


Abstract
Many carcinogens damage both DNA and protein constituents of chromatin, and it is unclear how cells respond to this compound injury. We examined activation of the main DNA damage-responsive kinase ATM and formation of DNA double-strand breaks (DSB) by formaldehyde (FA) that forms histone adducts and replication-blocking DNA-protein crosslinks (DPC). We found that low FA doses caused a strong and rapid activation of ATM signaling in human cells, which was ATR-independent and restricted to S-phase. High FA doses inactivated ATM via its covalent dimerization and formation of larger crosslinks. FA-induced ATM signaling showed higher CHK2 phosphorylation but much lower phospho-KAP1 relative to DSB inducers. Replication blockage by DPC did not produce damaged forks or detectable amounts of DSB during the main wave of ATM activation, which did not require MRE11. Chromatin-monitoring KAT5 (Tip60) acetyltransferase was responsible for acetylation and activation of ATM by FA. KAT5 and ATM were equally important for triggering of intra-S-phase checkpoint and ATM signaling promoted recovery of normal human cells after low-dose FA. Our results revealed a major role of the KAT5-ATM axis in protection of replicating chromatin against damage by the endogenous carcinogen FA.
INTRODUCTION
Preservation of genome stability is important for the maintenance of normal physiology of cells and essential for survival of species. Human cells possess several evolutionary conserved mechanisms of DNA repair preventing genetic alterations following DNA damage from endogenous and exogenous sources. DNA injury triggers a rapid activation of stress signaling that coordinates protective responses that include DNA repair, cell cycle checkpoints, gene expression and metabolic changes (1,2). One of the central regulators of genotoxic stress-associated signaling is ATM kinase. ATM phosphorylates proteins at Ser/Thr in the SQ/TQ sequence and belongs to a family of the phosphatidylinositol-3-kinase (PI3K) related protein kinases (3). Formation of DNA double-stranded breaks (DSB) by ionizing radiation is a canonical activator of ATM-dependent signaling. DSB are also produced by many cancer drugs, endogenous oxidants and generated physiologically during V(D)J recombination that occurs during development of T and B-lymphocytes. Despite its phosphorylation of multiple DSB repair-related proteins, ATM is required for repair of only about 10% of radiation-induced DSB (4). ATM activity appears to be critical for repair of DSB located in heterochromatin (5) or containing blocked ends (6). The lack of a functional ATM enzyme leads to the human autosomal recessive disorder ataxia telangiectasia (AT). AT syndrome is characterized by progressive neurodegeneration, immunodeficiency, acute sensitivity to ionizing radiation, cell cycle checkpoint defects, genome instability and predisposition to cancer (7). Although many of these symptoms can be attributed at a various degree to a compromised DSB repair, there is accumulating evidence that ATM is also involved in cellular processes other than DSB repair (3,8). Phosphoproteomic studies have found that ATM affected phosphorylation status of several hundreds of proteins, many of which are involved in chromatin metabolism (9–11).
In addition to elimination of DNA damage and replication errors, maintenance of genome stability also requires normal nuclear architecture and chromatin structure (12,13). Mutations altering lamin A processing and nuclear morphology (14,15) or manipulations of activity of histone modifying enzymes (16) all lead to genetic abnormalities. DNA groups that are commonly susceptible to damage by genotoxic agents are either not very different chemically from similar groups in proteins or even less reactive than protein groups. High lysine content of histones creates an abundance of amino groups that are chemically more reactive than DNA amino groups and more accessible than dG-NH2 (the most reactive DNA amino group) located in the sterically restricted minor groove. Thus, for a large group of carcinogens that form amino group-based adducts, a main bulk of chemical modifications is expected to occur in the protein component of chromatin, raising questions whether it is a biologically significant injury and whether cells can sense and respond to chromatin damage that is separate from DNA damage. In the case of ATM activation by ionizing radiation, chromatin binding by KAT5 (TIP60) lysine acetyltransferase triggers acetylation of ATM potentiating its activity (17,18). However, DSB sensing by the MRN complex also causes direct stimulation of ATM kinase (19–21) and promotes its acetylation by KAT5 (18), making it difficult to separate a chromatin-dependent branch of ATM activation from DNA damage. ATM kinase can also be triggered by global chromatin decondensatinon induced by hypotonic conditions (22), histone hyperacetylation (22,23) or depletion of the heterochromatin protein HP1α (23). Similar to radiation, sensing of chromatin decondensation involves binding of KAT5 to exposed H3K9me3, which is followed by ATM acetylation and kinase stimulation (23). Although the mechanism of ATM activation by experimentally induced chromatin decondensation is well characterized, it is less clear what common stressors can cause chromatin injury triggering ATM activation and what cellular processes this signaling would regulate.
One important group of human carcinogens that readily forms adducts with histone amino groups are aldehydes. Aldehydes are continuously produced in tissues as products of cellular metabolism and during lipid peroxidation. Formaldehyde (FA) has recently been classified as a multi-tissue carcinogen (24), with epidemiological data pointing to the bone marrow as its particularly sensitive target (25,26). In addition to tobacco smoking (27) and its other numerous external sources of human exposure (24), FA is also the most abundant endogenous aldehyde released during several normal biochemical processes including demethylation of histone lysines (28). Mouse models with the loss of the main aldehyde-detoxifying enzyme Aldh2 together with the inactivation of Fanconi anemia pathway showed a strikingly severe phenotype in the bone marrow, demonstrating a high endogenous production of toxic aldehydes (29,30). The maternal detoxification of endogenous aldehydes is also important for protection of the embryo from DNA damage and developmental abnormalities (31).
In the process of investigation whether blockage of replication forks by FA-induced DNA-protein crosslinks (DPC) can be associated with DSB production, we found and report here a surprisingly strong activation of ATM signaling by FA-chromatin damage in replicating cells. ATM activity was induced by KAT5-dependent acetylation and it was important for the establishment of intra-S checkpoint and suppression of cytotoxic effects of FA. Our data point to aldehydes as one of the continuous endogenous sources of chromatin damage that requires repair and is monitored by the KAT5-ATM axis. We did not find evidence for detectable DNA cleavage during the initial replication arrest by FA, demonstrating a high stability of DPC-blocked helicase complexes in normal human cells.
MATERIALS AND METHODS
Chemicals
KU55933 (ATMi-1) was obtained from Biovision, KU60019 (ATMi-2) and VE821 were from Selleckchem, NU9056 (KAT5i) from Tocris and imatinib (c-ABLi) from Enzo. Formaldehyde and other chemicals were from Sigma.
Cells and treatments
Human H460, A549, IMR90 and WI38 cells were obtained from the American Type Culture Collection. Seckel syndrome fibroblasts (GM18366) were purchased from Coriell Biorepository. H460 and A549 were cultured in RPMI-1640/F12K-10% serum media, respectively. IMR90, WI38 and Seckel syndrome fibroblasts were propagated in DMEM medium containing 10% serum. Immortalized cells were grown in 95% air/5% CO2, whereas normal human fibroblasts were kept in 5% O2 and 5% CO2. Stock solutions of FA were prepared in deionized water and added to cells in complete growth media. ATM, ATR and KAT5 inhibitors were added 1 h before FA treatments. IMR90 cells were put into a quiescent state by growing them to full confluence and then maintaining for 2 days in 0.5% serum.
shRNA and siRNA
For stable expression of shRNA, cells were infected with the constructs based on pSUPER-RETRO vector. The targeting sequences were 5′-GACTTTGGCTGTCAACTTTCG-3′ for ATM and 5′-GATGCCATTGAGGAATTAG-3′ and 5′-GAACCUGGUCCCAGAGGAG-3 for MRE11 vectors #1 and #2, respectively. Packaging, infection and selection conditions were as described previously (32). For KAT5 knockdown, ON-TARGETplus (CGUAAGAACAAGAGUUAUU) and control siRNAs were purchased from Dharmacon. Cells were seeded to obtain a 30% confluence on the day of transfection. Transfections were performed twice with 50 nM siRNA (final concentration) using Lipofectamine RNAimax (Invitrogen). Cells were treated with FA 48 h after transfection.
Western blotting and immunoprecipitation
Cells were seeded at approximately 50% confluence one day before FA or other treatments. Protein extracts were prepared by lysing cells for 10 min in a 2% SDS buffer (2% SDS, 50 mM Tris-HCl pH 6.8, 10% glycerol) supplemented with Halt protease and phosphatase inhibitors (Thermo Scientific) and 1 mM paramethylsulfoxide (Sigma). Samples were heated at 100°C for 10 min, cooled to room temperature and lysates were collected after centrifugation at 10000xg for 10 min. Chromatin was isolated as described previously (38). Proteins were separated by standard SDS-PAGE and electrotransferred to ImmunoBlot PVDF membrane (Bio-Rad). For detection of ATM crosslinks, a published procedure was followed and protein samples were not heated before loading on gels (33). Primary antibodies used were pT68-CHK2 (2661), CHK2 (2662), pS317-CHK1 (2344) and pS15-p53 (9284) from Cell Signaling; pS1981-ATM (ab81292), pT21-RPA (ab109394) and KAT5 (ab23886) from Abcam; pS824-KAP1 (A300–767A) and pS4/8-RPA (A300–245A) from Bethyl; γ-tubulin (T6557) from Sigma; ATM (sc-23921), cyclin A (H-432) and lamin B (sc-6216) from Santa Cruz; MRE11 (611366) and MSH2 (556349) from BD Biosciences; RPA32 (NA19L) from Calbiochem; AcK (acetyl-lysine antibody; 05–515) and γ-H2AX (05–636) from Millipore. All antibodies were used at 1:1000 dilution, except for AcK (1:500). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse IgG (12–349, Millipore; 1:5000 dilution), rabbit anti-goat IgG (2922, Santa Cruz; 1:500 dilution) and goat anti-rabbit IgG (7074, Cell Signaling; 1:2000 dilution).
In the immunoprecipitation experiments, cells were lysed for 30 min on ice in RIPA buffer (150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0, protease inhibitors, 10 μM trichostatin A and 10 mM nicotinamide). After centrifugation (12000xg, 4°C, 10 min), supernatants were incubated with 1.2 μg/mg cellular protein anti-ATM antibodies (PC116, Calbiochem) and 20 μl of protein A/G plus beads (Santa Cruz) overnight at 4°C. Beads were washed 3 times in RIPA buffer and boiled in the SDS loading buffer with 100 mM DTT to release captured proteins. ATM immunoprecipitates were run on 6% SDS-PAGE, transferred to PVDF membrane and sequentially probed with anti-AcK antibodies and after stripping, with anti-ATM antibodies.
Immunofluorescence
Cells were seeded on human fibronectin-coated coverslips, treated with FA and fixed with 4% paraformaldehyde for 15 min at room temperature, followed by permeabilization in PBS-0.2% Triton X-100 for 10 min. S-phase cells were labeled by incubation with at 10 μM 5-ethynil-2′-deoxyuridine (EdU) for 15 min before FA treatments. Slides were blocked with 3% BSA for 1 h followed by EdU staining using Click-iT EdU-Alexa Fluor 488 Imaging kit (Invitrogen). Immunostaining was done with the same antibodies as used in western blotting. The secondary antibodies (1:500 dilution, Life Technologies) were added for 1 h at 4°C. Primary and secondary antibodies were diluted in PBS containing 1% BSA and 0.5% Tween-20. Coverslips were then mounted on glass slides using Vectashield fluorescence mounting media with DAPI (H-1200). Cells were imaged on a Zeiss LSM710 confocal microscope at X630 magnification.
Fluorescence-activated cell sorting (FACS)
S-phase cells were labeled with 10 μM EdU for 1 h before treatments with FA. Cells were collected by trypsinization and fixed overnight in 80% ethanol at 4°C. Following two washes with PBS, cells were extracted with 0.5% Triton X-100 in PBS for 30 min at room temperature and centrifuged at 150xg for 5 min. Pellets were washed twice with PBS and then resuspended in 150 μl EdU Click-iT reaction cocktail from Invitrogen (Click-iT EdU-Alexa Fluor 488 Flow Cytometry Assay) and incubated for 30 min at room temperature in the dark. Cells were washed once with PBS, resuspended in 500 μl PBS containing 4 μg/ml propidium iodide, and incubated for 30 min at room temperature protected from light. Cell pellets were washed once with 2 ml PBS and resuspended in 0.5 ml PBS for flow cytometry analysis (FACSCalibur, BD Biosciences). Data were analyzed by CellQuest Pro software.
Pulsed field gel electrophoresis (PFGE)
Measurements of DSB in IMR90 using Bio-Rad CHEF MAPPER system were performed as previously described with minor modifications (34). Cells were treated in complete media with FA (200 μM, 300 μM), hydroxyurea (2 mM, 5 mM) or bleomycin (30 μg/ml) for 3 h and collected by trypsinization. A 0.5 ml solution containing 3.0 × 106 cells in a cell suspension buffer (10 mM Tris pH 7.2, 50 mM EDTA, 20 mM NaCl) was mixed with an equal volume of 2.0% Ultra Pure LMP agarose and immediately poured into CHEF Mapper XA System Plug Molds (3.0 × 105 cells per one plug). Solidified agarose plugs were digested with proteinase K (10 mM Tris, pH 8.0, 100 mM EDTA, 1.0% N-lauroylsarcosine, 0.2% sodium deoxycholate, 1 mg/ml proteinase K) at room temperature overnight with gentle mixing followed by 5X washes in 20 mM Tris pH 8.0, 50 mM EDTA for 1 h each. DNA separation was performed at 14°C in 1% agarose gels and 0.5X TBE buffer. Gels were stained for 1 h in 3X GelRed staining solution in 0.1 M NaCl.
Clonogenic survival
Toxicity of FA treatments in H460 cells, which were used in western blotting studies, was assessed by the colony formation assay. Similar to experiments with collection of protein extracts, cells were seeded at approximately 50% confluence and treated with FA on the next day. Cells were trypsinized, counted and seeded at low density onto 60 mm dishes (200–400 cells/dish) and grown for 7–8 days to form visible colonies. Cells were fixed with methanol and stained with Giemsa. Groups with 30 or more cells were counted as colonies.
Cytotoxicity in normal human cells
IMR90 fibroblasts do not form colonies in sparsely seeded conditions and cytotoxic effects of FA in the populations of these cells were examined using the CellTiter-Glo luminescent cell viability assay (Promega). For the determination of cytotoxicity of FA treatments in our biochemical studies, IMR90 cells were seeded at the same (∼50%) density and exposed to FA one day later. At the end of exposures, cells were trypsinized, counted and seeded at 1000 cells/well into 96-well optical bottom plates. Viability measurements were performed after growing cells for 72 h. In experiments assessing the impact of ATM inhibition on FA cytotoxicity, IMR90 cells were seeded at 500 cells/well in quadruplicates into 96-well optical bottom plates and treated with FA and ATM inhibitors one day later. Cell viability was determined at 72 h post-FA.
RESULTS
Activation of ATM signaling by FA
In the initial experiments, we examined the ability of FA to stimulate ATM kinase activity by treating cells for different times and collecting protein lysates at various post-exposure intervals. We thought that some limited ATM activation could occur at late times, potentially in connection with DNA cleavage at the sites of DPC-arrested transcription or DPC-stalled replication forks. Human H460 cells, which have been found to retain normal activation of ATM by ionizing radiation (35), and IMR90 normal human fibroblasts were tested for phosphorylation of three canonical targets of ATM kinase, such as its autophosphorylation at Ser1981 (22), Thr68 in CHK2 kinase (23) and Ser821 in KAP1 (5). We found that all three readouts of ATM-related signaling were strongly elevated in both cell types, reaching peaks during 2-h long incubations with FA (Figure 1A). Despite the treatment of cells with FA in the presence of serum, which is expected to limit the amount of the immediately reactive FA, ATM and CHK2 phosphorylation increases were already evident after short 30 min exposures. The appearance of phospho-KAP1 was delayed by about 0.5–1 h relative to CHK2 phosphorylation (Figure 1A), suggesting that CHK2 could be a preferred target of FA-activated ATM. The 2- and 3-h long FA treatments causing maximal ATM activation in H460 cells were only moderately cytotoxic in the colony-formation assay (Figure 1B). A long-term viability of IMR90 cells also remained relatively high after FA exposures that induced robust increases in ATM-associated phosphorylation (Figure 1C). Removal of FA resulted in the gradually diminished levels of ATM-related phosphorylation (Figure 1D), indicating that ATM activation was associated with the formation of the initial injury, not late secondary genetic lesions. Although all three phosphorylation sites that we examined are commonly used for monitoring of ATM activation in cells, there are examples of the same targets being phosphorylated by two other DNA damage-responsive kinases, ATR and DNAPK (36,37). We found that the addition of two selective inhibitors of ATM kinase eliminated phosphorylation of ATM, CHK2 and KAP1 (Figure 1E), confirming the ATM-specificity of the FA-induced responses. In agreement with its reported ATR dependence (38), p53-Ser15 phosphorylation by FA was unaffected by ATM inhibition.
Figure 1.
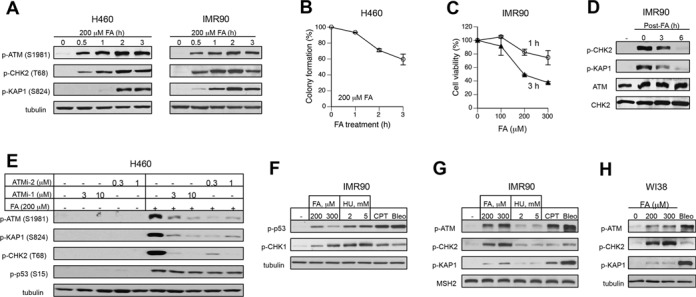
Activation of ATM pathway by FA. (A) ATM signaling in H460 and IMR90 cells treated with 200 μM FA for different time intervals. (B) Clonogenic survival of H460 cells treated with 200 μM FA for 1, 2 or 3 h. Means ± SD from two experiments each including 3 dishes/dose. (C) Cytotoxicity of 1-h and 3-h long FA treatments in IMR90 cells. Cell viability was measured at 72 h post-FA using the CellTiter-Glo assay. Means ± SD from two experiments with 3 dishes/dose. (D) ATM responses in IMR90 treated with 200 μM FA for 3 h and collected at the indicated recovery times. (E) Effects of ATM inhibitors on FA-induced protein phosphorylation (ATMi-1–KU55933, ATMi-2–KU60019). H460 cells were preincubated with inhibitors for 1 h and collected immediately after 3-h long FA treatments. (F) CHK1 and p53 phosphorylation in IMR90 cells treated for 3 h with FA, hydroxyurea (HU), camptothecin (CPT, 1 μM) or bleomycin (Bleo, 30 μg/ml). (G) ATM-related phosphorylation in IMR90 treated as in panel F. MSH2 was used as a loading control. (H) ATM signaling in WI38 normal cells treated with FA and bleomycin as IMR90 in panel F.
The observed increases in the phosphorylation levels of ATM targets by FA were clearly high relative to background levels in cells. However, these comparisons do not provide information regarding the overall magnitude of ATM activation, as sensitive western blot systems can allow detection of even relatively small responses when background levels are very low. To provide biological references for the extent of ATM activation by FA, we analyzed side-by-side ATM signaling triggered in normal human fibroblasts by other genotoxic stressors. We included a classic replication stressor hydroxyurea (HU), which causes a shortage of dNTPs and the resulting stalling of replication forks, the topoisomerase I poison camptothecin (CPT), which produces replication-associated DSB, and the radiomimetic bleomycin, which induces DNA strand breakage via oxidative reactions following its duplex intercalation. The selected doses of the reference stressors were roughly comparable to FA in their potency to activate ATR kinase (monitored by phosphorylation of its target Ser317 in CHK1 kinase) and more potent inducers of Ser15 phosphorylation in the p53 transcription factor (Figure 1F). Ser15-p53 is a common site for phosphorylation by several stress kinases (39), acting as a marker of the overall cellular stress. A lower Ser15 phosphorylation by 300 μM relative to 200 μM FA reflects bell-shaped dose dependence for this response in normal human cells (38). Despite their similarity as replication stressors and comparable levels of ATR activation under our conditions, FA was dramatically stronger inducer of ATM-related phosphorylation than HU (Figure 1G). ATM autophosphorylation by our highest dose of FA was similar to that by the DSB producer CPT but lower relative to bleomycin. Analysis of the downstream targets showed that FA-stimulated ATM preferentially targeted CHK2 versus KAP1, which was the opposite for the DSB inducers CPT and bleomycin. Surprisingly, CHK2 phosphorylation by FA was much stronger than that by either CPT or bleomycin. Consistent with the results in IMR90 cells, we also found a strikingly higher CHK2 phosphorylation and a dramatically lower phospho-KAP1 induction by FA in comparison to bleomycin in WI38 normal human cells (Figure 1H). Overall, the comparisons with other stressors showed that FA was a very potent activator of ATM, which, in contrast to the DSB-producing agents, preferentially targeted soluble CHK2 kinase over chromatin-bound KAP1.
S-phase specificity of ATM signaling
Our initial immunostaining experiments showed that phospho-ATM was present only in a fraction of FA-treated cells, pointing to a possibility of a cell cycle-specific response. Costaining of H460 cells for EdU incorporation and phospho-ATM or phospho-CHK2 revealed that FA-induced activation of ATM occurred exclusively in S-phase (Figure 2A). The ATM-specificity of the immunostaining signals was verified by the absence of binding of the employed antibodies to cells with ATM knockdown (Figure 2B). Scoring of phospho-CHK2 staining in H460 cells and IMR90 normal fibroblasts confirmed that ATM activation by FA was limited to EdU-incorporating cells (Figure 2C,D). FA failed to trigger ATM and CHK2 phosphorylation in growth-arrested IMR90 cells despite their higher ATM protein levels and normal CHK2 protein content (Figure 2E). We further found that inhibition of DNA replication by the DNA polymerase inhibitor aphidicolin strongly suppressed ATM-dependent phosphorylation in FA-treated cells (Figure 2F). Taken together, these results indicate that ATM activation by FA is specific to S-phase cells and requires DNA replication at the time of FA injury. Activation of ATR kinase by FA also occurs in S-phase (38), however, replication stress-induced stimulation of ATR did not affect ATM, as evidenced by its normal activity in IMR90 cells with suppressed ATR activity (Figure 2G). Blocking of ATR activity by a different inhibitor in H460 cells also had no significant effect on FA-induced ATM phosphorylation (Figure 2H). ATM activation in Seckel cells containing hypomorphic ATR mutations was also similar to normal cells (Figure 2I). Inhibition of ATM signaling by aphidicolin was unrelated to ATR activity, as shown by the inability of the three tested ATR inhibitors to increase ATM-dependent phosphorylation in response to FA (Figure 2J). An elevated background level in cells treated with ATRi2 (VE821) accounts for a somewhat higher p-CHK2 signal in FA samples treated with this inhibitor.
Figure 2.
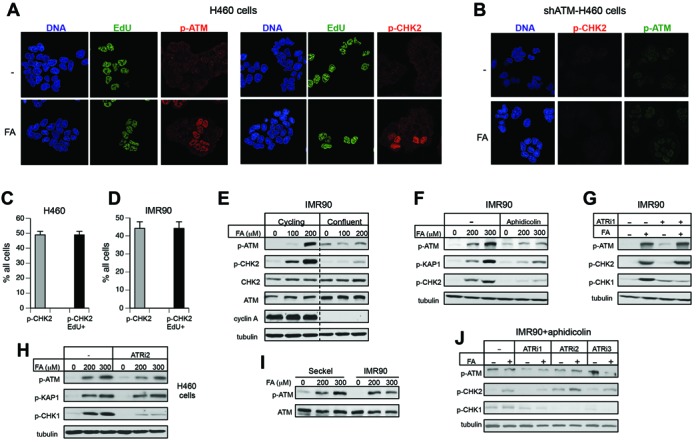
Replication-associated activation of ATM by FA. (A) FA-induced phospho-ATM and phospho-CHK2 localized to EdU-incorporating cells. H460 were treated for 3 h with 300 μM FA. (B) Absence of phospho-ATM and phospho-CHK2 immunostaining in H460 cells with shRNA-depleted ATM. (C) S-phase specificity of FA-induced phospho-CHK2 in H460 and (D) IMR90 cells (300 μM FA, 3 h). Labels: p-CHK2—percentage of cells displaying immunostaining for p-CHK2, p-CHK2/EdU+—percentage of cells that showed both p-CHK2 and EdU staining. Data are Means ± SD for three experiments with each scoring >100 cells. (E) Lack of ATM and CHK2 phosphorylation by FA in growth-arrested IMR90 cells. Data for cycling and confluent cells are from the same blots after removal of intervening lanes. (F) Inhibition of FA-induced ATM signaling in IMR90 cells by aphidicolin (2 μM, added 60 min before FA exposure for 1.5 h). (G) Independence of ATM signaling on the ATR kinase in IMR90 cells. ATRi1 (3 mM caffeine) was added for 1 h before FA exposure for 3 h. (H) Normal ATM and KAP1 phosphorylation in H460 cells in the presence of the ATR inhibitor VE821 (ATRi2, 10 μM). Cells were preincubated for 1 h with ATRi2 and then treated with FA for 3 h. (I) ATM phosphorylation in IMR90 and Seckel syndrome fibroblasts treated for 3 h with FA. (J) ATR inhibitors did not restore ATM activation by FA in the presence of aphidicolin (ATRi1 - 3 mM caffeine, ATRi2 - 10 μM VE821, ATRi3 - 0.5 μM AZ20). Cells were preincubated with 2 μM aphidicolin and ATR inhibitors for 1 h before FA exposure for 2 h.
Nongenotoxic mechanism of ATM activation by FA
DSB, the most well known activator of ATM, are initially detected and bound by the MRE11-RAD50-NBS1 (MRN) complex, which is followed by recruitment and stimulation of ATM kinase (19–21). We did not found significant changes in the levels of phospho-ATM, phospho-CHK2 or phospho-KAP1 either immediately after FA treatments or at 4 h post-exposure in IMR90 cells with shRNA-depleted MRE11 (Figure 3A). Control experiments showed that, as expected, MRE11 knockdown inhibited ATM activation in response to the DSB producer bleomycin (Figure 3B). Direct measurements of DSB by PFGE found no detectable DNA breakage in cells by FA whereas, as expected, bleomycin caused extensive formation of DSB (Figure 3C). The replication stressor HU also failed to generate DSB (Figure 3C), which is consistent with its very weak activation of ATM signaling (Figure 1G) and the stability of HU-stalled replication forks over several hours (40,41). In agreement with PFGE results, bleomycin caused a massive formation of γ–H2AX but only very faint bands of this genotoxic stress marker were seen in FA samples (Figure 3D). A large production of DSB by bleomycin versus none by FA also correlated well with the extent of KAP1 phosphorylation whereas CHK2 phosphorylation was remarkably higher by FA (Figure 3D). Further evidence supporting a non-DSB mode of ATM activation by FA included the lack of ATM-chromatin binding (Figure 3E) and the absence of a biochemical marker of S/G2 phase processed DSB (42), Ser4/Ser8-phosphorylated RPA32 (Figure 3F). Since FA is a potent inhibitor of DNA replication (38,43), another possible cause for ATM activation by FA could be the presence of damaged replication forks. Examination of phospho-Thr21-RPA32, which is produced in response to excessive ssDNA accumulation and in cells with damaged forks, showed the expected phosphorylation responses by the S-phase DSB producer CPT and by the radiomimetic bleomycin (Figure 3G). HU induced a clear but weaker phosphorylation of Thr21-RPA32 whereas FA caused no responses even in the overexposed blots (Figure 3G). Enhancement of FA-induced replication stress by the addition of the proteasome inhibitor MG132 (43) did not alter ATM signaling although, as expected, it increased Ser15-p53 phosphorylation (Figure 3H). Overall, DPC-arrested replication was not associated with the appearance of cleaved or damaged replication forks, pointing to a non-genotoxic mechanism of ATM activation by FA.
Figure 3.
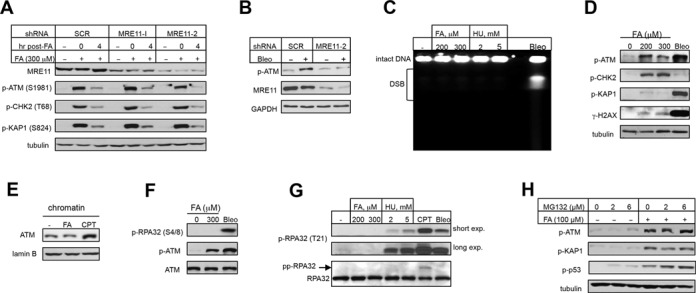
ATM signaling by FA is unrelated to DSB or replication stress. IMR90 cells were treated with FA and other stressors for 3 h. (A) MRE11 depletion did not affect ATM signaling by FA. (B) Inhibition of bleomycin-induced ATM autophosphorylation by MRE11 depletion (5 μg/ml bleomycin, 20 min). (C) Detection of DSB by PFGE in cells treated with FA, HU or bleomycin (Bleo, 30 μg/ml). (D) Protein phosphorylation in IMR90 treated with FA and bleomycin (Bleo, 30 μg/ml). (E) Amount of chromatin-bound ATM in cells treated with 200 μM FA or 2 μM CPT. (F) RPA32-S4/8 phosphorylation in cells treated with bleomycin (Bleo, 30 μg/ml) or FA. (G) RPA32-T21 phosphorylation in IMR90 treated with FA, HU, CPT (1 μM) and bleomycin (Bleo, 30 μg/ml). pp-RPA32—hyperphosphorylated form of RPA32. (H) Effect of the proteasome inhibitor MG132 on ATM, KAP1 and p53 phosphorylation by FA.
ATM crosslinking by FA
ATM can also be activated by its covalent dimerization via disulfide linkages, as it was discovered for hydrogen peroxide (33). FA is well known for its protein crosslinking properties, which involves reactions with both protein amino- and SH-groups. Therefore, we next investigated whether FA was able to cause ATM crosslinking with the resulting stimulation of its kinase activity. Analysis of cellular proteins by westerns under non-reducing/non-heated denaturing conditions preserving FA bonds showed dose-dependent losses of monomeric ATM in FA-treated cells, as well as the expected disappearance of ATM band following H2O2 treatment (Figure 4A, top blot). A longer exposure of the same blot revealed the presence of SDS-resistant ATM dimers and higher multimers in both FA- and H2O2-treated samples (Figure 4A, bottom blot). Probing of non-reducing/non-heated blots for S1981-ATM autophosphorylation detected kinase activity in ATM dimers for H2O2 (in agreement with findings in Ref.(33)) but exclusively in monomers for FA (Figure 4B). Thus, high FA doses were clearly able to cause intermolecular crosslinking of ATM, however, unlike hydrogen peroxide-induced dimerization, this chemically forced association blocked ATM kinase activity. FA concentrations (0.9 and 1.2 mM) producing the most extensive ATM crosslinking during 1.5 h exposures were highly cytotoxic based on the colony formation measurements (Figure 4C). However, viability of the cells (assayed by Trypan blue staining) collected immediately after the 1.5-h long treatment was high: 100.7 ± 4.2%, 100.1 ± 2.4% and 84.0 ± 2.3% for 0.3, 0.9 and 1.2 mM FA, respectively. While still causing losses of ATM monomers, 0.9 and 1.2 mM FA doses applied for 0.5 h were only moderately toxic in the clonogenic assay (Figure 4C). Thus, although crosslinking and chemical inactivation of ATM by FA is a high-dose effect, it can be compatible with a long-term survival of cells. Nakano et al. (44) have found Ser19-CHK2 phosphorylation after 12 h recovery but not immediately after a 3-h long FA treatment of human cells. It is possible that Ser19 was not targeted during the chromatin-dependent phase of ATM activation. Alternatively, the lack of early phospho-Ser19-CHK2 in this study could have reflected chemical inactivation of ATM due to the use of serum-free media for FA treatments. The susceptibility of ATM to direct damage by FA also suggests that under conditions of high exposure to this or related aldehydes, such as in heavy smokers, cellular responses to DSB and oxidative stress could be partially impaired. A very large size of ATM (350.7 kDa) can potentially make it vulnerable to chemical inactivation during chronic exposures to even moderate doses of FA or other reactive carbonyls.
Figure 4.
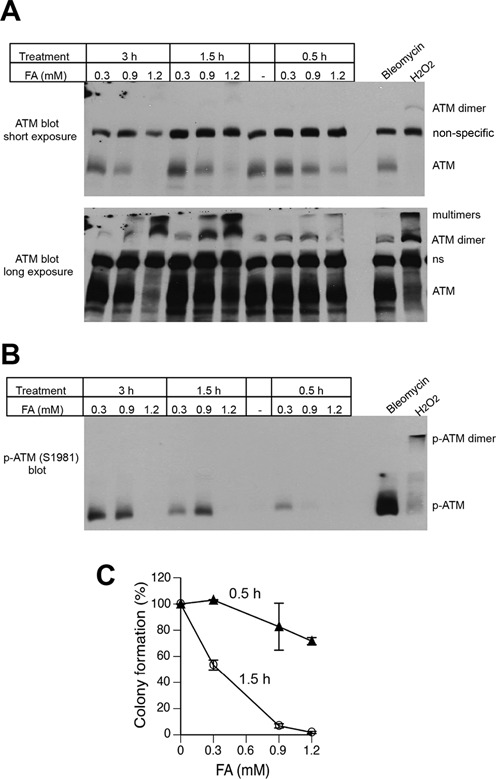
Formation of inactive ATM dimers and multimers by high FA doses. H460 cells were treated with FA for 0.5–3 h or bleomycin (30 μg/ml) and H2O2 (8 mM) for 30 min. Proteins were denatured by the addition of 2% SDS without boiling and separated under nonreducing conditions at 4°C. (A) Western blot with anti-ATM antibodies. Short (top panel) and long (bottom panel) exposures of the same blot are shown. A non-specific band between ATM monomer and dimer was used as a loading control. (B) Western blot with anti-phospho-ATM antibodies. (C) Clonogenic survival of H460 cells treated with FA for 0.5 or 1.5 h. Data are Means ± SD, n=3.
KAT5-dependence of ATM activation by FA
In addition to the DSB sensing MRE11-RAD50-NBS1 complex (19–21), ionizing radiation-induced activation of ATM is strongly potentiated by its KAT5 (Tip60)-mediated acetylation (17,18). This acetylation-dependent ATM stimulation requires binding of KAT5 to exposed histone H3K9me3. Acetylation by KAT5 was necessary for ATM activation by chromatin decondensation via histone hyperacetylation or unmasking of histone H3K9me3 by HP1α depletion (23). We reasoned that high chemical reactivity of FA with histones could also cause a significant chromatin injury triggering ATM activation. We found that ATM was acetylated in FA-treated but not control cells (Figure 5A) and a blockage of KAT5 activity abolished the ability of FA to induce ATM acetylation (Figure 5B). Next, we directly tested a functional importance of KAT5 in ATM activation. A pharmacological inhibition of KAT5 activity severely suppressed phosphorylation of ATM and its target proteins by FA in two normal human cell lines (Figure 5C,D). Knockdown of KAT5 by siRNA also blocked ATM-related phosphorylation in FA-treated cells (Figure 5E). c-ABL-mediated tyrosine phosphorylation of KAT5 is required for its chromatin-dependent ATM activation (23). Consistent with this mechanism, we found that inhibition of c-ABL kinase activity abolished ATM activation by FA (Figure 5F).
Figure 5.
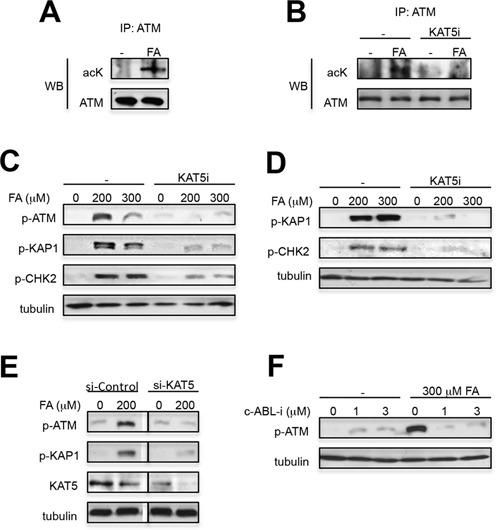
KAT5-dependent activation of ATM by FA. (A) ATM acetylation in A549 treated with 300 μM FA for 3 h. Immunoprecipitated ATM was probed with anti-acetyl-lysine (acK) antibodies. (B) Absence of ATM acetylation in FA-treated cells with inactive KAT5. Cells were treated with FA in the absence or presence of KAT5i (25 μM NU9056 added 1 h before FA) and analyzed for ATM acetylation as in panel A. (C) Suppression of ATM activation by KAT5 inhibitor (KAT5i—25 μM NU9056, added for 1 h before FA) in WI38 and (D) IMR90 cells. WI38 cells were treated with FA for 1 h and IMR90 for 3 h. (E) Loss of ATM signaling in IMR90 cells with KAT5 knockdown by siRNA. Cells were treated with FA for 3 h. Images for si-Control and si-KAT5 are from the same blot after removal of intervening bands. (F) Suppression of ATM autophosphorylation by c-ABL inhibition. IMR90 cells were treated for 3 h with imatinib before addition of FA for 3 h.
Biological significance of ATM in FA-treated cells
To gain insight into a potential role of ATM activation in S-phase cells, we measured EdU incorporation in replicating cells in the presence of ATM inhibitors. We found that the loss of ATM kinase activity resulted in significantly higher levels of DNA synthesis in FA-treated cells analyzed immediately (Figure 6A) or after 2 h recovery (Figure 6B), indicating that ATM signaling triggered the activation of intra-S checkpoint (suppression of DNA replication). Consistent with their involvement in the same signaling pathway, ATM and KAT5 had similar effects on S-checkpoint and a simultaneous inhibition of both proteins did not cause further changes in FA-resistant DNA synthesis over ATMi or KAT5i alone (Figure 6C). Analysis of EdU incorporation at 24 h recovery time showed that FA-treated cells with inactive ATM had slower rates of DNA synthesis, indicating elevated levels of the remaining damage (Figure 6D,E). Finally, we found that ATM was also important for a long-term viability of normal human cells treated with low-dose FA (Figure 6F).
Figure 6.
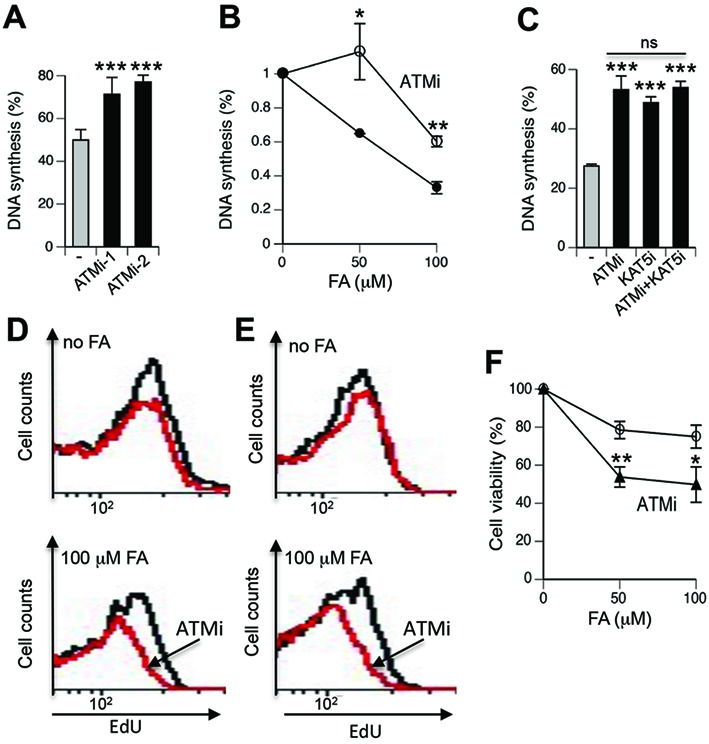
ATM role in cellular responses to FA. IMR90 cells were treated with FA for 3 h and inhibitors were added for 1 h before exposures. DNA synthesis was determined by FACS measurements of peak EdU incorporation in S-phase cells after subtraction of peak EdU fluorescence of non-S-phase cells. Data were normalized to EdU intensity in the corresponding FA-untreated controls. (A) DNA synthesis in cells treated with 50 μM FA in the presence of ATM inhibitors KU55933 (ATMi-1, 10 μM) and KU60019 (ATMi-2, 2 μM). Data are Means ± SD for five biological replicates, ***−P < 0.001 by two-tailed, unpaired t-test. (B) DNA synthesis at 2 h post-FA exposure in the absence and presence of KU55933 (ATMi). Means ± SD for three biological replicates, *−P < 0.05, **−P < 0.01 by two-tailed, unpaired t-test. (C) KAT5 and ATM are important for activation of intra-S checkpoint in response to FA. ATMi - 3 μM KU55933, KAT5i - 10 μM NU9056. Data are Means ± SD for three biological replicates, ***−P < 0.001 relative to FA-alone samples (two-tailed, unpaired t-test), ns—nonsignificant differences among inhibitor-treated groups. (D) Representative FACS profiles of EdU incorporation at 24 h post-FA exposure by S-phase IMR90 and (E) WI38 cells (ATMi - 10 μM KU55933, red lines). Three other biological replicates showed similar profiles. (F) Impact of ATM inhibition on viability of FA-treated IMR90 cells (ATMi - 6 μM KU55933). Viability measurements were recorded at 72 h post-FA using the CellTiter-Glo assay. Means ± SD are shown (n = 3, *−P < 0.05, **−P < 0.01 by two-tailed, unpaired t-test with the Bonferroni correction).
DISCUSSION
We found a surprisingly strong and rapid activation of the DNA damage-responsive ATM pathway in human cells by even mildly toxic doses of FA. ATM activity was triggered specifically in S-phase and required DNA replication at the time of FA injury. ATM signaling is typically associated with DSB-producing agents (1–3), and some investigators have suggested cleavage of replication forks blocked by FA-induced DPC (44). However, we obtained a diverse set of evidence indicating that DSB were not the cause of the main wave of ATM activation by FA. Although FA-provoked ATM signaling included phosphorylation of two canonical DSB-associated targets, Ser824-KAP1 and Thr68-CHK2, the extent of phosphorylation of these proteins by FA was very different from that by DSB-producing agents. Under similar levels of ATM activation, as measured by it autophosphorylation, Thr68-CHK2 phosphorylation was remarkably stronger and phosphorylation of Ser824-KAP1 was dramatically weaker by FA in comparison to the DSB inducers bleomycin and camptothecin. KAP1 phosphorylation by ATM is associated with the presence of DSB whereas Thr68-CHK2 phosphorylation can occur in response to ATM activation via oxidative dimerization in the absence of DSB (33). The induction of γ-H2AX by FA was also very low, which together with a weak KAP1 phosphorylation point to a little if any production of DSB. A limited formation of phospho-KAP1 may reflect its origin from a soluble pool of KAP1, which can potentially be increased by FA-induced chromatin damage. Our direct measurements of DSB by PFGE found no detectable levels of these lesions in FA-treated cells, which is consistent with the lack of DNA cleavage and a high stability of DPC-stalled replication forks in Xenopus egg extracts (45). Finally, we determined that ATM activation by FA was not affected by the knockdown of the MRN complex that recruits ATM to DSB and stimulates its activity (19–21). In disagreement with an earlier report (46), a recent study using a panel of DT40 mutants has found no hypersensitivity to FA in homologous recombination-deficient cells (47), which also argues against a significant formation of DSB or severely damaged replication forks. Blockage of in vitro replication by DPC was principally caused by stalling of the replicative helicase complex (45), which prevents accumulation of ssDNA. Consistent with the absence of extensive tracks of ssDNA, FA-treated cells showed no increases in chromatin foci of the ssDNA-binding protein RPA (38). DNA monoadducts or dNTP depletion by hydroxyurea permit continuing DNA unwinding but stall DNA polymerase elongation, generating long stretches of breakage-prone ssDNA and unstable forks (48). Thus, our data and mechanistic considerations of replication inhibition by DPC suggest that, at least initially, the arrest of replication forks by blocking duplex unwinding is probably less dangerous to the genome integrity than stalling of DNA polymerases under conditions of ongoing helicase activity.
We found that stimulation of ATM signaling by FA was dependent on KAT5 (Tip60) lysine acetyltransferase, which controls a chromatin-dependent mode of ATM activation (18,23). c-ABL-phosphorylated bromodomain of KAT5 binds histone H3K9me3 and this association triggers ATM acetylation and its kinase activation (23). H3K9me3 is normally bound by heterochromatic protein-1 (HP1) and recognition of this histone modification by KAT5 requires its unmasking via dissociation of HP1. Previous approaches for stimulation of ATM by chromatin changes included HP1α knockdown and drug-induced or mechanically forced chromatin decondensation (22,23), which all exposed H3K9me3. KAT5 activity can also be stimulated by its binding to K3K36me3 (18,23), however, it is currently unclear what processes control accessibility to this histone form in chromatin. ATM activation by FA occurred specifically in S-phase and required ongoing replication at the time of FA injury. It is well established that DNA replication is associated with chromatin opening and involves disassembly of nucleosomes in the front of replication forks (49). Thus, replication-induced chromatin decondensation also creates accessible H3K9me3, however, in the absence of histone/chromatin injury, this unmasking of H3K9me3 would be transient. Histone lysines are the main targets for FA conjugation in chromatin, which is expected to block the addition of acetyl and methyl groups to the FA-modified amino groups. In agreement with this suggestion, in vitro modifications of histone H4 lysines with FA inhibited their subsequent acetylation by PCAF (50). Newly deposited histones undergo additional Lys modifications to convert them into mature nucleosomal forms and the presence of FA-Lys adducts can directly or indirectly interfere with this process, resulting in the immature decondensed chromatin structure with exposed H3K9me3 and/or H3K36me3. The inability of cells to properly mature chromatin-incorporated histones results in nuclear and replication defects (49,51). In addition to hydrolytically labile Lys monoadducts, FA also forms more stable histone-histone crosslinks (52,53). Intermolecular histone crosslinking, particularly between components of separately loaded/removed H2A-H2B and H3-H4 dimers, is expected to interfere with the orderly nucleosome disassembly in front of the replication forks generating disorganized chromatin structure behind the forks and creating problems with the restoration of normal chromatin on replicated DNA. Our results on the inhibition of ATM signaling by the DNA polymerase inhibitor aphidicolin are consistent with KAT5 sensing of disorganized/decondensed chromatin on replicated DNA. Ongoing duplex unwinding by the MCM helicase complex in the presence of aphidicolin results in the accumulation of ssDNA, which does not permit nucleosome assembly and therefore, cannot recruit and activate KAT5. We found that FA-histone damage triggered KAT5/ATM-dependent intra-S checkpoint, which provides additional time for repair of chromatin damage and restoration of its structure. Cytotoxic and genotoxic effects of FA have been traditionally attributed to its ability to form DPC, which are genetic lesions. Our results showed that FA-histone damage even at mildly toxic doses also represents a significant injury in replicating chromatin and its repair is promoted by ATM signaling (Figure 7). Thus, the role of ATM in the preservation of the nuclear and genomic integrity via activation of S-checkpoint extends beyond DNA damage and includes histone/chromatin injury by FA in replicating cells.
Figure 7.
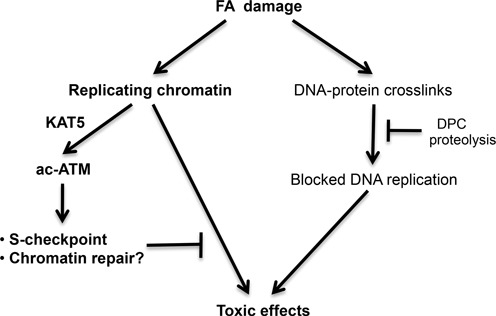
Model depicting cellular responses to FA damage. Activation of ATM was dependent on its acetylation by KAT5 that responds to chromatin perturbations via detection and binding to unmasked H3K9me3 and H3K36me3 (18,23). The initial inhibition of DNA replication by FA includes a physical blockage of replicative helicase by DPC in the leading strand (45) and a chromatin injury-associated S-phase checkpoint mediated by KAT5-activated ATM. ATM-coordinated responses to chromatin damage are important for prevention of long-term toxicity by FA.
It is likely that in addition to FA, ATM is also responsive to histone/chromatin damage by other endogenous aldehydes which all target histone lysines. Hematopoietic stem cells and progenitors are particularly sensitive to injury by endogenous aldehydes (29–31) and bone marrow failure in individuals with mutated ATM (7) could be in part caused by the inability of these cells to adequately deal with aldehyde-induced chromatin damage. Administration of a commonly used antioxidant N-acetylcysteine to Atm-/- mice inhibited bone marrow degeneration and the development of leukemia (54,55), which is viewed as evidence for the importance of ATM in protection of bone marrow stem cells against oxidative stress. However, SH-containing compounds are also excellent scavengers of aldehydes, as evidenced, e.g. by a complete rescue of FA toxicity in cultured cells by mercaptoethanol (47). N-acetylcysteine was protective against FA-induced tissue injury in vivo (56) and its use would be expected to alleviate effects of ATM deficiency on chromatin damage by endogenous aldehydes.
FUNDING
National Institute of Environmental Health Sciences [ES020689]. Funding for open access charge: National Institute of Environmental Health Sciences [ES020689].
Conflict of interest statement. None declared.
REFERENCES
- 1.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 4.Riballo E., Kuhne M., Rief N., Doherty A., Smith G.C., Recio M.J., Reis C., Dahm K., Fricke A., Krempler A., et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 5.Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Lobrich M., Jeggo P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez-Quilon A., Serrano-Benitez A., Lieberman J.A., Quintero C., Sanchez-Gutierrez D., Escudero L.M., Cortes-Ledesma F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014;5:3347. doi: 10.1038/ncomms4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKinnon P.J. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 8.Ditch S., Paull T.T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem. Sci. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R. 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 10.Mu J.J., Wang Y., Luo H., Leng M., Zhang J., Yang T., Besusso D., Jung S.Y., Qin J. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 2007;282:17330–17334. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 11.Bensimon A., Schmidt A., Ziv Y., Elkon R., Wang S.Y., Chen D.J., Aebersold R., Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 12.Misteli T., Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009;10:243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukas J., Lukas C., Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell. Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 14.Krishnan V., Chow M.Z., Wang Z., Zhang L., Liu B., Liu X., Zhou Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12325–12330. doi: 10.1073/pnas.1102789108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh M., Hunt C.R., Pandita R.K., Kumar R., Yang C.R., Horikoshi N., Bachoo R., Serag S., Story M.D., Shay J.W., et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol. Cell. Biol. 2013;33:1210–1222. doi: 10.1128/MCB.01676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black J.C., Allen A., Van Rechem C., Forbes E., Longworth M., Tschop K., Rinehart C., Quiton J., Walsh R., Smallwood A., et al. Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol. Cell. 2010;40:736–748. doi: 10.1016/j.molcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Sun Y., Xu Y., Roy K., Price B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell Biol. 2007;27:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y., Jiang X., Xu Y., Ayrapetov M.K., Moreau L.A., Whetstine J.R., Price B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J.H., Paull T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 20.Lee J.H., Paull T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 21.Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 22.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 23.Kaidi A., Jackson S.P. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498:70–74. doi: 10.1038/nature12201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.National Toxicology Program. Final Report on Carcinogens Background Document for Formaldehyde. Rep. Carcinog. Backgr. 2010;10–5981:i512. [PubMed] [Google Scholar]
- 25.Hauptmann M., Stewart P.A., Lubin J.H., Beane Freeman L.E., Hornung R.W., Herrick R.F., Hoover R.N., Fraumeni J.F. Jr, Blair A., Hayes R.B. Mortality from lymphohematopoietic malignancies and brain cancer among embalmers exposed to formaldehyde. J. Natl. Cancer Inst. 2009;101:1696–1708. doi: 10.1093/jnci/djp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwilk E., Zhang L., Smith M.T., Smith A.H., Steinmaus C. Formaldehyde and leukemia: an updated meta-analysis and evaluation of bias. J. Occup. Environ. Med. 2010;52:878–886. doi: 10.1097/JOM.0b013e3181ef7e31. [DOI] [PubMed] [Google Scholar]
- 27.Hecht S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 28.Walport L.J., Hopkinson R.J., Schofield C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012;16:525–534. doi: 10.1016/j.cbpa.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 30.Garaycoechea J.I., Crossan G.P., Langevin F., Daly M., Arends M.J., Patel K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 31.Oberbeck N., Langevin F., King G., de Wind N., Crossan G.P., Patel K.J. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol. Cell. 2014;55:807–817. doi: 10.1016/j.molcel.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds M., Peterson E., Quievryn G., Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J. Biol. Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 33.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 34.Reynolds M.F., Peterson-Roth E.C., Bespalov I.A., Johnston T., Gurel V.M., Menard H.L., Zhitkovich A. Rapid DNA double-strand breaks resulting from processing of Cr-DNA cross-links by both MutS dimers. Cancer Res. 2009;69:1071–1079. doi: 10.1158/0008-5472.CAN-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang D., Zaugg K., Mak T.W., Elledge S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 36.Stiff T., Walker S.A., Cerosaletti K., Goodarzi A.A., Petermann E., Concannon P., O'Driscoll M., Jeggo P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25:5775–5782. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomimatsu N., Mukherjee B., Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009;10:629–635. doi: 10.1038/embor.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong V.C., Cash H.L., Morse J.L., Lu S., Zhitkovich A. S-phase sensing of DNA-protein crosslinks triggers TopBP1-independent ATR activation and p53-mediated cell death by formaldehyde. Cell Cycle. 2012;11:2526–2537. doi: 10.4161/cc.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruse J.P., Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lomonosov M., Anand S., Sangrithi M., Davies R., Venkitaraman A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altmeyer M., Toledo L., Gudjonsson T., Grofte M., Rask M.B., Lukas C., Akimov V., Blagoev B., Bartek J., Lukas J. The chromatin scaffold protein SAFB1 renders chromatin permissive for DNA damage signaling. Mol. Cell. 2013;52:206–220. doi: 10.1016/j.molcel.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 42.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortega-Atienza S., Green S.E., Zhitkovich A. Proteasome activity is important for replication recovery, CHK1 phosphorylation and prevention of G2 arrest after low-dose formaldehyde. Toxicol. Appl. Pharmacol. 2015;286:135–141. doi: 10.1016/j.taap.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakano T., Katafuchi A., Matsubara M., Terato H., Tsuboi T., Masuda T., Tatsumoto T., Pack S.P., Makino K., Croteau D.L., et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009;284:27065–27076. doi: 10.1074/jbc.M109.019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duxin J.P., Dewar J.M., Yardimci H., Walter J.C. Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell. 2014;159:346–357. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ridpath J.R., Nakamura A., Tano K., Luke A.M., Sonoda E., Arakawa H., Buerstedde J.M., Gillespie D.A., Sale J.E., Yamazoe M., et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;67:11117–11122. doi: 10.1158/0008-5472.CAN-07-3028. [DOI] [PubMed] [Google Scholar]
- 47.Rosado I.V., Langevin F., Crossan G.P., Takata M., Patel K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011;18:1432–1434. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- 48.Toledo L.I., Altmeyer M., Rask M.B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 49.MacAlpine D.M., Almouzni G. Chromatin and DNA replication. Cold Spring Harb. Perspect. Biol. 2013;5:a010207. doi: 10.1101/cshperspect.a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu K., Boysen G., Gao L., Collins L.B., Swenberg J.A. Formaldehyde-induced histone modifications in vitro. Chem. Res. Toxicol. 2008;21:1586–1593. doi: 10.1021/tx8000576. [DOI] [PubMed] [Google Scholar]
- 51.Tessarz P., Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 52.Jackson V. Studies on histone organization in the nucleosome using formaldehyde as a reversible cross-linking agent. Cell. 1978;15:945–954. doi: 10.1016/0092-8674(78)90278-7. [DOI] [PubMed] [Google Scholar]
- 53.O'Connor P.M., Fox B.W. Isolation and characterization of proteins cross-linked to DNA by the antitumor agent methylene dimethanesulfonate and its hydrolytic product formaldehyde. J. Biol. Chem. 1989;264:6391–6397. [PubMed] [Google Scholar]
- 54.Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I., Nomiyama K., Hosokawa K., Sakurada K., Nakagata N., et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 55.Reliene R., Schiestl R.H. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair (Amst) 2006;5:852–859. doi: 10.1016/j.dnarep.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 56.Skrzydlewska E., Elas M., Ostrowska J. Protective Effects of N-Acetylcysteine and Vitamin E Derivative U83836E on Proteins Modifications Induced by Methanol Intoxication. Toxicol. Mech. Methods. 2005;15:263–270. doi: 10.1080/15376520590968815. [DOI] [PubMed] [Google Scholar]