCG WT transformed with pMBC357 was used in two of the ChIP-seq experiments. M. bovis BCG Δcmr transformed with pMBC357 was used as the multicopy cmr complementation strain.
Expression and purification of Cmr
Recombinant His-tagged cmr (full length) and the HTH+ and HTH− truncated cmr were PCR amplified from Mtb H37Rv DNA template, using primers listed in Supplementary Table S3. The amplified DNA products were cloned into pET28a+ (Novagen) between EcoRI and HindIII restriction sites to generate pMBC370, pMBC787 and pMBC788 respectively, all with a N-terminal His6x tag. C-terminal His-tagged Cmr (pMBC1760) was expressed by cloning cmr ORF (open reading frame) into the NcoI and XhoI sites in the pet28a+ vector. The plasmids were sequence verified, and maintained in E. coli BL21 (DE3) strain.
Bacterial cultures were grown to an optical density at 600nm (OD600) of 0.4–0.6, and the expression of full length Cmr was induced overnight with 1 mM isopropyl-β-thiogalactopyranoside (IPTG) at room temperature. The expression of the truncated proteins was induced overnight at 15°C with 1 mM IPTG. Protein expression was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis using anti-His monoclonal antibody (Clontech).
Protein purification was carried out as described previously (9), with modifications. For purification, a 500 ml culture was pelleted, washed with 1 mM Tris–HCl, and resuspended in 12.5 ml lysis buffer containing 50 mM Tris–HCl (pH 8.0), 0.02% glycerol, 1 mM dithiothreitol and 1% protease inhibitor cocktail (Sigma). Bacteria were lysed by three freeze-thaw cycles and sonication for 5 min (at 4°C), followed by three additional freeze-thaw cycles. The lysate was cleared by centrifugation at 13 000 rpm for 15 min at 4°C. The His-tagged proteins were purified using HisTrap affinity column (GE Lifesciences) per the manufacturer's instructions, and the eluted protein was dialyzed against phosphate-buffered saline (PBS) with 10% glycerol. Protein concentration was measured with a NanoOrange Protein Quantitation Kit (Molecular Probes) and diluted to 0.3 mg/ml before being stored in aliquots at −70°C.
Full length Cmr with C-terminal His6x tag was also expressed and purified as above. Both N- and C-terminal His-tagged Cmr proteins showed equal dimerization and DNA binding ability, thereby confirming that the His-tag does not interfere with the protein folding or function (data not shown).
Preparation of polyclonal antibody and western blot analysis
Two New Zealand white rabbits were immunized subcutaneously with 1 mg purified N-terminal His-tagged Cmr emulsified 1:1 with Titermax Gold (Sigma) and boosted thrice biweekly with the same amount of protein and adjuvant. The specificity of serum was confirmed by western blotting with His-Cmr. Purified His-Cmr was mixed with SDS-PAGE sample buffer, and run on a 12% SDS-PAGE gel. Proteins were blotted onto polyvinylidene difluoride (PVDF) membranes and sequentially probed with anti-Cmr serum and with a peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch Laboratories). Peroxidase detection was carried out with the enhanced chemiluminescence (ECL) western blotting detection reagents and analysis system (Amersham Biosciences).
Mycobacterial protein extraction and western blot analysis
Liquid cultures of M. bovis BCG WT and mutant strains were grown for 7 days in the required conditions. The bacteria were washed with cold PBS containing 0.25% protease inhibitor cocktail. The pellet was resuspended in lysis buffer containing 0.3% SDS, 200 mM DTT, 28 mM Tris–HCl, 22 mM Tris Base and 1% protease inhibitor cocktail. Protein was extracted by vigorous sonication and freeze/thaw cycles. Cell debris was removed by centrifugation. The supernatant was used as total protein lysate. NanoOrange® Protein quantitation kit (Molecular probes) or Micro BCA™ Protein Assay kit (Life Technologies) was used to quantify the protein. Thirty microgram total protein mixed with SDS-PAGE sample buffer was loaded in each well of denaturing SDS-PAGE gel (15% resolving gel, 5% stacking gel). The gel was run at 100V until the dye front reached the bottom of the gel, and the proteins were blotted onto a PVDF membrane using a standard wet-transfer protocol. Equal loading of samples was visualized using a duplicate SDS-PAGE gel stained with standard Coomassie Brilliant Blue staining protocol.
To detect Cmr, the membrane was sequentially probed with Rabbit-anti-Cmr polyclonal serum (precleared with M. bovis BCGΔcmr lysate), followed by Goat Anti-rabbit PolyHRP antibody (Thermo Scientific Pierce Protein Research Products). To detect Rv2623, the membrane was sequentially probed with Mouse-monoclonal-anti-Rv2623 antibody (generously provided by Dr John Chan, Albert Einstein College of Medicine, NY), followed by Goat Anti-mouse-HRP antibody (Jackson ImmunoResearch Laboratories). Mouse-anti-KatG antibody (BEI Resources) was used to detect KatG, used as loading control. Peroxidase detection was carried out with SuperSignal™ West Pico Chemiluminescent Substrate (Life Technologies).
Cross-linking of Cmr by glutaraldehyde
Cross-linking of purified His-tagged Cmr was performed as previously described in (36). His-Cmr was diluted to 310 nM in cross-linking buffer (50 mM sodium phosphate, pH 7.4, 20% glycerol, 5 mM MgCl2), and was then incubated with glutaraldehyde at a final concentration of 7 mM for 1 h at room temperature. The reaction was quenched by the addition of SDS-PAGE sample buffer, and 8 μl of cross-linking reaction was separated on a 12% SDS-PAGE gel. The protein was transferred onto a PVDF membrane and visualized by western blot with anti-Cmr serum.
Electrophoretic mobility shift assays (EMSA)
PCR forward primers (Supplementary Table S3) were labeled with [γ- 33P]-ATP (MP Biomedicals or Perkin Elmer) using T4 DNA polynucleotide kinase (New England Biolabs). DNA probes were generated using labeled forward primer and unlabeled reverse primer in PCR reaction. About 0.05 pmol DNA probe was used in each 10 μl binding reaction mixture. Briefly, 0.3 μM His-Cmr (N- or C-terminal tagged) and DNA probes were incubated at room temperature for 30 min in DNA binding buffer [10 mM Tris–HCl (pH 8.0), 50 mM KCl, 1 mM EDTA, 50 μg/ml bovine serum albumin, 1 mM dithiothreitol, 0.05% non-ionic P-40 detergent, 20 μg/ml poly(dI-dC) and 10% glycerol], with or without 100 μM cAMP. Samples were loaded on an 6, 8 or 12% non-denaturing polyacrylamide gel, depending on the size of the DNA probe, and run for 2–3 h at 14 V/cm in 0.5× Tris-borate-EDTA buffer at 4°C. Gels were transferred to Whatman paper, vacuum-dried, exposed overnight on a phosphor screen, scanned with Storm 860 PhosporImager (Molecular Dynamics) and analyzed with ImageQuant software (Molecular Dynamics). About 200–500× excess competitor DNA was used wherever mentioned.
SELEX from agarose gel
This procedure was carried out as previous reported in (37). M. bovis BCG genomic DNA was digested to completion with Sau3AI and ligated to a linker sequence (5′-CGA ATTCAGGAAACAGCTATGTTAATTAA-3′) prepared with a Sau3AI-compatible sticky end as previously reported. The DNA was mixed with purified His-Cmr for 30 min at room temperature, electrophoresed on a 1% agarose gel before excising a gel slice that contained DNA fragments >1 kb. DNA was extracted from the gel slice, amplified by PCR with KM1040 and cloned into TA vector (Invitrogen). The clones were then amplified by PCR with 33P-labeled KM1040 (5′-CGAATTCAGGAAACAGCTATG), and further screened by electrophoretic mobility shift assays (EMSA) with purified His-Cmr in DNA binding buffer containing cAMP. DNA sequences were obtained for the EMSA-positive clones and referenced to the H37Rv genome to identify Cmr's potential binding sites in Mtb.
SELEX with magnetic beads
Mycobacterium bovis BCG genomic DNA was digested and ligated to linkers as mentioned above. A SELEX procedure was carried out as previously reported (9) using extract from Cmr expressing bacterial strain. A crude extract from 100 ml of His-Cmr expressing culture was applied to 100 μl His Mag agarose beads (Novagen), which were then washed and equilibrated with DNA binding buffer, per the manufacturer's directions. Five micrograms of DNA fragments in 100 μl of DNA binding buffer was added to the Cmr-bound beads for 15 min at room temperature. The beads were washed three times to remove non-specific DNA before eluting Cmr–DNA complexes in a volume of 100 μl. A total of 10 μl of this eluted protein–DNA complex was heated for 10 min at 95°C and used as a template for PCR with primer KM1040. The amplification product was used to bind again with the immobilized Cmr protein. The final elution was PCR amplified with KM1040 and cloned with TA cloning kit (Invitrogen). Individual E. coli DH5α transformants were picked into Luria broth containing 25 μg/ml kanamycin, grown overnight and then used as templates for PCRs with primer KM1040 to amplify the captured insert fragments. PCR products of 200–300 bp were selected for further electrophoretic mobility shift assay (EMSA) analysis. The clones that bound with Cmr were DNA sequenced and referenced to the H37Rv genome as described above.
DNase I footprinting
A 187-bp DNA fragment containing the clone 1C1 was PCR amplified with primers KM1410 and KM1411 (Supplementary Table S3). The forward primer KM1410 was end labeled with [γ-32P] ATP using T4 polynucleotide kinase (New England Biolabs) to generate a single end-labeled probe. Binding reactions were performed in a final volume of 100 μl containing ∼0.25 pmol DNA probe, 1.5 or 3 μM Cmr protein and 20 μg/ml poly (dI-dC) in binding buffer (10 mM Tris–HCl [pH 8.0], 50 mM KCl, 1 mM EDTA, 50 μg/ml bovine serum albumin, 1 mM dithiolthreitol, 0.05% NP-40 detergent, 100 μM cAMP). Binding reactions were performed for 30 min at room temperature and were then subjected to DNase I digestion in 10 mM MgCl2, 5 mM CaCl2 for 1 min. Digestion was terminated by addition of 200 μl stop solution (1% SDS, 200 mM NaCl, 20 mM EDTA and 20 μg/ml salmon sperm DNA). Samples were extracted with phenol–chloroform and DNA was precipitated with ethanol, dried and resuspended in formamide loading dye. A dideoxynucleotide sequencing ladder was generated using a Thermo Sequenase cycle sequencing kit (USB) with the same end-labeled primer as was used for footprinting.
RNA extraction
Mycobacterium bovis BCG cultures were grown in required condition for 7 days in 5 ml mycomedium. Immediately after the flasks were taken out of the incubator, the cultures were treated with 0.5 ml of 5 M guanidine isothiocyanate (GTC). The cultures were pelleted by centrifugation at 13 000 rpm for 10 min at 4°C. Total bacterial RNA was extracted using TRIzol® reagent (Life Technologies) as per the manufacturer's instructions. Briefly, bacterial pellets were resuspended in 1 ml TRIzol® reagent, and cells were disrupted mechanically with a bead beater (BioSpec Products) with three rounds of bead beating, 70 s each, in a mixture of 0.1 mm zirconia–silica beads (BioSpec Products). After brief centrifugation, the supernatant was transferred to fresh microcentrifuge tubes and incubated at room temperature for 5 min to allow dissociation of nucleoprotein complex. About 300 μl of 24:1 mixture of chloroform: isoamyl alcohol was added to the supernatant, and mixed vigorously for 15–20 s. The samples were centrifuged at 13 000 rpm for 30 min at 4°C. The upper aqueous phase was removed and nucleic acid was extracted by isopropanol extraction. The nucleic acid pellet was washed with ethanol and resuspended in RNase free water. DNA contamination was removed using RNase-free DNase protocol in solution following manufacturer's specification (Qiagen).
Semi-quantitative RT-PCR
cDNA was prepared for use in reverse transcriptase PCRs (RT-PCRs). A 0.5 μg aliquot of RNA was incubated with 0.125 μg of random primers RPA00, RPT00, RPC00 and RPG00 at 100°C for 1 min, followed by 65°C for 5 min. A mixture of 10 mM dNTP, 0.1 M DTT, RNase Out (Invitrogen), 5 μl first-strand buffer (Invitrogen) and RT Superscript III (Invitrogen) was added to the RNA-random primer mixture and incubated at 25°C for 10 min, 42°C for 1 h and 70°C for 15 min. RNA levels for genes of interest were examined through a series of cDNA dilutions (0–10−6) to ensure that the quantitation was in the linear range of the PCR. When analyzing strongly induced genes, such as Rv2623, cDNA template levels are optimized for each growth condition. polA (Rv1629) or 16S rRNA expression was used as normalization controls. Before quantitation of individual genes, polA or 16S PCR products from an experiment were normalized to one another, by adjusting the volume of template used, to ensure equal levels of starting cDNA in each reaction. These PCRs were also performed using total RNA without reverse transcriptase (RT), to ensure the absence of DNA contamination.
ChIP-Seq
Mycobacterium bovis BCG WT, M. bovis BCG (p0805:Rv1264cat) liquid cultures were started from frozen seed stocks and grown shaking for 7 days, in mycomedium, in ambient conditions (21% O2, 0% CO2). Duplicate flasks of M. bovis BCG (pMBC357) cultures were started from frozen seed stocks, and grown shaking for 3 days at 37°C, in hypoxia supplemented with CO2 conditions (1.3% O2, 5% CO2) in a controlled atmospheric incubator. On day 3, dibutyryl-cAMP (dbcAMP) was added to one culture flask for a final concentration of 10 mM and returned to the incubator. All cultures were started in 225-cm2 tissue culture flasks in mycomedium at a shallow depth of 2 mm. dbcAMP is a cell permeable compound, which gets cleaved upon internalization by bacteria to produce cAMP (33).
A total of 50 ml culture volume of mid-log phase M. bovis BCG (pMBC357) cells was cross-linked with 1% formaldehyde, for 30 min at room temperature, on a slow rocking platform. Cross-linking was quenched by addition of 250 mM glycine and incubation for 15 min at room temperature with slow rocking. The cells were harvested, washed with cold PBS and resuspended in 0.5–0.6 ml of Buffer I (20 mM HEPES pH 7.9, 50 mM KCl, 0.5 M DTT, 10% glycerol) with protease inhibitor cocktail (Sigma). Bacterial cells were lysed and DNA sheared by sonication for 25 min (Covaris). The salt concentration of the cleared cell lysate was adjusted to a final concentration of 10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% NP-40 (IPP150 buffer). Immunoprecipitation was carried out by incubation of lysate with 10 μl of Cmr antiserum, on nutator at 4°C. A total of 50 μl of Protein A agarose beads were rinsed with IPP150 buffer, and added to the lysate–antiserum mixture. Protein A agarose–lysate–antiserum mixture was incubated at 4°C for 30 min and room temperature for 1.5 h. The beads were washed at least five times with IPP150 buffer followed by two washes with TE buffer (10 mM Tris HCl pH 8.0, 1 mM EDTA). DNA from agarose beads was eluted by incubation with 150 μl elution buffer (50 mM Tris HCl pH 8.0, 10 mM EDTA, 1% SDS) at 65°C for 15 min. A second elution was carried out by incubation of pellet with 100ul of TE buffer+ 1% SDS at 65°C for 5 min. Both elutions were pooled and de-crosslinked by incubating with 1 mg/ml Proteinase K at 37°C for 2 h and 65°C overnight. DNA was purified using PCR purification kit (QIAGEN).
Sequencing was performed as described previously (8). Briefly, sequencing was performed on the Illumina platform, using a GAIIx (Boston University, sequencing core). Coverage along the genome was calculated using Bowtie2 (38) and SamTools (39). Enriched regions were called using log-normal distributions as described in (40). The criteria for a region to be considered ‘enriched’ were a minimum region length of 150 nt and at least 60 nt shift between its forward and reverse peaks. Region coverage was normalized using mean coverage of an experiment, correcting for the number of reads amongst experiments. Exact binding sites were determined as described by (40), and motif was determined using MEME (41,42). Regions from the BCG genome were mapped to the Mtb H37Rv genome by sequence similarity using BLAST.
The following method was used to assign the targets of a binding site. An intergenic site was assigned two potential regulatory targets—the immediate upstream and downstream genes, regardless of the gene orientation. An ‘intragenic’ binding site (in the open reading frame of genes) was assigned an additional potential regulatory target—the gene the binding site is located in. The same assignment method was used for genes in an operon too. We used Tuberculist functional annotations (43) to assign the functional classification to each gene within the Cmr ChIP-seq data. The percentage of potentially regulated genes (based on the target assignment discussed above) was calculated for each functional classification. This percentage was calculated for enriched regions, and individual binding sites within enriched regions separately. The expectation of functional classification for the Mtb H37Rv genome was also calculated. In order to determine significant differences between the genome and the type of ChIP-seq site, hypergeometric calculations, with a correction factor of 11, were performed using Excel. P < 0.05 was considered significant.
RESULTS
Purified His-Cmr forms a dimer
N-terminal His-tagged Cmr was overexpressed in E. coli, purified using a His-affinity column (Figure 1A), and the purified protein was confirmed to be His-Cmr by western blot using monoclonal anti-His antibody (Figure 1B). The majority of purified His-Cmr appeared as a 31 kDa band, consistent with the monomeric form of protein (Figure 1A). However, a weaker band that migrated as a dimer-sized protein was also detected in these purified protein samples by western blot using anti-Cmr serum (Figure 1C). Many transcription factors form oligomers when bound to their DNA ligands. CRP proteins from E. coli and Mtb are dimeric (9,36,44), so we cross-linked the purified recombinant protein with glutaraldehyde to evaluate the oligomeric state of Cmr protein. A band slightly above the 60 KDa migration point, corresponding in size to a Cmr dimer, was detected by western blot using anti-Cmr serum on a glutaraldehyde-treated sample (Figure 1C). This result supports a homodimeric state for Cmr.
Figure 1.
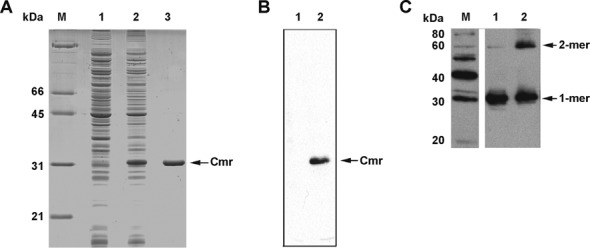
Overexpression, purification and dimerization of Cmr. (A) Cmr was overexpressed in Escherichia coli pMBC370, N-terminal 6x-His tagged Cmr protein expression strain and purified with a HisTrap column. Coomassie brilliant blue stained SDS-PAGE gel showing induction of protein expression. Lanes: M, Molecular weight marker; 1, uninduced pMBC370 lysate; 2, IPTG induced pMBC370 lysate; 3, purified His-Cmr. (B) Western blot using anti-His monoclonal antibody, showing overexpression of Cmr in induced E. coli pMBC370. Lanes: 1, uninduced bacterial cell lysate; 2, induced bacterial lysate. (C) Glutaraldehyde cross-linking of purified Cmr analyzed by western blotting using rabbit anti-Cmr antiserum. Lanes: M, Magic Mark XP Western protein standard; 1, purified Cmr; 2, glutaraldehyde cross-linked Cmr. The molecular weight of Cmr is 26.7 kDa. Note that the overexpressed protein contains a His tag of about 4.4 kDa. Therefore, the recombinant protein migrates at about 31 kDa.
Identification and confirmation of Cmr binding motif
The Cmr encoding genes in Mtb (cmr or Rv1675c) and M. bovis BCG (BCG_1713c) are identical. We combined SELEX-based affinity capture and computational methods to identify genomic Cmr binding sites in TB complex bacteria, using M. bovis BCG as a model system. Cmr-bound genomic DNA fragments were recovered using gel electrophoresis or agarose bead-based SELEX approaches (see ‘Materials and Methods’ section). Nine DNA sequences that were confirmed by EMSA to strongly bind His-Cmr were chosen for further characterization (Supplementary Figure S1, Table S1). The specificity of interaction for each confirmed DNA fragment with Cmr was further established by using competitor DNA in EMSA experiments. On addition of excess unlabeled specific competitor (identical DNA fragment as the probe), the band shifts corresponding to the DNA–protein complexes were lost. In contrast, these Cmr–DNA interactions were unaffected by addition of excess unlabeled non-specific competitor DNA (Supplementary Figure S1B, S1C).
EMSA was also performed to define a 40-bp DNA region within probe 1C1 that is sufficient for Cmr binding (Figure 2A and B). Specific binding was confirmed using an excess of this unlabeled 40-bp 1C1 region as competitor DNA in the binding reactions (Figure 2C). Similar results were obtained for clones 2C5 and 2G5 (Supplementary Figure S2), so we further defined the Cmr binding site using a DNase I footprinting assay. A 34-bp region within the 40-bp 1C1 binding sequence, which includes the 16-bp palindrome TGTCAccggtgACACT was protected from DNase I digestion in the presence of His-Cmr (Figure 2D). The importance of this DNA sequence for Cmr binding was further established by partially replacing it in a 42-bp DNA fragment from clone 1C1. The palindrome-substituted 1C1 DNA (Mt) fragment could neither bind with Cmr, nor outcompete WT probe for Cmr binding (Figure 2E). This indicates that sequence within this 16-bp palindrome is essential for 1C1 DNA binding with Cmr. Alignment of the combined DNA binding sequences from all SELEX experiments resulted in a consensus sequence consisting of two 5-nt palindromic half sequences separated by a 6-nt spacer (TGTCANNNNNNC/TGACA), with 70% identity to the 1C1 probe (Supplementary Table S1).
Figure 2.
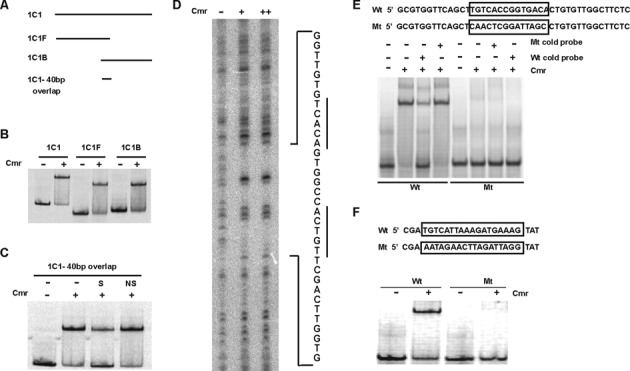
Mapping Cmr's DNA binding sequence in SELEX fragment 1C1. (A) Schematic of DNA fragments used to map Cmr binding site in 1C1. 1C1, full-length fragment (185 bp); 1C1F, forward fragment (101 bp); 1C1B, back fragment (124 bp). There is a 40 bp overlap between 1C1F and 1C1B, shown by 1C1–40 bp overlap. (B) EMSA, showing in vitro DNA binding ability of Cmr with 1C1, 1C1F and 1C1B as probes, in the absence or the presence of 300 nM His-Cmr recombinant protein, as indicated (C) EMSA showing DNA binding ability of His-Cmr with the 40 bp overlapping DNA fragment between 1C1F and 1C1B, as probe. Lane denoted ‘S’ was supplemented with 500× unlabeled 1C1 40 bp fragment as specific competitor DNA; Lane denoted ‘NS’ was supplemented with a 40 bp DNA fragment upstream of Rv1624c, as non-specific competitor DNA. (D) DNase I footprinting assay with His-Cmr using 1C1 DNA as template. 1C1 DNA was digested in the absence of Cmr (lane indicated ‘−’) or in the presence of 1.5 μM (lane ‘+’) or 3 μM Cmr (lane ‘++’). Sequence of the region protected from DNase I digestion, in the presence of Cmr, is shown adjacent to the figure. The palindromic half sites in the sequence are underlined. (E) Confirmation of Cmr's DNA binding motif recognition specificity, using mutational analyses. Sequence mutation of Cmr binding motif in 1C1 40 bp overlap region. The boxed WT (Wt) sequence was replaced as boxed in the mutant sequence (Mt). EMSA showing DNA binding ability of His-Cmr with WT and mutated motif sequences in 1C1 overlap region, in the absence (−) or presence (+) of 300 nM Cmr. 500× cold competition (Wt or Mt DNA) was used wherever indicated. (F) Sequence mutation of Cmr-binding motif in Rv1265 promoter. The underlined sequence in Rv1265 WT promoter (Wt) was replaced as shown in the mutant sequence (Mt). EMSA showing binding ability of His-Cmr with Rv1265 WT and mutated promoter sequences in the absence (−) or presence (+) of 900 nM Cmr.
We previously showed that Cmr regulates expression of Rv1265 within macrophages and binds the DNA sequence upstream of the Rv1265 ORF (32). Replacement of a DNA sequence in the Rv1265 upstream binding region that is similar to the essential palindromic sequence in the 1C1 fragment also abolished Cmr binding in this region (Figure 2F). These experiments further established the biological importance of these 16-bp palindromic DNA sequences for Cmr binding and provided additional evidence for direct regulation of Rv1265 by Cmr (32).
The predicted HTH region of Cmr is essential for DNA binding
HTH domains are common as DNA binding motifs in many transcription factors and DNA binding proteins. We used a HTH prediction program (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_hth.html) to predict the HTH sequence of Cmr as 195LAQRTLAAMLGAQRPSINKILK216. We expressed truncated versions of Cmr that included or lacked the predicted HTH region (Figure 3A, Supplementary Figure S3) and compared their DNA binding abilities to that of full-length Cmr using EMSA. The truncate that retained the HTH region (HTH+) bound with the DNA probes, while the polypeptide that lacked the HTH region (HTH−) failed to bind DNA (Figure 3B). This binding of Cmr to DNA probes was specific, as excess unlabeled competitor probe was able to compete away the binding (Figure 3C) and indicates that the predicted HTH region is necessary for Cmr's DNA binding ability.
Figure 3.
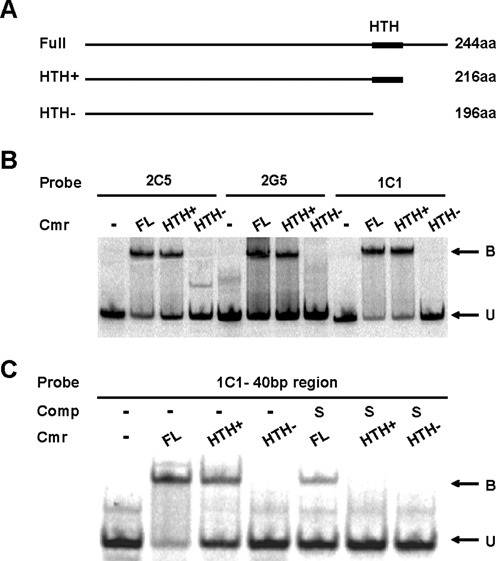
DNA binding using truncated His-Cmr proteins. (A) Schematic of full length and truncated Cmr protein. Amino acids 217–244 deleted in HTH+. Amino acids 195–244 deleted in HTH− (B) EMSA showing DNA binding ability of purified full-length and truncated Cmr proteins with SELEX fragments 2C5, 2G5 and 1C1 fragment as DNA probes. A total of 300 nM of full-length, HTH+ or HTH− Cmr was used wherever indicated. ‘B’ indicates bound protein–DNA complex, and ‘U’ indicates unbound DNA. (C) EMSA showing DNA binding ability of purified full-length and truncated Cmr proteins with the 40 bp overlap region of 1C1 as probe. A total of 300 nM of full-length, HTH+ or HTH− Cmr was used wherever indicated. Lanes denoted ‘S’ also contains 500× of unlabeled 1C1 40 bp overlap DNA as competitor DNA.
Genome wide ChIP-seq and functional classification analysis
We next performed Chromatin Immunoprecipitation with high-throughput sequencing (ChIP-seq) to characterize Cmr's DNA binding profile in vivo. For clarity, we will refer to experiments using recombinant purified protein as in vitro, whereas ChIP-seq experiments are referred to as in vivo, because they identify transcription factor binding to DNA sequences within viable bacterial cells. Experiments were performed in the presence or absence of excess cAMP, because Cmr was identified previously as the regulator of a subset of genes responsive to cAMP treatment (32).
Preliminary studies were performed using M. bovis BCG WT and M. bovis BCG (pRv0805:Rv1264cat) strains. Both strains express basal levels of native Cmr, and the latter strain also produces excess cAMP endogenously (32). These experiments yielded 26 distinct regions that were enriched for Cmr binding. However, previously known regulatory targets of Cmr such as groEL2 (Rv0440), Rv1265 (32) and strong SELEX candidates like 1C1, identified in this study were not enriched for Cmr binding in these experiments. Several of the enriched regions from this ChIP-seq experiment were tested for in vitro binding with Cmr using EMSA, but none showed strong binding (Supplementary Figure S4). In addition, MEME (http://meme.nbcr.net/meme/cgi-bin/meme.cgi) was not able to detect a binding motif with high confidence among these enriched DNA regions. Together, these results suggested that the binding regions obtained with M. bovis BCG expressing basal levels of Cmr were not sufficiently representative of Cmr's direct DNA binding profile in vivo.
Transcription factor expression can vary widely in response to environmental stimuli and/or growth phase (45,46), and the Cmr expression level in WT M. bovis BCG grown in the same conditions as the ChIP-seq experiment was very low (Supplementary Figure S5). We reasoned that the environmental conditions used in our experiment might not be optimal for detecting Cmr expression and/or target binding using an immunoprecipitation-based method like ChIP-seq. Other investigators have increased transcription factor expression to achieve comprehensive ChIP-seq analyses of all possible DNA interactions (45,47), and found that inducing transcription factor expression to saturating levels improves the occupancy of strong sites while augmenting the identification of weaker affinity binding sites (8).
To improve our ChIP-seq sensitivity, we performed additional experiments with an overexpression strain, M. bovis BCG (pMBC357) in the absence or presence of exogenous dibutyryl cAMP (dbcAMP) to elevate cAMP levels within M. bovis BCG, as previously described (33). Experiments using bacteria that overexpressed native Cmr protein revealed ∼200 enriched regions that were not detected with basal Cmr expression levels. The highest magnitude of enrichment observed in experiments with elevated Cmr levels was 42-fold compared with 17-fold in the basal expression strain. In contrast to the basal expression results, Cmr binding regions identified using the overexpression model included known Cmr binding sites and regulatory targets, as detailed below.
The binding enrichment data from all experiments was cross-referenced to the Mtb H37Rv genome for analysis, and the BRACIL deconvolution method was used to detect individual binding sites within the enriched regions. BRACIL is a post peak calling method that is designed to specifically detect multiple sites within a ChIP-seq enriched region with very high sensitivity (40). Application of this deconvolution algorithm to the Cmr enriched regions revealed multiple Cmr binding sites clustered within some enriched regions. The number of enriched regions and deconvolved binding sites from each experiment are listed in Supplementary Table S2. Consolidating throughout all experiments, 368 Cmr binding sites were identified, of which 67.7% mapped within open reading frames (intragenic binding sites) and 32.3% mapped to intergenic regions. A detailed distribution of these sites in context of their exact placement in intragenic or intergenic regions is presented in Supplementary Figure S6A.
The normalized binding peak heights of Cmr across the genome are plotted in Figure 4A. Functional classifications of the genes associated with Cmr binding sites were compared to the relative percentages of each group within the Mtb H37Rv genome, using Tuberculist as a reference (Figure 4B). The functional classification percentages were calculated separately using either binding regions or individual deconvolved binding sites, to address the issue of site clustering observed at many loci. Genes associated with Cmr binding sites were significantly enriched for ‘Cell wall’ and ‘Stable RNAs’ categories, when all binding sites were considered, suggesting a key biological role for Cmr in regulating these gene classes in Mtb. Conversely, genes associated with ‘Insertion sequences’, ‘Intermediary metabolism’ and ‘Conserved hypothetical proteins’ were underrepresented for associated Cmr binding sites.
Figure 4.
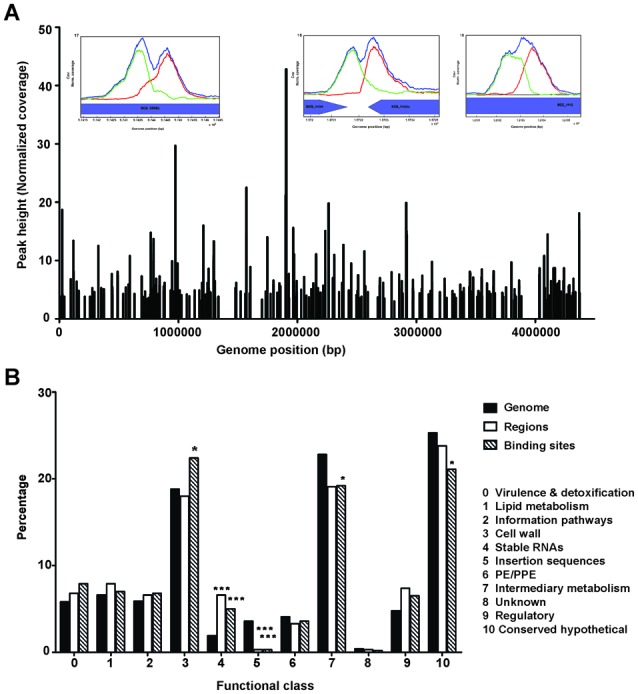
Coverage and functional classification of Cmr binding sites. (A) Genome-wide distribution of Cmr binding sites obtained using ChIP-seq. Inset figures show bimodal peak plots at representative binding sites in/around Rv0846c (BCG_0898c), Rv1373c (BCG_1434) and Rv1057 (BCG_1115). (B) Functional classification of the ChIP-seq binding sites in various categories, as annotated in Tuberculist (http://tuberculist.epfl.ch/). ‘Genome’ is shown as a reference for the overall distribution of genes as annotated in the H37Rv genome. ‘Regions’ represents the percentages calculated using enriched regions to account for the effects of site clustering. ‘Binding sites’ refers to the distribution of all sites without accounting for the redundancy due to site clustering. Statistical significance was determined using hypergeometric calculations, with a correction factor of 11. *P < 0.05, **P < 0.005, ***P < 0.0005. Note that the Tuberculist annotations do not include all non-coding RNAs identified recently, and therefore have not been included in this analysis.
Comparison of in vitro and in vivo DNA binding motifs
Surprisingly, one of the strongest in vitro SELEX candidates, 1C1, occurred as a relatively low enrichment region in vivo in the ChIP-seq experiment. Therefore, the DNA motifs obtained using the affinity capture experiments and the ChIP-seq analyses were compared to identify possible differences between Cmr's in vitro and in vivo binding profiles. A Cmr binding motif for each group was determined using MEME (Figure 5A and B), which refined the consensus sequence determined by the detailed molecular analyses (Figure 2). Nucleotides at positions 3T and 14A were conserved strongly in both motifs, while two additional positions 2G and 15C appeared to be critical only in the model generated using the ChIP-seq hits (Figure 5B). We replaced each of these conserved nucleotide positions with its complementary purine or pyrimidine base (i.e. 2G to C, 3T to A, 14A to T and 15C to G) individually within a 22-bp sequence to determine the importance for Cmr binding of each of the designated motif positions. Nucleotides 8T and 9G served as negative controls because they were not conserved in either motif model.
Figure 5.
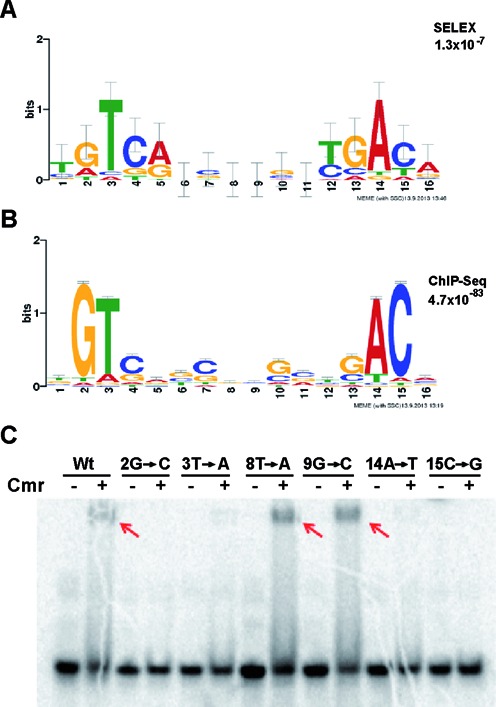
Comparison of Cmr's DNA binding motif obtained via modified-SELEX and ChIP-seq. Both the motifs were generated using http://meme.nbcr.net/meme/cgi-bin/meme.cgi. (A) Motif generated using SELEX hits, listed in Supplementary Table S1. (B) Motif generated using ChIP-seq binding sites. (C) EMSA used to test the critical nucleotide positions in the motif. A 22-bp DNA fragment containing the 16-bp DNA binding motif in 1C1 was used as WT sequence (WT). The indicated positions were replaced with their alternative purine or pyrimidine base to maintain the AT/GC content of the DNA fragment used. DNA–protein binding reactions were run on a 12% native PAGE gel.
Cmr was unable to bind DNA when the highly conserved nucleotides 2G, 3T, 14A or 15C were mutated, but substitution of non-conserved positions 8T and 9G did not affect Cmr-DNA binding (Figure 5C). These results show that all four of the highly conserved nucleotides contribute to Cmr binding in vitro. The identification of additional critical positions in the ChIP-seq generated motif may be explained by the larger sample size used to generate the motifs, or by differences between in vivo versus in vitro environments.
Characterization of top scoring Cmr binding peaks from ChIP-seq
We further characterized the 10 highest scoring Cmr binding regions to gain an overview of Cmr's role in Mtb biology. ChIP-seq can detect direct (protein of interest bound to DNA) as well as indirect transcription factor binding (protein of interest bound to another protein that interacts with DNA). Mtb H37Rv DNA sequences were used in these in vitro EMSA experiments to assess Cmr binding to these specific regions of the Mtb H37Rv genome, shown schematically in Figure 6A. The Mtb sequences corresponding to all ten regions bound with Cmr by EMSA (Figure 6B), confirming direct binding of Cmr with DNA in all enriched regions. In vitro binding of Cmr to the ChIP-seq binding sites was shown to be specific using competition assays (Supplementary Figure S7), but no obvious effect of cAMP was observed in these in vitro experiments (Figure 6B).
Figure 6.

In vitro binding of ChIP-seq enriched regions using EMSA. Genomic context of the top 10 ChIP-seq enriched regions for Cmr in Mycobacterium bovis BCG genome were mapped to the Mtb H37Rv genome by sequence similarity using BLAST. (A) Cmr binding sites in the top 10 enriched regions mapped to the H37Rv genome are shown schematically (maps not drawn to scale). Cmr binding sites (red boxes), primers (black arrows) used to amplify DNA probes (blue) are shown. Primer sequences are listed in Supplementary Table S3 in supplementary material. (B) EMSA showing in vitro binding of Cmr to probes shown in (A). A total of 300 nM Cmr and 10 mM final concentration of cAMP was added to the DNA binding reaction wherever indicated. EMSAs have been pooled from multiple independent experiments, and therefore the probe sizes between gel images are not comparable.
The highest enrichment for Cmr binding was observed at a site within the open reading frame of BCG_1714 (ortholog of Rv1676), located 150 bp upstream of the Cmr translational start site. Cortes et al. (48) re-annotated the translational start site of Rv1676 to 129 nucleotides downstream of the present Tuberculist annotation, thus making the Cmr binding site in this region intergenic to Rv1675c and Rv1676. Additional sequences that failed to show the signature bimodal peak in this region were not considered for further analysis. The second top-scoring peak, with 24-fold enrichment, occurred within the open reading frame of BCG_0898c (ortholog of Rv0846c), a gene shown to have multicopper oxidase activity. Cmr binding to this region in Mtb could have regulatory effects on either Rv0846c or the divergently expressed lpqS, a lipoprotein, also shown to belong to the copper sensitivity regulon (49). The third ranking peak occurred in the intergenic region between orthologs of convergent genes BCG_1434 (Rv1373) and BCG_1435c (Rv1374c), 57 bp upstream of the small non-coding RNA MTS1082 (MTB000076, ncRv1374). A cluster of Cmr binding sites with strong enrichment was also found in the open reading frame of the Rv1057 ortholog, BCG1115, the gene encoding for the only beta-propeller protein in Mtb (50). Binding sites for CRPMt (31), LexA (51), MprA (52), EspR (10) and TrcR (53) have been previously reported in the region upstream of Rv1057. The density and diversity of these potential regulatory targets suggests a complex regulatory role for Cmr with interplay between multiple transcription factors.
Autoregulation of cmr expression
The binding site upstream of cmr bound specifically with Cmr by EMSA (Figures 6B and 7A, Supplementary Figure S7), and we reasoned that it may contribute to cmr regulation. A cmr knockout strain (32), in which 180 bp at the 5′ end of the cmr open reading frame remain intact (Figure 7B and ‘Materials and Methods section’ for details), allowed the use of semi-quantitative RTPCR to study cmr expression by using primers that amplify this short uninterrupted region of cmr. The transcripts spanning this region are present in both the WT and the knockout strains. We observed increased expression of cmr in the knockout strain compared to the WT, indicating repression of cmr expression (Figure 7C and D). Complementation of the BCGΔcmr strain with a WT cmr copy restored the expression to WT levels, after taking into account the doubling of transcripts for the amplified region in this strain (Figure 7B).
Figure 7.
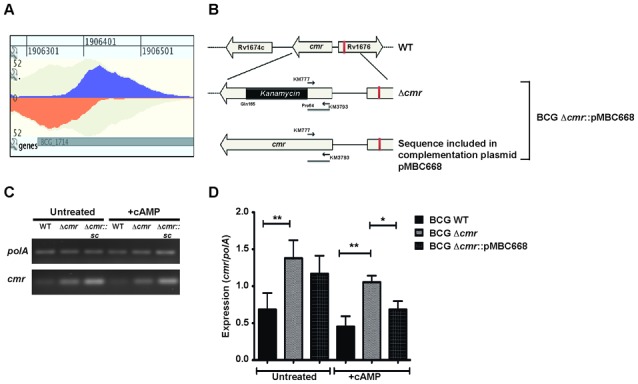
Autoregulation of cmr expression. (A) Bimodal peak plot from GenomeView showing Cmr binding site upstream of Rv1675c (BCG_1713c). The peak is centered 150 bp upstream of cmr translational start site and 60 bp into the open reading frame of divergent gene, Rv1676. In vitro binding of Cmr to this region is shown in Figure 6B. (B) Schematic map showing the genomic organization of the cmr locus in Mycobacterium bovis BCG and Mtb H37Rv WT, Δcmr and complementation strains. Black arrows indicate the primers used in semi-quantitative RTPCR. (C) Semi-quantitative RTPCR analyses showing relative expression of cmr in M. bovis BCG WT, BCGΔcmr and complementation strain, BCGΔcmr::pMBC668, under −/+ exogenous cAMP treatment conditions. The cDNA across samples was normalized to polA expression levels. A representative image from three biologically independent experiments is shown. (D) Quantitation of relative expression of cmr normalized to polA expression. Data presented as means ± S.E.M of three independent biological experiments, with technical duplicates. The asterisks above the bars indicate statistically significant difference calculated using two tailed, paired t-test. *P < 0.05, **P < 0.005.
Effect of cAMP on Cmr binding in vivo
We found strong concordance of Cmr binding enrichment between the untreated and dbcAMP treated ChIP-seq samples, but a small number of sites showed differential enrichment in response to cAMP. Eight binding sites within six regions had increased peak heights (>1.5×) in the presence of dbcAMP compared to untreated conditions (Figure 8A), while 18 binding sites distributed among six loci were decreased in binding enrichment (>1.5×). All but one of these 18 decreased enrichment binding sites are within or adjacent to members of the DosR regulon, including Rv2623, Rv2030c and Rv1739c (Figure 8A and B). Nine additional Cmr binding sites were also associated with DosR regulon members, but cAMP did not affect binding at these sites. The peak height ratios between +/− dbcAMP treatments for all ChIP-seq binding sites, analyzed on the basis of their association with the DosR regulon, is shown in Figure 8B. These results suggest the potential role of Cmr and/or cAMP in the regulation of DosR regulon members.
Figure 8.
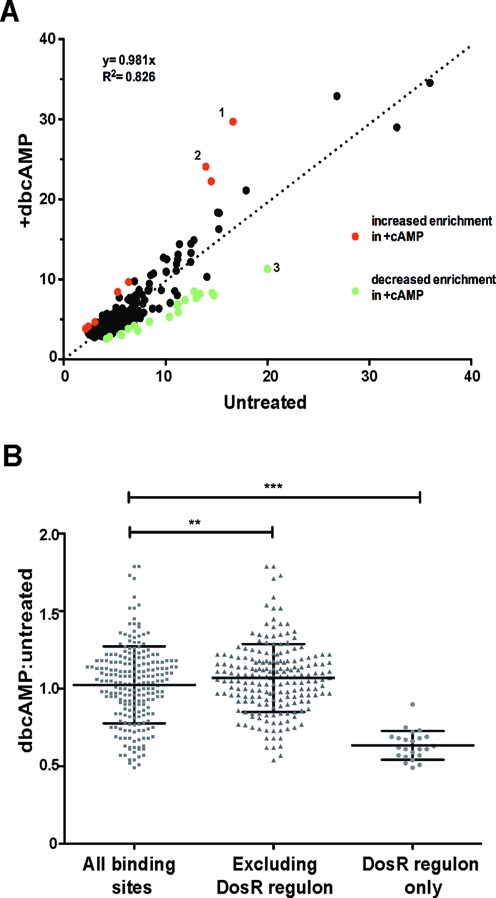
Effect of dbcAMP on Cmr's in vivo DNA binding profile. (A) Normalized peak height at all Cmr binding sites in untreated (x-axis) and +dbcAMP treatment (y-axis) is plotted on the graph. Binding sites that show greater than 1.5-fold increase or decrease in enrichment in +dbcAMP condition are shown in red and green respectively. Sites in Rv0846c (denoted 1,2) and Rv2623 (denoted 3) show the highest differential enrichment between −/+dbcAMP treatments (B) dbcAMP: untreated ratio of normalized peak height at binding sites plotted on y-axis. Differential ratio of binding at all ChIP-seq binding sites shown as gray squares; excluding members of DosR regulon shown as gray triangles and DosR regulon members exclusively shown as gray circles, along x-axis. Statistical significance was calculated using two-tailed unpaired Student's t-test. **P = 0.0085, ***P < 0.0001.
Effect of cAMP on Cmr binding to clusters of binding sites within dosR regulon genes
Clusters of relatively low affinity binding sites were identified at many loci, including M. bovis BCG orthologs of DosR regulon members Rv2623, Rv2030c, Rv1738 and Rv2626c. Cmr bound to most of these DNA regions in vitro, but distant binding sites were also needed for maximal Cmr binding in some cases. For example, a cluster of six Cmr binding sites was distributed across a 1-kb region at the Rv2030c-acr-acg locus. Moderate in vitro binding was observed using DNA probe (a) and (b) that contained only one binding site (Figure 9A). However, specific binding significantly improved when longer probes (c) and (d), containing multiple binding sites were used (Figure 9A, Supplementary Figure S7). Similarly, specific in vitro binding with Cmr was observed in the Rv2623 region, using a 580 bp DNA fragment that included all four Cmr binding sites and two DosR binding sites previously identified in this region, but not when the individual binding sites were tested independently (Figure 9B, Supplementary Figure S7). Specific in vitro binding with Cmr was also observed at the Rv1739c locus, which contains just two closely spaced binding sites (Figure 9C).
Figure 9.
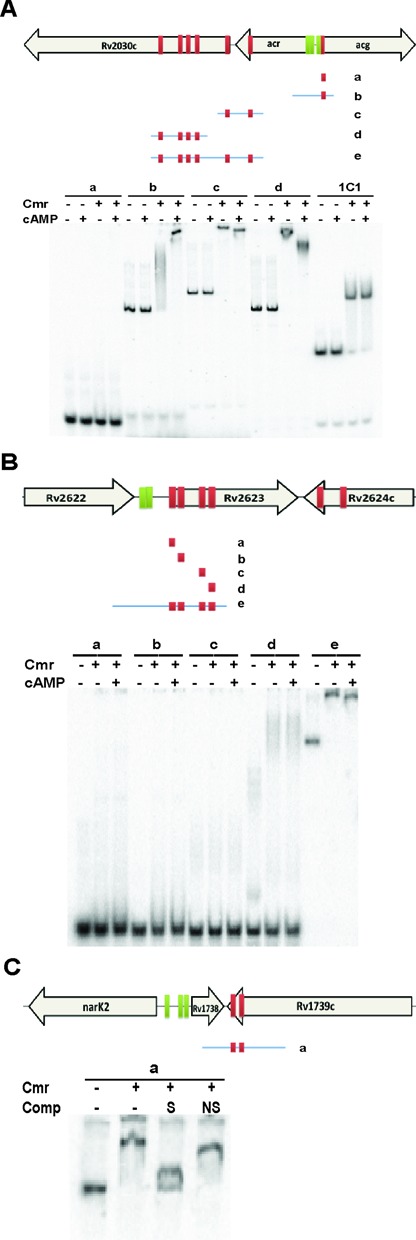
Effect of cAMP on in vitro binding of Cmr to clustered binding sites adjacent to genes belonging to DosR regulon. Schematic maps (not drawn to scale) and EMSA showing multiple Cmr binding sites in the (A) Rv2030c, (B) Rv2623 and (C) Rv1738. Cmr binding sites (red boxes), DosR binding sites (green boxes), and probes used in EMSA (blue lines with Cmr binding sites) are shown. A total of 1 μM Cmr and 1 mM cAMP was used in this set of EMSA experiments, wherever indicated. About 500× cold DNA identical to probe was used as specific competition (S). About 500× annealed oligos from an unrelated region (listed in Supplementary Table S3) was used as non-specific competition (NS).
In contrast to the results using smaller DNA probes (Figure 6B), cAMP affected the mobility of multi-site DNA probes in the Rv2030c and Rv2623 regions, bound with Cmr (Figure 9A and B). This differential mobility of DNA-protein co-complexes in the presence of cAMP suggests that cAMP alters the binding of Cmr to these DNA regions, which could have relevance for gene regulation in vivo.
While high-affinity binding sites are more likely to regulate target genes, many weak- or low-affinity binding sites also have regulatory functions (54). To investigate the possible regulation of Rv2623 by Cmr, we used semi-quantitative RT-PCR to measure the expression of Rv2623 in M. bovis BCG WT, Δcmr and Δcmr::pMBC668 (single copy complementation strain) at the RNA and protein levels. Rv2623 expression is highly induced by DosR in shallow standing cultures relative to the low levels of basal expression that occur in ambient shaking cultures (55). We observed increased Rv2623 mRNA expression in the Δcmr strain relative to WT in non-inducing conditions (Figure 10A), and decreased Rv2623 mRNA expression in the Δcmr strain relative to WT under inducing conditions (Figure 10B). Similarly, Rv2623 protein expression was decreased in the Δcmr background relative to WT under inducing conditions (Figure 10C and D). Western blot analysis was not sensitive enough to detect Rv2623 protein levels under non-inducing conditions (not shown). Rv2623 expression was restored at both the mRNA and protein levels in the complementation strain (Figure 10), demonstrating that Cmr directly regulates the expression of Rv2623 in Mtb.
Figure 10.
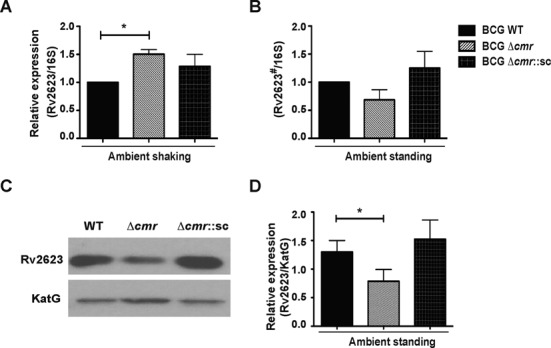
Cmr's role in Rv2623 expression. Semi-quantitative RTPCR showing expression of Rv2623 normalized to 16S rRNA expression in Mycobacterium bovis BCG WT, BCGΔcmr and single copy integrating complementation strain BCG Δcmr::pMBC668 (referred to as BCG Δcmr::sc) strains grown in (A) non-inducing (shaking) or (B) inducing (standing) ambient air conditions. Rv2623 expression is highly induced in ambient standing conditions. Note that amplification of the template during semi-quantitative RT PCR makes it possible to detect Rv2623 expression in non-inducing conditions, but the cDNA requires at ∼100-fold dilution for measurement of changes in Rv2623 expression levels under inducing conditions. The ‘#’ denotes the use of 100-fold diluted cDNA in the inducing condition compared to that used in the non-inducing condition. The data are presented as average ± S.E.M of four independent biological experiments. (C) Representative western blot image showing the expression of Rv2623 protein in M. bovis BCG WT, Δcmr and Δcmr::pMBC668 in ambient standing conditions. KatG protein expression is shown as loading control. Baseline Rv2623 protein expression is undetectable in non-inducing conditions. (D) Bar graph showing the quantitation of Rv2623 expression compared to expression of KatG. Data presented as means ± S.E.M of three independent biological experiments. The asterisks above the bars indicate statistically significant difference, analyzed using two-tailed, paired t-test. *P < 0.05.
DISCUSSION
This study provides a comprehensive characterization of Cmr (Rv1675c), as a cAMP associated transcription factor in TB complex mycobacteria. Molecular analyses established the DNA binding motif recognized by Cmr, while ChIP-seq was employed to define genome-wide Cmr binding sites. Use of the BRACIL deconvolution method (40) allowed us to predict closely spaced Cmr binding sites within ChIP-seq enriched regions, providing a high level of resolution for downstream Cmr binding site analyses. We identified over 300 Cmr binding sites, including a number of clustered Cmr binding sites that are proximal to genes within the DosR regulon. cAMP affected Cmr binding to a subset of these clustered sites in vivo and in vitro, but not to single Cmr binding sites when tested individually. Cmr based regulation of members of the dormancy regulon, including Rv2623, suggests a role for Cmr in latency related adaptation of Mtb in the host environment.
Comparison with other CRP family transcription factors
Cmr is a CRP/FNR family transcription factor, but shares only ∼24% sequence identity with CRPMt over 206 amino acid residues (50–244 in Cmr and 19–224 in CRPMt). The motif recognized by Cmr is very similar to the CRPMt binding motif (9,30,31), but there was little overlap between the ChIP-seq binding sites for Cmr with the genome-wide CRP binding sites in Mtb. This result indicates that Cmr is able to distinguish among subtly different DNA binding motifs and controls a regulon that is distinct from those of its CRP-family counterparts. While the DNA binding motif recognized by Cmr (Figure 5) is nearly identical to the motif recognized by CO-responsive transcription factor, CooA in Rhodospirillum rubrum (56), CO had no effect on Cmr's DNA binding ability (data not shown). Both Cmr and CRPMt bind DNA via the HTH domain at the C terminal end of the proteins. Two of the three residues in the HTH domain (Arg188, Glu189 and Lys193) that are directly involved in DNA binding (9,24,57) are conserved between CRPMt and Cmr. However, Glu189 in CRPMt is replaced by a proline residue at the corresponding position in Cmr, which may contribute to binding specificity.
Escherichia coli CRP is one of the best studied CRP-family transcription factors, and it has been shown to both positively and negatively autoregulate its own expression (58,59). While no autoregulation has been reported for CRPMt, we show that Cmr autorepresses its own expression, likely by binding to a site centered 150 nt upstream of its translational start site (Figure 7). This Cmr binding site was the most highly enriched by ChIP-seq in this study. We were interested to note that the next highest Cmr enrichment mapped to the intragenic locus of Rv0846c, a mycobacterial multicopper oxidase which belongs to a copper sensitivity regulon (49). One of the strongest CRPMt binding sites occurs upstream of the putative copper transporter gene, ctpB (Rv0103c) (30,31), and we previously established a role for CRPMt in copper resistance in M. bovis BCG using a Δcrp mutant that showed increased sensitivity to copper treatment (30). This suggests an important role for CRP-family transcription factors in metal homeostasis in Mtb.
Intergenic and intragenic binding sites
Approximately 29% of Cmr binding sites occur in the linear or divergent intergenic regions, and ∼3% of sites are located in convergent intergenic regions (Supplementary Figure S6A). This total 32% of intergenic binding is much higher than that observed for CRP in Mtb, where only 17% of all binding sites were found to be intergenic (30). The Cmr binding site to TSS (transcription start site) distance analysis shows 57% of the mapped sites cluster in the −100 to +100 region around the TSS (Supplementary Figure S6B). ChIP-seq also revealed that 16% of Cmr binding sites occurs in the first 10% of annotated open reading frames. Taken together, this promoter proximal localization is consistent with canonical transcription factor binding behavior and suggestive of extended regulatory regions for Cmr. Nonetheless, many binding sites (51% of total binding sites) occur well into the intragenic regions, and their potential contributions to gene regulation cannot be dismissed. With the advent of techniques like ChIP-seq and ChIP-exo, the notion of regulatory elements being confined to intergenic regions (60,61) is being increasingly challenged. Galagan et al. (8) noted recently that a high frequency of intragenic binding sites has been observed for almost all transcription factors studied at the genomic level, and suggests the presence of more long range regulatory interactions than previously thought. Recent studies to map the flagellar regulatory network in E. coli showed that more than half of the ChIP-seq binding sites map to intragenic regions (46), while we previously established the contribution of intragenic CRPMt binding site in the regulation of Rv0249c-Rv0250c (30). Intragenic binding of transcription factors has also been observed in eukaryotes (62,63). It has been suggested that transcription factor binding in the exons of a gene has implications for its evolution, because its codon usage would be directed in a way that must also accommodate transcription factor binding requirements (64).
In addition to its role as a classical transcription factor, the clusters of closely spaced binding sites within some Cmr binding regions raise the possibility that Cmr can also contribute to gene regulation through a mechanism that involves localized genome reorganization. The role of nucleoid-associated proteins (NAPs) in restructuring the genome, thereby modulating the accessibility of transcription factors to certain binding sites, can have a substantial effect on the transcriptome. The impact of genome remodeling on gene regulation is increasingly being appreciated in Mtb with studies characterizing NAPs such as Lsr2 (Rv3597c) (65,66), EspR (Rv3849) (10), HNS (Rv3852) (67), miHF (Rv1388) (68) and hupB (Rv2986c) (69). Further exploration of the possibility that Cmr has multiple roles on the continuum from canonical transcription factor to NAP is warranted, and the DosR regulon may be especially relevant in this regard.
Clustering of Cmr binding sites near members of the DosR regulon
We were particularly interested to note a correlation between clustered Cmr binding sites and their proximity to DosR-regulated genes. The DosR (DevR) dormancy regulon is comprised of 48 genes distributed among nine groups across the Mtb genome. Hypoxia, nitric oxide treatment or growth in shallow standing cultures without agitation has been shown to induce expression of these genes (70–72). In vitro studies showed dosR to be important for Mtb survival in hypoxic environments (70) and upregulation of DosR regulon members is also associated with the increased virulence and spread of the W-Beijing Mtb lineage of Mtb (73–75).
We observed 26 closely spaced Cmr binding sites distributed in the vicinity of seven of the DosR regulon gene clusters. In particular, deconvolution of the Cmr enriched regions revealed six Cmr binding sites at the acr/acg locus and four proximal to Rv2623. In vitro binding of Cmr to these individual binding sites from these regions, as measured by EMSA, was very poor, but robust binding was observed when multiple binding sites were present (Figure 9). The need for longer DNA sequences spanning multiple binding sites suggests cooperative binding of Cmr to these regions. Closely spaced transcription factor binding sites could be a way to modulate the strength of the regulatory signal, and the use of cooperative binding of transcription factors to interspersed strong and weak binding sites is an additional way to fine tune expression levels in response to multiple signals (76).
Multiple DosR binding motifs are located upstream of Rv2623, divergent genes acr and acg, and divergent genes Rv1738 and narK2. Extensive work characterizing the regulatory regions of DosR regulon members has indicated complex control of this regulon both DosR-dependent and DosR-independent manners (55,77–80). For example, MprA binding motifs upstream of DosR regulon members, Rv1812c-Rv1813c (81) and components within the Rv0081- Rv0088 gene cluster are co-regulated by DosR, MprA and Rv0081 (82). In this study, we establish Cmr as another co-regulator of Rv2623 expression. Rv2623 is an ATPase with a role in the establishment of persistent Mtb infection (75), which suggests that Cmr also contributes to latency-related adaptation of Mtb. The multitude of DosR and Cmr binding sites in the region upstream of known DosR regulon members warrants further investigation into the potential of cooperativity between Cmr and/or DosR binding.
cAMP effect on Cmr's DNA binding
Although Cmr was first established as a regulator of genes differentially expressed in an exogenous cAMP model (32), the relationship of Cmr with cAMP has been difficult to define. Cmr binding of individual sites was not affected by cAMP using EMSA, and physical interactions between Cmr and cAMP have not been observed (unpublished results). Nonetheless, we observed differential enrichment of Cmr at a subset of binding sites in the presence of cAMP in vivo (Figure 8A), most of which are located proximal to members of the DosR regulon (Figure 8B). The mobility of these Cmr–DNA complexes was affected by the addition of cAMP in vitro (Figure 9), consistent with an effect of cAMP on the conformation of the Cmr–DNA complex. Both E. coli CRP and CRPMt have been shown to bend their DNA targets upon binding, and we propose that the presence of cAMP affects Cmr's ability to shape its target DNA upon binding as well. The mechanism by which cAMP mediates its effect on Cmr–DNA complex formation is not clear. However, a localized DNA conformation effect mediated by Cmr and cAMP could have important biological implications for the regulation of Rv2623, acr and acg, where the concentration of clustered Cmr binding sites is greatest. In E. coli, cAMP induces a conformational change in CRP that is required for specific DNA binding (83–85). Binding of cAMP to CRPMt has been demonstrated, and shown to improve its DNA binding in vitro (9,24). However, with the exception of a few DNA loci (26,27), cAMP is not necessary for DNA binding by CRPMt (9,24). Similarly, cAMP is not necessary for DNA binding by Cmr (Figure 6B). We speculate that cAMP could be involved in DNA binding by Cmr in regions where multiple Cmr binding sites are present, affecting the affinity of Cmr to particular binding sites. Six residues in E. coli CRP were found to be involved in cAMP binding (57), four of which are conserved in CRPMt (24). However, none of these residues are conserved in Cmr. This suggests that the mechanism of cAMP binding to Cmr is different than that in E. coli CRP and CRPMt. The effect of cAMP on Cmr and CRPMt's DNA binding ability to particular DNA sequences is particularly intriguing to us and currently under investigation.
Comparison of native and overexpression ChIP-seq
Our preliminary ChIP-seq experiment with basal levels of Cmr expression yielded 26 enriched regions, 17 of which were also detected with low levels of enrichment in the ChIP-seq experiment using the overexpression strain. However, we were not able to validate any of these enriched regions as bona fide Cmr binding sites, as they failed to yield a common motif or show robust direct binding with Cmr in gel shift assays (Supplementary Figure S4). In contrast, DNA sequences obtained from the overexpression ChIP studies contained a strong motif, showed robust direct binding with Cmr and contributed to Cmr-dependent gene regulation in WT bacteria. Nonetheless, it remains possible that at least some enriched regions from the basal ChIP-seq experiment contain legitimate binding sites for which as yet unknown in vivo cofactors facilitate interactions with Cmr at low concentrations, but are absent in the presence of high levels of Cmr.
Transcription factor expression can vary widely in response to environmental stimuli and/or growth phase (45,46). Overexpression of transcription factors allows an overall view of all possible binding sites across the genome instead of a snapshot of binding sites in particular physiological environments, with the calculated risk of identifying some false positives. Definite regulatory function for a binding site can be assigned only by additional experiments, and a key result of this study is the biological validation of potential regulatory targets of Cmr identified using the ChIP-seq experiments. Our demonstration that Cmr regulates cmr and Rv2623 (Figures 7 and 10), which were only identified when Cmr was overexpressed, illustrates the usefulness of transcription factor overexpression as a tool to identify potential regulatory targets of transcription factors that are expressed transiently or at low levels in the chosen assay conditions.
Future studies should be directed toward understanding the regulatory role of Cmr in Mtb, conditions that affect Cmr expression, the relationship between Cmr, cAMP and other possible interacting partners, and the significance of its genome wide binding sites, both intragenic and intergenic. The overlapping regulons of multiple transcription factors highlights the complexity of gene regulation in Mtb, and its ability to integrate diverse environmental cues to adapt within the host.
Supplementary Material
Acknowledgments
We gratefully appreciate the valuable technical assistance provided by Damen Schaak, Dr Zhuo Ma, Eric Smith, Dr. Sahadevan Raman and Dr Sang Tae Park. We thank Dr John Chan and Dr Siva Chavadi for generously providing anti-Rv2623 antibody used in this study. We thank members of the McDonough lab for their helpful discussions. We acknowledge the Wadsworth Center Molecular Genetics Core for DNA sequencing support and BEI Resources for reagents.
Footnotes
Present addresses:
Guangchun Bai, Center for Immunology and Microbial Disease, Albany Medical College, Albany, NY 12208, USA.
Anna Lyubetskaya, Broad Institute, Boston, MA 02142, USA.
Antonio Gomes, Department of Systems Biology, Columbia University, New York, NY 10032, USA.
Michaela Gazdik, Intermountain Healthcare, Salt Lake City, UT 84111, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [HHSN272200800059C to J.E.G; R01A1063499, R01AI045658 to K.A.M]. Funding for open access charge: NIH [R01AI063499].
Conflict of interest statement. None declared.
REFERENCES
- 1.WHO. Global Tuberculosis Report 2014. Geneva: World Health Organization; 2014. [Google Scholar]
- 2.Corbett E.L., Watt C.J., Walker N., Maher D., Williams B.G., Raviglione M.C., Dye C. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. intern. Med. 2003;163:1009–1021. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- 3.Espinal M.A. The global situation of MDR-TB. Tuberculosis (Edinb) 2003;83:44–51. doi: 10.1016/s1472-9792(02)00058-6. [DOI] [PubMed] [Google Scholar]
- 4.Bloom B.R., Murray C.J. Tuberculosis: commentary on a reemergent killer. Science. 1992;257:1055–1064. doi: 10.1126/science.257.5073.1055. [DOI] [PubMed] [Google Scholar]
- 5.Dye C., Scheele S., Dolin P., Pathania V., Raviglione M.C. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 6.Barry C.E. 3rd, Boshoff H.I., Dartois V., Dick T., Ehrt S., Flynn J., Schnappinger D., Wilkinson R.J., Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E. 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 8.Galagan J.E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I., et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499:178–183. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai G., McCue L.A., McDonough K.A. Characterization of Mycobacterium tuberculosis Rv3676 (CRPMt), a cyclic AMP receptor protein-like DNA binding protein. J. Bacteriol. 2005;187:7795–7804. doi: 10.1128/JB.187.22.7795-7804.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blasco B., Chen J.M., Hartkoorn R., Sala C., Uplekar S., Rougemont J., Pojer F., Cole S.T. Virulence regulator EspR of Mycobacterium tuberculosis is a nucleoid-associated protein. PLoS Pathog. 2012;8:e1002621. doi: 10.1371/journal.ppat.1002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Av-Gay Y., Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000;8:238–244. doi: 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]
- 12.Bishai W. The Mycobacterium tuberculosis genomic sequence: anatomy of a master adaptor. Trends Microbiol. 1998;6:464–465. doi: 10.1016/s0966-842x(98)01414-0. [DOI] [PubMed] [Google Scholar]
- 13.McDonough K.A., Rodriguez A. The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 2012;10:27–38. doi: 10.1038/nrmicro2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia D., Merey G., Nakayama S., Zheng Y., Zhou J., Luo Y., Guo M., Roembke B.T., Sintim H.O. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 2013;42:305–341. doi: 10.1039/c2cs35206k. [DOI] [PubMed] [Google Scholar]
- 15.Yang J., Bai Y., Zhang Y., Gabrielle V.D., Jin L., Bai G. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol. Microbiol. 2014;93:65–79. doi: 10.1111/mmi.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong Y., Zhou X., Fang H., Yu D., Li C., Sun B. Cyclic di-GMP mediates Mycobacterium tuberculosis dormancy and pathogenecity. Tuberculosis (Edinb) 2013;93:625–634. doi: 10.1016/j.tube.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 17.McCue L.A., McDonough K.A., Lawrence C.E. Functional classification of cNMP-binding proteins and nucleotide cyclases with implications for novel regulatory pathways in Mycobacterium tuberculosis. Genome Res. 2000;10:204–219. doi: 10.1101/gr.10.2.204. [DOI] [PubMed] [Google Scholar]
- 18.Shenoy A.R., Sreenath N.P., Mahalingam M., Visweswariah S.S. Characterization of phylogenetically distant members of the adenylate cyclase family from mycobacteria: Rv1647 from Mycobacterium tuberculosis and its orthologue ML1399 from M. leprae. Biochem. J. 2005;387:541–551. doi: 10.1042/BJ20041040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai G., Knapp G.S., McDonough K.A. Cyclic AMP signalling in mycobacteria: redirecting the conversation with a common currency. Cell Microbiol. 2011;13:349–358. doi: 10.1111/j.1462-5822.2010.01562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai G., Schaak D.D., McDonough K.A. cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol. Med. Microbiol. 2009;55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agarwal N., Lamichhane G., Gupta R., Nolan S., Bishai W.R. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature. 2009;460:98–102. doi: 10.1038/nature08123. [DOI] [PubMed] [Google Scholar]
- 22.Nambi S., Basu N., Visweswariah S.S. cAMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 2010;285:24313–24323. doi: 10.1074/jbc.M110.118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shenoy A.R., Visweswariah S.S. New messages from old messengers: cAMP and mycobacteria. Trends Microbiol. 2006;14:543–550. doi: 10.1016/j.tim.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Rickman L., Scott C., Hunt D.M., Hutchinson T., Menéndez M.C., Walan R., Hinds J., Colston M.J., Green J., Buxton R.S. A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol. Microbiol. 2005;56:1274–1286. doi: 10.1111/j.1365-2958.2005.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai G., Schaak D.D., Smith E.A., McDonough K.A. Dysregulation of serine biosynthesis contributes to the growth defect of a Mycobacterium tuberculosis crp mutant. Mol. Microbiol. 2011;82:180–198. doi: 10.1111/j.1365-2958.2011.07806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agarwal N., Raghunand T.R., Bishai W.R. Regulation of the expression of whiB1 in Mycobacterium tuberculosis: role of cAMP receptor protein. Microbiology. 2006;152:2749–2756. doi: 10.1099/mic.0.28924-0. [DOI] [PubMed] [Google Scholar]
- 27.Stapleton M.R., Smith L.J., Hunt D.M., Buxton R.S., Green J. Mycobacterium tuberculosis WhiB1 represses transcription of the essential chaperonin GroEL2. Tuberculosis (Edinb) 2012;92:328–332. doi: 10.1016/j.tube.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spreadbury C.L., Pallen M.J., Overton T., Behr M.A., Mostowy S., Spiro S., Busby S.J., Cole J.A. Point mutations in the DNA- and cNMP-binding domains of the homologue of the cAMP receptor protein (CRP) in Mycobacterium bovis BCG: implications for the inactivation of a global regulator and strain attenuation. Microbiology. 2005;151:547–556. doi: 10.1099/mic.0.27444-0. [DOI] [PubMed] [Google Scholar]
- 29.Stapleton M., Haq I., Hunt D.M., Arnvig K.B., Artymiuk P.J., Buxton R.S., Green J. Mycobacterium tuberculosis cAMP receptor protein (Rv3676) differs from the Escherichia coli paradigm in its cAMP binding and DNA binding properties and transcription activation properties. J. Biol. Chem. 2010;285:7016–7027. doi: 10.1074/jbc.M109.047720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knapp G.S., Lyubetskaya A., Peterson M.W., Gomes A.L., Ma Z., Galagan J.E., McDonough K.A. Role of intragenic binding of cAMP responsive protein (CRP) in regulation of the succinate dehydrogenase genes Rv0249c-Rv0247c in TB complex mycobacteria. Nucleic Acids Res. 2015;43:5377–5393. doi: 10.1093/nar/gkv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahramanoglou C., Cortes T., Matange N., Hunt D.M., Visweswariah S.S., Young D.B., Buxton R.S. Genomic mapping of cAMP receptor protein (CRPMt) in Mycobacterium tuberculosis: relation to transcriptional start sites and the role of CRPMt as a transcription factor. Nucleic Acids Res. 2014;42:8320–8329. doi: 10.1093/nar/gku548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazdik M.A., Bai G., Wu Y., McDonough K.A. Rv1675c (cmr) regulates intramacrophage and cyclic AMP-induced gene expression in Mycobacterium tuberculosis-complex mycobacteria. Mol. Microbiol. 2009;71:434–448. doi: 10.1111/j.1365-2958.2008.06541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gazdik M.A., McDonough K.A. Identification of cyclic AMP-regulated genes in Mycobacterium tuberculosis complex bacteria under low-oxygen conditions. J. Bacteriol. 2005;187:2681–2692. doi: 10.1128/JB.187.8.2681-2692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sassetti C.M., Boyd D.H., Rubin E.J. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sassetti C.M., Boyd D.H., Rubin E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 36.Bai G., Gazdik M.A., Schaak D.D., McDonough K.A. The Mycobacterium bovis BCG cyclic AMP receptor-like protein is a functional DNA binding protein in vitro and in vivo, but its activity differs from that of its M. tuberculosis ortholog, Rv3676. Infect. Immun. 2007;75:5509–5517. doi: 10.1128/IAI.00658-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen J.D., Gray D.M. Selection of genomic sequences that bind tightly to Ff gene 5 protein: primer-free genomic SELEX. Nucleic Acids Res. 2004;32:e182. doi: 10.1093/nar/gnh179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes A.L., Abeel T., Peterson M., Azizi E., Lyubetskaya A., Carvalho L., Galagan J. Decoding ChIP-Seq peaks with a double-binding signal refines binding peaks to single-nucleotide and predicts cooperative interaction. Genome Res. 2014;24:1686–1697. doi: 10.1101/gr.161711.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 43.Lew J.M., Kapopoulou A., Jones L.M., Cole S.T. TubercuList–10 years after. Tuberculosis (Edinb) 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Kolb A., Busby S., Buc H., Garges S., Adhya S. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- 45.Davies B.W., Bogard R.W., Mekalanos J.J. Mapping the regulon of Vibrio cholerae ferric uptake regulator expands its known network of gene regulation. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12467–12472. doi: 10.1073/pnas.1107894108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitzgerald D.M., Bonocora R.P., Wade J.T. Comprehensive mapping of the Escherichia coli flagellar regulatory network. PLoS Genet. 2014;10:e1004649. doi: 10.1371/journal.pgen.1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones C.J., Newsom D., Kelly B., Irie Y., Jennings L.K., Xu B., Limoli D.H., Harrison J.J., Parsek M.R., White P., et al. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog. 2014;10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cortes T., Schubert O.T., Rose G., Arnvig K.B., Comas I., Aebersold R., Young D.B. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013;5:1121–1131. doi: 10.1016/j.celrep.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi X., Festa R.A., Ioerger T.R., Butler-Wu S., Sacchettini J.C., Darwin K.H., Samanovic M.I. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. MBio. 2014;5 doi: 10.1128/mBio.00876-13. doi:10.1128/mBio.00876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haydel S.E., Clark-Curtiss J.E. The Mycobacterium tuberculosis TrcR response regulator represses transcription of the intracellularly expressed Rv1057 gene, encoding a seven-bladed beta-propeller. J. Bacteriol. 2006;188:150–159. doi: 10.1128/JB.188.1.150-159.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smollett K.L., Smith K.M., Kahramanoglou C., Arnvig K.B., Buxton R.S., Davis E.O. Global analysis of the regulon of the transcriptional repressor LexA, a key component of SOS response in Mycobacterium tuberculosis. J. Biol. Chem. 2012;287:22004–22014. doi: 10.1074/jbc.M112.357715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pang X., Samten B., Cao G., Wang X., Tvinnereim A.R., Chen X.L., Howard S.T. MprAB regulates the espA operon in Mycobacterium tuberculosis and modulates ESX-1 function and host cytokine response. J. Bacteriol. 2013;195:66–75. doi: 10.1128/JB.01067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pang X., Cao G., Neuenschwander P.F., Haydel S.E., Hou G., Howard S.T. The beta-propeller gene Rv1057 of Mycobacterium tuberculosis has a complex promoter directly regulated by both the MprAB and TrcRS two-component systems. Tuberculosis (Edinb) 2011;91(Suppl. 1):S142–S149. doi: 10.1016/j.tube.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galagan J., Lyubetskaya A., Gomes A. ChIP-Seq and the complexity of bacterial transcriptional regulation. Curr. Top. Microbiol. Immunol. 2013;363:43–68. doi: 10.1007/82_2012_257. [DOI] [PubMed] [Google Scholar]
- 55.Vasudeva-Rao H.M., McDonough K.A. Expression of the Mycobacterium tuberculosis acr-coregulated genes from the DevR (DosR) regulon is controlled by multiple levels of regulation. Infect. Immun. 2008;76:2478–2489. doi: 10.1128/IAI.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aono S., Takasaki H., Unno H., Kamiya T., Nakajima H. Recognition of target DNA and transcription activation by the CO-sensing transcriptional activator CooA. Biochem. Biophys. Res. Commun. 1999;261:270–275. doi: 10.1006/bbrc.1999.1046. [DOI] [PubMed] [Google Scholar]
- 57.Schultz S.C., Shields G.C., Steitz T.A. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 58.Aiba H. Autoregulation of the Escherichia coli crp gene: CRP is a transcriptional repressor for its own gene. Cell. 1983;32:141–149. doi: 10.1016/0092-8674(83)90504-4. [DOI] [PubMed] [Google Scholar]
- 59.Hanamura A., Aiba H. A new aspect of transcriptional control of the Escherichia coli crp gene: positive autoregulation. Mol. Microbiol. 1992;6:2489–2497. doi: 10.1111/j.1365-2958.1992.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 60.Lee D.J., Minchin S.D., Busby S.J. Activating transcription in bacteria. Annu. Rev. Microbiol. 2012;66:125–152. doi: 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 61.Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 62.MacQuarrie K.L., Fong A.P., Morse R.H., Tapscott S.J. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet. 2011;27:141–148. doi: 10.1016/j.tig.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hughes T.R., de Boer C.G. Mapping yeast transcriptional networks. Genetics. 2013;195:9–36. doi: 10.1534/genetics.113.153262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stergachis A.B., Haugen E., Shafer A., Fu W., Vernot B., Reynolds A., Raubitschek A., Ziegler S., LeProust E.M., Akey J.M., et al. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013;342:1367–1372. doi: 10.1126/science.1243490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gordon B.R., Imperial R., Wang L., Navarre W.W., Liu J. Lsr2 of Mycobacterium represents a novel class of H-NS-like proteins. J. Bacteriol. 2008;190:7052–7059. doi: 10.1128/JB.00733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J.M., Ren H., Shaw J.E., Wang Y.J., Li M., Leung A.S., Tran V., Berbenetz N.M., Kocincova D., Yip C.M., et al. Lsr2 of Mycobacterium tuberculosis is a DNA-bridging protein. Nucleic Acids Res. 2008;36:2123–2135. doi: 10.1093/nar/gkm1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Werlang I.C., Schneider C.Z., Mendonca J.D., Palma M.S., Basso L.A., Santos D.S. Identification of Rv3852 as a nucleoid-associated protein in Mycobacterium tuberculosis. Microbiology. 2009;155:2652–2663. doi: 10.1099/mic.0.030148-0. [DOI] [PubMed] [Google Scholar]
- 68.Mishra A., Vij M., Kumar D., Taneja V., Mondal A.K., Bothra A., Rao V., Ganguli M., Taneja B. Integration host factor of Mycobacterium tuberculosis, mIHF, compacts DNA by a bending mechanism. PLoS One. 2013;8:e69985. doi: 10.1371/journal.pone.0069985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prabhakar S., Annapurna P.S., Jain N.K., Dey A.B., Tyagi J.S., Prasad H.K. Identification of an immunogenic histone-like protein (HLPMt) of Mycobacterium tuberculosis. Tuber. Lung Dis. 1998;79:43–53. doi: 10.1054/tuld.1998.0004. [DOI] [PubMed] [Google Scholar]
- 70.Voskuil M.I., Schnappinger D., Visconti K.C., Harrell M.I., Dolganov G.M., Sherman D.R., Schoolnik G.K. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ohno H., Zhu G., Mohan V.P., Chu D., Kohno S., Jacobs W.R. Jr, Chan J. The effects of reactive nitrogen intermediates on gene expression in Mycobacterium tuberculosis. Cell Microbiol. 2003;5:637–648. doi: 10.1046/j.1462-5822.2003.00307.x. [DOI] [PubMed] [Google Scholar]
- 72.Purkayastha A., McCue L.A., McDonough K.A. Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr aene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect. Immun. 2002;70:1518–1529. doi: 10.1128/IAI.70.3.1518-1529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reed M.B., Gagneux S., Deriemer K., Small P.M., Barry C.E. 3rd. The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J. Bacteriol. 2007;189:2583–2589. doi: 10.1128/JB.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu Y., Coates A.R. Mycobacterium tuberculosis acg gene is required for growth and virulence in vivo. PLoS One. 2011;6:e20958. doi: 10.1371/journal.pone.0020958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drumm J.E., Mi K., Bilder P., Sun M., Lim J., Bielefeldt-Ohmann H., Basaraba R., So M., Zhu G., Tufariello J.M., et al. Mycobacterium tuberculosis universal stress protein Rv2623 regulates bacillary growth by ATP-Binding: requirement for establishing chronic persistent infection. PLoS Pathog. 2009;5:e1000460. doi: 10.1371/journal.ppat.1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oehler S., Eismann E.R., Kramer H., Muller-Hill B. The three operators of the lac operon cooperate in repression. EMBO J. 1990;9:973–979. doi: 10.1002/j.1460-2075.1990.tb08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Florczyk M.A., McCue L.A., Purkayastha A., Currenti E., Wolin M.J., McDonough K.A. A family of acr-coregulated Mycobacterium tuberculosis genes shares a common DNA motif and requires Rv3133c (dosR or devR) for expression. Infect. Immun. 2003;71:5332–5343. doi: 10.1128/IAI.71.9.5332-5343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hutter B., Dick T. Analysis of the dormancy-inducible narK2 promoter in Mycobacterium bovis BCG. FEMS Microbiol. Lett. 2000;188:141–146. doi: 10.1111/j.1574-6968.2000.tb09185.x. [DOI] [PubMed] [Google Scholar]
- 79.Chauhan S., Tyagi J.S. Interaction of DevR with multiple binding sites synergistically activates divergent transcription of narK2-Rv1738 genes in Mycobacterium tuberculosis. J. Bacteriol. 2008;190:5394–5403. doi: 10.1128/JB.00488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chauhan S., Tyagi J.S. Cooperative binding of phosphorylated DevR to upstream sites is necessary and sufficient for activation of the Rv3134c-devRS operon in Mycobacterium tuberculosis: implication in the induction of DevR target genes. J. Bacteriol. 2008;190:4301–4312. doi: 10.1128/JB.01308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bretl D.J., He H., Demetriadou C., White M.J., Penoske R.M., Salzman N.H., Zahrt T.C. MprA and DosR coregulate a Mycobacterium tuberculosis virulence operon encoding Rv1813c and Rv1812c. Infect. Immun. 2012;80:3018–3033. doi: 10.1128/IAI.00520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He H., Bretl D.J., Penoske R.M., Anderson D.M., Zahrt T.C. Components of the Rv0081-Rv0088 locus, which encodes a predicted formate hydrogenlyase complex, are coregulated by Rv0081, MprA, and DosR in Mycobacterium tuberculosis. J. Bacteriol. 2011;193:5105–5118. doi: 10.1128/JB.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gaston K., Bell A., Kolb A., Buc H., Busby S. Stringent spacing requirements for transcription activation by CRP. Cell. 1990;62:733–743. doi: 10.1016/0092-8674(90)90118-x. [DOI] [PubMed] [Google Scholar]
- 84.Joung J.K., Le L.U., Hochschild A. Synergistic activation of transcription by Escherichia coli cAMP receptor protein. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3083–3087. doi: 10.1073/pnas.90.7.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reznikoff W.S. Catabolite gene activator protein activation of lac transcription. J. Bacteriol. 1992;174:655–658. doi: 10.1128/jb.174.3.655-658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.