


Abstract
The centromeres of many eukaryotic chromosomes are established epigenetically on potentially variable tandem repeats; hence, these chromosomes are at risk of being acentric. We reported previously that artificially created acentric chromosomes in the fission yeast Schizosaccharomyces pombe can be rescued by end-to-end fusion with functional chromosomes. Here, we show that most acentric/functional chromosome fusion events in S. pombe cells harbouring an acentric chromosome I differed from the non-homologous end-joining-mediated rearrangements that result in deleterious dicentric fusions in normal cells, and were elicited by a previously unidentified homologous recombination (HR) event between chromosome end-associated sequences. The subtelomere repeats associated with the non-fusogenic ends were also destabilized in the surviving cells, suggesting a causal link between general subtelomere destabilization and acentric/functional chromosome fusion. A mutational analysis indicated that a non-canonical HR pathway was involved in the rearrangement. These findings are indicative of a latent mechanism that conditionally induces general subtelomere instability, presumably in the face of accidental centromere loss events, resulting in rescue of the fatal acentric chromosomes by interchromosomal HR.
INTRODUCTION
The eukaryotic genome is divided into several linear DNA molecules, each of which is individually packaged as a chromosome. The chromosomes move independently during mitosis and meiosis by virtue of centromeres that allow elaborate microtubule interactions and precise chromosome segregation (1–3). The existence of a single centromere is a prerequisite for the proper behaviour and function of chromosomes, and dicentric or acentric chromosome formation has a detrimental on faithful genome inheritance (3,4). However, because of the unsteady nature of telomeres and centromeres, the occasional formation of dicentric and acentric chromosomes is inevitable.
Telomere unsteadiness stems largely from the sequence repetition. Telomeres at the ends of chromosomes are composed of tandem repeats, and telomerase reverse transcriptase (Trt1 in fission yeast (5)) successively extends the telomere repeat (tel) to compensate for incomplete terminal DNA replication by canonical DNA polymerases (6,7). The resulting tel array acts as a platform for a number of protein complexes that play a role in chromosome end protection; for example, the shelterin complex that includes the subunits TRF1/2 and POT1 (6) (Taz1 and Pot1 in fission yeast (8,9)), which bind to double-stranded and single-stranded tel DNAs, respectively, and the DNA repair complex comprising Ku70 and Ku80 (7) (Pku70 and Pku80 in fission yeast (10–12)). Accordingly, the existence of appropriate amounts of tel DNA is sufficient to protect DNA ends against nucleolytic activities and fusogenic rearrangements (6,7). However, even in the presence of telomerase activity, stochastic shortening of tel DNA to a critical length is unavoidable, and is caused mainly by an abrupt deletion within the tel DNA rather than gradual erosion (13–15). This stochastic deprotection allows dicentric chromosome fusion and can lead to catastrophic consequences, such as those postulated to be involved in carcinogenesis (15,16).
Dicentric fusions can be mediated by non-homologous end-joining (NHEJ), a major DNA double-stranded break (DSB) repair pathway in cells (13–17). Cellular DSB repair pathways are classified broadly into two types, namely NHEJ-type and homologous recombination (HR)-type (18–20). Canonical NHEJ re-joins two broken DNA ends directly and is catalysed by a series of protein complexes, including the DNA ligase IV/XRCC4 complex (Lig4/Xrc4 in fission yeast (10,21)) and the Ku70/Ku80 complex (19,20). By contrast, HR generally requires a reference DNA molecule to exchange its DNA strand with that of the broken end (19,20). Hence, specialized enzymes such as Rad51 recombinase (formally called Rhp51 in fission yeast (22)) and Rad52 recombination mediator (formally called Rad22 in fission yeast (23)) are required for most HR-type pathways, although the specific requirements of each pathway vary (18–20,24). For example, single-strand annealing (SSA), an HR-related DSB repair pathway, specifically anneals long single-stranded homologous regions (>30 nucleotides) near the broken ends and requires Rad52 and the end-trimming endonuclease XPF/ERCC1 (Rad16/Swi10 in fission yeast (25,26)), but not Rad51 (18–19,24). Among such a wide variety of cellular DSB repair pathways, NHEJ pathways must be selected to establish dicentric fusions according to the environmental conditions created by the deprotected chromosomal ends. In fact, the alternative NHEJ pathway that utilizes a 5–25 nucleotide microhomologous region at the vicinity of DNA ends (19,24,27) has been proposed as the actual mechanism involved in spontaneous dicentric fusions in human cell lines (13–15), whereas canonical NHEJ has been proposed in the case of fission yeast (17).
Like telomeres, centromeres are also inherently unsteady (3,4). Although the protein constituents of centromeres are highly conserved across species, the centromere-specific sequences that are localized commonly in every chromosome lack conservation across species, suggesting that the centromeres are established epigenetically (1–3). The discovery of ectopic neocentromeres at centromere-irrelevant DNA sequences in various organisms stresses the epigenetic characteristics of centromeres further (3,28–29). Hence, centromere maintenance during cell division relies on the epigenetic inheritance mechanism, failure of which results in acentric chromosome formation (4,30). In addition, centromeres are constantly exposed to the risk of internal deletion and loss from the chromosome, because most centromeric DNAs comprise tandem repeats that are structurally unstable (3,28–29,31). A few reports have described the events involved in centromere loss; however, these studies have been hampered by experimental limitations, and centromere loss can be overlooked without the occurrence of secondary stabilization events (28). Analyses of the evolutionary transition of chromosome configuration have suggested that the loss and gain of centromeres is one of the key events for chromosomal rearrangements (4,32–33).
Notably, centromere instability is used to circumvent deleterious dicentric fusions that arise from telomere instability (16,29,34). In previous studies, loss of one of the two centromeres was shown to stabilize fusogenic chromosomes and enable their discovery through cytogenetic analyses (34). Epigenetic inactivation and genetic deletion have been identified as the centromere loss mechanisms (16,29). Based on the results of genetic experiments (30,35–37), it is largely believed that the centromere loss occurs after the formation of otherwise deleterious dicentric chromosomes (16,29); however, this notion does not negate the other possibility that stabilized dicentrics are generated through the primary formation of an acentric chromosome, followed by its end-to-end fusion with a functional chromosome (16,34). Considering the inherent instabilities and dynamics of centromeres and telomeres, both types of sequential events should be possible in nature and may not necessarily be mutually exclusive. In fact, the two events are equally envisioned, at least for evolutionary considerations (32,33). However, poor elucidation of the fate of acentric chromosomes in living cells prevents further discussion of the formation of stable chromosome fusions and the possible use of linear chromosome ends to circumvent accidentally-formed acentric chromosomes (34).
In our previous study, we reported the formation of acentric chromosomes in fission yeast using conditional expression of Cre recombinase in cells in which the endogenous centromere of chromosome I (cen1) was flanked by two loxP sites (loxP-cen1) (38). Most of the cells died after cen1 excision (Δcen1), but the acentric chromosome I was maintained in a fraction of yeast survivors either by formation of a neocentromere on the chromosome, or by end-to-end fusion with the remaining chromosomes II or III (38). Here, we examined the individual features of the end-to-end fusions found in the fission yeast Δcen1 survivors in detail. Surprisingly, most of the acentric/functional fusions differed from the spontaneous dicentric-forming NHEJ-mediated fusions. Instead, the data suggest that the induction of HR-based rearrangements promoted chromosome end-to-end fusion to stabilize the acentric chromosome in the Δcen1 cells.
MATERIALS AND METHODS
Yeast cell manipulations
The yeast strains used in this study are listed in Supplementary Table S1. YES and EMM2 were used as basic media. Gene disruption and gene tagging were performed using a PCR-based method, as described previously (39). The cen1 deletion assays were performed in various mutant backgrounds, as described previously (40). The 19 Δcen1-f survivors analysed in this study corresponded to all of the fusion survivors sampled unbiasedly over the course of two cen1 deletion experiments performed in a wild-type background (38). Naming of the survivors followed the conventions described previously (40).
The presence of subtelomere repeats in the subtelomere-associated sequence (SAS)-distal regions of chromosome III was demonstrated using pulsed-field gel electrophoresis (PFGE) and Southern blotting. To this end, the pFA6a-RTboundarySN-LEU2 plasmid, which harboured a ScaI/NcoI sub-fragment of SAS and the Saccharomyces cerevisiae LEU2 gene, was linearized by PCR amplification of the entire plasmid, and then integrated into the SAS regions of the loxP-cen3 strain (40). In each case, the plasmid was integrated into the same locus multiple times.
The frequencies of lithium chloride (LiCl)-resistant cells were determined as described previously (41), with some modifications. Wild-type and Δrad51 cells harbouring pREP81 (42) were cultured in EMM2 medium containing adenine, uracil, histidine and lysine, and then plated onto agar plates at a density of 2 × 107 cells per plate in the same medium containing 20 mM LiCl. The LiCl-resistant colonies were counted after incubating the plates at 33°C for approximately 7 days.
PFGE
DNA plug preparation, restriction enzyme digestion and PFGE were performed as described previously (38,40). The PFGE conditions were as follows: (i) NotI-digested samples: 0.8% agarose gel with 0.5X TBE buffer at 6 V/cm and a 120° angle, with a pulse time of 83–167 s for 20 h; (ii) SfiI-digested DNA: 0.8% agarose gel with 0.5X TBE buffer at 6 V/cm and a 120° angle, with a pulse time of 40–120 s for 20 h; and (iii) BamHI-digested DNA: 1.0% agarose gel with 0.5X TAE buffer at 6 V/cm and a 120° angle, with a pulse time of 2.0–1.8 s for 11 h.
Southern blotting
Gel-separated genomic DNAs were alkaline-transferred onto 0.45 μm nylon membranes (Whatman/GE Healthcare, Piscataway, NJ, USA) and incubated with probes labelled with [α-32P]-dCTP or alkaline phosphatase using the Random Primer DNA Labelling Kit (version 2; Takara Bio, Shiga, Japan) or the AlkPhos Direct Labelling and Detection System (GE Healthcare), respectively. The DNA probes were as follows: TAS1, an 807 bp EcoRI-/ApaI-digested fragment obtained from p282 (a derivative of pNSU70, kindly provided by Dr Ishikawa, Kyoto University, Kyoto, Japan); TAS2, a 3477 bp NsiI-digested fragment obtained from p282; TAS3, a 734 bp EcoRV-/HindIII-digested fragment obtained from p282; tel/STE1’, a 300 bp ApaI-/SacI-digested fragment obtained from pITN1 (provided by Dr Ishikawa); and Padh1, a 774 bp PacI-/NdeI-digested fragment obtained from pBS-AS-ura4PB-kanloxNco (38). A tel-specific probe was prepared by T4 DNA polynucleotide kinase-mediated phosphorylation of the 5′-end of the tel oligonucleotide with [γ-32P]-ATP. Hybridization of the membranes with radiolabelled probes was performed in ULTRAhyb hybridization buffer (Ambion Life Technologies, Grand Island, NY, USA), according to the manufacturer's instructions. Radioisotope signals were detected using the BAS 2500 Image Analysis System (Fujifilm, Tokyo, Japan), and non-radioisotope hybridization signals were detected using the ImageQuant LAS 4000 image analyser (GE Healthcare).
Genomic PCR
Genomic DNA was recovered from the yeast cells by vigorous shaking with glass beads and used as a template for genomic PCRs (gPCRs), which were performed using Ex Taq or Gflex polymerase (Takara Bio) and the primers shown in Supplementary Table S2. The PCR conditions were as follows: 14206/31268: initial denaturation at 94°C for 2 min, followed by 22 cycles of 94°C for 10 s, 55°C for 30 s and 68°C for 4 min, and then a final extension at 68°C for 10 min; 7926/31268: initial denaturation at 94°C for 2 min, followed by 30 cycles of 94°C for 10 s, 55°C for 30 s and 72°C for 2 min, and then a final extension at 72°C for 10 min; Asp(1L)/Asp(2L): initial denaturation at 94°C for 2 min, followed by 25 cycles of 94°C for 10 s, 55°C for 30 s and 72°C for 1 min, and then a final extension at 72°C for 10 min; TAS1_TAIL-II/TAS3_TAS2-F1: initial denaturation at 94°C for 2 min, followed by 35 cycles of 94°C for 10 s, 55°C for 30 s and 72°C for 2 min 40 s, and then a final extension at 72°C for 10 min; TAS1_TAS2-F1/subtelrD-R4: initial denaturation at 94°C for 2 min, followed by 30 cycles of 94°C for 10 s, 55°C for 15 s and 72°C for 3 min, and then a final extension at 72°C for 2 min; c-rDNA-t/60241: initial denaturation at 94°C for 1 min, followed by 30 cycles of 98°C for 10 s, 55°C for 15 s and 68°C for 10 min, and then a final extension at 68°C for 10 min; and SAS-Rv/TAS1-Rv1: initial denaturation at 94°C for 2 min, followed by 30 cycles of 98°C for 10 s, 55°C for 15 s and 68°C for 20 min, and then a final extension at 68°C for 10 min.
Quantitative PCR
Genomic DNA was recovered from the yeast cells by vigorous shaking with glass beads and used as a template for quantitative PCRs (qPCRs), which were performed using the StepOnePlus Real Time PCR System (Applied Biosystems, Foster City, CA, USA) and Power SYBR Green PCR Master Mix (Applied Biosystems). The primers were designed using Primer Express software (Applied Biosystems) and the sequences are listed in Supplementary Table S2. For subtelomeric element 2 (STE2) copy number quantification, the difference between the copy numbers of STE2 and act1+ was multiplied by a constant value of 0.775 to make the average number of STE2 copies in loxP-cen1 isolates equal to 15.
Direct cloning of telomere-associated sequence 2
The telomere-associated sequence 2 (TAS2) NsiI fragments of the wild-type yeast genome were cloned directly into a vector via ligation-independent cloning. The NsiI-digested and size-fractionated wild-type genomic DNA was mixed with PCR-amplified Bluescript vector harbouring terminal sequences that matched the ends of the TAS2 NsiI fragments, and ligation-independent cloning was performed using the In-Fusion HD Cloning Kit (Takara Bio), according to the manufacturer's instructions. The primers used for vector preparation are listed in Supplementary Table S2. The cloned plasmid harbouring the longest TAS2 fragment was subjected to exonuclease digestion using the Kilo-Sequence Deletion Kit (Takara Bio), according to the manufacturer's instructions. In this way, a series of terminal deletion constructs was prepared to determine the entire TAS2 DNA sequence, regardless of the presence of direct repeats.
Statistical analysis
Before performing a statistical test to verify two experimental results, an F-test was conducted to determine whether the variance of the two groups was similar. The results of the F-tests indicated unequal variance; therefore, Welch's t-tests were used for statistical comparisons. Fisher's exact test was also employed for statistical analyses of the ratios of HR-type and NHEJ-type telomere-fusion survivors in given backgrounds because a 2 × 2 contingency table could be built. Data processing and plotting were performed using R software, version 3.0.0 (http://www.r-project.org/).
To evaluate the joint probability of telomere-fusion survivor emergence in wild-type and mutant strains, we developed a Bayesian generalized linear mixed model that allowed us to estimate the posterior distributions of strain effects on yeast survivorship as random effects by fitting to the experimental data. The model is essentially equivalent to the mixed linear logistic model including two random effects, namely replications and strains. The posterior distributions were estimated using Gibbs sampling software JAGS 3.4.0 (http://mcmc-jags.sourceforge.net/). Additional details are provided in the Supplementary Methods.
Fluorescence microscopy
Methanol fixation and subsequent rehydration of cells were performed as described previously (38). Wide-field fluorescence images of the rehydrated cells were obtained using a Ti-E microscope (Nikon Corp., Tokyo, Japan) equipped with a PlanApo × 100 (NA = 1.45) oil-immersion objective lens and an electron-multiplying charge-coupled device (iXon+; Andor Technology, Belfast, United Kingdom). Images were collected every 0.3 μm along the z-axis. The stack was then three-dimensionally blind-deconvoluted using the AutoQuant Modules of NIS Elements AR software (version 4.0; Nikon), followed by maximum intensity projection to generate two-dimensional images.
RESULTS
Most end-to-end fusion events in Δcen1 survivors are accompanied by a loss of subtelomeric sequences from the fusogenic ends
The fusions in the Δcen1 survivors were either between chromosomes I and II (denoted as Δcen1-f(1;2)) or between chromosomes I and III (denoted as Δcen1-f(1;3)) (38). First, we investigated the Δcen1-f(1;2) rearrangements (Figure 1A). A total of 15 Δcen1-f(1;2) survivors were obtained from two Δcen1 screens (Supplementary Figure S1). PFGE and Southern blot analyses of the NotI-digested chromosomal DNAs of the survivors revealed that the new NotI fragments encompassing the fusogenic ends mostly lacked a detectable level of tel repeats, TAS1, TAS2 and sometimes TAS3 (Figure 1A–F). These repetitive elements were commonly located at the terminal regions of the original chromosomes I and II (Figure 1A and Supplementary Figure S2). Therefore, the vast majority of Δcen1-f(1;2) events appeared to be associated with a substantial loss of terminal sequences from both the acentric donor chromosome end and the normal recipient chromosome end. This property differs from that of the previously reported spontaneous end-to-end fusion in fission yeast that occurs at or just adjacent to the tel repeat (17).
Figure 1.
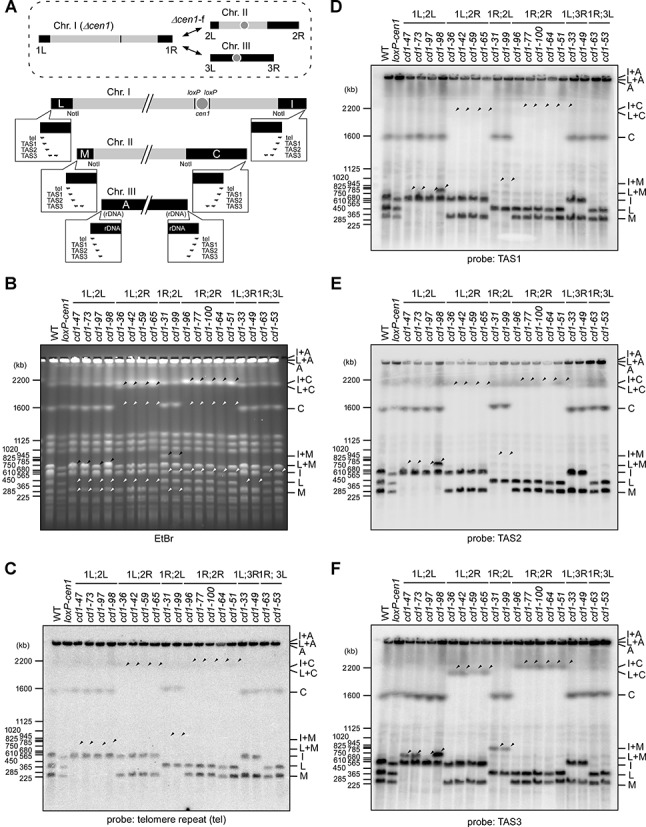
Properties of the fusion ends in the Δcen1 survivors. (A) A schematic diagram of fission yeast chromosomes with emphasis on the terminal NotI fragments (black boxes). Their relative positions in the chromosomes are indicated in the upper image representing the Δcen1-f rearrangements (dashed circle). The original fragments were detectable by Southern blot probes corresponding to the tel repeat and subtelomere repeats (TAS1, TAS2 and TAS3). The positions and orientations of the repeats are indicated by half arrows in the balloons enlarging the chromosome ends. Nomenclature of the NotI fragments follows previous conventions (64). The cen1 region is flanked by loxP sites to represent the loxP-cen1 construct. (B–F) PFGE analyses of NotI-digested chromosomes of telomere-fusion survivors collected from two individual cen1 deletion experiments. The PFGE samples were subjected to ethidium bromide (EtBr) staining (B) and Southern blotting with the tel (C), TAS1 (D), TAS2 (E) and TAS3 (F) probes. The white arrowheads indicate the disappearance of NotI bands in the survivors due to the fusion rearrangement, and the black arrowheads indicate the newly-generated fusion bands. The identities of the bands with altered migration are indicated at the right-hand side of the gels. A number of the fusion bands failed to hybridize with the tel or subtelomere probes. WT, wild-type; loxP-cen1, cen1 deletion strain.
The majority of Δcen1-fusions are mediated through specific sequence homologies
The disappearance of subtelomeric sequences from both fusogenic ends is reminiscent of the intrachromosomal end-to-end fusion caused by artificial telomere loss, such as that seen in the survivors of Δtrt1 or Δpot1 mutations (43,44). The rearrangements in these survivors occur between a pair of inversely homologous sequences located around the degenerated TAS3 elements at the telomere-distal regions (Supplementary Figure S2A) (43); therefore, we investigated whether the fusion points of the Δcen1-f(1;2) survivors were also related to sequence homology. To this end, a series of gPCRs were performed using outward-facing primers that generated a product only when fusion occurred at a site external to each primer (Figure 2A). Using the same primer sets as those used for investigation of the Δpot1 survivors (43), the same pattern of gPCR products was obtained for 13 of the 15 Δcen1-f(1;2) survivors (Figure 2B). These results suggested that their fusion point could be identical to the reported fusion point in the Δpot1 survivors (43), and this proposal was confirmed by sequencing of the gPCR products (Supplementary Figure S3A). Moreover, a Δcen1-f(1L;2L) survivor (cd1-97) that lacked TAS3 in its fusogenic NotI fragment (Figure 1) yielded a specific gPCR product when using primers that bound to a pair of highly homologous L-asparaginase (Asp) genes (SPAC977.12 and SPBPB8B6.05c; 98% identity at the nucleotide sequence level) located 70 kb and 110 kb upstream of the 1L and 2L ends, respectively (Figure 2A and B). Therefore, DNA sequence homology is most likely a preferable property for the observed Δcen1-f(1;2) fusions, which contrasts with the spontaneous end-to-end fusions generated by NHEJ (17).
Figure 2.
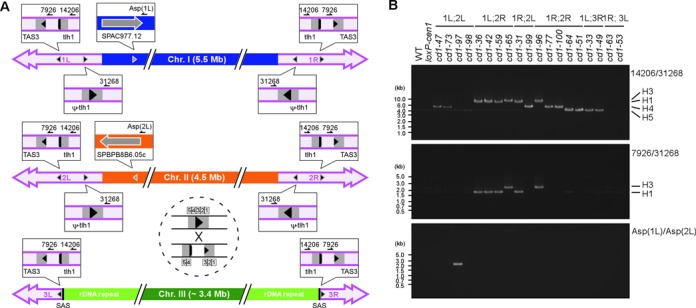
Chromosome fusion-point mapping in the Δcen1 survivors. (A) A schematic diagram of candidate fusion points along the fission yeast chromosomes. The approximate positions and relative orientations of the fusion points in each chromosome are represented by grey arrowheads, and the individual details are shown in the enlarged balloons. A crossover event is exemplified within the dashed circle. The positions of the primers used for gPCR are indicated by half arrows in the balloons. The following nearby genes are also shown: tlh1, telomere-linked helicase (typically SPAC212.11); and ψ-tlh1, a gene sharing DNA sequence homology with tlh1 (typically SPAC212.06c). The homology segment at the telomere-proximal region is drawn as though it is segregated into two parts; however, as indicated in the dashed circle, the region actually comprises five separate parts (H1–H5) (43), with the largest interval between H3 and H4. SAS is a chromosome III-specific sequence demarcating the subtelomere and rDNA repeats (see Supplementary Figure S5). (B) A gPCR analysis demonstrating the fusion between subtelomere segments (14206/31268 and 7926/31268) and L-asparaginase genes (Asp(1L)/Asp(2L)). The position of each primer is indicated in (A). Note that the size variation among the subtelomeric gPCR products reflects the actual crossover position, which is indicated at the right-hand side of the gels, and that the 7926/31268 gPCR was effective only when the crossover occurred at either H4 or H5.
NHEJ contributes to a minor fraction of fusion events in Δcen1 survivors
Of the 15 Δcen1-f(1;2) survivors analysed, 14 utilized sequence homology for their fusion event. By contrast, the cd1-98 isolate did not produce a gPCR product indicative of homology-directed fusion (Figure 2B) and retained the TAS1, TAS2 and TAS3 sequences in its fusogenic NotI fragment (Figure 1D–F). This finding suggested that the Δcen1-f(1L;2L) rearrangement in cd1-98 was a consequence of NHEJ-mediated spontaneous fusion; therefore, we performed gPCRs using several PCR primers that were designed to hybridize to different telomere-proximal regions in an outward orientation. When using a primer that hybridized within TAS1 and another that hybridized to a region between TAS2 and TAS3, cd1-98 yielded a 5 kb gPCR product (Supplementary Figure S3B and data not shown). DNA sequencing of the product revealed that the rearrangement was a head-to-head fusion among the direct repeats located in TAS1 and TAS2 (STE1’ repeat; see below for nomenclature), and that NHEJ accounted for the event (Supplementary Figure S3C). Therefore, we concluded that the fusion events in the Δcen1-f(1;2) survivors included both inverse homology-directed rearrangement (HR-type) and end-joining between non-homologous sequences (NHEJ-type). Because the HR-type rearrangement has never been shown to be involved in stochastically-occurring spontaneous end-to-end fusions in fission yeast (17), it may be a specific response to Δcen1 chromosome generation. By contrast, the NHEJ-type rearrangement can happen in wild-type cells and it may be selected in the screen simply by removing cen1. Given that the rate of generation of NHEJ-type Δcen1-f(1;2) survivors represents the spontaneous fusion frequency in the presence of functional telomeres, the HR-type Δcen1-f(1;2) rearrangement was estimated to occur 14 times more frequently than the NHEJ-type.
Chromosome III-participating fusions are also classified as HR- and NHEJ-type events
Next, we analysed the Δcen1-f(1;3) survivors and compared their fusion spectrum with that of the Δcen1-f(1;2) survivors. The existence of ribosomal DNA (rDNA) repeats at both termini and the total absence of NotI sites complicated the structural analyses of chromosome III; therefore, we used SfiI digestion for the PFGE analysis. The NotI and SfiI digestion patterns of the chromosomes collectively identified two survivors (cd1-33 and cd1-49) showing a Δcen1-f(1L;3R) fusion and two survivors (cd1-53 and cd1-63) showing a Δcen1-f(1R;3L) fusion (Figure 1 and Supplementary Figure S4). Furthermore, PFGE, Southern blotting and gPCR analyses revealed that the two Δcen1-f(1L;3R) events were the result of fusion between the inversely homologous regions in the subtelomeres, while the two Δcen1-f(1R;3L) events were the result of NHEJ-mediated fusion between the tel repeat and rDNA repeat (Figure 2B and Supplementary Figure S4B). These observations suggest that the fusions of both Δcen1-f(1;2) and Δcen1-f(1;3) occurred as a result of either an HR- or NHEJ-type rearrangement. In the Δcen1-f(1;3) survivors, the efficiency of the HR-type fusion was equal to that of the NHEJ-type fusion, although the efficiencies of both were lower than that of the HR-type fusion in the Δcen1-f(1;2) survivors; this observation was explained by our finding that the proximal half of the subtelomere segment including the homologous regions used for the HR-type fusion was omitted from both ends of the original chromosome III (Supplementary Figure S5). Consistent with the extent of homology, the Δcen1-f(1;2) fusions occurred more frequently than the Δcen1-f(1;3) fusions.
Subtelomere regions of Δcen1 survivors are generally destabilized
The establishment of previously unidentified HR-type fusions presumably requires some kind of trigger. Despite the resemblance to the fusion properties of the Δtrt1 or Δpot1 survivors, telomere shortening associated with Δtrt1 or Δpot1 mutants is unlikely to be a trigger of the HR-type fusions because centromere disruption must have no influence on the functions of Pot1 and Trt1 at the telomeres. No obvious telomere shortening was detected via Southern blot analyses of the Δcen1-f survivors or cells undergoing acentric chromosome formation (Supplementary Figure S6); however, we cannot rule out the possibility that telomere shortening occurred in only a small fraction of the Δcen1 cells and led to Δcen1-f rearrangements. Nevertheless, the Southern blot analyses did reveal an altered band pattern in the Δcen1 survivors when probes specific for the telomere-distal TAS2 and TAS3 regions were employed (Figure 3A–C). One or two of the alterations could be attributable to chromosome end-to-end fusions because HR-type fusions obliterated the terminal portion of the fusogenic ends (Supplementary Figure S7A). However, numerous other alterations occurred independently of the fusion, which is indicative of structural alterations in the fusion-irrelevant subtelomeres of the survivors (Figure 3A–C, see also Supplementary Figure S6B). Such general alterations were barely detected in the subtelomeres of normally growing wild-type cells (Supplementary Figures S7B–D).
Figure 3.
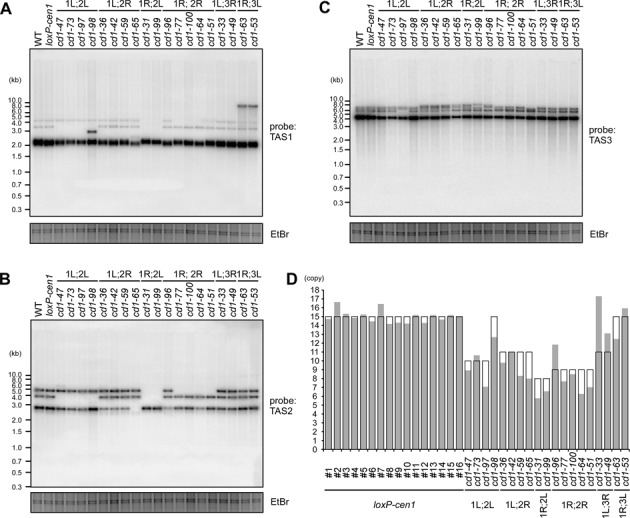
Alteration of subtelomere structures in the Δcen1 fusion survivors. (A–C) Southern blot analyses of genomic DNAs from the indicated telomere-fusion survivors. The DNAs were digested with NsiI and analysed by Southern blotting with the subtelomeric TAS1 (A), TAS2 (B) and TAS3 (C) probes. EtBr-stained images of the gels are shown as loading controls. The fusogenic end-dependent and survivor-specific band pattern alterations were observed by Southern blotting. In addition, the band intensity ratio was variable, even between survivors exhibiting the same band pattern. These findings suggest general subtelomere destabilization in the survivors. (D) The predicted and experimentally determined STE2 copy numbers in the indicated survivors and 16 independent loxP-cen1 clones of different ages (clones #1–16) are shown in the open and grey columns, respectively. Details of the STE2 copy number prediction are shown in Supplementary Figure S7A. For experimental STE2 copy number determination, the STE2 qPCR Ct value was normalized to that of act1+ in the same genome as a single-copy control, and was adjusted further by multiplying by a constant factor to ensure that the average STE2 copy number in the loxP-cen1 clones was equal to 15. The difference between the predicted and measured STE2 copy numbers was significantly larger in the survivors than the parental loxP-cen1 clones (P = 0.00017 by a Welch's two-tailed t-test; see also Supplementary Figure S9B).
To elucidate the cause of the structural alterations, the copy numbers of subtelomeric repeats in the Δcen1 survivors were examined. For descriptive purposes, we redefined the internal repeats in the database-registered standard subtelomeric clone pNSU70 as the following: the previously reported STE1 (∼88 bp) (45,46), a repeat consensus duplicated more than 20 times in the vicinity of the TAS1/TAS2 region (45,47); the STE1’ (∼206 bp), a repeat encompassing a previously reported 64-mer repeat (11) and duplicated three times within and outside TAS1; and the STE2 (∼482 bp) and STE2’ (∼205 bp), two degenerated repeat units alternately duplicated six and three times, respectively, in the vicinity of TAS2/TAS3 (Supplementary Figures S2A–D). Notably, the copy numbers and compositions of these STE repeats differed among the individual subtelomeric clones in the database (Supplementary Figures S2B and S2D). However, the genome sequences suggested that the TAS2 variations observed in the Southern blot analyses were explained by the STE2 copy number; specifically, two and three copies of STE2 in the database clones (Supplementary Figures S2B and S2D) accounted for the two smaller TAS2 bands (Figure 3B and Supplementary Figures S7A and S7C), and four copies of STE2 in a novel TAS2 clone that was cloned directly from the wild-type genome (Supplementary Figure S2E) accounted for the largest TAS2 band (Figure 3B and Supplementary Figures S7A and S7C). Because Southern blotting of various Δcen1-f survivors allowed us to deduce the size of the TAS2 band at each subtelomere of wild-type cells (Supplementary Figure S7A, see also Supplementary Figure S2B), we were also able to predict the total STE2 copy number in the wild-type and fusion survivor clones (Figure 3D and Supplementary Figures S2A). However, qPCR analyses revealed that the actual STE2 copy number in each survivor differed significantly from the predicted number (Figure 3D), supporting the supposition that the survivors underwent induction of non-reciprocal HR between different subtelomeres and/or tandem repeat destabilization within individual subtelomeres.
HR-type Δcen1 fusion is not mediated by XPF/ERCC1-dependent SSA and is increased by Rad51 deficiency
To elucidate the mechanism involved in the homology-directed Δcen1 fusions, we performed experiments using a loxP-cen1 strain harbouring assorted DNA repair mutations (Figure 4 and Supplementary Figures S1 and S8). The results obtained did not indicate a clear dependency of Δcen1-f rearrangements on any single canonical DSB repair pathway, but rather stressed a possible involvement of non-canonical HR rearrangement.
Figure 4.
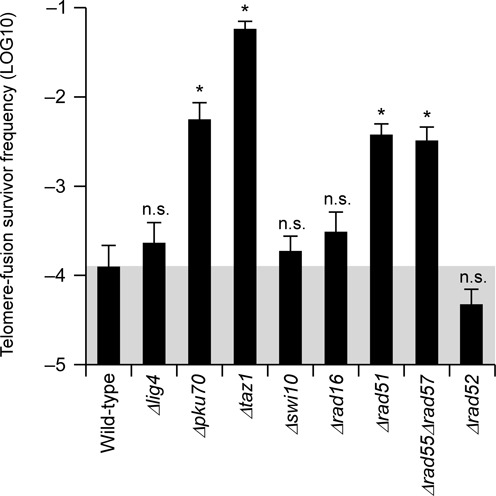
The effects of various mutations on the frequency of telomere-fusion survivor generation. The frequencies of telomere-fusion survivor generation upon cen1 deletion in the indicated mutant backgrounds are shown. Data are represented as the mean ±SEM of n = 6 replicates. *, the 95% Bayesian confidence interval (BCI) does not include zero; n.s., the 95% BCI includes zero (not significant).
Lig4 and Pku70. Despite the virtual disappearance of NHEJ-type survivors (Supplementary Figures S8A and S8B), the efficiencies of Δcen1-f rearrangements in NHEJ-deficient Δlig4 and Δpku70 mutants (2.3 × 10−4 and 5.6 × 10−3, respectively; Figure 4) were not significantly lower than that in wild-type cells (1.3 × 10−4; Figure 4). This result supports the minor contribution of NHEJ to Δcen1-f rearrangements. Notably, the numbers of HR-type Δcen1-f survivors were increased significantly in the Δpku70 mutant (Figure 4 and Supplementary Figures S8A and S8B). This result may be related to the reported instability of subtelomere repeats, most prominently STE1 and STE1’, in this mutant (10–12).
Taz1. Loss of the shelterin subunit Taz1 promotes NHEJ-mediated chromosome end-to-end fusion (48). In line with this finding, the Δcen1-f rearrangement frequency was approximately 460-fold higher in Δtaz1 mutant cells (5.8 × 10−2; Figure 4) than wild-type cells. However, only 44% of the augmented survivors were classified as NHEJ-type, and the remainder were HR-type (Supplementary Figures S8A and S8B); therefore, the net increase in the HR-type fusion frequency caused by the Δtaz1 mutation was approximately 300-fold. Given the induction of HR-mediated STE1 destabilization in Δtaz1 cells (47), the observed increase in HR-type fusions in these cells may also be attributable to HR-mediated subtelomere instability.
Swi10 and Rad16. The sites used for most HR-type Δcen1-f rearrangements were identical to those used in the end-to-end fusion events caused by the Δpot1 and Δtrt1 mutations, which are known to be established through Swi10/Rad16-mediated SSA (43). However, the Δcen1-f spectra of the Δswi10 and Δrad16 mutants and their frequencies (1.9 × 10−4 and 3.1 × 10−4, respectively; Figure 4 and Supplementary Figure S8) were similar to those of the wild-type cells. Therefore, unlike telomerase deficiency-provoked fusions, Swi10/Rad16-dependent SSA does not seem to contribute to Δcen1-f rearrangements in any major fashion.
Rad51. Rad51 recombinase-mediated crossover-type HR (18,19) is another plausible mechanism involved in the HR-type Δcen1-f rearrangements. However, the Δrad51 mutation did not interfere with the generation of Δcen1-f; rather, it enhanced the fusion frequency by 30-fold (3.8 × 10−3; Figure 4). A similar enhancement was observed when Rad51 activity was downregulated by mutating Rad55 and Rad57 (49) (3.3 × 10−3; Figure 4). A fusion spectrum analysis indicated that the increased number of mutant survivors was explained by HR-type events (Supplementary Figures S8A and S8B). Taken together, these results suggest that Rad51 deficiency creates a favourable condition for HR-type fusions.
Rad52. The HR mediator protein Rad52 facilitates the Rad51-mediated crossover reaction (18). However, unlike the results observed for the Δrad51 mutant, the frequency of Δcen1-f rearrangements was reduced slightly but was not abolished or increased in the Δrad52 mutant (4.7 × 10−5; Figure 4 and Supplementary Figure S8). Together with the SSA-independency of the Δcen1-f rearrangements, identified using the Δswi10 and Δrad16 mutants, the differential effects of the Δrad51 and Δrad52 mutations indicate that the Δcen1-f mechanism is a previously unidentified HR-type rearrangement, the details of which remain enigmatic.
Rad51 deficiency promotes subtelomeric genome alterations
STE2 copy number reduction was observed not only in the HR- and NHEJ-type fusion survivors (Figure 3D), but also the neocentromere survivors (Supplementary Figure S9). In addition, extensive alterations of rDNA repeat numbers were detected in some Δcen1-f survivors (Supplementary Figures S4 and S5), as well as some neocentromere survivors, especially those harbouring immature neocentromeres (40). These observations suggest that genome destabilization at chromosomal termini-associated repeats is a general phenomenon in cells that encounter an acentric chromosome.
Notably, Rad51 deficiency increased the efficiency of HR-type fusions but not those of NHEJ-type fusions or neocentromere formation (Supplementary Figure S1 and S9), questioning the general relationship between subtelomere destabilization and Δcen1-f events. However, Southern blot and STE2 qPCR analyses revealed that the subtelomeric TAS2 regions were destabilized even in Δrad51 mutant cells (Figure 5A). In addition, a different analysis revealed a Δrad51-dependent increase in homologous rearrangement susceptibility at the terminally-located Asp genes of chromosomes 1L and 2R (Figure 5B). A previous study reported multiple formation of a 180 kb linear extrachromosome harbouring the sod2+ gene, which encodes a Li+/ H+ antiporter, in LiCl-resistant fission yeast cells (41). This extrachromosome consisted of 1L and 2L terminal fragments that recombined at the Asp genes (50), suggesting that the formation mechanism is common between the LiCl-resistant extrachromosome and the HR-type fusion of cd1-97. The spontaneous emergence of LiCl-resistant cells in the Δrad51 background was 90-fold higher than that in wild-type cells (Figure 5B), indicating that Rad51 deficiency increases both the instability of terminally-located repeats and the frequency of HR-type Δcen1-f rearrangement. It is likely that the Δrad51 mutation promotes terminal genome instability as the initial trigger for fusion rearrangement, whereas NHEJ-type fusion and neocentromere formation occur independently of this initial influence.
Figure 5.
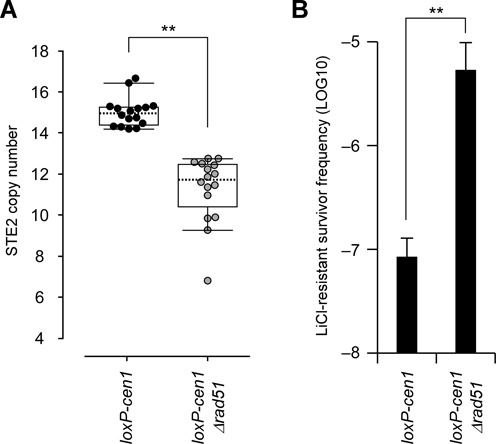
Negative contributions of Rad51 to end-to-end fusion and subtelomere destabilization. (A) The effects of the Δrad51 mutation on the STE2 copy numbers in 16 independent colonies of the indicated strains were determined by qPCR, as described in Figure 3D. Loss of Rad51 reduced the STE2 copy number significantly (**P < 0.01 by a Welch's two-tailed t-test). (B) The frequencies of LiCl-resistant colonies in the indicated strains. The Δrad51 mutation increased the frequency of resistant colonies significantly (**P < 0.01 by a Welch's two-tailed t-test). Data are represented as the mean ±SEM of n = 5 replicates.
The numbers of DSB foci are increased upon acentric chromosome formation
The results described above suggested an enhancement of HR-related events in cells undergoing centromere deletion. We examined this possibility directly by cytological observation of nuclear Rad52 foci formation. The recombination mediator Rad52 generates nuclear foci upon DSB formation (51); hence, nuclear foci formation can be used as a sensitive indicator of intra-nuclear DSBs. Microscopic cellular localization analyses revealed that the formation of green fluorescent protein-tagged Rad52 (Rad52-GFP) foci was increased upon centromere deletion (Figures 6A and B). Intriguingly, simultaneous expression of Rad52-GFP and Taz1-mCherry revealed that the increased numbers of Rad52 foci did not necessarily overlap with telomeric regions in the nuclei (Figure 6B). This observation suggests that DSBs are induced upon centromere deletion, but perhaps not in a chromosome end-restricted manner. The genome alterations that were biased towards chromosome termini in the survivors may have been due to differential behaviours of the DSB repair machineries.
Figure 6.
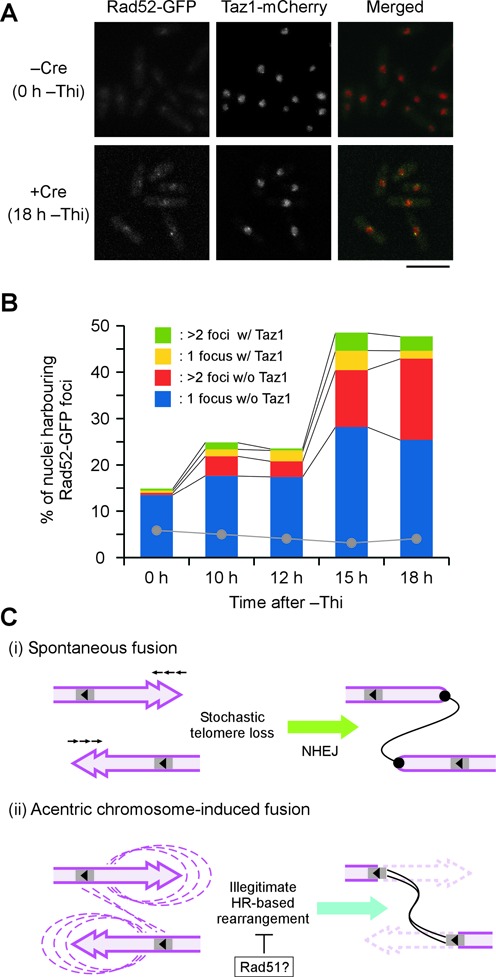
Illegitimate chromosomal rearrangement involving DSB formation in response to acentric chromosome generation. (A) Increase in the number of Rad52 foci during the course of cen1 deletion. Rad52-GFP and Taz1-mCherry were visualized in loxP-cen1 cells before (−Cre) and after (+Cre) the induction of Cre recombinase expression by depletion of thiamine from the medium (−Thi). Scale bar, 10 μm. (B) Quantification of the Rad52-GFP foci-harbouring nuclei observed in (A) and the colocalisation of Rad52 and Taz1 during cen1 deletion. The Rad52 foci frequencies in cells that lacked the loxP-cen1 construct but underwent the same Cre induction treatment are indicated by grey dots. (C) Schematic diagram of two different mechanisms leading to chromosome end-to-end fusion. (i) Spontaneous fusion occurs between chromosome ends exhibiting stochastic telomere loss and is mediated by NHEJ. (ii) Acentric chromosome-induced fusion stems from illegitimate HR-based rearrangement, which generally destabilizes subtelomere regions. Fusion takes place when the rearrangement occurs between inversely located homologous sequences. Rad51 may suppress the reaction. Spontaneous fusion (i) occurs constantly, whereas acentric chromosome-induced fusion (ii) is inducible.
DISCUSSION
This study describes an unusual mechanism of end-to-end fusion between an acentric and a functional chromosome in fission yeast. The mechanism is distinct from NHEJ, which mediates end-to-end fusion events that occur under normal physiological conditions and results in the formation of dicentric chromosomes. By contrast, acentric chromosome-saving fusion is established through DNA sequence homologies that are found in the vicinity of the telomeres (Figure 6C). Although the details of the latter HR-type fusion mechanism remain to be clarified, it is tempting to speculate that the occurrence of so-called undesirable fusion and beneficial fusion events are the consequences of different pathways being influenced by the specific cellular circumstances.
Principal mechanisms involved in the formation and stabilization of fusion chromosomes
Chromosome end-to-end fusion is traditionally regarded as an undesirable or harmful event that creates dicentric chromosomes and triggers the ‘breakage-fusion-bridge’ cycle of massive genome instability (15–16,29). This notion is based on the fact that two centromeres residing in a single chromosome impose uncoordinated chromosomal behaviours in an independently manner. The rearrangement is tolerated when the two centromeres are located so close that they act cooperatively, or when one of the two centromeres is lost from the chromosome (16,29). We showed previously that chromosome end-to-end fusion can act in a positive manner to rescue otherwise fatal acentric chromosomes that are created artificially by chromosome engineering in fission yeast (38). The final consequence of this rearrangement is reminiscent of the centromere loss-mediated dicentric chromosome stabilization; simply the sequence of the two events, end-to-end fusion and centromere loss, can be reversed between them. However, we revealed here that the mechanism involved in the majority of acentric-rescuing fusions, the HR-type fusions, differs from that of dicentric-forming NHEJ-mediated rearrangements (Figure 6C). This observation suggests that the events that take place during the formation and stabilization of fusion chromosomes are more complicated than a simple synchronization of two inherent instabilities associated with centromeres and telomeres.
Centromere loss has been classified mechanistically into two types, namely epigenetic inactivation and structural deletion (16,29). Intriguingly, epigenetic inactivation appears to account for the majority of centromere loss events that occur in constitutional dicentric chromosomes in normal cells (30,35–37), whereas recent reports have identified a higher occurrence of structural centromere deletion in stabilized dicentrics in malignant cancer cells (16). The structures of the genome and chromosomes are unstable in cancer cells, and centromere deletion has been observed on some occasions (52). It might be more plausible to infer that dicentric fusions in cancer cells are formed, at least in part, by HR-type rearrangements that occur in response to centromere deletion, as observed here in fission yeast. By the same token, in normal cells, epigenetic centromere inactivation is a preferential response to NHEJ-mediated random dicentric fusions (36,37). It will be interesting to study the establishment of these chromosome rearrangements in more detail.
A recent study of more than 80 natural isolates derived from Schizosaccharomyces pombe strains revealed extensive karyotype diversification, and several break points causing translocations and gross chromosome rearrangements were mapped at close proximities to telomeres (53,54). The signals and mechanisms contributing to these rearrangements are unclear, but they are unlikely to be associated with telomere dysfunction or chromosome end-to-end fusion. In one of the isolates, extensive rearrangements around the centromeres were observed (54). Centromere-driven induction of rearrangements at the subtelomere regions can therefore occur in nature and may participate in the acquisition of biodiversity.
Illegitimate recombination for end-to-end fusion
Our mutational analysis indicated that HR-type fusions following acentric chromosome formation are elicited by a previously unknown DNA repair pathway (Figure 6C). Contradictory to the general idea that Rad51 and Rad52 act cooperatively to complete the strand-exchange reaction (18,24), the HR-mediated fusion was increased specifically in Δrad51 mutant but not Δrad52 mutant cells. In view of the reported enhancement of SSA activity in budding yeast lacking Rad51 (55), SSA seemingly accounts for the HR-type fusion; however, Δcen1-f rearrangements showed no dependency on the Rad16/Swi10 nuclease, which is essential for SSA in fission yeast. It should be noted that the end-to-end fusion examined here was a rare event. The results of our mutational analysis may represent the rate of stochastic errors that occurred in each mutant rather than the DNA repair deficiency itself, although the survivor spectra in the mutants were mostly relevant to that of wild-type cells (Supplementary Figure S8). In the future, it will be important to determine if any DSB repair factor(s) has the ability to promote HR-type end-to-end fusion directly.
In budding yeast, non-conservative HR-mediated translocation and meiotic non-allelic HR are reportedly inhibited by Rad51 but not Rad52 (55,56). Hence, Rad51 recombinase might be so efficient at promoting legitimate recombinatorial repair that DNA lesions, including those at subtelomere regions, are rapidly returned to their former uninjured states without prompting unfaithful rearrangements (such as Δcen1-f rearrangements) when Rad51 is active (Figure 6C). In agreement with this proposal, deletion of Rad51 increases the number of gross chromosome rearrangements originating from the centromere repeats in fission yeast (57). Furthermore, the minimum homology length required for break-induced replication, yet another HR-related rearrangement, is far shorter when Rad51 is absent from budding yeast (58). Rad51 deficiency may provide cells with a greater opportunity to pursue less efficient and more challenging recombination reactions.
Subtelomere destabilization and chromosome rearrangement
We observed a general destabilization of subtelomere repeats in the fusion-irrelevant normal chromosome ends of Δcen1-f survivors (Figure 3). This type of subtelomere instability is reminiscent of the reported phenotypes of Δpku70, Δpku80 and Δtaz1 cells (10–12,47,59). However, in these mutants, subtelomere destabilization results mainly from the tandem expansion of STE1 and/or STE1’. By contrast, in the Δcen1-f survivors, shrinkage of STE2 was the prominent phenotype, and STE1/STE1’ may have undergone partial shrinkage at best, with rare expansion (Figure 3A and Supplementary Figure S6C). Therefore, the type of subtelomere destabilization may depend on the strain backgrounds and/or chromosomal domains.
Considering that chromosome end tips form the major working site for the Ku complex and Taz1/shelterin, it makes sense that deficiencies of these molecules would influence the end-proximal regions. By contrast, the destabilization in the Δcen1-f survivors presumably arose from the acentric chromosome, the influence of which may not be confined to the end-proximal region; hence, Ku and shelterin should rather be proficient in stabilizing the site of action in acentric chromosome-harbouring cells. We observed an increase in the number of Rad52 foci upon acentric chromosome generation, which is indicative of enhanced DSB formation (51) (Figure 6A and B), but these foci showed no significant overlap with Taz1 signals (Figure 6B). Therefore, the DSB-associated triggers of chromosome rearrangement may not necessarily be linked intimately to the chromosome ends. Recombination near the telomere regions has been studied extensively in budding yeast and human cells as a mechanism to compensate for DNA end shortening that occurs in the absence of telomerase (60). In the case of budding yeast, two types of recombinatorial reactions have been characterized based on differential target sites and trans-acting factors, namely, type-I recombination at the tel repeat and type-II recombination at the subtelomeric Y’ repeat (60). Therefore, differential recombinatorial influences towards the chromosomal ends and subtelomeric regions may be a phenomenon that is conserved across eukaryotes (6,60).
In summary, this study describes a novel chromosome-altering response that occurs upon the formation of acentric chromosomes in fission yeast cells. Although the molecular pathway integrating subtelomere destabilization with end-to-end fusion is unknown, the chromosome-altering response is probably not unique to the experimental system used here. Accumulating evidence has suggested that the subtelomeric regions of organisms, from yeast cells to humans, are generally unstable, even in the presence of telomerase, and are implicated largely in chromosome and genome evolution (32,61–62). Multiple types of rearrangements have been envisioned within the complex subtelomere regions (63).Having such versatile regions near the ends of chromosomes might be key to making the best use of a linear chromosome configuration for genome stability as well as flexibility.
Supplementary Material
Acknowledgments
We are grateful to Dr Fuyuki Ishikawa (Kyoto University) and Dr Hiroshi Iwasaki (Tokyo Institute of Technology) for providing the S. pombe subtelomere clones and h− smt-0 strains, respectively. We thank Dr Hiroshi Iwasaki, Dr Yasuhiro Tsutsui (Tokyo Institute of Technology), Dr Miki Shinohara (Osaka University), and Dr Akira Shinohara (Osaka University) for helpful discussions. We also thank Dr Hiroshi Iwasaki, Dr Yasuhiro Tsutsui, and Dr Junko Kanoh (Osaka University) for critical reading of the manuscript.
Footnotes
Present address: Yuki Ogiyama, Institute of Human Genetics, CNRS UPR 1142, 34396 Montpellier, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Japanese Ministry of Education, Culture, Sports, Science and Technology [22125004, 24370003, 26650124 and 15H05977 to K.I.]; Toray Science Foundation and the Mochida Memorial Foundation for Medical and Pharmaceutical Research. Y.Oh. is a Research Fellow of the Japan Society for the Promotion of Science [24–534]. Funding for open access charge: The Japanese Ministry of Education, Culture, Sports, Science and Technology [22125004, 24370003, 26650124 and 15H05977 to K.I.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Cheeseman I.M. The kinetochore. Cold Spring Harb. Perspect. Biol. 2014;6:a015826. doi: 10.1101/cshperspect.a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamagishi Y., Sakuno T., Goto Y., Watanabe Y. Kinetochore composition and its function: lessons from yeasts. FEMS Microbiol. Rev. 2014;38:185–200. doi: 10.1111/1574-6976.12049. [DOI] [PubMed] [Google Scholar]
- 3.Fukagawa T., Earnshaw W.C. The centromere: chromatin foundation for the kinetochore machinery. Dev. Cell. 2014;30:496–508. doi: 10.1016/j.devcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalitsis P., Choo K.H. The evolutionary life cycle of the resilient centromere. Chromosoma. 2012;121:327–340. doi: 10.1007/s00412-012-0369-6. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura T.M., Morin G.B., Chapman K.B., Weinrich S.L., Andrews W.H., Lingner J., Harley C.B., Cech T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 6.Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 7.Nandakumar J., Cech T.R. Finding the end: recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell. Biol. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper J.P., Nimmo E.R., Allshire R.C., Cech T.R. Regulation of telomere length and function by a Myb-domain protein in fission yeast. Nature. 1997;385:744–747. doi: 10.1038/385744a0. [DOI] [PubMed] [Google Scholar]
- 9.Baumann P., Cech T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 10.Baumann P., Cech T.R. Protection of telomeres by the Ku protein in fission yeast. Mol. Biol. Cell. 2000;11:3265–3275. doi: 10.1091/mbc.11.10.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kibe T., Tomita K., Matsuura A., Izawa D., Kodaira T., Ushimaru T., Uritani M., Ueno M. Fission yeast Rhp51 is required for the maintenance of telomere structure in the absence of the Ku heterodimer. Nucleic Acids Res. 2003;31:5054–5063. doi: 10.1093/nar/gkg718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyoshi T., Sadaie M., Kanoh J., Ishikawa F. Telomeric DNA ends are essential for the localization of Ku at telomeres in fission yeast. J. Biol. Chem. 2003;278:1924–1931. doi: 10.1074/jbc.M208813200. [DOI] [PubMed] [Google Scholar]
- 13.Capper R., Britt-Compton B., Tankimanova M., Rowson J., Letsolo B., Man S., Haughton M., Baird D.M. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–2508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letsolo B.T., Rowson J., Baird D.M. Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements. Nucleic Acids Res. 2010;38:1841–1852. doi: 10.1093/nar/gkp1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muraki K., Nyhan K., Han L., Murnane J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front Oncol. 2012;2:135. doi: 10.3389/fonc.2012.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackinnon R.N., Campbell L.J. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011;2011:643628. doi: 10.4061/2011/643628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almeida H., Godinho Ferreira M. Spontaneous telomere to telomere fusions occur in unperturbed fission yeast cells. Nucleic Acids Res. 2013;41:3056–3067. doi: 10.1093/nar/gks1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardo B., Gomez-Gonzalez B., Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell. Mol. Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyman C., Kanaar R. DNA double-strand break repair: all's well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 21.Li J., Yu Y., Suo F., Sun L.L., Zhao D., Du L.L. Genome-wide Screens for Sensitivity to Ionizing Radiation Identify the Fission Yeast Nonhomologous End Joining Factor Xrc4. G3 (Bethesda) 2014;4:1297–1306. doi: 10.1534/g3.114.011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muris D.F., Vreeken K., Schmidt H., Ostermann K., Clever B., Lohman P.H., Pastink A. Homologous recombination in the fission yeast Schizosaccharomyces pombe: different requirements for the rhp51+, rhp54+ and rad22+ genes. Curr. Genet. 1997;31:248–254. doi: 10.1007/s002940050202. [DOI] [PubMed] [Google Scholar]
- 23.Ostermann K., Lorentz A., Schmidt H. The fission yeast rad22 gene, having a function in mating-type switching and repair of DNA damages, encodes a protein homolog to Rad52 of Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:5940–5944. doi: 10.1093/nar/21.25.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Symington L.S., Rothstein R., Lisby M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014;198:795–835. doi: 10.1534/genetics.114.166140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carr A.M., Schmidt H., Kirchhoff S., Muriel W.J., Sheldrick K.S., Griffiths D.J., Basmacioglu C.N., Subramani S., Clegg M., Nasim A., et al. The rad16 gene of Schizosaccharomyces pombe: a homolog of the RAD1 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 1994;14:2029–2040. doi: 10.1128/mcb.14.3.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodel C., Kirchhoff S., Schmidt H. The protein sequence and some intron positions are conserved between the switching gene swi10 of Schizosaccharomyces pombe and the human excision repair gene ERCC1. Nucleic Acids Res. 1992;20:6347–6353. doi: 10.1093/nar/20.23.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McVey M., Lee S.E. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall O.J., Chueh A.C., Wong L.H., Choo K.H. Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am. J. Hum. Genet. 2008;82:261–282. doi: 10.1016/j.ajhg.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stimpson K.M., Sullivan B.A. Epigenomics of centromere assembly and function. Curr. Opin. Cell Biol. 2010;22:772–780. doi: 10.1016/j.ceb.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Mythreye K., Bloom K.S. Differential kinetochore protein requirements for establishment versus propagation of centromere activity in Saccharomyces cerevisiae. J. Cell Biol. 2003;160:833–843. doi: 10.1083/jcb.200211116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaco I., Canela A., Vera E., Blasco M.A. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008;181:885–892. doi: 10.1083/jcb.200803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon J.L., Byrne K.P., Wolfe K.H. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011;7:e1002190. doi: 10.1371/journal.pgen.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schubert I., Lysak M.A. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 2011;27:207–216. doi: 10.1016/j.tig.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Therman E., Trunca C., Kuhn E.M., Sarto G.E. Dicentric chromosomes and the inactivation of the centromere. Hum. Genet. 1986;72:191–195. doi: 10.1007/BF00291876. [DOI] [PubMed] [Google Scholar]
- 35.Higgins A.W., Gustashaw K.M., Willard H.F. Engineered human dicentric chromosomes show centromere plasticity. Chromosome Res. 2005;13:745–762. doi: 10.1007/s10577-005-1009-2. [DOI] [PubMed] [Google Scholar]
- 36.Stimpson K.M., Song I.Y., Jauch A., Holtgreve-Grez H., Hayden K.E., Bridger J.M., Sullivan B.A. Telomere disruption results in non-random formation of de novo dicentric chromosomes involving acrocentric human chromosomes. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001061. doi:10.1371/journal.pgen.1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato H., Masuda F., Takayama Y., Takahashi K., Saitoh S. Epigenetic inactivation and subsequent heterochromatinization of a centromere stabilize dicentric chromosomes. Curr. Biol. 2012;22:658–667. doi: 10.1016/j.cub.2012.02.062. [DOI] [PubMed] [Google Scholar]
- 38.Ishii K., Ogiyama Y., Chikashige Y., Soejima S., Masuda F., Kakuma T., Hiraoka Y., Takahashi K. Heterochromatin integrity affects chromosome reorganization after centromere dysfunction. Science. 2008;321:1088–1091. doi: 10.1126/science.1158699. [DOI] [PubMed] [Google Scholar]
- 39.Krawchuk M.D., Wahls W.P. High-efficiency gene targeting in Schizosaccharomyces pombe using a modular, PCR-based approach with long tracts of flanking homology. Yeast. 1999;15:1419–1427. doi: 10.1002/(SICI)1097-0061(19990930)15:13<1419::AID-YEA466>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogiyama Y., Ohno Y., Kubota Y., Ishii K. Epigenetically induced paucity of histone H2A.Z stabilizes fission-yeast ectopic centromeres. Nat. Struct. Mol. Biol. 2013;20:1397–1406. doi: 10.1038/nsmb.2697. [DOI] [PubMed] [Google Scholar]
- 41.Jia Z.P., McCullough N., Martel R., Hemmingsen S., Young P.G. Gene amplification at a locus encoding a putative Na+/H+ antiporter confers sodium and lithium tolerance in fission yeast. EMBO J. 1992;11:1631–1640. doi: 10.1002/j.1460-2075.1992.tb05209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maundrell K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- 43.Wang X., Baumann P. Chromosome fusions following telomere loss are mediated by single-strand annealing. Mol. Cell. 2008;31:463–473. doi: 10.1016/j.molcel.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura T.M., Cooper J.P., Cech T.R. Two modes of survival of fission yeast without telomerase. Science. 1998;282:493–496. doi: 10.1126/science.282.5388.493. [DOI] [PubMed] [Google Scholar]
- 45.Tomita K., Cooper J.P. Fission yeast Ccq1 is telomerase recruiter and local checkpoint controller. Genes Dev. 2008;22:3461–3474. doi: 10.1101/gad.498608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain D., Hebden A.K., Nakamura T.M., Miller K.M., Cooper J.P. HAATI survivors replace canonical telomeres with blocks of generic heterochromatin. Nature. 2010;467:223–227. doi: 10.1038/nature09374. [DOI] [PubMed] [Google Scholar]
- 47.Rog O., Miller K.M., Ferreira M.G., Cooper J.P. Sumoylation of RecQ helicase controls the fate of dysfunctional telomeres. Mol. Cell. 2009;33:559–569. doi: 10.1016/j.molcel.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 48.Ferreira M.G., Cooper J.P. The fission yeast Taz1 protein protects chromosomes from Ku-dependent end-to-end fusions. Mol. Cell. 2001;7:55–63. doi: 10.1016/s1097-2765(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 49.Akamatsu Y., Tsutsui Y., Morishita T., Siddique M.S., Kurokawa Y., Ikeguchi M., Yamao F., Arcangioli B., Iwasaki H. Fission yeast Swi5/Sfr1 and Rhp55/Rhp57 differentially regulate Rhp51-dependent recombination outcomes. EMBO J. 2007;26:1352–1362. doi: 10.1038/sj.emboj.7601582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Albrecht E.B., Hunyady A.B., Stark G.R., Patterson T.E. Mechanisms of sod2 gene amplification in Schizosaccharomyces pombe. Mol. Biol. Cell. 2000;11:873–886. doi: 10.1091/mbc.11.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du L.L., Nakamura T.M., Moser B.A., Russell P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol. Cell Biol. 2003;23:6150–6158. doi: 10.1128/MCB.23.17.6150-6158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garsed D.W., Marshall O.J., Corbin V.D., Hsu A., Di Stefano L., Schroder J., Li J., Feng Z.P., Kim B.W., Kowarsky M., et al. The architecture and evolution of cancer neochromosomes. Cancer Cell. 2014;26:653–667. doi: 10.1016/j.ccell.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 53.Brown W.R., Liti G., Rosa C., James S., Roberts I., Robert V., Jolly N., Tang W., Baumann P., Green C, et al. A Geographically Diverse Collection of Schizosaccharomyces pombe Isolates Shows Limited Phenotypic Variation but Extensive Karyotypic Diversity. G3 (Bethesda) 2011;1:615–626. doi: 10.1534/g3.111.001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown W.R., Thomas G., Lee N.C., Blythe M., Liti G., Warringer J., Loose M.W. Kinetochore assembly and heterochromatin formation occur autonomously in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1903–1908. doi: 10.1073/pnas.1216934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manthey G.M., Bailis A.M. Rad51 inhibits translocation formation by non-conservative homologous recombination in Saccharomyces cerevisiae. PLoS One. 2010;5:e11889. doi: 10.1371/journal.pone.0011889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shinohara M., Shinohara A. Multiple pathways suppress non-allelic homologous recombination during meiosis in Saccharomyces cerevisiae. PLoS One. 2013;8:e63144. doi: 10.1371/journal.pone.0063144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamura K., Okamoto A., Katou Y., Yadani C., Shitanda T., Kaweeteerawat C., Takahashi T.S., Itoh T., Shirahige K., Masukata H., et al. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008;27:3036–3046. doi: 10.1038/emboj.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ira G., Haber J.E. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian L., Moser B.A., Nakamura T.M. Recombination-based telomere maintenance is dependent on Tel1-MRN and Rap1 and inhibited by telomerase, Taz1, and Ku in fission yeast. Mol. Cell Biol. 2008;28:1443–1455. doi: 10.1128/MCB.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lundblad V. Telomere maintenance without telomerase. Oncogene. 2002;21:522–531. doi: 10.1038/sj.onc.1205079. [DOI] [PubMed] [Google Scholar]
- 61.Brown C.A., Murray A.W., Verstrepen K.J. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr. Biol. 2010;20:895–903. doi: 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Linardopoulou E.V., Williams E.M., Fan Y., Friedman C., Young J.M., Trask B.J. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437:94–100. doi: 10.1038/nature04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo Y., Hermetz K.E., Jackson J.M., Mulle J.G., Dodd A., Tsuchiya K.D., Ballif B.C., Shaffer L.G., Cody J.D., Ledbetter D.H., et al. Diverse mutational mechanisms cause pathogenic subtelomeric rearrangements. Hum. Mol. Genet. 2011;20:3769–3778. doi: 10.1093/hmg/ddr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan J.B., Grothues D., Smith C.L. Alignment of Sfi I sites with the Not I restriction map of Schizosaccharomyces pombe genome. Nucleic Acids Res. 1991;19:6289–6294. doi: 10.1093/nar/19.22.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.