

Cancer and neurodegeneration include a group of diseases that are mechanistically distinct but may share common therapeutic targets. Autophagy is a common quality control mechanism shared by mitotic and post-mitotic cells and it can be exploited to accelerate clearance of unwanted oncogenes and reduce accumulation of toxic proteins in cancer and neurodegeneration, respectively. Tyrosine kinase inhibition is a therapeutically relevant strategy that can induce autophagy, leading to normal cell survival. This article provides insights into how tyrosine kinase inhibition is clinically used to arrest mitotic cell division and tumor growth, and may promote survival of post-mitotic neurons in neurodegeneration.
Neurodegenerative diseases include a group of genetic and sporadic disorders associated with neuronal death and progressive nervous system dysfunction. Cancer is also a collection of related genetic diseases, in which cells begin to divide without stopping and spread into surrounding tissues. Unlike neurodegeneration, in which no regeneration happens when damaged or aging post-mitotic neurons die, damaged cells survive when they should die in cancer, resulting in uncontrolled mitotic cell division to form tumors. Cancerous tumors are malignant as they spread or invade nearby tissues by cellular contiguity or metastasize via blood and/or humoral transport. In neurodegeneration, the spread of disease by contiguity is supported by the hypotheses that toxic or prion-like proteins, like oncogenes, propagate along neuroanatomical pathways (Polymenidou and Cleveland, 2012), leading to progressive spread of disease and cell death. The spread of toxic or misfolded proteins in neurodegeneration may be similar to the spread of metastatic cancer, as both pathologies spread from the place where they originate. In neurodegeneration, failure of cellularquality control mechanisms leads to inadequate protein degradation via the proteasome or autophagy (Ciechanover and Kwon, 2015), resulting in intracellular accumulation of toxic and pathological proteins. Consequently, these proteins are secreted from a pre-synaptic neuron and can traverse the synaptic cleft and enter a contiguous post-synaptic neuron (Figure 1). Secreted proteins may not penetrate an adjacent cell via the synapse but they may be re-routed into the cell and recycled via the endosomal system to fuse with autophagic vacuoles like the autophagosome or the lysosome (Jerram et al., 1996; Mellman, 1996; Luzio et al., 2000). Microglia, the brain resident immune cells may also phagocytose and destroy toxic proteins (Kettenmann et al., 2011).
Figure 1.
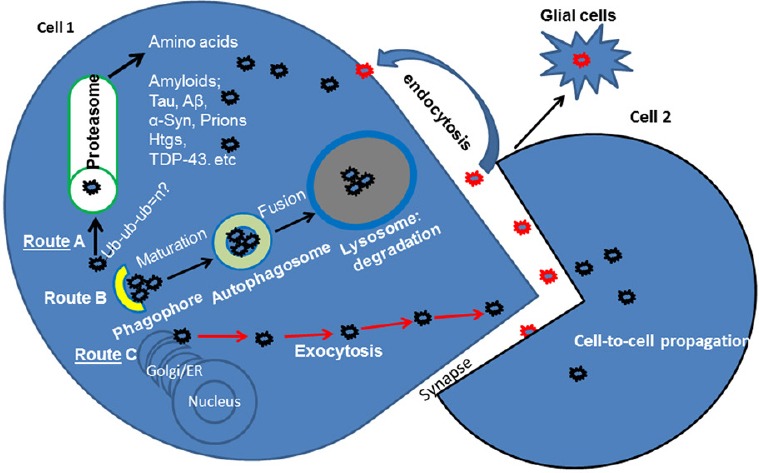
Protein propagation and secretion in neurodegeneration.
Intracellular (cell 1) accumulation of pathogenic or misfolded proteins triggers quality control clearance mechanisms. Route A–Protein can be ubiquitinated (Ub: Ubiquitin) and degraded via the proteasome. Route B–Autophagy is a major bulk degradation pathway. Accumulating proteins are first sequestered into a phagophore, which matures into a double-membrane autophagosome and fuses with the lysosome that contains digestive enzymes. If proteins are not degraded due to failure of the proteasome or autophagy, they may be secreted (Route C via exocytosis) into the synapse. Toxic proteins in the synaptic cleft may cross to (cell 2) an adjacent cell, resulting in trans-synaptic protein propagation. Extracellular or secreted proteins may be re-routed via the endosomal system and re-internalized into Cell 1 or destroyed by immune responses via activated microglia. Aβ: Amyloid beta; α-Syn: alpha-Synuclein; ER: endoplasmic reticulum; TDP-43: transactive response DNA binding protein 43 kDa.
Accumulation of toxic proteins, including alpha-synuclein (Lewy bodies), beta-amyloid plaques, tau tangles, Huntington, prions and TDP-43 are major culprits in neurodegeneration. These toxic proteins trigger progressive apoptotic cell death leading to loss of many CNS functions, including mentation, cognition, movement, gastrointestinal motility, sleep and many others. The discoveries of toxic protein propagation from cell to cell (Polymenidou and Cleveland, 2012), leading to progression of neurodegeneration triggered a series of pre-clinical and clinical studies to limit protein propagation via antibodies (active and passive immune therapies) that can capture the protein and destroy it en route to healthy neurons. This approach is fraught with difficulties, including failure to stop neurocognitive decline and brain edema. Manipulation of autophagy is a novel therapeutic approach that focuses on degradation of neurotoxic proteins at the manufacturing site in order to prevent their secretion and propagation. This novel strategy essentially leads to unclogging the cell's disposal machine and degradation of toxic proteins, thus preserving neuronal survival via bulk digestion. Preservation of neuronal survival maintains the level of neurotransmitters that are necessary for cognitive, motor and other CNS functions, leading to alleviation of symptoms as well as arrest of neurodegeneration. As neurons are post-mitotic cells, pulsatile autophagy may promote protein degradation and provide an effective disease-modifying therapy for neurodegenerative diseases.
There are more than 100 types of cancer, including carcinoma, sacrcoma, lymphoma, multiple myeloma, melanoma and brain and spinal cord tumors. Autophagy is a double-edged sword in cancer, either preventing accumulation of damaged proteins and organelles to suppress tumors, or promoting cell survival mechanisms that lead to tumor growth and proliferation (Yang et al., 2011). Regulation of autophagy continues to evolve in order to maximize its therapeutic advantage in cancer. Leukemia and many other cancer treatments have been revolutionized by manipulation of autophagy, which leads to bulk degradation of unwanted or toxic molecules (Macintosh and Ryan, 2013). For example in leukemia, genetic mutations and DNA damage can lead to large numbers of abnormal white blood cells (leukemia cells and leukemic blast cells) to accumulate in the blood and bone marrow, crowding out normal blood cells. Autophagy can lead to the degradation of the products of cancer-causing genes (oncogenes), tumor suppressor genes, damaged DNA and essential components of the cytosol, thereby controlling abnormal mitotic division and limiting tumor growth. Autophagy can also lead to self-cannibalization via promotion of programmed cell death, or apoptosis. Activation of the tumor suppressor p53 in response to DNA damage leads the cell to arrest proliferation, initiate DNA repair, and promote survival. However, if the DNA damage cannot be resolved by p53, it can trigger apoptotic death (Crighton et al., 2006; Yee et al., 2009). Cell division and apoptosis are mediated by signaling mechanisms via the endosomal (early and recycling) system (Mellman, 1996). Tyrosine kinases are activated via phosphorylation, triggering various signaling mechanisms that mediate cell division and/or apoptosis (Cadena and Gill, 1992; Nakada et al., 2013). Tyrosine kinase inhibition via de-phosphorylation leads to signaling via the late endosomal-lysosomal pathway (Figure 2), thus increasing autophagic degradation (Jerram et al., 1996; Mellman, 1996; Luzio et al., 2000), leading to arrest of tumor growth.
Figure 2.
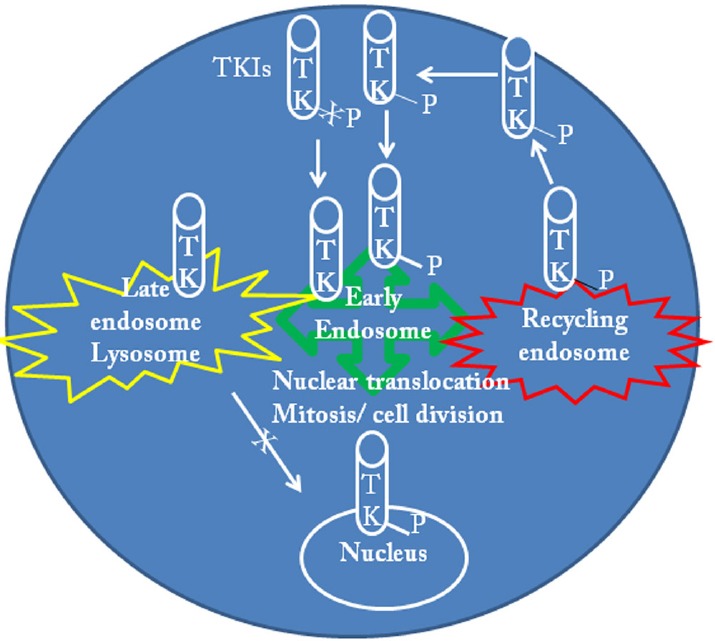
The role of tyrosine kinases (TK) in the modulation of the endosome-lysosome pathways.
Receptor and non-receptor TK are activated via phosphorylation and play a critical role in the endosomal system via translocation of transcription factors that can include mitotic cell division in normal and cancer cells. Networks of early and recycling endosomes modulate cell division and differentiation. Breakpoint Cluster Region-Abelson (BCR-ABL) is constitutively activated in chronic myeloid leukemia causing abnormal mitotic division and differentiation of leukemia and leukemic blast cells. ABL inhibition via de-phosphorylation reduces mitotic cell division and shifts signal transduction pathways away from the nucleus to activate the late endosome-lysosome system and degrade oncegenes. Activation of the lysosomal pathway (autophagy) can also degrade essential cytosolic compartments (i.e., mitochondria) and lead to apoptosis. In post-mitotic neurons, where ABL and TKs are activated autophagy is inhibited. Inhibition of TKs activates autophagy and leads to degradation of toxic cytosolic proteins. Pulsatile or ON/OFF autophagy can lead to protein clearance without degradation of cytosolic compartments, avoiding self-cannibalization. Manipulation of autophagy via TK inhibition provides a potentially common drug target for both cancer and neurodegeneration.
Tyrosine kinase inhibitors (TKIs) have significantly improved the life quality and expectancies in many cancers, including chronic myeloid leukemia (CML) (Holyoake and Helgason, 2015; Jabbour et al., 2015). CML is characterized by the translocation of chromosomes 9 and 22 to form the “Philadelphia” chromosome resulting in the expression of a constitutively active Breakpoint Cluster Region-Abelson (BCR-ABL) tyrosine kinase. This oncogenic protein activates intracellular signaling pathways and induces cell proliferation. Our laboratory investigated TKIs that activate autophagy and are FDA-approved for CML, thus significantly reducing research and development efforts and cost by re-purposing (reviewed by Moussa, 2015). In neurodegeneration, the non-receptor tyrosine kinase ABL is activated. Nilotinib and bosutinib are second generation BCR-ABL and SRC (short for Sacoma)-ABL inhibitors, respectively, that are therapeutically used for individuals with CML. Afraction of nilotinib and bosutinib crosses the blood-brain barrier (BBB), inhibits ABL and facilitates autophagic amyloid clearance, leading to neuroprotection and improved cognition and motor behavior (Hebron et al., 2013a; Lonskaya et al., 2013b). Mice treated with a much lower dose of these drugs (< 25% of the typical CML dose) show significant motor and cognitive improvement and degradation of alpha-Synuclein, beta-amyloid, tau and TDP-43 without any evidence of increased inflammation. There was also significant reversal of neurotransmitter alterations, including dopamine and glutamate in several models of neurodegeneration, including Alzheimer's disease (AD) and other dementias, Parkinson's disease (PD) and movement disorders and amyotrophic lateral sclerosis (ALS). We are currently testing Nilotinib in a phase I/II clinical trial for PD, PD with dementia and lewy body dementia (LBD). Saracatinib, a tyrosine kinase SRC inhibitor is also in a phase II clinical trial for mild to moderate AD (Wadman, 2013).
Several blood-borne tyrosine kinases are amenable to manipulation in neurodegenerative diseases as well as cancer. In addition to ABL and SRC inhibitors, other tyrosine kinases are potential therapeutic targets in neurodegeneration. For example, colony stimulating factor 1 receptor (CSF1R), c-KIT and platelet derived growth factor receptors (PDGFRα/β) are members of the PDGFR family of cell surface receptor tyrosine kinases and are altered in neurodegeneration. CSF1R is expressed on mononuclear phagocytes and regulates microglia activity (Sasmono et al., 2003; Erblich et al., 2011). The tyrosine kinase macrophage colony-stimulating factor (M-CSF or CSF-1) increases parenchymal microglia number, reduces Aβ levels and improves cognition in AD transgenic mice (Boissonneault et al., 2009). C-KIT also regulates microglia and the veterinary c-KIT inhibitor Masitinib is currently in a phase III clinical trial for mild to moderate AD (Folch et al., 2015). Discoidin domain receptors (DDR1/2) are members of the DDR family of collagen-activated, cell surface receptor tyrosine kinases. Both receptors mediate cell division and differentiation and may lead to regulation of myeloid-derived glial cells (Kamohara et al., 2001; Seo et al., 2008; Roig et al., 2010), providing protective mechanisms in neurodegeneration. Other tyrosine kinases like sterile alpha motif and leucine zipper containing kinase (ZAK) andarginase 2 are potential drug targets that may provide beneficial effects for protein clearance in neurodegenerative diseases (Schlatterer et al., 2011, 2012; Lonskaya et al., 2012, 2013a, b; Hebron et al., 2013a, b).
Many non-specific tyrosine kinase inhibitors are approved by American and European regulatory agencies for the treatment of cancers, including leukemia (Wu et al., 2015). However, the safety and tolerability of TKIs vary and careful consideration must be given to different disease indications that may manifest with various comorbidities. CNS penetration of TKIs is generally low (below 3–5% plasma levels) and their CNS bioavailability is poor (Hebron et al., 2013a; Lonskaya et al., 2013b). However, low penetration and short CNS availability may be advantageous by inducing slow and pulsatile ON/OFF autophagy to clear neurotoxic protein accumulation without causing degradation of essential cytosolic components and inducing apoptosis in post-mitotic neurons (Hebron et al., 2013a; Lonskaya et al., 2013b). As modulators of myeloid cells (Zhang and Li, 2013), TKIs may also positively regulate neuronal death and produce neuro-restorative effects via increased production of necessary growth factors and proliferation of myeloid-derived glia. Autophagic toxic protein clearance and production of growth factors may restore loss of neurotransmitters, leading to improved motor and cognitive functions. Tyrosine kinase inhibition provides a double-edge sword via manipulation of autophagy to inhibit cell division and tumor growth in cancer on one hand, and promote toxic protein degradation and neuronal survival in neurodegeneration on the other hand.
These studies were supported by Georgetown University funding to CEHM. Georgetown University and one or more authors have intellectual property interest related to technology in this paper.
References
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009;132:1078–1092. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- Cadena DL, Gill GN. Receptor tyrosine kinases. FASEB J. 1992;6:2332–2337. doi: 10.1096/fasebj.6.6.1312047. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM a p53-induced modulator of autophagy is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia disrupted brain development and olfactory deficits. PLoS One. 2011;6:e26317. doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Petrov D, Ettcheto M, Pedros I, Abad S, Beas-Zarate C, Lazarowski A, Marin M, Olloquequi J, Auladell C, Camins A. Masitinib for the treatment of mild to moderate Alzheimer's disease. Expert Rev Neurother. 2015;15:587–596. doi: 10.1586/14737175.2015.1045419. [DOI] [PubMed] [Google Scholar]
- Hebron M, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of á-synuclein in Parkinson's disease models. Hum Mol Genet. 2013a;22:3315–3328. doi: 10.1093/hmg/ddt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron ML, Lonskaya I, Moussa CE. Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy. 2013b;9:1249–1250. doi: 10.4161/auto.25368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoake TL, Helgason GV. Do we need more drugs for chronic myeloid leukemia? Immunol Rev. 2015;263:106–123. doi: 10.1111/imr.12234. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Kantarjian H, Cortes J. Use of second- and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: an evolving treatment paradigm. Clin Lymphoma Myeloma Leuk. 2015;15:323–334. doi: 10.1016/j.clml.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerram AH, Smith PF, Darlington CL. A dose-response analysis of the behavioral effects of (+)MK-801 in guinea pig: comparison with CPP. Pharmacol Biochem Behav. 1996;53:799–807. doi: 10.1016/0091-3057(95)02075-6. [DOI] [PubMed] [Google Scholar]
- Kamohara H, Yamashiro S, Galligan C, Yoshimura T. Discoidin domain receptor 1 isoform-a (DDR1alpha) promotes migration of leukocytes in three-dimensional collagen lattices. FASEB J. 2001;15:2724–2726. doi: 10.1096/fj.01-0359fje. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Lonskaya I, Desforges NM, Hebron ML, Moussa CE. Ubiquitination increases parkin activity to promote autophagic alpha-synuclein clearance. PLoS One. 2013a;8:e83914. doi: 10.1371/journal.pone.0083914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonskaya I, Hebron ML, Algarzae NK, Desforges N, Moussa CE. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson's disease. Neuroscience. 2012;232C:90. doi: 10.1016/j.neuroscience.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med. 2013b;5:1247–1262. doi: 10.1002/emmm.201302771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Rous BA, Bright NA, Pryor PR, Mullock BM, Piper RC. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci. 2000;113:1515–1524. doi: 10.1242/jcs.113.9.1515. [DOI] [PubMed] [Google Scholar]
- Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol. 2013;23:344–351. doi: 10.1016/j.semcancer.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Ann Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Moussa CE. Parkin is dispensable for mitochondrial function but its ubiquitin ligase activity is critical for macroautophagy and neurotransmitters: therapeutic potential beyond Parkinson's Disease. Neurodegener Dis. 2015 doi: 10.1159/000430888. DOI:10.1159/000430888. [DOI] [PubMed] [Google Scholar]
- Nakada M, Kita D, Teng L, Pyko IV, Watanabe T, Hayashi Y, Hamada J. Receptor tyrosine kinases: principles and functions in glioma invasion. Adv Exp Med Biol. 2013;986:143–170. doi: 10.1007/978-94-007-4719-7_8. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209:889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig B, Franco-Pons N, Martorell L, Tomas J, Vogel WF, Vilella E. Expression of the tyrosine kinase discoidin domain receptor 1 (DDR1) in human central nervous system myelin. Brain Res. 2010;1336:22–29. doi: 10.1016/j.brainres.2010.03.099. [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- Schlatterer SD, Tremblay MA, Acker CM, Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis. 2011;25:119–133. doi: 10.3233/JAD-2011-102025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatterer SD, Suh HS, Conejero-Goldberg C, Chen S, Acker CM, Lee SC, Davies P. Neuronal c-Abl activation leads to induction of cell cycle and interferon signaling pathways. J Neuroinflammation. 2012;9:208. doi: 10.1186/1742-2094-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo MC, Kim S, Kim SH, Zheng LT, Park EK, Lee WH, Suk K. Discoidin domain receptor 1 mediates collagen-induced inflammatory activation of microglia in culture. J Neurosci Res. 2008;86:1087–1095. doi: 10.1002/jnr.21552. [DOI] [PubMed] [Google Scholar]
- Wadman M. NIH gambles on recycled drugs. Nature. 2013;499:263–264. doi: 10.1038/499263a. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009;16:1135–1145. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li S. Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 2013;4:186–196. doi: 10.1007/s13238-013-2115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]