

Gut Microbes in Health and Disease
The human intestinal tract is home to at least 1000 distinct species of bacteria, which collectively number over 100 trillion organisms. This diverse ecosystem is shaped by early life events but can evolve over time through interactions between its constituents, host genes, and exogenous factors, such as diet and environmental exposures. Until recently, characterization of the gut microbiome relied mostly on conventional culture-based microbiological techniques, which was a major hindrance since the vast majority of bacteria in the gut are not readily amenable to cell culture. However, advances in next generation genomic technologies now allow us to identify and classify gut bacterial composition in an unprecedented manner. In this approach, in-depth sequencing of the variable regions of bacterial 16s rRNA genes is used to determine the diversity and proportion of bacterial taxa within the microbial community. While a major step forward, the evolutionary resolution provided by 16s sequencing is still limited with current platforms since bacterial composition with this approach is typically identified only down to the genus level. Overcoming these challenges will not only improve our ability to identify and quantitate distinct species of bacteria, but may have implications for understanding the pathological mechanisms through which specific bacteria affect both human health and disease processes.
Metagenomic analyses has revealed that the human gut is mostly comprised of a common core of bacteria from two major phyla, Firmicutes and Bacteriodetes, with the remainder of the gut microbiota being remarkably diverse. This diversity often includes less abundant representation from the phyla Proteobacteria, Verrumicrobia, Actinobacteria, Fusobacteria, and Cyanobacteria, as well as the domain Archaea 1. For the most part, metagenomic analyses on the human gut microbiota have used fecal samples but it is clear from animal studies that certain anaerobic organisms, such as Akkermansia muciniphila, reside primarily in the mucosal layers of the gut and are not readily detected in analyses of only feces. Indeed, the microbial composition throughout the gut varies considerably, both with respect to the anatomic location along the intestinal tract and within a given site, according to the micro-environment. For example, at a given site of intestinal mucosa/lumen, distinct microbes can uniquely reside deep within the crypts, versus on the surface of the mucosal villi, versus within the fecal material. Thus, without invasive procedures to get samples from distinct anatomical regions through the intestines, obtaining a more complete picture of the full spectrum of gut microbiota, at least in humans, poses significant challenges.
It has become widely appreciated that our gut symbionts play integral roles in human health since perturbations of this bacterial community or the products they can produce have been associated with increased susceptibility to a variety of diseases (see Figure) 2-4. The first indications of these associations were for colitis and inflammatory bowel disease, but altered gut microbial composition or function as a potential contributor to disease risk has now been established in the development of obesity and related metabolic abnormalities 5-8, atherosclerosis 9, 10, and even neurobehavioral conditions, such as autism 11.
Figure. Schematic illustration of some of the many phenotypes and diseases where gut micorbe involvement is reported.
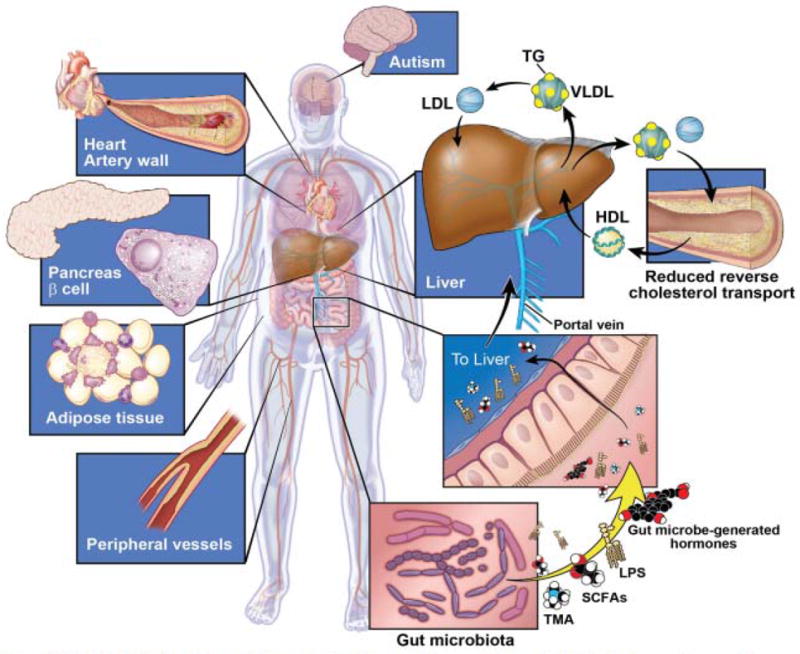
Multiple lines of evidence support a role for altered gut microbial composition or function as a contributor to the development of obesity and related metabolic abnormalities (ie, type 2 diabetes), peripheral and coronary artery disease, and even neurobehavioral conditions, such as autism. Recent observations of significant associations between proportions of specific intestinal bacteria taxa with high-density lipoprotein (HDL) cholesterol and triglyceride (TG) levels in subjects independent of body mass index suggest a role for gut microbes in modifying host lipid metabolism. Gut microbe effects may be mediated through multiple mechanisms, including elaboration of lipopolysaccharide (LPS) or other bioactive metabolites that act fundamentally as hormones, since they can circulate within the host and act at distant sites. Gut microbial production of short chain fatty acids (SCFAs) and secondary bile acids are two such examples. Evidence shows that gut bacteria also generate intermediate precursors (eg, trimethylamine [TMA]) form certain dietary nutrients that can then be further metabolized by the host to generate biologically active products (eg, trimethylamine N-oxide), which then can exert direct effects on lipid metabolism and contribute to disease development or progression. Biological mechanisms impacted by gut microbial metabolites can involve reverse cholesterol transport, hepatic cholesterol and sterol metabolism, intestinal lipid transport, bile acid composition and pool size, glucose and insulin metabolism, energy havest/expenditure, as well as others. LDL indicates low-density lipoprotein; and VLDL, very low-density lipoprotein.
Association of Gut Microbiota with Lipid Metabolism
Early studies comparing germ free versus conventionally raised mice first supported a role for gut microbes in both affecting host energy metabolism and modulating lipid levels 12; however, the design of these early studies did not permit identification of candidate microbes involved in promoting the observed phenotypic changes in conventionalized (microbe colonized) mice. In this issue of Circulation Research, Fu and colleagues13 now provide some of the first evidence in humans that variation in gut bacteria are associated with blood lipid levels, as well as clues toward discovering the microbes involved. Using a subset of the LifeLines population-based cohort, the fecal microbial composition of 893 subjects was determined by assessing genetic variation of bacterial 16S rRNA genes.
This effort represents one of the largest gut microbial composition studies in humans to date and the results revealed several interesting observations. First, microbial richness and diversity, as indicated by operational taxonomic units (OTUs), was inversely correlated with BMI and triglyceride (TG) levels and positively associated with high-density lipoprotein cholesterol (HDL) levels. Second, while some associations were shared across BMI and lipids, there were also microbial taxa whose proportions were primarily associated with lipids alone, and some of these were newly identified associations with TG and HDL levels. Third, the correlations between microbial taxa and lipids did not appear to be modified by BMI or host genetic factors, at least based on a risk score calculated from host genetic variants identified from genome-wide association studies (GWAS) for lipids and BMI. Lastly, incorporation of the results from gut microbial composition analysis in a risk model that included age, sex, and previous validated genetic risk factors (for lipids and BMI) significantly increased the percent of variance explained for BMI, TG, or HDL each by ~5%.
Overall, the associations reported in the LifeLines cohort are consistent with prior studies. For example, the number of OTUs, or microbial richness, was inversely related to BMI and TG levels and positively associated with HDL. These observations reinforce the concept that increased diversity of the gut microbiota is associated with more favorable cardiovascular and metabolic profiles. Previously described associations between obesity and certain taxa of bacteria 14, 15, such as Akkermansia, Christensenellaceae, and Tenericutes, were also confirmed. Importantly, with this new and largest yet study of its kind, the authors were able to identify additional significant and novel associations between the proportions of Eggerthella, Pasteurellaceae, and Butyricimonas and host TG and/or HDL levels. Surprisingly, there were only weak relationships noted between microbial variation and levels of total cholesterol or low-density lipoprotein cholesterol (LDL), suggesting that gut bacteria affect specific aspects of lipid metabolism and distinct classes of lipoproteins. Taken together, the observations by Fu and colleagues13 provide exciting insights and suggest new avenues worth pursuing both for validation studies and as follow up investigations to understand the underlying mechanism(s) and/or determine whether these associations are causal. It should be noted, however, that some of the associations observed between bacterial groups and lipid levels were at the level of phyla, while others were, at best, to the genus level. It will thus be difficult to narrow down the responsible microbial species, at least based on currently used methods.
Another concept addressed by Fu et al. was the role of host genetic factors in determining variation in gut microbiota. While the collective effect of ~250 previously validated variants for lipid levels and/or BMI were confirmed for these clinical phenotypes, the authors did not find any evidence for genetic effects, either alone or as a risk score, on microbiome composition. This may be explained, in part, by the fact that the only variants tested were, understandably, selected based on their main effects on lipids and BMI. One might speculate that host genetic variants that are involved in regulating bacterial abundance within the host may be more likely to be associated with observed changes in proportions of gut microbiota taxa; however, linking these taxa proportions to changes in host lipid levels would pose its own unique challenges. It is also probable that, even though the present study represents the largest gut microbiome study reported to date, its sample size is insufficient to have permitted identification of genetic associations. Third, the negative findings of a link between host genetic variants that impact lipids and BMI also being associated with gut microbiota proportions does not necessarily indicate there is a lack of host genetic variants influencing microbial composition. Given the enormous genetic diversity of the gut microbiome, the current study may simply have been underpowered to detect such associations. By comparison, a recent genetic analysis with ~100 inbred mouse strains, all of which were maintained on equivalent diet and housing conditions, identified seven host loci that were associated with common bacterial genera. In particular, one of these host genetic loci was associated with the proportion of Akkermansia muciniphila, a taxa whose proportion was also associated with gonadal fat mass and plasma TG levels 16. Similar genetic associations likely exist in humans as well, but their identification will require larger sample sizes and/or broader interrogation of genes, or new discovery approaches.
Biological Mechanisms through which Gut Microbes May Affect Lipid Metabolism
As with any gut microbiota study that is associative in nature, a major challenge is elucidating the underlying biological mechanisms involved in the associations and proving whether the associations are due to a causal relationship. Given the known clinical correlation between obesity and dyslipidemia, it is possible that the associations with TG and HDL levels reported by Fu et al. are mediated through effects on BMI. The data however suggest this not to be the case since statistical analyses showed that the proportions of bacterial taxa identified were independently associated with TG and HDL following adjustments for BMI. Moreover, a statistical model that included addition of microbial OTUs to age, sex, BMI, and genetic factors significantly increased the percent of the variation in lipid levels that could be explained compared to a model without inclusion of the fecal microbial composition data.
Evidence from other prior studies also supports the notion that the gut microbiome may mechanistically impact host lipid levels. For example, certain facultative and anaerobic bacteria in the large bowel produce secondary bile acids from the pool of bile salts secreted into the intestine. A small fraction of these bacterially derived bile acids are absorbed into the bloodstream and can modulate hepatic and/or systemic lipid and glucose metabolism through nuclear or G protein-coupled receptors (GPCRs), such as FXR or TGR5, respectively 17-19. Notably, several of the bacteria identified by Fu et al. are known to be involved in bile acid metabolism, suggesting that gut microbe associated alterations in bile acid composition and/or pool size may at least account for part of the biological basis for the associations between the identified fecal taxa proportions and plasma lipid levels.
Another potential mechanism through which gut microbes could affect lipid metabolism may involve fermentation of nondigestable carbohydrates to short chain fatty acids (SCFAs) by a subset of anaerobic bacteria found in the cecum and proximal colon. These SCFAs, such as acetate, propionate, and butyrate, are known to regulate intestinal immune homeostasis and serve as an energy source for colonic epithelial cells. However, SCFAs are also absorbed from the gut and can have potent effects on energy expenditure and insulin sensitivity in peripheral metabolic tissues through different GPCRs, such as GPR41 and GPR43 20, 21.
It is also possible that gut bacteria generate intermediate precursors that are further metabolized by the host to products that exert direct effects on lipid levels. In this regard, studies by our group recently identified trimethylamine N-oxide (TMAO) as a metabolite that increases atherosclerosis in mice and cardiovascular risk in humans, in part through perturbations of reverse cholesterol transport, cholesterol and sterol metabolism, and/or the quantity and composition of bile acids 9, 22-25. In this regard, TMAO is derived secondarily through hepatic oxidation of TMA, which is first produced through gut microbe-mediated metabolism of dietary choline and L-carnitine 9, 22.
The Challenge of Proving Causality
Fecal microbial composition studies are associative, and thus hypothesis generating. Ultimately, proof of causality for identified microbial associations with host phenotypes requires additional experimentation, such as manipulating gut bacterial composition and observing changes in physiological parameters that were identified in the initial associations. In both animal models and humans, intestinal microbial transplantation studies have provided evidence for a causal role of gut bacteria in modulating metabolic and cardiovascular phenotypes, as well as treating various intestinal diseases, most notably Clostridium difficile infection. For example, studies in mice have elegantly demonstrated that transfer of gut microbes can transmit obesity phenotypes, as well as efficiency of energy extraction from food 7. A similar strategy provided evidence that atherosclerosis susceptibility in mice could also be transmitted to a host by gut microbial transplantation 10. Such approaches have yet to be implemented in humans for treating cardiometabolic traits, but it is of interest that fecal transplantation studies in humans have shown that transfer of gut microbes from lean donors through a duodenal infusion into recipients with metabolic syndrome can increase insulin sensitivity 26, underscoring the therapeutic potential of interventions that alter gut microbial composition or function.
Targeting the Gut Microbiota for Therapeutic Applications
An important clinical implication from studies on gut microbiota is how to leverage findings for therapeutic purposes. Selective manipulation of the gut microbial ecosystem might provide new avenues to treat and/or prevent cardiometabolic diseases, but this will first require a better understanding of which specific bacteria, or alternatively, which bacterial metabolites, are the appropriate targets for intervention and manipulation 4. The simplest point of intervention may be to limit consumption of dietary constituents that either foster the growth of undesirable bacteria or serve as substrates for microbe-dependent generation of products that disrupt lipid homeostasis or other metabolic processes.
Another viable therapeutic strategy may be the use of prebiotics or probiotics to produce a desired change in microbial composition and/or function that favorably impacts host global metabolism4. Prebiotic therapy consists of ingestion of select nutrients or dietary constituents (nonmicrobial compositions) that provide a growth advantage of beneficial bacteria, whereas probiotic therapy involves the ingestion of one or more live bacterial strains, attempting to take advantage of the mutualism of microbes. Alternatively, therapeutic intervention could rely on the use of broad or class-specific antibiotics to eliminate bacterial species or their products associated with dyslipidemia and other metabolic disturbances. However, this approach is not a sustainable long-term option. For example, many gut microbial products are beneficial to the host and even infrequent antibiotic treatment, particularly in very young children whose gut microbiota has yet to be fully established, can adversely impact host global metabolism via changes in the gut microbial community 27 and facilitate the emergence of antibiotic-resistant bacterial strains.
Conclusions
For many years, the community of bacteria living in our gut was largely ignored. However, emerging evidence clearly demonstrates that our microbial symbionts play multiple fundamentally important roles in maintaining normal metabolic homeostasis. These discoveries have broad implications for elucidating bacterially-mediated pathophysiological mechanisms that alter lipid metabolism and other related metabolic traits. From a clinical perspective, this newly recognized endocrine organ system can be targeted for therapeutic benefit or prevention of metabolic diseases. The ability to manipulate the gut microbiome for improved health and prevention of diseases is still in the early phases of development, but recent rapid advances in gut microbiome studies highlight both the potential and promise of targeting intestinal microbes for therapeutic gain.
Acknowledgments
Work in the authors’ laboratories is supported, in part, by NIH grants P01ES022845, R01HL103866, R01DK106000, and U.S. EPA Grant RD83544101. The funders had no role in preparation, review, or approval of the manuscript.
Footnotes
Disclosures:
None
References
- 1.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 4.Brown JM, Hazen SL. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu Rev Med. 2015;66:343–359. doi: 10.1146/annurev-med-060513-093205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103:12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 8.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ, Hazen SL. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, Patterson PH, Mazmanian SK. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Boren J, Oresic M, Backhed F. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. doi: 10.1194/jlr.M002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu J, Bonder MJ, Cenit MC, Tigchelaar E, Maatman A, Dekens JAM, Brandsma E, Marczynska J, Imhann F, Weersma RK, Franke L, Poon TW, Xavier RJ, Gevers D, Hofker MH, Wijmenga C, Zhernakova A. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circulation Research. 2015;117:xxx–xxx. doi: 10.1161/CIRCRESAHA.115.306807. in this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Org E, Parks BW, Joo JW, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, Drake TA, Eskin E, Lusis AJ. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015 Aug 10; doi: 10.1101/gr.194118.115. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 18.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. Tgr5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, Seeley RJ. Fxr is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ. The orphan g protein-coupled receptors gpr41 and gpr43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 21.Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, Takahashi T, Miyauchi S, Shioi G, Inoue H, Tsujimoto G. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor gpr43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shih DM, Wang Z, Lee R, Meng Y, Che N, Charugundla S, Qi H, Wu J, Pan C, Brown JM, Vallim T, Bennett BJ, Graham M, Hazen SL, Lusis AJ. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56:22–37. doi: 10.1194/jlr.M051680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, Sacks J, Rong X, Vallim TA, Chou J, Ivanova PT, Myers DS, Brown HA, Lee RG, Crooke RM, Graham MJ, Liu X, Parini P, Tontonoz P, Lusis AJ, Hazen SL, Temel RE, Brown JM. The tmao-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015 doi: 10.1016/j.celrep.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916. e917. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 27.Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol. 2015;11:182–190. doi: 10.1038/nrendo.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]