

Abstract
Purpose
This open-label, multicenter study was designed to allow access to vemurafenib for patients with metastatic melanoma, bridging the time between end of enrollment in the phase III registration trial (December 2010) and commercial availability following US Food and Drug Administration approval of vemurafenib for the treatment of unresectable or metastatic BRAFV600-mutated melanoma (August 2011).
Patients and Methods
Eligible patients had metastatic melanoma with a BRAFV600 mutation (detected by the cobas 4800 BRAF V600 Mutation Test). Unlike previous vemurafenib trials, patients with poor performance status (PS) and treated brain metastases were permitted. Enrolled patients received oral vemurafenib 960 mg twice daily.
Results
Of 374 patients enrolled at 29 US sites (December 2010 to October 2011), 371 patients received vemurafenib and were followed up for a median of 2.8 months (the study had a prespecified end upon vemurafenib approval and commercial availability). At baseline, most patients (75%) had stage M1c disease, and 19% had an Eastern Cooperative Oncology Group PS of 2 or 3; 72% of patients had received prior systemic therapy for metastatic melanoma, 27% received prior ipilimumab, and 29% radiotherapy for prior brain metastases. Because reassessment data to confirm response were not available for most patients, point estimates of objective response rate (ORR) are reported. Among 241 efficacy-evaluable patients, the ORR was 54% (median time to response, 1.9 months). The ORR in non–central nervous system sites in patients with previously treated brain metastases (n = 68) was 53%. The ORR in prior ipilimumab-treated patients (n = 68) was 52%. For patients with PS of 0 or 1 (n = 210) and 2 or 3 (n = 31), the ORRs were 55%, and 42%, respectively. The safety profile observed was consistent with that reported in previous studies. The number of patients with grade 3 or 4 treatment-related adverse events was higher in patients with PS 2 or 3 than in those with PS 0 or 1 (10% vs. 5%, respectively). Adverse events requiring a dose reduction (at least 1 level) occurred in 11% of patients, and 9 patients (2%) experienced events leading to vemurafenib withdrawal, including 2 with repeated QT interval prolongation.
Discussion
This study confirmed the established rapid and high tumor response rate achievable with vemurafenib in BRAFV600 mutation–positive metastatic melanoma. Several groups not included in previous studies, including patients with previously treated brain metastases, Eastern Cooperative Oncology Group PS 2 to 3, or previous ipilimumab treatment had benefitted from vemurafenib similar to the overall population. No new safety signals were detected.
Keywords: Melanoma/drug therapy, melanoma/genetics, BRAF protein, human, neoplasm metastasis/drug therapy, brain neoplasms/secondary, brain neoplasms/drug therapy, vemurafenib/therapeutic use, vemurafenib/adverse effects, clinical trial, humans
The incidence of melanoma has continued to increase in the United States and worldwide during recent years.1,2 In the United States alone, melanoma was predicted to occur in about 76,250 persons in 2012 and to be responsible for an estimated 9180 deaths.3
Until recently, there were few treatment options for patients with metastatic melanoma, and those that were available—including chemotherapy, radiotherapy, and immunotherapy—were associated with low response rates and had no impact on survival rates.4 However, during the last 2 years, ipilimumab and vemurafenib have both proven to extend survival in metastatic melanoma and are now considered part of standard treatment.5, 7 Dabrafenib, a BRAF inhibitor, and trametinib, a MEK inhibitor, were also recently approved.
Vemurafenib, an orally available selective inhibitor of on-cogenic BRAF kinase, has demonstrated high response rates in previously treated patients with BRAFV600-positive metastatic melanoma.6 Furthermore, vemurafenib was associated with improved overall survival, progression-free survival, and response rate compared with standard chemotherapy in patients with previously untreated BRAFV600-mutated metastatic melanoma.7 After enrollment had reached its goal in this study, the safety monitoring board determined that both the overall survival and progression-free survival endpoints had met the prespecified criteria for statistical significance in favor of vemurafenib and subsequently recommended that patients in the dacarbazine arm be allowed to cross over into the vemurafenib arm. Based on these data, vemurafenib was approved by the US Food and Drug Administration (FDA) for the treatment of patients with un-resectable or metastatic melanoma with BRAFV600 mutation, as detected by an FDA-approved test. Vemurafenib has also now received approval in more than 50 countries. The present study provided access to vemurafenib treatment for patients with cobas test–positive BRAFV600-mutant metastatic melanoma who had no other treatment options, bridging the gap between the end of enrollment in the phase III registration trial7 in December 2010 and commercial availability after approval of the drug by the FDA in August 2011. Secondary objectives were to assess the safety of vemurafenib in this patient population and to determine the objective tumor response according to Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST 1.1), in patients with measurable disease.
Patients and Methods
Study Design
The primary objective of this study was to provide vemurafenib treatment for patients in medical need. This dictated an open-label, noncomparative design. The trial was conducted in multiple centers across the United States and consisted of a 28-day screening period, a treatment phase (starting on day 1), and 1 poststudy follow-up visit 30 days after the last dose of vemurafenib. The secondary objectives of the study were to assess (1) the safety of vemurafenib in patients with metastatic melanoma and (2) the objective response rate (ORR) of vemurafenib in patients with measurable disease.
End of enrollment/study was defined as the day when vemurafenib became commercially available following FDA approval for use in patients with metastatic melanoma harboring a BRAFV600 mutation. Patients already enrolled by that date could receive up to 1 additional 28-day cycle of therapy.
The study was conducted in accordance with the principles of the Declaration of Helsinki. The protocol, informed consent form, and accompanying patient information materials were approved by the institutional review board at each participating site before study initiation. All patients were required to provide a signed informed consent form before study entry and before participating in any study-related procedures.
Patients
Patients eligible for entry into the study were at least 16 years of age and had histologically confirmed metastatic or locally unresectable BRAFV600 mutation–positive melanoma. Presence of the BRAFV600 mutation was determined by the cobas 4800 BRAF V600 Mutation Test performed by a central laboratory. Additional entry criteria included adequate recovery from the most recent systemic or local treatment of cancer, adequate organ function, an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 to 3, and measurable or nonmeasurable disease (as defined by RECIST 1.1).
Patients were excluded from the study if they were receiving concurrent antitumor therapy or had a concurrent condition (e.g., active infection, laboratory abnormalities) or history of a prior condition that placed the patient at unacceptable risk from the study drug or confounded the ability to interpret data from the study. In particular, those with a history of congenital long QT syndrome were not eligible. Also excluded were those with a mean QTc interval of 470 milliseconds or greater at baseline or ongoing cardiac dysrhythmia of grade 2 or greater, pregnant or breast-feeding women, and patients unwilling to practice effective birth control.
A protocol amendment subsequently provided broader access to those patients with high medical need with brain metastases or concurrent malignancies not requiring current antitumor therapy.
Treatment
Patients received oral vemurafenib 960 mg (provided as four 240-mg film-coated tablets) twice daily, beginning on day 1 and continuing until one of the following occurred: disease progression, development of an intolerable adverse event (AE) considered probably associated with study treatment, withdrawal of consent from the study, study termination by the sponsor (following FDA approval of vemurafenib), or death. Doses were to be taken at least 1 hour before or 2 h after a meal; missed doses or those partially absorbed due to emesis were not repeated. Patients were strongly advised to avoid prolonged sun exposure and to use sunscreen and lip balm of at least SPF 30 during treatment and for at least 5 days thereafter. One cycle of therapy was defined as 28 days of treatment. Patients returned to the treatment center at the end of each cycle for clinical assessment and to receive a further 28 days' supply of study treatment.
Treatment interruption or dosage modification was required based on safety assessments detailed in the next section (assessments) and included QTc interval greater than 500 milliseconds or greater than 60-millisecond increase from baseline, as measured on 3 separate electrocardiographs (ECGs) performed at 10-minute intervals (temporary cessation of treatment, followed by resumption at 1 dose-level reduction [720 mg twice daily] after resolution of QT and electrolyte parameters), and grade 4 hema-tologic or grade 3 to 4 nonhematologic toxicities (treatment was interrupted until recovery to baseline or grade 1 and then resumed at 1 dose-level reduction). Up to 2 dose-level reductions were permitted for recurrent toxicities (720 mg then 480 mg twice daily); thereafter, treatment was to be permanently discontinued. There was no dose reduction or interruption for skin cancer, as this was considered a known and manageable AE of this therapy. A protocol amendment to strengthen the cardiac safety requirements subsequently disallowed resumption of treatment with further dosage reduction following repeat episodes of increased QTc interval.
Temporary discontinuation of vemurafenib for up to 8 weeks was allowed for any reason, including emergent toxicities (described above) or to allow nonprotocol cancer therapies such as surgery or radiotherapy.
Assessments
Assessments of medical history, patient characteristics and demographics, and eligibility for study enrollment were performed at screening. Physical, gynecologic, dermatologic, and ECG assessments were made at baseline and at regular intervals during study treatment, as described below. Blood counts, biochemical analyses, and ECOG PS were determined at each visit.
Adverse events, graded by the National Cancer Institute Common Terminology Criteria (NCI-CTC) for Adverse Events (version 4.0), were assessed on day 1 of each cycle and during the 30-day safety follow-up or at study termination. A limited physical examination was performed to focus on organ systems that were related to a potential AE as suggested by a patient's interim medical history and/or existing clinical and preclinical data for vemurafenib and included a complete skin examination (each cycle) and head and neck examination (every 3 cycles).
Regular skin examinations, after cycles 1, 4, 7, 10, and every 3 cycles thereafter, were performed by the investigator and dermatologist to monitor for cutaneous squamous cell carcinoma (cuSCC). A complete skin examination by a dermatologist was also required at follow-up visit, but only if not performed within the previous 3 months.
Standard 12-lead ECG was required monthly for the first 3 months and every 3 months thereafter, or more often as clinically indicated, with a focus on QTc interval. Treatment cessation and subsequent resumption as a result of prolonged QTc interval are described above.
Tumor assessments for patients with measurable disease were performed at screening and then every 4 to 8 weeks (for lesions assessable by physical examination) or 8 to 16 weeks (for imaging studies) from the time of the first dose (±5 days) regardless of dosing delays. The assessment of objective response was based on RECIST 1.1 after initial documentation of response. The same radiographic procedure was used to define measurable disease sites at baseline and throughout the study.
Statistical Analysis
The treated population and safety population were defined as all enrolled patients who received at least 1, or a partial, dose of vemurafenib. The efficacy-evaluable population was defined as all treated patients who had measurable disease at baseline and at least 1 postbaseline tumor assessment. With the protocol-defined end of this study when vemurafenib was approved and commercially available, the limited number of patients with sufficient follow-up time for 2 postbaseline tumor scans at least 4 weeks apart did not allow for a reliable estimate of the confirmed response rate.
The final analysis was performed on patient data collected through 30 days after the last vemurafenib dose for the last patient enrolled into the study. Continuous data were summarized in a descriptive statistical manner, using mean, SD, median, minimum, and maximum. Discrete data were summarized using frequencies and percentages. Median durations of follow-up were calculated as median with range. The ORR was defined as the number of patients with a best overall response of complete (CR) or partial response (PR) using RECIST 1.1, divided by the number of efficacy-evaluable patients. The ORR and exact 2-sided 95% confidence intervals (CIs) were calculated by the Clopper-Pearson method. The safety analysis was performed in all patients who received at least 1 dose of study drug. Descriptive statistics were reported for safety endpoints.
Results
Among 745 patients screened at 29 sites in the United States between December 2010 and October 2011, 374 patients were enrolled into the study, with 371 receiving study treatment (Fig. 1). The major reason for screening failure was a negative result for BRAFV600 mutation by cobas test, as expected since the frequency of the mutation was anticipated to be 40% to 50% of the overall screened population. A total of 259 patients (69%) switched from study treatment to receive the commercial product after FDA approval; this was classified as a discontinuation from the study, and follow-up of individual patients ceased once they had switched. Other reasons for study treatment discontinuation included disease progression (13%), death (7%), withdrawal of consent (5%), and AEs (2%). The transition of the majority of patients to commercially available vemurafenib accounted for the median follow-up duration of less than 3 months for the entire cohort. This final analysis was based on data accumulated up until the data-lock date of April 5, 2012.
Figure 1.
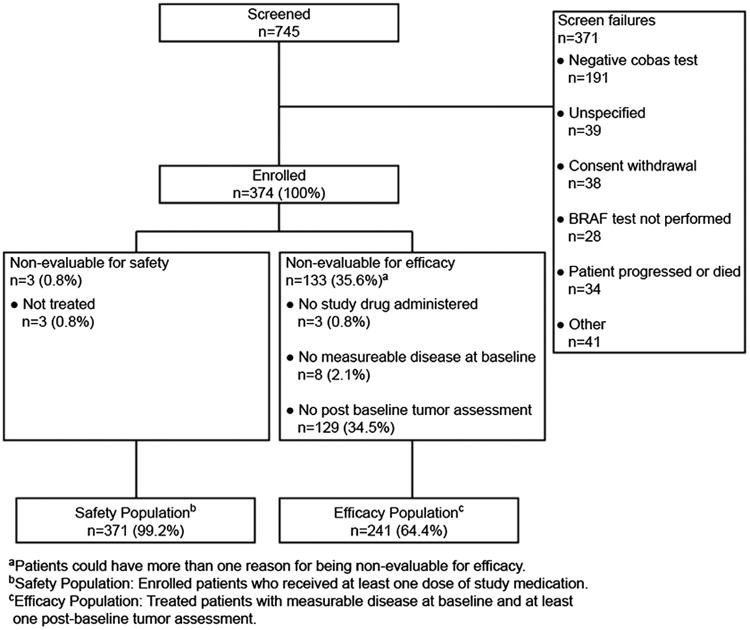
Patient flow.
Baseline patient demographic and disease characteristics for the treated population are shown in Table 1. Overall, 19% of patients had poor functional status (ECOG PS 2 or 3), and 75% had stage M1c disease. Mean (SD) patient age at baseline was 53.5 (SD, 13.8) years (range, 17–87 years). The majority (72%) of patients had previously received systemic therapy for meta-static melanoma, including 102 patients who received at least 1 dose of ipilimumab. Compared with the ipilimumab-naive patient cohort (n = 269), the incidence of patients younger than 65 years (77% vs. 82%), as well as patients with ECOG PS 2 or 3 (17% vs. 25%), was slightly higher in the ipilimumab-pretreated cohort. One hundred nine patients (29%) had prior radiotherapy for brain metastases.
Table 1. Baseline Patient Demographic and Disease Characteristics.
| Characteristic | Vemurafenib (n = 371) |
|---|---|
| Age, mean (SD), y | 53.5 (13.8) |
| Male, n (%) | 229 (62) |
| ECOG PS, n (%) | |
| 0 | 162 (44) |
| 1 | 138 (37) |
| 2 | 59 (16) |
| 3 | 12 (3) |
| Metastatic melanoma stage, n (%) | |
| M1a | 33 (9) |
| M1b | 35 (9) |
| M1c | 277 (75) |
| Unresectable stage III | 24 (7) |
| Unknown | 2 (<1) |
| Brain metastases, n (%) | 109 (29) |
| Prior cancer therapy | |
| Systemic therapy, n (%) | 266 (72) |
| 1 Therapy | 75 (20) |
| 2 Therapies | 64 (17) |
| ≥3 Therapies | 127 (34) |
| No. prior systemic therapies, mean (SD) | 2.9 (2) |
| Radiotherapy, n (%) | 175 (47) |
Safety
The 371 patients who received at least 1 dose of study drug were included in the safety population. The median duration of follow-up for safety observations was 2.8 months (range, 0.1–10.1 months).
Because of the FDA's rapid approval of vemurafenib, at which time patients were switched to the commercial product, overall exposure to vemurafenib study treatment was relatively short, with a median number of 2 treatment cycles (range, 1–10 cycles) or median treatment duration of 8.4 weeks (range, 0.4–44 weeks). Complete dosage information was not available for 25 patients. Of the 346 patients with complete dosage records, 135 patients (39%) completed at least 12 weeks of vemurafenib treatment, and 316 patients (85%) were receiving the full dosage of vemurafenib at the time of their last administration. Overall, 62 patients (17%) missed at least 1 dose because of AEs, and 42 patients (11%) required dose reductions because of AEs (with 83% requiring only 1 reduction).
A total of 346 patients (93%) experienced a treatment-emergent AE of any grade (regardless of causality), with treatment-related AEs reported in 308 patients (83%) (Table 2). The most common treatment-related AEs were rash of any kind (37%), arthralgia (35%), photosensitivity reaction (26%), and fatigue (20%). The majority of treatment-related AEs were grade 1 or 2 in severity. At least 1 treatment-related grade 3 AE was reported by 76 patients (21%), and 6 patients (2%) reported at least 1 treatment-related grade 4 AE.
Table 2. Summary of Treatment-Related AEs Occurring in at Least 5% of Treated Patients (Vemurafenib; n = 371).
| AE by Preferred Term | All Grades | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Total patients with ≥1 AE, n (%) | 308 (83) | 155 (42) | 76 (21) | 6 (2) |
| Total no. of AEs | 1353 | 310 | 126 | 7* |
| Rash (all forms)* | 137 (37) | 45 (12) | 12 (3) | 0 |
| Arthralgia | 129 (35) | 31 (8) | 17 (5) | 0 |
| Photosensitivity reaction† | 98 (26) | 19 (5) | 2 (<1) | 0 |
| Fatigue ‡ | 74 (20) | 23 (6) | 2 (<1) | 0 |
| Nausea | 37 (10) | 3 (<1) | 2 (<1) | 0 |
| Alopecia | 32 (9) | 1 (<1) | 0 | 0 |
| Myalgia | 28 (8) | 5 (1) | 3 (<1) | 0 |
| Diarrhea | 26 (7) | 3 (<1) | 2 (<1) | 0 |
| Pyrexia | 25 (7) | 9 (2) | 0 | 0 |
| Dry skin | 24 (7) | 3 (<1) | 0 | 0 |
| Hyperkeratosis | 24 (7) | 5 (1) | 0 | 0 |
| Peripheral edema | 23 (6) | 5 (1) | 2 (<1) | 0 |
| Squamous cell carcinoma of the skin | 22 (6) | 3 (<1) | 10 (3) | 0 |
| Palmar-plantar erythrodysesthesia syndrome | 22 (6) | 5 (1) | 5 (1) | 0 |
| Decreased appetite | 21 (6) | 4 (1) | 0 | 0 |
Grade 4 treatment-related AEs (each in 1 patient): respiratory failure, neutropenia, hyperbilirubinemia, increased blood creatine phosphokinase, prolonged ECG QT, decreased neutrophil count, confusional state.
Includes dermatitis, dermatitis bullous, erythema, folliculitis, generalized erythema, rash, rash erythematosus, rash follicular, rash generalized, rash macular, rash maculopapular, rash maculovesicular, rash morbilliform, rash papular, rash papulosquamous, rash pruritic, rash pustule, and rash vesicular.
Includes photosensitivity reaction and sunburn.
Includes asthenia, fatigue, lethargy, listless, malaise, and sluggishness.
Analysis of ECG findings showed that 24 patients (7%) had QTc interval increases to more than 480 milliseconds, 11 patients (3%) had QTc intervals of more than 500 milliseconds, and 19 patients (5%) experienced an increase in QTc interval from baseline of at least 60 milliseconds. None of these QTc interval prolongations was associated with any significant clinical finding (e.g., arrhythmia). Treatment-related photosensitivity reactions and/or sunburn were reported by 26% of patients. Among other toxicities requiring cautionary monitoring with vemurafenib, 24 treatment-related eye disorders were reported in 19 patients. Only 2 of them were of NCI-CTC grade 3 (photophobia and uveitis [1 event each]), with all other different forms of NCI-CTC grade 1 or 2 disorders including scleral disorders (3 events), photophobia, blurred vision, pain, dry eye, ocular hyperemia, periorbital edema (2 events each), uveitis (1 event), or single other reported events. Eight cases of grade 3 or 4 treatment-related liver function test abnormalities were reported: elevated change to abnormal alkaline phosphatase (4 cases, all grade 3), elevated change to abnormal alanine aminotransferase (1 case, grade 3), elevated change to abnormal aspartate amino-transferase (2 cases, all grade 3), and hyperbilirubinemia (1 case, grade 4). The pattern and severity of reported treatment-related AEs were similar for patients who were exposed to prior ipilimumab treatment compared with ipilimumab-naive patients (Supplemental Table 1, http://links.lww.com/PPO/A12).
Treatment-related serious AEs were reported by 28 patients (8%), including nausea, vomiting, pyrexia, prolonged QT interval, and respiratory failure each in 2 patients. Nine patients experienced 10 treatment-emergent AEs leading to vemurafenib discontinuation. The most common reasons were arthralgia (2 cases) and prolonged QT interval (2 cases). Other AE-related reasons for treatment withdrawal were neutropenia, abdominal pain, death, raised blood creatine phosphokinase, pain in extremity, and central nervous system metastases.
A total of 43 patients (12%) died during the study; 13 of them died during the follow-up period after discontinuation of study. Of the 30 documented deaths within the study period, 22 resulted from disease progression, and 8 were attributed to AEs, of which 2, both ipilimumab-naive patients, were considered to be related to study drug. Of the treatment-related deaths, 1 patient with a history of central nervous system metastases and bleeding died of an intracranial hemorrhage after the first dose of vemurafenib, and the second patient died because of multiorgan failure. The reported death within 30 days from the last vemurafenib dosage was 11% for patients with prior ipilimumab exposure and 6% for patients who were ipilimumab naive.
Squamous cell carcinoma of the skin was reported as a treatment-related AE in 22 patients. An expanded analysis based on AE preferred terms—Bowen disease, keratoacanthoma, lip neoplasm, lip neoplasm malignant stage unspecified, squamous cell carcinoma of the skin, and treatment-related secondary malignancy—revealed that a total of 31 patients (8%) reported at least 1 cuSCC-related AE (55 cases in total), with 9 of these patients reporting multiple cuSCCs. A greater exposure to study drug was observed in a post hoc analysis in these 31 patients compared with those who did not develop cuSCC, with median total cumulative vemurafenib doses of 160.3 and 107.5 g, respectively, a median number of drug cycles of 3.0 versus 2.0, and mean treatment duration of 13.3 weeks versus 10.5 weeks. Representative examples typical of skin toxicities experienced by patients treated with vemurafenib in this study are illustrated in Figure 2.
Figure 2.
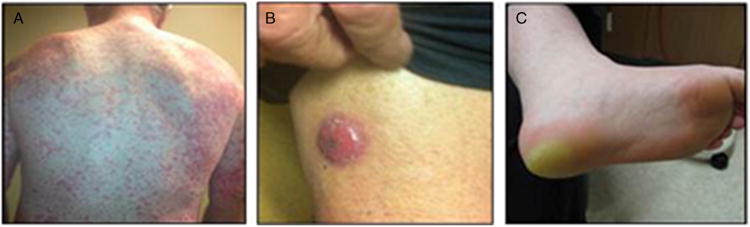
Examples of skin toxicities experienced by patients treated with vemurafenib. A shows a patient with a maculopapular rash, which developed approximately 3 weeks after initiation of vemurafenib therapy. B shows squamous cell carcinoma of the skin, which was detected 4 to 6 weeks after initiation of vemurafenib therapy. C shows a case of hand-foot skin reaction with hyperkeratosis, which was also reported as palmar-plantar erythrodysesthesia syndrome.
Efficacy
A total of 241 patients had measurable disease at baseline and sufficient follow-up time for at least 1 postbaseline tumor assessment and were therefore included in the efficacyevaluable population. Of those, 129 patients (54% [95% CI, 47%–60%]) had an unconfirmed objective response to treatment (PR or CR; Table 3), including 1 patient (<1%) with a CR, and a further 73 patients (30%) with stable disease. Because of limited follow-up time, further efficacy evaluations required for confirmation of response were possible in only a limited number of patients, not allowing for analysis of a reliable estimate for the confirmed ORR (Fig. 3). The median time to objective response (unconfirmed) was 1.9 months (range, 1.9–2.7 months).
Table 3. Efficacy of Vemurafenib for All Patients, and for Patients With Previously Treated Brain Metastases, ECOG PS Subgroups, and Prior Ipilimumab Treatment.
| Patient subgroups | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| Response, n (%) | All Patients (n = 241) | Previously Treated Brain Metastases (n = 68) | ECOG PS 0 or 1 (n = 210) | ECOG PS 2 or 3 (n = 31) | Prior Ipilimumab Treatment (n = 68) | Ipilimumab Naive (n = 173) |
| Unconfirmed ORR | 129 (54) | 36 (53) | 116 (55) | 13 (42) | 35 (52) | 94 (54) |
| (95% CI) | (47.0–60.0) | (40.4–65.2) | (48.2–62.1) | (24.5–60.9) | (39–63.8) | (46.6–61.9) |
| CR | 1 (<1) | 0 | 1 (<1) | 0 | 0 | 1 (<1) |
| PR | 128 (53) | 36 (53) | 115 (55) | 13 (42) | 35 (52) | 93 (54) |
| SD | 73 (30) | 19 (28) | 64 (31) | 9 (29) | 25 (37) | 48 (28) |
| PD | 29 (12) | 11 (16) | 23 (11) | 6 (19) | 7 (10) | 22 (13) |
| Unable to evaluate | 10 (4) | 2 (3) | 7 (3) | 3 (10) | 1 (2) | 9 (5) |
| Median (95% CI) time to response, mo | 1.9 (1.9–2.7) | 1.9 (1.9–3.7) | 1.9 (1.9–2.7) | 1.9 (1.9–NA) | 2.1 (1.9–3.7) | 1.9 (1.9–2.5) |
NA indicates not assessed; PD, progressive disease; SD, stable disease.
Figure 3.
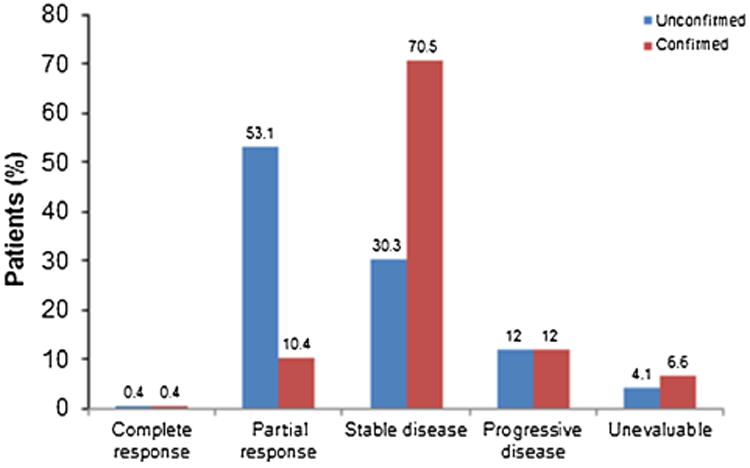
Unconfirmed and confirmed response rates in the efficacy-evaluable population (n = 241).
Response rates to vemurafenib were also assessed and compared in several patient subsets. The ORR, outside the brain, in the 68 patients who had previously treated brain metastases, was similar to the ORR for all patients (53% [95% CI, 40%–65%] and 54% [95% CI, 25%–61%], respectively). In the 31 patients who had poor PS (ECOG PS 2 or 3) at the time of study entry, the ORR was 42%. The median time to response was similar across all subgroups analyzed (1.9 months; Table 3). The outcome observed for patients who were exposed to prior ipilimumab was comparable to those who were naive to prior ipilimumab (Table 3).
Discussion
This study granted access to vemurafenib treatment for more than 370 patients across the United States with metastatic melanoma and no standard treatment options and bridged the time between the end of enrollment in the clinical development studies and FDA approval of vemurafenib. Even with limited follow-up time and a broader patient population, treatment with vemurafenib produced an unconfirmed ORR of 54%, with a generally favorable toxicity profile.
The efficacy of vemurafenib has been remarkably consistent in the major studies reported to date. In the phase II study (BRIM-2) in which patients with previously treated BRAFV600-mutant metastatic melanoma received vemurafenib, a confirmed ORR of 53% was reported.6 In the pivotal phase III (BRIM-3) study, the confirmed ORR with vemurafenib therapy was 48% in patients with previously untreated metastatic melanoma, compared with 5% for dacarbazine.7 In a follow-up analysis presented at ASCO 2012, confirmed ORR was 57% for vemurafenib and 9% for dacarbazine.8 Longer-term follow-up in that study revealed that vemurafenib was also associated with a significant improvement in survival relative to dacarbazine. At the February 2012 data cutoff, median survival time was 13.6 months for vemurafenib and 9.7 months for dacarbazine, and 12-month overall survival rates were 56% and 44%, respectively (hazard ratio, 0.70; 95% CI, 0.57–0.87; P < 0.001).8 As planned, the present expanded access study was halted after marketing authorization of vemurafenib, before a similar survival analysis could be conducted.
The relatively liberal entry criteria for this expanded access study allowed treatment of 3 patient groups not eligible or available for previous vemurafenib trials: (1) patients with poor PS (ECOG PS 2 and 3), (2) patients with previously treated brain metastases, and (3) patients previously treated with ipilimumab. All of these groups benefited from receiving vemurafenib, with overall response rates similar to those seen in the overall group. In addition, vemurafenib was generally well tolerated by patients in all of these groups, although grade 3/4 treatment-related AEs were more frequent in patients with poor PS (10% vs. 5%). Of note, a previous observation of grade 3 rash in 4 (31%) of 13 patients previously treated with ipilimumab9 was not confirmed in this much larger study (3% incidence in 102 patients treated, all of whom had received their last dose of ipilimumab >30 days before starting vemurafenib).
The documentation of a 42% ORR in patients with poor PS is arguably the most important clinical finding derived from this study. In the phases II and III trials of vemurafenib, such patients were excluded. It had been noted, however, that patients with disease-related symptoms at baseline experienced rapid improvement in symptoms leading to speculation that vemurafenib might serve as an effective palliative therapy for patients with even more advanced disease-related symptoms causing poor PS. This study provides the first documentation of the feasibility and efficacy of vemurafenib treatment in patients with poor PS, although the duration of benefit remains to be defined.
Although this trial did not address the efficacy of vemurafenib in treating uncontrolled brain metastases, the 53% response rate of systemic metastases in patients with previously treated brain metastases is also an important new observation. Many patients with brain metastases also have systemic metastases that impact the disease course, and effective therapy in this situation has not been previously available. Preliminary data suggest that vemurafenib may also have direct activity against brain metastases10; further information regarding this question will be supplied by an ongoing phase II trial (NCT01378975).
Safety monitoring was an important objective of this open-label study, and vemurafenib was shown to be generally well tolerated with a safety profile consistent with previous clinical trials and no new safety signals detected. Most treatment-related AEs were grade 1 or 2 in severity, and only 9 patients withdrew from study treatment as a result of treatment-emergent AEs. Overall, 11% of the patients receiving vemurafenib required dose modification because of treatment-emergent AEs.
The incidence of most AEs reported in the current study was lower than previously reported, most likely related to the shorter duration of treatment and follow-up. For example, the reported incidence of rash of any type across phases I to III trials of vemurafenib ranges from 64% to 75% (40% in the present study), photosensitivity from 35% to 63% (28% in the present study), and palmar-plantar erythrodysesthesia from 8% to 10% (6% in the present study).11 The rate of cuSCC-related events (8% of patients) was also lower than reported in previous clinical trials (18%–26%),6,7 also attributable to the limited follow-up time.
Monitoring of cardiac safety in the present study was ensured through regular assessment of ECG and electrolytes, as well as a planned schedule of treatment interruption/dosage modification or treatment cessation in response to emergent QTc prolongation events. A small proportion (<10%) of patients exhibited abnormal ECG findings requiring a review of study treatment, and 2 patients (0.5%) required permanent treatment discontinuation as a result of recurrent QTc prolongation. These events were not associated with any clinical symptoms or arrhythmias. Thus, ECG monitoring remains an important precaution for patients treated with vemurafenib, and specific guidance for this is detailed in the vemurafenib prescribing information.12
Despite limitations due to the nonrandomized design and short treatment duration, the results of this expanded access study confirm the previously reported rapid and high tumor response rate achieved by vemurafenib in the treatment of metastatic BRAFV600 mutation–positive melanoma. In addition, this study provides new data suggesting the efficacy of vemurafenib in several groups of patients with metastatic melanoma not addressed in previous trials. No new safety signals were detected, and the results provide further experience on the established toxicity profile to inform clinicians on managing vemurafenib treatment in their patients. The development of vemurafenib is an important first step in improving treatment for patients with metastatic BRAFV600 mutation–positive melanoma. Continued improvement in this area requires a better understanding of resistance mechanisms, identification of other critical (and targetable) pathways, and, if possible, more durable ways of blocking the BRAF/MEK/ERK pathway. Novel BRAF and MEK inhibitors, as well as various combinations, are in development, and further advances in therapy are likely in the future.
Supplementary Material
Acknowledgments
The authors thank acknowledge Drs Robert Weaver and James Khatcheressian for their contributions to this study.
Source of Funding: Support for third-party writing assistance for this manuscript was provided by Roche/Genentech, Inc. L.F. has received grant support and has served on the board of Merck; has consulted for Roche-Genentech, BMS, GSK, Genomic Health, and Merck; and has received payment for lectures for Roche/Genentech. O.H. has received grant support from Genentech and is a consultant and has received payment for speaker bureaus for Genentech. G.L. is a speaker for Roche/Genentech. S.H. is a speaker for Novartis and BMS. R.G. has received grant support from Roche/Genentech and has been a past consultant and received past support for travel for Roche/Genentech. C.L.C. has consulted and been a speaker for Roche/Genentech. A.P. has received grant support and financial support for being a consultant and providing review activities. B.C. has been a past consultant for BMS and Prometheus Pharmaceuticals and is awaiting grants from Prometheus Pharmaceuticals and the National Institutes of Health. D.L. has attended advisory boards for Roche/Genentech, has received support for study expenses, and has received grants for ongoing clinical trials. P.B.C. has received grant and travel support and has consulted for Genentech/Roche. K.M. has received grant support from Roche/Genentech. A.R. has consulted for Roche/Genentech, with the honoraria paid to his institution, and received financial support for travel. D.M. has received financial support for past consultancy. K.F. received grant support from and consults for Roche/Genentech. L.C. has received grant support from Roche/Genentech, with funds going to his institution, is a consultant for GSK and Merck, is on a speaker bureau; is a consultant for Roche/Genentech, Merck, BMS, and Prometheus; and has received research funding from Roche/Genentech and BMS for study protocols, with funds going to his institution. F.S.H. has received financial support for travel to meetings, served as a nonpaid consultant to Roche/Genentech, and received clinical trial support from Genentech/Roche. B.-M.D. is an employee of Genentech Inc and has received stock/stock options. R.L. is a previous employee of Roche/Genentech and received stock/stock options. J.H. received grant support from Roche/Genentech.
Footnotes
Conflicts of Interest: F.K. and L.S. have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
Supplemental digital contents are available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.journalppo.com).
References
- 1.Linos E, Swetter SM, Cockburn MG, et al. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129:1666–1674. doi: 10.1038/jid.2008.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacKie RM, Hauschild A, Eggermont AMM. Epidemiology of invasive cutaneous melanoma. Ann Oncol. 2009;20(Suppl. 6):vi1–vi7. doi: 10.1093/annonc/mdp252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Cancer Society. Cancer Facts and Figures 2012. [Accessed July 15, 2013]; Available at: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf.
- 4.Garbe C, Peris K, Hauschild A, et al. Diagnosis and treatment of melanoma: European consensus-based interdisciplinary guideline. Eur J Cancer. 2010;46:270–283. doi: 10.1016/j.ejca.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 5.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. published correction appears in N Engl J Med 2010;363:1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman PB, Hauschild A, Robert C, et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman PB, Hauschild A, Robert C, et al. Updated overall survival results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib with dacarbazine in previously untreated patients with BRAFV600E-mutated melanoma. J Clin Oncol. 2012;30(suppl; abstr 8502) [Google Scholar]
- 9.Harding JJ, Pulitzer M, Chapman PB. Vemurafenib sensitivity skin reaction after ipilimumab. N Engl J Med. 2012;366:866–868. doi: 10.1056/NEJMc1114329. [DOI] [PubMed] [Google Scholar]
- 10.Rochet NM, Kottschade LA, Markovic SN. Vemurafenib for melanoma metastases to the brain. N Engl J Med. 2011;365:2439–2441. doi: 10.1056/NEJMc1111672. [DOI] [PubMed] [Google Scholar]
- 11.Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated melanoma patients. Oncologist. 2013;18:314–322. doi: 10.1634/theoncologist.2012-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.South San Francisco, CA: Genentech, Inc; 2013. [Accessed July 15, 2013]. ZELBORAF™ (Vemurafenib) Tablet, Oral. prescribing information. Available at: http://www.gene.com/download/pdf/zelboraf_prescribing.pdf. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.