

The mitogen-activated protein kinase (MAPK) pathways are a group of conserved intracellular signalling pathways present in most cells including neurons and glia. These pathways respond to a variety of stimuli including growth factors, cytokines and oxidative stress to generate appropriate cellular responses such as modulation of gene expression, cell proliferation, differentiation and survival as well as the stress response (Korhonen and Moilanen, 2014). The details of the cascades have been discussed elsewhere (Dickinson and Keyse, 2006; Korhonen and Moilanen, 2014) but usually follow a three-tier structure; an MAP3K activates an MAPK kinase (MAPKK), which in turn activates an MAPK, that can in turn regulate the activity of its cellular target by phosphorylation of specific amino acid residues. Three groups of MAPK have been identified; the extracellular signal-regulated kinases of which extracellular signal-regulated kinase (ERK)1/2 and ERK5 form two distinct classes, c-JUN N-terminal kinases (JNK1, JNK2 and JNK3), and the p38 kinases (p38 α, β, γ and δ) (Wancket et al., 2012). The ERK1/2 and ERK5 pathways respond to both mitotic and stress signals provided by growth factors, hormones and inflammatory cytokines. The other MAPK, p38 and JNK, are activated in response to growth factors, cytokines, viral proteins, lipopolysaccharides as well as cellular stress conditions. Collectively, the MAPK pathways play important roles in regulating cellular ageing, cell division, cell survival/apoptosis, neuronal activity, insulin signalling, inflammation and the immune response (Dickinson and Keyse, 2006; Korhonen and Moilanen, 2014).
In addition to transcriptional and translational regulation, MAPK activity is tightly controlled at the post-transcriptional level by phosphorylation and de-phosphorylation of key threonine and tyrosine amino acids. The dephosphorylating enzymes include tyrosine, serine/threonine and the dual-specificity phosphatases (tyrosine and threonine, dual-specificity phosphatase (DuSP)), which are often referred to as MAPK phosphatases (MKPs) (Korhonen and Moilanen, 2014). In mammalian cells, the ten conserved MKPs are the principal phosphatases responsible for dephosphorylation and thus inactivation of MAPKs (Dickinson and Keyse, 2006). While MKPs vary in cellular localization, expression patterns and substrate specificity, they share a highly conserved C-terminal catalytic domain, a kinase interaction motif which confers individual substrate specificity, and two Cdc25/rhodanese-homology domains near the N-terminus (Figure 1) (Wancket et al., 2012).
Figure 1.
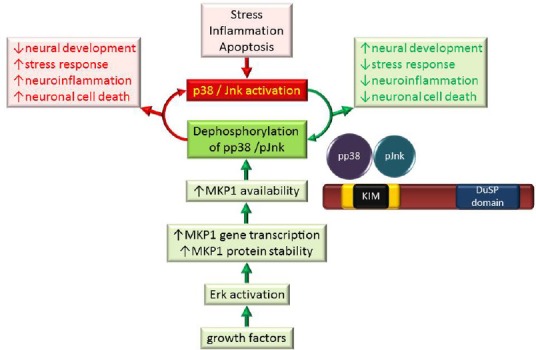
MKP1 attenuates the detrimental effects of p38 and Jnk kinases.
Cellular stress, inflammation or apoptosis may lead to the phosphorylation and activation of p38 and Jnk MAP kinases which contribute to decreased neural development, increased stress response, increased neuroinflammation and increased neuronal cell death in the nervous system (red box). Growth factor signalling leads to Erk activation which stimulates gene transcription and increases MKP1 protein stability and availability. The resulting increase in MKP1 activity leads to a decrease in p38 and Jnk activity, contributing to improvements in neural development, decreased neuroinflammation and reduced neuronal cell death (green box). Activated MAP kinases are bound by MKP1's kinase interaction motif (KIM) with decreasing preference (p38 > Jnk > Erk) and dephosphorylated by the dual specificity phosphatase (DuSP) domain. Erk: Extracellular signal-regulated kinase; Jnk: c-Jun N-terminal kinase; MAP: mitogen-activated protein; MKP1: mitogen-activated protein kinase phosphatase 1; p38: 38 kDa protein; pJnk: p-c-Jun N-terminal kinase; pp38: 38-kDa phosphorylated protein.
MKP1, the original member of the family, is a stress- and growth factor-inducible protein with the capacity to bind and dephosphorylate ERK, and subsequently p38 and JNK kinases (for review see (Collins et al., 2015)). Studies have shown that under physiological conditions, where MKP1 levels are relatively low, MKP1 has a weak binding affinity for ERK kinases and exerts little effect on their function. MKP1 has also been shown to have a higher binding affinity for p38 and JNK kinases than ERK kinases under physiological conditions (Dickinson and Keyse, 2006; Wancket et al., 2012). However, when MKP1 levels are high, it also inhibits ERK kinases, although to a lesser extent than p38 and JNK, and thus provides a negative feedback loop on many cellular processes. The interplay between the MAPK pathways and MKP1also influences MKP1 gene expression, protein stability and catalytic activity (reviewed in Wancket et al., 2012; Lawan et al., 2013; Korhonen and Moilanen, 2014).
Recent studies have yielded important insights into the physiological functions of MKP1, particularly its role in regulating responses in immunological and metabolic diseases, cancer and more recently in the nervous system (reviewed by Wancket et al., 2012; Lawan et al., 2013). For example, MKP1 has been shown to inhibit MAPK-mediated inflammatory responses in macrophages and dendritic cells and to protect mice from endotoxic insult (Collins et al., 2015). In this regard, MKP1 knock-out mice show higher levels of inflammation, autoimmunity and metabolic defects than their wild-type littermates. Conversely a pathophysiological involvement of MKP1 has been suggested in obesity and metabolic syndrome (reviewed by Lawan et al., 2013; Korhonen and Moilanen, 2014; Collins et al., 2015). Increased expression of MKP1 has also been reported in various cancers, suggesting that it may be involved in the inhibition of apoptotic pathways through its regulation of MAPK, and could ultimately lead to excess cell proliferation (for review see Wu, 2007). Emerging data also shows that MKP1 participates positively in the regulation of neuronal development, plasticity and survival as well as learning and memory (reviewed by Collins et al., 2015). Thus, while researchers are exploring ways of preventing up-regulation of MKP1 in cancer cells as it impedes pro-apoptotic drug therapies, the opposite is required to preserve the integrity of neuronal cells. Current research therefore positions MKP1 as a potential new drug target not only for the regulation of the immune response, metabolic function and cell proliferation in tumour cells, but also for diseases of the nervous system.
There is accumulating evidence to suggest that MKP1 may be an important regulator of neural cell development. It is expressed in the developing nervous system of mice and rats as well as in cultured embryonic neuronal precursor cells where its activity and expression can be stimulated by various agents such as retinoic acid or brain-derived neurotrophic factor (Collins et al., 2015). MKP1 has been specifically implicated in neuronal axonal development in rat cortical and ventral midbrain dopaminergic neurons (Jeanneteau et al., 2010; Collins et al., 2013). These studies demonstrated that MKP1 overexpression during embryonic development or in cultured neurons resulted in increased dendritic morphological complexity (Jeanneteau et al., 2010; Collins et al., 2013). Conversely, inhibition of MKP1 by expression of siRNA resulted in decreased dendritic arborisation complexity (Jeanneteau et al., 2010). It has further been shown that MKP1 interaction with the JNK pathway is essential for axonal development in cortical neurons and that the activity of p38 is essential for dopaminergic neurons development (Jeanneteau et al., 2010; Collins et al., 2013). Treatment with the dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA) led to a reduction in dendritic complexity which was accompanied by a selective decrease in MKP1 expression. Moreover, in the same 6-OHDA-treated neurons, overexpression of MKP1 prevented the decrease in neurite length (Collins et al., 2013). As the morphological development of neural cells is critical for successful generation and integration of cells aimed at cell replacement therapy for neurodegenerative diseases such as Huntington's disease (HD) and Parkinson's disease (PD), the results from these studies study suggest that MKP1 may be a promising growth promoting mediator of neuronal processes and a potential neuroprotective protein. Further work will be needed to determine how its interaction with the MAPK signalling may be modulated to maximize its beneficial effects in vivo.
While extensive research supports a role for MAPKs in neuronal cell survival and/or intrinsic apoptotic (mitochondrial) cell death following injury or degenerative stimuli, little, and sometimes conflicting, information is available on the involvement of MKP1 in these processes. Some evidence shows that in neuronal models where the intrinsic apoptotic pathway is triggered by stimuli such as hypoxia, ischemia, or oxidative death, either up-regulation or knockdown of MKP1 can increase cell sensitivity to insult, or even trigger cell death. Conversely, neuroprotection has generally been associated with higher endogenous expression and activity levels of MKP1 (reviewed by Collins et al., 2015). A down-regulation of Mkp1 mRNA expression has been reported in human post-mortem HD brain samples, as well as in the striatum and cortex of R6/2 mice (a transgenic mouse model of HD) and in cultured neurons expressing expanded polyglutamine fragments of huntingtin (Taylor et al., 2013). Mkp1 expression was also transiently increased at the onset of neurodegeneration in the substantia nigra of 6-OHDA treated rats (a model of PD) where it returned to basal levels as cell death progressed (Collins et al., 2014). Thus for the most part it appears that MKP1 plays a protective role in neurons, but variations in tissue type, the nature, timing and duration of insult and injury, as well as the transient nature of MAPK signalling may account for the discrepancies reported. Therefore, while a substantial number of reports point to an up-regulation of MKP1 as a potential therapeutic strategy to reduce neuronal loss as a result of apoptotic activation through injury, hypoxia, ischemia or degeneration (Collins et al., 2015), much work has yet to be done to establish distinct mechanisms of endogenous MKP1 activity in the various cell death paradigms.
In addition to exerting direct roles in neuronal development and survival, a role for MKP1 has been established in a variety of neuroinflammatory processes involving glial cells (reviewed in Collins et al., 2015). Neuroinflammation has been well documented in PD and other neurodegenerative disorders. For example, evidence of the accumulation of activated microglial cells and increased expression of pro-inflammatory cytokines in damaged brain areas due to toxic insults or neuronal death has been consistently reported (reviewed by Collins et al., 2012; Ward et al., 2015). A substantial body of evidence indicates that MKP1 is expressed in glial cells, and that its regulation may impact upon neuroinflammatory processes (reviewed by Collins et al., 2015). Indeed, MKP1 overexpression in cultured glial cells has been shown to block MAPK activity and the release of pro-inflammatory cytokines (Ndong et al., 2012). Several compounds, including dexamethasone, costunolide, dehydroglyasperin C, anandamide, and the peroxisome proliferator-activated receptor (PPAR) agonists 5,8,11,14-eicosatetraynoic acid (ETYA) and 15-Deoxy-Δ12,14-Prostaglandin J2 (15d-PGL2) have all been reported to reduce microglial activation and the associated release of pro-inflammatory cytokine through the promotion of glial MKP1 expression and activity (Collins et al., 2015). While their effect may be indirect through a reduction of the neuroinflammatory processes, these molecules could prove beneficial in neurodegenerative diseases by reducing glial activation and maintaining brain homeostasis.
Increasing evidence suggests that MKP1 plays definite roles in a number of central nervous system (CNS) diseases. The complexity of the cellular mechanisms underlying its action in the brain is also starting to be revealed, although much work remains to fully understand the specific role that MKP1 plays in disease states, especially in the clinical setting. It is also clear that MKP1 is a key player in the regulation of glial cell-mediated neuroinflammatory processes, and its manipulation represents a promising target to limit neuronal damage which occurs as a result of neuroinflammation. A number of molecules have been shown to stimulate MKP1 activity in CNS disease models and several others are being developed for other pathologies such as arthritis, asthma, and chronic obstructive pulmonary disease, where MKP1 is involved (Doddareddy et al., 2012). The manipulation of MKP1 expression and activity alone or in conjunction with inhibition of specific MAPK pathways may broaden the therapeutic spectrum in CNS disorders. Considering the current lack of therapeutic molecules targeting the neurodegenerative process in neurological disorders, and based on the central regulatory role of MKP1 in the pathophysiology of these disorders, the development of therapeutic strategies targeting MKP1 should be a high priority.
The authors acknowledge support from Science Foundation Ireland under grant No. SFI/IA/1537. The authors declare no conflicts of interest.
References
- Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology. 2012;62:2154–2168. doi: 10.1016/j.neuropharm.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Collins LM, Downer EJ, Toulouse A, Nolan YM. Mitogen-activated protein kinase phosphatase (MKP)-1 in nervous system development and disease. Mol Neurobiol. 2015;51:1158–1167. doi: 10.1007/s12035-014-8786-6. [DOI] [PubMed] [Google Scholar]
- Collins LM, O’Keeffe GW, Long-Smith CM, Wyatt SL, Sullivan AM, Toulouse A, Nolan YM. Mitogen-activated protein kinase phosphatase (MKP)-1 as a neuroprotective agent: promotion of the morphological development of midbrain dopaminergic neurons. Neuromolecular Med. 2013;15:435–446. doi: 10.1007/s12017-013-8230-5. [DOI] [PubMed] [Google Scholar]
- Collins LM, Gavin AM, Walsh S, Sullivan AM, Wyatt SL, O’Keeffe GW, Nolan YM, Toulouse A. Expression of endogenous Mkp1 in 6-OHDA rat models of Parkinson's disease. Springerplus. 2014;3:205. doi: 10.1186/2193-1801-3-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- Doddareddy MR, Rawling T, Ammit AJ. Targeting mitogen-activated protein kinase phosphatase-1 (MKP-1): structure-based design of MKP-1 inhibitors and upregulators. Curr Med Chem. 2012;19:163–173. doi: 10.2174/092986712803414196. [DOI] [PubMed] [Google Scholar]
- Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci. 2010;13:1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen R, Moilanen E. Mitogen-activated protein kinase phosphatase 1 as an inflammatory factor and drug target. Basic Clin Pharmacol Toxicol. 2014;114:24–36. doi: 10.1111/bcpt.12141. [DOI] [PubMed] [Google Scholar]
- Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol Life Sci. 2013;70:223–237. doi: 10.1007/s00018-012-1041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndong C, Landry RP, DeLeo JA, Romero-Sandoval EA. Mitogen activated protein kinase phosphatase-1 prevents the development of tactile sensitivity in a rodent model of neuropathic pain. Mol Pain. 2012;8:34. doi: 10.1186/1744-8069-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DM, Moser R, Regulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos Mde F, Kim J, Jones L, Goldstein DR, Ferrante RJ, Luthi-Carter R. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition. J Neurosci. 2013;33:2313–2325. doi: 10.1523/JNEUROSCI.4965-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wancket LM, Frazier WJ, Liu Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology physiology and disease. Life Sci. 2012;90:237–248. doi: 10.1016/j.lfs.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Dexter DT, Crichton RR. Ageing neuroinflammation and neurodegeneration. Front Biosci. 2015;7:189–204. doi: 10.2741/S433. [DOI] [PubMed] [Google Scholar]
- Wu GS. Role of mitogen-activated protein kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev. 2007;26:579–585. doi: 10.1007/s10555-007-9079-6. [DOI] [PubMed] [Google Scholar]