

Abstract
The signaling mechanisms underlying ischemia-induced nerve cell apoptosis are poorly understood. We investigated the effects of apoptosis-related signal transduction pathways following ischemic spinal cord injury, including extracellular signal-regulated kinase (ERK), serine-threonine protein kinase (Akt) and c-Jun N-terminal kinase (JNK) signaling pathways. We established a rat model of acute spinal cord injury by inserting a catheter balloon in the left subclavian artery for 25 minutes. Rat models exhibited notable hindlimb dysfunction. Apoptotic cells were abundant in the anterior horn and central canal of the spinal cord. The number of apoptotic neurons was highest 48 hours post injury. The expression of phosphorylated Akt (p-Akt) and phosphorylated ERK (p-ERK) increased immediately after reperfusion, peaked at 4 hours (p-Akt) or 2 hours (p-ERK), decreased at 12 hours, and then increased at 24 hours. Phosphorylated JNK expression reduced after reperfusion, increased at 12 hours to near normal levels, and then showed a downward trend at 24 hours. Pearson linear correlation analysis also demonstrated that the number of apoptotic cells negatively correlated with p-Akt expression. These findings suggest that activation of Akt may be a key contributing factor in the delay of neuronal apoptosis after spinal cord ischemia, particularly at the stage of reperfusion, and thus may be a target for neuronal protection and reduction of neuronal apoptosis after spinal cord injury.
Keywords: nerve regeneration, ischemic spinal cord injury, cell apoptosis, neurological function, serine-threonine protein kinase, extracellular signal-regulated kinase, c-Jun N-terminal kinase, neural regeneration
Introduction
Ischemia/reperfusion (I/R) injury can result in nerve cell apoptosis (Coselli et al., 2002; Safi et al., 2003; Yang et al., 2012). However, the precise mechanism of such apoptosis is not fully understood. Several reports have suggested that stress-responsive mitogen-activated protein kinase pathways might play a protective role in ischemic spinal cord injury (Chang and Karin, 2001; Badrian et al., 2006). There are three major forms of kinase: extracellular signal-regulated kinases (ERKs) and c-Jun N-terminal kinases (JNKs), and serine-threonine kinase (Akt). Activation of ERK by I/R injury is believed to confer a survival advantage to neurons (Shackelford and Yeh, 2003; Kilic et al., 2005). Activation of JNK is associated with neuronal death (Willaime-Morawek et al., 2003). JNK is active in the normal spinal cord, and its activity is reduced by ischemia; however, levels of JNK after reperfusion depend on the duration of ischemia (Shackelford and Yeh, 2001). Akt mediates growth factor-induced neuronal survival (Crowder and Freeman, 1998) and its activation in the early stages of reperfusion may be one of the factors responsible for the delay in neuronal death after spinal cord ischemia (Sakurai et al., 2001). Furthermore, the spatiotemporal expression of Akt, JNK, and ERK protein kinases has been described after ischemic brain injury (Kitagawa et al., 1999), but their expression has not been examined in ischemic spinal cord. Therefore, it is possible that Akt, JNK and ERK exert beneficial or harmful effects via phosphorylated Akt (p-Akt), JNK and ERK affecting the induction of apoptosis, with the existence of different expression time windows. This study examined the spatiotemporal expression of phosphorylated Akt, JNK and ERK after permanent aortic occlusion in rats.
Materials and Methods
Ethics statement
Rats received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23, revised 1996). All procedures were approved by the Institutional Animal Care and Use Committee of the Capital Medical University, Beijing, China. Precautions were taken to minimize suffering and the number of animals used in each experiment.
Animals
Ninety clean male Sprague-Dawley rats aged 13 weeks and weighing 280–350 g (310 ± 20 g) were provided by the Experimental Animal Center of Beijing Institute of Heart, Lung and Blood Vessel Diseases in China (license No. SYXK (Jing) 2005-0026). All rats were neurologically intact prior to anesthesia and surgery. Rats were randomly divided into sham group (n = 10) and I/R group (n = 80).
Establishment of ischemic spinal cord injury models
Rats were anesthetized with an intraperitoneal injection of 3% (w/v) sodium pentobarbital (30–50 mg/kg). After endotracheal intubation, an Inspira Advanced Safety Ventilator (Harvard Apparatus, Holliston, MA, USA) was connected. The tidal volume was set at 15 mL/kg, and the respiratory frequency was 80–100 breaths/min with an inspiratory to expiratory ratio of 1:1. The rectal temperature was monitored, and body temperature was maintained at 36.5–37.5°C with an infrared heat lamp and a heating pad. The left carotid artery was cannulated with a 20 gauge catheter (B. Braun Medical Inc., Bethlehem, PA, USA) to measure the mean proximal aortic pressure, which was maintained at 65 ± 3 mmHg throughout the procedure. A 24-gauge catheter was inserted into the tail artery to monitor the mean distal arterial pressure. The carotid artery cannula was connected to a heated (37.5°C) blood collection circuit that was primed with heparinized normal saline at 4 U/mL. The mean proximal aortic pressure, mean distal arterial pressure, and temperature were recorded using a Powerlab/8SP Polygraph (AD Instruments, Sydney, Australia). Spinal cord ischemia was induced by inserting a 2F Fogarty balloon catheter (Edwards Life Sciences, Irvine, CA, USA) via the left femoral artery into the descending thoracic aorta, 10 cm from the femoral arteriotomy, so that the tip of the catheter balloon lay 3–4 mm caudal to the left subclavian artery (Yang et al., 2012). All rats received 200 U of heparin sodium through the carotid artery cannula. The aortic occlusion was confirmed by an immediate and sustained loss of detectable pulse pressure and a decrease in mean distal arterial pressure. At the end of the 25-minute ischemic period the balloon was deflated, animals received 200 U of protamine sulfate, catheters were removed and spinal cord blood flow was restored. The surgical incisions were closed and the rats were returned to their cages to recover. Rats in the sham group underwent the same surgical procedure as those in the I/R group but without aortic catheter occlusion and were euthanized with an intraperitoneal injection of sodium pentobarbital 25 minutes after surgery for subsequent histological examination.
The I/R rats were divided into eight groups according to time points after I/R: 0, 1, 2, 4, 12, 24, 48 and 72 hours (n = 10 per group). At 0, 1, 2, 4, 12, 24, 48 and 72 hours after reperfusion, the rats were euthanized for histological examination. Rats in the 4-, 12-, 24-, 48- and 72-hour groups also underwent neurological assessment prior to euthanasia.
Assessment of neurological function
Hindlimb function was scored using the Basso, Beattie, and Bresnahan open-field locomotor scale (Basso et al., 1995). Scores ranged from 0 (no detectable hindlimb movement) to 21 (normal hindlimb locomotion).
Sample preparation
Rats were euthanized with an intraperitoneal injection of sodium pentobarbital (30–50 mg/kg). Lumbar (L3–5) spinal cord segments were dissected and post-fixed in 4% (w/v) paraformaldehyde for 2–4 days. The cord was embedded in paraffin, and serial sections (5 µm thick) were cut for Nissl staining and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) to detect cellular apoptosis.
Nissl staining
After dewaxing and rehydration, spinal cord sections were immersed in 0.5% (w/v) cresyl violet. Neurons containing Nissl substance in the cytoplasm, loose chromatin, and prominent nucleoli were considered normal neurons. Damaged neurons were identified as cells with reduced cytoplasmic Nissl substance, cavitations around the nucleus, and pyknotic homogenous nuclei. A pathologist who was unaware of the study groups and the neurological outcomes examined each spinal cord section and counted the total number of normal motor neurons in half the gray matter of each section. The number of normal motor neurons in each animal was obtained by averaging counts from three different Nissl-stained slides. A pathologist unaware of the neurological outcomes examined the tissue and counted the total number of normal motor neurons in the gray matter of each section.
TUNEL staining
To detect DNA fragmentation in cell nuclei, TUNEL assay was performed using an apoptosis kit (Roche Diagnostics, Mannheim, Germany) in accordance with the manufacturer's instructions. To determine the number of motor neurons undergoing apoptosis, two independent and blinded pathologists counted the numbers of positive or negative motor neurons in TUNEL-stained sections using a fluorescence microscope (ECLIPSE90i, Nikon, Tokyo, Japan). For each slide, color images of 10 separate fields were captured randomly and digitized in half the gray matter of each section. Cells with clear nuclear labeling were defined as TUNEL-positive cells and the number of TUNEL-positive cells in the gray matter of each section was counted.
Western blot assay
The homogenates of spinal cord were used for western blot assay. Forty micrograms of protein per animal was resolved on a 10% sodium dodecyl sulfate polyacrylamide gel and transferred onto polyvinylidene difluoride membranes. The samples were electrophoresed in a 10% polyacrylamide gel. Protein samples were boiled in 2.5% sodium dodecyl sulfate and 5% β-mercaptoethanol. A total of 20 µg protein samples and markers (MagicMark XP Western Standard; Invitrogen, Carlsbad, CA, USA) of each group were electrophoresed at 20 mA for 90 minutes. Electrophoresis buffer was prepared by 25 mM tris(hydroxymethyl)aminomethane, 250 mM glycine and 0.1% sodium dodecyl sulfate. Proteins were transferred onto polyvinylidene fluoride membrane (LC2002; Invitrogen) using transmembrane buffer and 10% methanol. The membrane was incubated with rabbit anti-p-ERK, p-Akt, p-JNK and GAPDH polyclonal antibodies (1:1,000; Beijing Biosynthesis Biotechnology Co., Ltd., Beijing, China) at room temperature for 1 hour, washed with PBS, and then incubated with horseradish peroxidase-labeled goat anti-rabbit IgG (1:1,000; Beijing Biosynthesis Biotechnology Co., Ltd.) at room temperature for 90 minutes. The samples were visualized with ECL Plus Kit (Amersham Bio-sciences, Piscataway, NJ, USA). Optical density values were analyzed using NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was conducted using SPSS for Windows 17.0 (SPSS Inc., Chicago, IL, USA). All results are presented as the mean ± SD. All data were compared using one-way analysis of variance, and correlations were explored using the Pearson linear correlation analysis. A value of P < 0.05 was considered statistically significant.
Results
Change in hindlimb function in rat models of ischemic spinal cord injury
In all rats, heart rate, blood pressure, and body temperature were stable and did not differ between groups before, during, or after surgery. Sham-operated rats did not show any neurological impairment. However, in the I/R groups all rats exhibited notable hindlimb dysfunction, which persisted for 72 hours after reperfusion (Figure 1).
Figure 1.
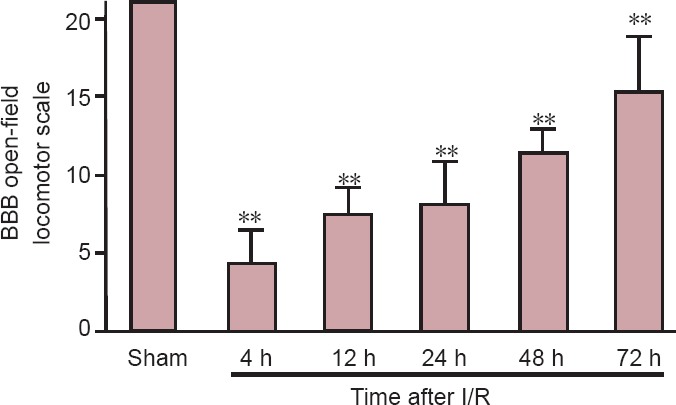
BBB open-field locomotor scale in rats with ischemic spinal cord injury.
Data are expressed as the mean ± SD, with 10 rats in each group at each time point. One-way analysis of variance was used. **P < 0.01, vs. sham group. BBB: Basso, Beattie, and Bresnahan; I/R: ischemia/reperfusion; h: hours.
Neuronal damage in the spinal cord of rats with ischemic spinal cord injury
Histological examination by Nissl staining of the spinal cord from the sham group at 25 minutes after surgery revealed that spinal cord tissue was well maintained and that neurons were morphologically normal with clear karyosomes and a uniformly stained cytoplasm. Compared with the sham group, there were fewer morphologically normal neurons in the I/R groups, and cells appeared swollen with a darkly stained and shrunken cytoplasm. Pyknosis and vacuolation were also observed in the I/R groups. The lowest number of morphologically normal neurons was observed at 48 hours after reperfusion (Figure 2).
Figure 2.

Histological changes in the spinal cord of rats with ischemic spinal cord injury.
(A) Nissl staining of spinal cord tissue sections from rats in the S and I/R groups at different time points after reperfusion (0–72 hours) (× 400). Arrows show normal neuronal cells. (B) Number of normal neurons in the anterior horn of the spinal cord. Data are expressed as the mean ± SD of 10 rats in each group at each time point. All data were compared using one-way analysis of variance. *P < 0.05, vs. S group; #P < 0.05, vs. I/R groups at all other time points. S: Sham; I/R: ischemia/reperfusion; h: hour(s).
Apoptosis in the spinal cord of rats with ischemic spinal cord injury
TUNEL-positive cells appeared brown and were mainly distributed in the anterior horn and central canal of the spinal cord. Very few TUNEL-positive cells were observed in sham-operated rats. In the I/R groups, significantly more apoptotic neurons were noted at all time points after reperfusion. The highest number of apoptotic neurons was observed at 48 hours after reperfusion (P < 0.05; Figure 3).
Figure 3.

Apoptosis in the spinal cord of rats with ischemic spinal cord injury.
(A) TUNEL staining of spinal cord from rats in the S and I/R groups at different time points after reperfusion (0–72 hours) (× 400). Positive reaction of TUNEL staining shows a brown color. (B) Number of apoptotic cells at different time points after reperfusion. Data are expressed as the mean ± SD of 10 rats in each group at each time point. One-way analysis of variance was used. *P < 0.05, vs. sham group; #P < 0.05, vs. I/R at all other time points. TUNEL: Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling; I/R: ischemia/reperfusion; S: sham; h: hour(s).
Akt, ERK and JNK phosphorylation in the spinal cord of rats with ischemic spinal cord injury
Western blot assay revealed that in sham-operated rats, p-Akt (serine-473) and p-ERK were constitutively expressed in motor neurons. In the I/R group, p-Akt and p-ERK expression levels peaked at 4 hours and 2 hours after reperfusion, respectively. p-Akt and p-ERK expression then decreased at 12 hours after reperfusion, finally returning to an elevated level at 24 hours after reperfusion, which was sustained until the end of the observation period. Significant differences in p-Akt and p-ERK expression were observed between the sham and I/R groups at all time points after reperfusion (P < 0.05). Furthermore, Akt expression at 4 hours and 12 hours was significantly different from the other time points in the I/R groups (P < 0.05). p-JNK expression after I/R injury was lower than that in sham-operated animals immediately after reperfusion (P < 0.05), but approached sham levels at 12 hours (P < 0.05, vs. all other time points). However, from 24 hours, expression returned to below sham levels until the end of the observation period (P < 0.05, vs. all other time points) (Figure 4).
Figure 4.

p-Akt (A), p-ERK (B) and p-JNK (C) protein expression in S and I/R groups at different time points after reperfusion (0–72 hours).
Data are expressed as the mean ± SD, with 10 rats in each group at each time point. The data were compared using oneway analysis of variance. *P < 0.05, vs. S group; #P < 0.05, vs. I/R groups at all other time points. p: Phosphorylated; Akt: serine-threonine kinase; ERK: extracellular signal-regulated kinase; JNK: c-Jun N-terminal kinase; S: sham; I/R: ischemia/reperfusion; h: hour(s).
Relationship between cell apoptosis and Akt, ERK and JNK phosphorylation in the spinal cord of rats with ischemic spinal cord injury
The result of Pearson linear correlation analysis showed that apoptosis negatively correlated with p-Akt expression (r = −0.352, P < 0.05); p-ERK and p-JNK expression did not show any correlation with apoptosis (r = −0.24, P = 0.056; r = 0.09, P = 0.47; Figure 5).
Figure 5.

Correlation between apoptosis and p-Akt, p-ERK or p-JNK expression.
Pearson linear correlation analysis shows that apoptosis is negatively correlated with p-Akt expression (r = −0.352, P < 0.05); p-ERK and p-JNK expression showed no correlation with apoptosis (r = −0.24, P = 0.056; r = 0.09, P = 0.47). p: Phosphorylated; Akt: serine-threonine kinase; ERK: extracellular signal-regulated kinase; JNK: c-Jun N-terminal kinase.
Discussion
ERKs, JNKs and Akt are all crucial to the survival and apoptosis of neuronal cells in the spinal cord (Maulik et al., 2008). Recently, it has been demonstrated that Akt mediates growth factor-induced neuronal survival (Yamauchi et al., 2006). JNK, a subfamily of mitogen-activated protein kinases, was considered a degenerative signal in the nervous system. In this study, our principal findings using this acute spinal cord injury rat model are: (1) levels of p-Akt and p-ERK increase at the beginning of reperfusion and then start to decrease after 12 hours with the respective peak of p-Akt and p-ERK expression being 4 hours and 2 hours after reperfusion; (2) p-JNK expression is lowest immediately after reperfusion but then increases and peaks at 12 hours; and (3) p-Akt expression correlates negatively with the number of apoptotic neurons in the first 72 hours of reperfusion. This confirms that the spinal cord injury was responsible for the high expression of ERK, Akt and JNK.
Akt mediates growth factor-induced neuronal survival (Crowder and Freeman, 1998), and a transient increase in p-Akt is observed in some models of cerebral ischemia (Noshita et al., 2001; Yano et al., 2001), similar to the increased neuronal expression of p-Akt observed in the present study after spinal cord ischemia. Yu et al. (2005) showed that p-Akt expression influences the number of motor neurons that survive at 1 day after spinal cord injury, and that inhibition of phosphatidylinositol 3-kinase reduces the expression of p-Akt. Here we have demonstrated that p-Akt expression in motor neurons increases in the first 12 hours after reperfusion, but decreases thereafter. Furthermore, neuronal apoptosis correlates negatively with Akt activation. The optimal time window for this approach was within 12 hours of reperfusion. As a result, the optimal time window for inhibiting apoptosis was within 12 hours of reperfusion.
While different models of cerebral or spinal cord ischemia have consistently implicated p-Akt as a mediator of neuroprotection, conflicting results have been obtained with p-ERK. In animal models of brain injury caused by ischemia or trauma, elevated p-ERK expression is involved in neuronal death; for example, ERK1/2 is activated upon deprivation of growth factors in neurons and renal epithelial cells, but inhibition of the ERK pathway blocks apoptosis (Zhuang and Schnellmann, 2006). In other studies, ERK was also shown to be involved in neuroprotective effects of pre- and post-conditioning in rat hippocampus and spinal cord (Choi et al., 2006; Jiang et al., 2009). Choi et al. (2006) demonstrated that ERK1/2 activation shows characteristic time- and cell-dependent patterns in a rat model of ischemic tolerance induction; basal levels of ERK1/2 phosphorylation were observed in CA1 neurons after 30 minutes of reperfusion, then in the CA3 and granule cells by 1 hour, and finally in dentate hilar neurons at 12 hours. By contrast, phosphorylation of ERK1/2 in mossy fibers and the CA1 dendritic field was sustained for at least 3 days. In the present study, ERK activation was suggestive of a negative correlation with apoptosis. Therefore, upregulation of p-ERK expression in the spinal cord may be required for sustained spinal cord protection.
Activation of JNK has been associated with induction of apoptosis, protection from cell death, proliferation, or differentiation in response to extracellular signals in various cell types (Minden and Karin, 1997; Ip and Davis, 1998; Leppä and Bohmann, 1999; Mielke and Herdegen, 2000). Leppä and Bohmann (1999) suggested that p-JNK might trigger the induction of the apoptotic effector p53. Activation of JNK was also implicated in neuronal cell death (Xia et al., 1995; Maroney et al., 1999). Xia et al. (1995) demonstrated that activation of JNK and concurrent inhibition of ERK are critical for the induction of apoptosis in neuronal cells, and considered that the dynamic balance between ERK and JNK-p38 pathways may be important in determining whether a cell survives or undergoes apoptosis. The findings from the sham group in our study show that JNK has a high basal activity in the spinal cord, supporting a previous report (Coffey et al., 2000); however, after 25 minutes of ischemia, activity of JNK was significantly lower than that in sham-operated animals at the start of reperfusion. However, JNK activity increased to sham levels by 12 hours, indicating that ischemia caused a decrease in JNK activity in this model. No evidence of a correlation between JNK activity and apoptosis was observed in this part of the spinal cord. Therefore, it is unlikely that inhibition of JNK will be neuroprotective in rat models of ischemic spinal cord injury.
In summary, Akt activation in spinal cord neurons correlates with local apoptosis in the first 72 hours of reperfusion after ischemia. The activation of Akt during the first 12 hours may be one of the factors responsible for the delay in neuronal apoptosis after spinal cord ischemia. Activated Akt is a strong candidate for use as a therapeutic agent in the treatment of ischemic spinal cord injury in the near future.
Footnotes
Funding: This research was supported by the National Natural Science Foundation of China, No. 81271387; the Research Special Fund of Public Welfare and Health Department of China, No. 201402009; the National Key Technology R&D Program in China, No. Z141107002514031.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded, stringently reviewed by international expert reviewers.
Copyedited by Paul P, de Souza M, Yu J, Qiu Y, Li CH, Song LP, Zhao M
References
- Badrian B, Casey TM, Lai MC, Rakoczy PE, Arthur PG, Bogoyevitch MA. Contrasting actions of prolonged mitogen-activated protein kinase activation on cell survival. Biochem Biophys Res Commun. 2006;345:843–850. doi: 10.1016/j.bbrc.2006.04.161. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim HY, Cha JH, Lee MY. Ischemic preconditioning-induced activation of ERK1/2 in the rat hippocampus. Neurosci Lett. 2006;409:187–191. doi: 10.1016/j.neulet.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coselli JS, LeMaire SA, Conklin LD, Köksoy C, Schmittling ZC. Morbidity and mortality after extent II thoracoabdominal aortic aneurysm repair. Ann Thorac Surg. 2002;73:1107–1115. doi: 10.1016/s0003-4975(02)03370-2. discussion 1115-1116. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Jiang X, Ai C, Shi E, Nakajima Y, Ma H. Neuroprotection against spinal cord ischemia-reperfusion injury induced by different ischemic postconditioning methods: roles of phosphatidylinositol 3-kinase-Akt and extracellular signal-regulated kinase. Anesthesiology. 2009;111:1197–1205. doi: 10.1097/ALN.0b013e3181bf1d93. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Kitagawa H, Warita H, Sasaki C, Zhang WR, Sakai K, Shiro Y, Mitsumoto Y, Mori T, Abe K. Immunoreactive Akt PI3-K and ERK protein kinase expression in ischemic rat brain. Neurosci Lett. 1999;274:45–48. doi: 10.1016/s0304-3940(99)00676-x. [DOI] [PubMed] [Google Scholar]
- Leppä S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O’Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA. CEP-1347 (KT7515) an inhibitor of JNK activation rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem. 1999;73:1901–1912. [PubMed] [Google Scholar]
- Maulik D, Ashraf QM, Mishra OP, Delivoria-Papadopoulos M. Activation of p38 mitogen-activated protein kinase (p38 MAPK) extracellular signal-regulated kinase (ERK) and c-jun N-terminal kinase (JNK) during hypoxia in cerebral cortical nuclei of guinea pig fetus at term: Role of nitric oxide. Neurosci Lett. 2008;439:94–99. doi: 10.1016/j.neulet.2008.02.037. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T. JNK and p38 stresskinases--degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- Noshita N, Lewen A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Safi HJ, Miller CC, Huynh TT, Estrera AL, Porat EE, Winnerkvist AN, Allen BS, Hassoun HT, Moore FA. Distal aortic perfusion and cerebrospinal fluid drainage for thoracoabdominal and descending thoracic aortic repair: ten years of organ protection. Ann Surg. 2003;238:372–381. doi: 10.1097/01.sla.0000086664.90571.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Hayashi T, Abe K, Itoyuama Y, Tabayashi K. Induction of phosphatidylinositol 3-kinase and serine-threonine kinase-like immunoreactivity in rabbit spinal cord after transient ischemia. Neurosci Lett. 2001;302:17–20. doi: 10.1016/s0304-3940(01)01609-3. [DOI] [PubMed] [Google Scholar]
- Shackelford DA, Yeh RY. Differential effects of ischemia and reperfusion on c-Jun N-terminal kinase isoform protein and activity. Brain Res Mol Brain Res. 2001;97:178–192. doi: 10.1016/s0169-328x(01)00245-5. [DOI] [PubMed] [Google Scholar]
- Shackelford DA, Yeh RY. Activation of extracellular signal-regulated kinases (ERK) during reperfusion of ischemic spinal cord. Brain Res Mol Brain Res. 2003;115:173–186. doi: 10.1016/s0169-328x(03)00206-7. [DOI] [PubMed] [Google Scholar]
- Willaime-Morawek S, Brami-Cherrier K, Mariani J, Caboche J, Brugg B. C-Jun N-terminal kinases/c-Jun and p38 pathways cooperate in ceramide-induced neuronal apoptosis. Neuroscience. 2003;119:387–397. doi: 10.1016/s0306-4522(02)00996-x. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Sakurai M, Abe K, Takano H, Sawa Y. Neuroprotective effects of activated protein C through induction of insulin-like growth factor-1 (IGF-1) IGF-1 receptor, and its downstream signal phosphorylated serine-threonine kinase after spinal cord ischemia in rabbits. Stroke. 2006;37:1081–1086. doi: 10.1161/01.STR.0000206280.30972.21. [DOI] [PubMed] [Google Scholar]
- Yang YW, Lu JK, Qing EM, Dong XH, Wang CB, Zhang J, Zhao LY, Gao ZF, Cheng WP. Post-conditioning by xenon reduces ischaemia-reperfusion injury of the spinal cord in rats. Acta Anaesth Scand. 2012;56:1325–1331. doi: 10.1111/j.1399-6576.2012.02718.x. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21:351–360. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Yu F, Sugawara T, Maier CM, Hsieh LB, Chan PH. Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol Dis. 2005;20:491–499. doi: 10.1016/j.nbd.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]