

Background: Two intracellular proteins, MMACHC and MMADHC, functionally interact for cobalamin trafficking.
Results: MMADHC crystal structure reveals protein-interacting regions and unexpected homology to MMACHC; mutations on either protein interfere with complex formation via different mechanisms.
Conclusion: Complex formation likely depends on prior cobalamin processing and can be broken by disease mutations.
Significance: MMACHC-MMADHC heterodimerization forms the essential trafficking chaperone delivering cobalamin to client enzymes.
Keywords: crystal structure, metabolic disease, protein-protein interaction, site-directed mutagenesis, small-angle x-ray scattering (SAXS), nitroreductase fold, vitamin B12
Abstract
Conversion of vitamin B12 (cobalamin, Cbl) into the cofactor forms methyl-Cbl (MeCbl) and adenosyl-Cbl (AdoCbl) is required for the function of two crucial enzymes, mitochondrial methylmalonyl-CoA mutase and cytosolic methionine synthase, respectively. The intracellular proteins MMACHC and MMADHC play important roles in processing and targeting the Cbl cofactor to its destination enzymes, and recent evidence suggests that they may interact while performing these essential trafficking functions. To better understand the molecular basis of this interaction, we have mapped the crucial protein regions required, indicate that Cbl is likely processed by MMACHC prior to interaction with MMADHC, and identify patient mutations on both proteins that interfere with complex formation, via different mechanisms. We further report the crystal structure of the MMADHC C-terminal region at 2.2 Å resolution, revealing a modified nitroreductase fold with surprising homology to MMACHC despite their poor sequence conservation. Because MMADHC demonstrates no known enzymatic activity, we propose it as the first protein known to repurpose the nitroreductase fold solely for protein-protein interaction. Using small angle x-ray scattering, we reveal the MMACHC-MMADHC complex as a 1:1 heterodimer and provide a structural model of this interaction, where the interaction region overlaps with the MMACHC-Cbl binding site. Together, our findings provide novel structural evidence and mechanistic insight into an essential biological process, whereby an intracellular “trafficking chaperone” highly specific for a trace element cofactor functions via protein-protein interaction, which is disrupted by inherited disease mutations.
Introduction
Vitamin B12 (referred hereafter as cobalamin, Cbl)5 is an essential cofactor for two human enzymes: cytosolic methionine synthase (MS, EC 2.1.1.13), which requires methyl-Cbl (MeCbl), and mitochondrial methylmalonyl-CoA mutase (MUT, EC 5.4.99.2), which requires adenosyl-Cbl (AdoCbl) (1). An intracellular pathway of at least seven human gene loci has evolved, encoding proteins responsible for the transport, processing and delivery of the appropriate Cbl form via a cytosol-targeted route to MS, and a mitochondrion-targeted route to MUT (2, 3). Mutations in any of the seven genes, depending on its relative position within the pathway, can result in three broad disease phenotypes collectively referred to as “intracellular Cbl disorders” (2, 4). They include (i) homocystinuria (HC) associated with reduced MeCbl production and MS deficiency (1, 5), (ii) methylmalonic aciduria (MMA) associated with reduced AdoCbl production and MUT deficiency (6, 7), or (iii) a combined HC and MMA defect (HC+MMA) (4). MMACHC and MMADHC are two proteins involved in the early Cbl processing steps that are shared by both mitochondrial and cytosolic targeting routes. MMACHC gene mutations are the most common cause of Cbl metabolic disorders, resulting in HC+MMA (8). Genetic defects of MMADHC, by contrast, uniquely cause any of the above three phenotypes (9, 10).
A possible explanation for the heterogeneous MMADHC phenotypes is the presence of distinct functional domains at the MMADHC protein level, responsible for trafficking to either mitochondrial MUT (of AdoCbl) or cytosolic MS (of MeCbl), or both. This hypothesis is supported by analysis of metabolic phenotypes from known disease mutations (11) and alanine-scanning mutagenesis (12). These studies dissect the 296-amino acid (aa) human MMADHC polypeptide functionally into the N-terminal 115 aa that is required for the mitochondrial route but dispensable for cytosolic trafficking, the C-terminal 180 aa that contribute to both routes, and two regions therein (aa 197–226 and 246–259) that have greater influence on the cytosolic than the mitochondrial route. Current knowledge points to a role of MMADHC in regulating Cbl delivery at the branch point between the mitochondrial and cytoplasmic target enzymes, albeit via an incompletely understood mechanism.
Furthermore, despite the MMADHC gene discovery seven years ago (10), few of the biochemical properties and functions of the MMADHC protein are known. Although previously postulated to contain Cbl and ATP binding motifs (10), MMADHC has been shown not to bind Cbl (13) or to hydrolyze ATP (14). Further biochemical exploration has been hampered in part by a lack of structural knowledge or sequence homologues. The inability of MMADHC to bind Cbl has implicated protein-protein interaction as the means of its Cbl-targeting role. This is supported by recent evidence from phage display (13, 15), bacterial two-hybrid studies (15), native PAGE (14), and surface plasmon resonance (13, 15) that MMADHC interacts directly with MMACHC in vitro (13, 15), and the interaction does not require the N-terminal 115 aa of MMADHC (14). Because MMACHC has been shown structurally (16, 17) and biochemically (18, 19) to bind various Cbl forms, and to process the upper Cbl ligand by reductive decyanation (19) or dealkylation (20) using the flavin cofactors FMN/FAD and GSH, respectively, it seems plausible that MMACHC-bound Cbl can partner with MMADHC to be ferried to the two destination enzymes.
To evaluate the importance and molecular basis of the MMACHC-MMADHC interaction, we have defined a minimal MMACHC interaction module of MMADHC, and demonstrated that experimental and patient missense mutations disrupt this interaction. We have further determined the structure of this interaction module from mouse MMADHC to 2.2 Å resolution, and established the 1:1 stoichiometry of the MMACHC-MMADHC heterodimer using small angle x-ray scattering (SAXS). Finally, based on the combined findings, we propose the first structural model of the MMACHC-MMADHC interaction.
Experimental Procedures
Recombinant Production of MMACHC and MMADHC
DNA fragments encoding human (Homo sapiens, IMAGE clone: 3826071) and mouse (Mus musculus, IMAGE clone: 3493526) MMADHC, harboring different N- and C-terminal boundaries, were amplified and subcloned into pNIC28-Bsa4 vector (GenBankTM accession number EF198106) in-frame with a tobacco etch virus protease cleavable N-terminal His6 tag. Constructs of human HsMMACHC in pNIC28-Bsa4 vector were prepared previously (16, 21). Site-directed mutations were constructed using the QuikChange mutagenesis kit (Stratagene) and confirmed by sequencing. Cloning and site-directed mutagenesis primers are available upon request. Proteins were expressed in Escherichia coli BL21(DE3)R3 and purified by affinity (Ni-Sepharose; GE Healthcare) and size-exclusion (Superdex 200; GE Healthcare) chromatography. For crystallization, MMADHC proteins were further purified by ion exchange chromatography (Resource Q; GE Healthcare). Selenomethionine (SeMet)-derivatized proteins were expressed using SelenoMethionine Medium Complete (Molecular Dimensions) and purified as above.
Crystallization and Structural Determination
Purified SeMet-derivatized and native MmMMADHCΔ128 were concentrated to 15–20 mg/ml, and crystals were grown by sitting drop vapor diffusion at 4 °C. The mother liquor conditions are summarized in Table 1. Crystals were cryo-protected in mother liquor containing ethylene glycol (25% v/v) and flash-cooled in liquid nitrogen. X-ray diffraction data were collected at the Diamond Light Source beamline I04 and processed using XIA2. Selenium atoms were located using SHELXC/D (22) for initial phase calculation in SHELXE, and subsequently for automated building with BUCCANEER (23). The structure was solved by selenium single-wavelength anomalous diffraction phasing and refined using PHENIX (24), followed by manual rebuilding in COOT (25). The SeMet MMADHC model was used to solve the native structure by molecular replacement using PHASER (26). The final refined model consists of aa 132–157, 169–236, and 246–296 of MmMMADHC. Atomic coordinates and structure factors for MmMMADHCΔ128 have been deposited in the Protein Data Bank with the accession code 5A4R. Data collection and refinement statistics are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics for MmMMADHC structure
| MmMMADHCΔ128 SeMeta | MmMMADHCΔ128 nativeb | |
|---|---|---|
| Data collection and processingc | ||
| Beamline | Diamond I04 | Diamond I04 |
| Wavelength (Å) | 0.9782 | 0.9782 |
| Unit cell parameters a, b, c (Å) | 73.5 88.7 65.7 | 74.2 89.5 64.7 |
| Unit cell parameters α, β, γ (°) | 90.00 90.00 90.00 | 90.00 90.00 90.00 |
| Space group | C 2 2 21 | C 2 2 21 |
| Resolution range (Å) | 44.33–2.61 (2.68–2.61) | 44.76–2.25 (2.31–2.25) |
| Observed/Unique reflections | 78,726/6293 (6117/471) | 51,530/10,530 (3778/762) |
| Rsym (%) | 12.2 (54.8) | 2.5 (59.8) |
| CC1/2 | 0.99 (0.96) | 1.0 (0.80) |
| I/σ(I) | 15.1 (4.7) | 31.6 (2.6) |
| Completeness (%) | Anomalous 92.1 (98.8) | 99.8 (99.6) |
| Multiplicity | Anomalous 6.7 (6.8) | 4.9 (5.0) |
| Refinement | ||
| Rcryst (%) | 22.9 | |
| Rfree (%) | 25.5 | |
| Wilson B factor (Å2) | 55.5 | |
| Average total B factor (Å2) | 66.2 | |
| r.m.s.d. bond length (Å2) | 0.0034 | |
| r.m.s.d. bond angle (°) | 0.87 | |
| Missing residues | 129–131, 160–168, 237–245 | |
| MolProbity analysis | ||
| Clashscore | 5.98 (98th percentile) | |
| Ramachandran favored (%) | 96.48% | |
| Ramachandran disallowed (%) | 0.00% | |
| Rotamer outliers (%) | 0.00% | |
| PDB code | 5A4R | |
a Crystallization condition: 28% PEG3350, 100 mm Bis-Tris, pH 6.5, 250 mm NaCl.
b Crystallization condition: 22% polyacrylic acid 5100, 100 mm HEPES, pH 7.5, 20 mm MgCl2.
c Data from highest resolution shell are shown in parentheses.
Small Angle X-ray Scattering
For SAXS, MMACHC and MMADHC were cleaved by tobacco etch virus protease to remove the N-terminal His6 tag, followed by reverse affinity purification. Purified proteins were then incubated separately or together with the appropriate ligands and further purified by size-exclusion chromatography. Concentrations of proteins used were 24 mg/ml (MMADHC), 18 mg/ml (MMACHC), and 17 mg/ml (complex). Scattering data were collected at the Diamond Light Source B21 beamline. Data collection was performed either in-line with size-exclusion chromatography (KW404 column, Shodex) or in batch mode where the peak fractions were collected and immediately subjected to SAXS by flowing sample through an in-vacuum quartz capillary of 1.6-mm diameter. Data were collected using a Pilatus 2M detector (DECTRIS, Baden, Switzerland) at a sample-detector distance of 3914 mm and a wavelength of λ = 1 Å. The range of momentum transfer of 0.1 < s < 5 nm−1 was covered (s = 4πsinθ/λ, where θ is the scattering angle). For the in-line mode, 1-s exposures were collected, whereas for batch samples, a comparison of eighteen 10-s exposures was performed. Radiation damage was checked for the batch mode by monitoring changes in radius of gyration in each frame, where no significant changes were observed. The data were radially averaged, and the scattering of the buffer was subtracted. The forward scattering I(0), radius of gyration Rg, pair distribution of the particle P(r), and maximum dimension Dmax were analyzed using Scatter (27) and the ATSAS suite of programs (28).
Solution Characterization of MMACHC and MMADHC
Blue native-PAGE was performed with 25 μm protein (MMACHC and/or MMADHC) alone or in the presence of 50 μm cobalamin (MeCbl, AdoCbl, CNCbl) and/or 8 mm ligand (GSH, FMN, FAD), which was loaded onto the native-PAGE gel system, after preincubation in the dark at room temperature for 1 h, and then run following the manufacturer's instructions (Life Technologies). All blue native-PAGE experiments were performed at least twice independently. Analytical gel filtration (16), FMN/FAD binding by intrinsic fluorescence quenching (17), and differential scanning fluorimetry (21, 29), were performed as described previously. For native mass spectrometry, 75 μl of sample containing ∼4 mg/ml MMADHC and MMACHC incubated in the presence of 0.5 mm MeCbl and 2 mm reduced GSH underwent four desalting steps using Micro Bio-Spin columns (Bio-Rad) that were pre-equilibrated with 50 mm ammonium acetate buffer (pH 6.5). Following desalting, samples were loaded into a 1.0-ml gas-tight positive displacement syringe (Hamilton) that was inserted into the syringe pump. Samples were then directly infused with a constant flow rate of 6 μl/min through a PEEK capillary tubing (inner diameter, 0.005 inches) into a Q-TOF 6530 mass spectrometer attached to a standard electrospray ionization source (Agilent Technologies). The mass spectrometer was operated in positive ion mode, using the 1-GHz detector mode with a scan range of 100–20,000 m/z and a fragmentor voltage of 430 V. Following data acquisition, results were evaluated by using the MassHunter Qualitative Analysis software (Agilent Technologies).
Results
MMADHC aa 154–296 Is an MMACHC Interaction Module
To better understand the required regions, ligand dependence, and stoichiometry of their in vitro interactions, we generated a series of human (Hs) MMADHC and MMACHC truncation proteins by recombinant expression (Fig. 1A). Using blue native-PAGE (BN-PAGE) (Fig. 1B), full-length MMACHC (HsMMACHCFL) alone migrated as two bands (lane 1), consistent with its known monomeric and dimeric forms, whereas the dimeric band became more prominent in the presence of MeCbl and GSH (lane 2, white dot). The HsMMADHC proteins with various N-terminal truncations migrated as single bands (lanes 3–8) corresponding to their respective monomers, corroborating previous investigations (13). Complex formation, as judged by the appearance of an additional band in BN-PAGE as compared with single-protein controls, was observed when HsMMACHCFL preincubated with MeCbl and GSH was added to HsMMADHCΔ61 or HsMMADHCΔ123, or to a lesser extent, HsMMADHCΔ153 (lanes 9–11, white asterisk). However,no complex band was observed with N-terminal truncation of HsMMADHC beyond aa 154 (HsMMADHCΔ157, HsMMADHCΔ167, or HsMMADHCΔ172; lanes 12–14) under the same conditions. The HsMMADHC-HsMMACHC complex bands migrated at a position intermediate of the estimated monomeric and dimeric HsMMACHC bands, suggesting a possible 1:1 heterodimeric complex. HsMMACHCΔC, which lacks the C-terminal Pro-rich region (aa 236–282; Fig. 1A), was sufficient for complex formation with HsMMADHCΔ123 (Fig. 1C, lane 2). Size-exclusion chromatography using MmMMADHCΔ128 and HsMMACHCΔC proteins further confirmed the regions required for interaction and the 1:1 stoichiometry of the complex (Fig. 1D). Therefore, the MMADHC region C-terminal to aa 154 and the MMACHC region without the Pro-rich C terminus is sufficient for direct protein-protein interaction. Notably, the MMADHC C-terminal region is the most evolutionarily conserved section of the polypeptide from human to Caenorhabditis elegans (Fig. 1E), suggesting selected retention of this interaction module in evolution.
FIGURE 1.

Interaction study of MMACHC and MMADHC. A, domain diagram showing recombinant human MMACHC and MMADHC constructs described in this figure. For reference, mouse construct MmMMADHCΔ128 used to generate the crystal structure is also shown. B, interaction of human MMACHC full-length and MMADHC truncated proteins by BN-PAGE. Lanes 1–8: single protein controls including MMACHCFL alone (lane 1) and with MeCbl and GSH (lane 2); and MMADHC truncations including MMADHCΔ61 (lane 3), MMADHCΔ123 (lane 4), MMADHCΔ153 (lane 5), MMADHCΔ157 (lane 6), MMADHCΔ167 (lane 7), and MMADHCΔ172 (lane 8). Lanes 9–14: combined protein incubation of MMACHCFL in presence of MeCbl and GSH with MMADHCΔ61 (lane 9), MMADHCΔ123 (lane 10), MMADHCΔ153 (lane 11), MMADHCΔ157 (lane 12), MMADHCΔ167 (lane 13), and MMADHCΔ172 (lane 14). C, interaction of MMACHC mutants and MMADHCΔ123 by BN-PAGE, in the presence of MeCbl and GSH. Lanes 1–6: MMACHCΔC alone (lane 1) and with MMADHCΔ123 (lane 2); MMACHCR161Q alone (lane 3) and with MMADHCΔ123 (lane 4); and MMACHCΔPNR alone (lane 5) and with MMADHCΔ123 (lane 6). For B and C, white dots indicate MMACHC homodimer, and white asterisks indicate MMACHC-MMADHC heterodimer. D, analysis of complex formation by size-exclusion chromatography including chromatographs of MmMMADHCΔ123 (gray line), HsMMACHCΔC + MeCbl + GSH (black line), and MmMMADHCΔ123 + HsMMACHCΔC + MeCbl + GSH (black dotted line). x axis: elution volume; left y axis: absorbance (milliabsorbance units (mAU)) for HsMMACHCΔC + MeCbl + GSH; right y axis: absorbance (mAU) for MmMMADHCΔ123 and MmMMADHCΔ123 + HsMMACHCΔC + MeCbl + GSH. Molecular weights of each peak are calculated from a calibration curve using molecular weight protein standards (Sigma-Aldrich). E, sequence alignment of MMADHC from H. sapiens (HS, NP_056517.1, human) and predicted MMADHC protein (Y76A2B.5) from C. elegans (Ce) using the BLAST server (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The boxed region represents the smallest interacting construct of HsMMADHC for HsMMACHC. F, BN-PAGE of HsMMACHCFL and HsMMADHCΔ123 under various conditions. White asterisks indicate MMACHC-MMADHC heterodimer. G, native mass spectrometry of HsMMACHCFL and HsMMADHCΔ123 incubated with MeCbl and GSH. Expected sizes: HsMMACHCFL, 31818.6 Da; HsMMADHCΔ123, 19727.7 Da; MeCbl, 1343.6 Da; and GSH, 307.3 Da.
In agreement with previous work (14), complex formation is favored by preincubation of proteins with GSH and MeCbl, conditions known to facilitate upper Cbl ligand (R-group) removal. We observed either significantly weaker or no interaction between HsMMACHCFL and HsMMADHCΔ123 when GSH was replaced by other reducing factors (FMN, FAD, DTT) or when MeCbl was replaced by other Cbl forms (AdoCbl, CNCbl) (Fig. 1F), conditions known to result in much slower (>10-fold) or no R-group removal (30, 31). We further investigated two HsMMACHC mutants with reduced MeCbl dealkylation activity (HsMMACHCΔPNR, missing aa 109–111 (16), or HsMMACHCR161Q (16, 32)), on their ability to complex with HsMMADHCΔ123. Upon incubation with MeCbl and GSH, the HsMMACHCR161Q mutant was less able than HsMMACHCFL to complex with HsMMADHCΔ123 (compare Fig. 1C, lane 4 with Fig. 1B, lane 10), whereas HsMMACHCΔPNR did not complex with HsMMADHCΔ123 at all (Fig. 1C, lane 6).
The MMACHC-MMADHC interaction is further studied by native mass spectrometry, where the complex of HsMMACHCFL and HsMMADHCΔ123, incubated in the presence of MeCbl and GSH, generated two peaks consistent with a 1:1 stoichiometry, one missing the 15 atomic mass units of the methyl group (Fig. 1G, 52,875 atomic mass units) and the other missing 16 atomic mass units, corresponding to the methyl group plus the hydrogen from GSH (Fig. 1G, 53,181 atomic mass units). Thus, R-group removal appears to be a prerequisite for MMACHC-MMADHC interaction.
MMADHC Contains a Nitroreductase-like Fold with Homology to MMACHC
We next characterized MMADHC by x-ray crystallography to elucidate the molecular properties of its MMACHC interaction module. To maximize the probability of crystallization, we adopted a cross-species approach that involved the study of recombinant human, M. musculus, and Xenopus laevis MMADHC proteins, coupled with an extensive survey of construct boundaries. From >40 purified proteins, we successfully crystallized M. musculus MMADHC aa 129–296 (MmMMADHCΔ128; Figs. 1A and 2A), a region bearing 93% identity with the human sequence. In our BN-PAGE assay, MmMMADHC(e.g. MmMMADHCΔ128) can substitute the equivalent human construct in stable complex formation with HsMMACHC (not shown). Despite repeated attempts, the equivalent HsMMADHC construct did not crystallize. The structure of MmMMADHCΔ128 (Fig. 2, A and B), determined to 2.6 Å resolution by single-wavelength anomalous dispersion phasing for the selenomethionine (SeMet) derivative, and to 2.2 Å by molecular replacement for the native protein (Table 1), is a mixed α/β domain composed of a central four-stranded antiparallel β-sheet (β1, β2, β6, β3) flanked by a 14-turn helix (αB) and a short β-turn (β4-β5) (Fig. 2C).
FIGURE 2.

Structure of the MMACHC interaction module of MMADHC. A, graphic representation of the MmMMADHCΔ128 structure in orthogonal views. Secondary structures are colored green for β-sheets and red for α-helices. The first (aa 132) and last (aa 296) residues observed in the structure are labeled with N and C, respectively. Dotted lines indicate disordered regions. Inset: view of the σ-weighted (2Fo − Fc) electron density map of MmMMADHCΔ128 aa region 190–217, contoured at σ = 1. B, surface representation of MmMMADHCΔ128 (same orientation as A, right panel) highlighting the three protrusions (Pr1–Pr3) in blue. C, topology diagram of the MmMMADHCΔ128 secondary structure with the same coloring and labeling as in A and B. Disordered regions are shown as dashed lines.
A structural comparison of MmMMADHCΔ128 with other proteins using DALI (33) revealed that the core structure is derived from the nitroreductase (NTR) fold (Fig. 3A), primarily found in flavoenzymes and oxidoreductases that utilize FMN/FAD cofactors and form homodimers by β-strand exchange, as well as MMACHC (Fig. 3B). Nevertheless, MmMMADHCΔ128 has extensively modified the NTR fold with three loop protrusions (Pr1–Pr3), rendering it functionally different from classical NTRs. Firstly, MmMMADHCΔ128 does not bind FMN/FAD in solution (data not shown) because the canonical binding site found in NTRs is disrupted by part of the MMADHC Pr2 loop protrusion (Fig. 4A). Secondly, although in crystallo MmMMADHCΔ128 reveals a symmetry-related dimer, its dimeric interface does not match that of the conventional NTRs (Fig. 4B). Indeed, MmMMADHCΔ128 is a monomer in solution by BN-PAGE, size-exclusion chromatography (not shown), and SAXS (see “A Structural Model of the MMACHC-MMADHC Complex”), consistent with HsMMADHC constructs migrating as monomers in BN-PAGE (Fig. 1B).
FIGURE 3.
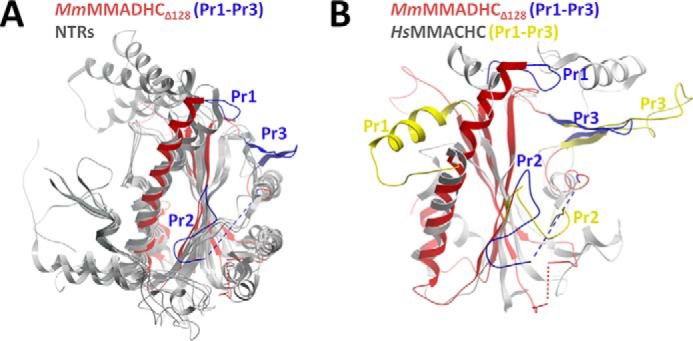
Structural comparison of MMADHC and proteins of the nitroreductase family. A, structural superposition of MmMMADHCΔ128 (red) and two other NTRs (Protein Data Bank (PDB): 2HAY, 2ISL; gray). B, superposition of MmMMADHCΔ128 (red) and MMACHC (PDB: 3SOM; gray), highlighting the absence of the HsMMACHC four-helix cap domain (aa 185–234) in MmMMADHCΔ128 as well as the different orientations of protrusions Pr1 and Pr2 (HsMMACHC: Pr1 aa 69–77; Pr2 aa 104–116; MmMMADHCΔ128: Pr1 aa 183–190, Pr2 aa 227–246) between the proteins. Protrusions (Pr1–Pr3) are colored blue for MmMMADHCΔ128 and yellow for HsMMACHC.
FIGURE 4.

Comparison of MMADHC to other NTR dimers. A, overlay of the MmMMADHCΔ128 structure (red) with nitroreductases from Desulfovibrio desulfuricans (PDB: 3E39), Streptococcus pyogenes (PDB: 2HAY), and Enterobacter cloacae (PDB: 1KQD). The canonical FMN/FAD binding site of NTRs (left, indicated by the green stick ligand), located at the conventional dimer interface, is not present in MMADHC (right) due to disruption by its protrusion loop Pr2. B, structural alignment of a single subunit (protomer A) of MmMMADHCΔ128 (MMADHC), HsMMACHCFL (MMACHC, PDB: 3SOM), and an NTR (PDB: 1KQD) in gray, with the second subunit (protomer B) of their crystallographic dimers color-coded (MMADHC in blue; NTR in yellow; MMACHC in red), demonstrating the lack of a conserved dimeric orientation and interface among the three structures.
To our surprise, the closest structural homologue of MMADHC is its interaction partner MMACHC (DALI (33) Z-score, 5.2), despite a lack of obvious sequence conservation. Like MMADHC, MMACHC contains three loop protrusions, at equivalent spatial positions, in the core NTR fold (16, 17) (Fig. 3B). MMACHC also lacks the classical FMN binding site present among NTRs, while retaining the ability to bind flavins in solution (17), likely involving a non-canonical binding site (16). The MMADHC and MMACHC structures superimpose only moderately and within the core NTR secondary structure elements (Cα-r.m.s.d. 3.0 Å, 96 aligned aa), beyond which they differ substantially (Fig. 3B). For example, MmMMADHCΔ128 does not contain the four-helix cap domain and differs in length and conformation of two protrusions as compared with MMACHC, all of which form part of the MMACHC Cbl binding pocket (16, 17). These extensive structural differences from MMACHC likely account for the reported lack of Cbl binding capability of MMADHC (13). We additionally found no sequence elements or homology consistent with ATPase activity.
Patient and Experimental MMADHC Mutations Knock Out Interaction with MMACHC
Our previous mutagenesis studies in patient fibroblasts (11, 12) revealed specific MMADHC regions responsible for the intracellular targeting of the two cobalamin cofactors. These mutations correlate with the MMA, HC, or HC+MMA phenotypes based on their amino acid location (Fig. 5A). Missense mutations causing decreased MeCbl production (HC phenotype) or both AdoCbl and MeCbl production (HC+MMA phenotype) in cells are found within three sequence stretches at the MMADHC C-terminal half (Fig. 5A, stretches 1–3). When mapped onto our MmMMADHCΔ128 structure, these three stretches cluster in spatial proximity and are surface-accessible (Fig. 5B). We investigated whether these mutations impact on the MMACHC-MMADHC interaction by introducing onto the HsMMADHCΔ123 construct a selection of HC and HC+MMA causing mutations from the three stretches (Stretch 1: p.T182N, p.M186A, p.W189A; Stretch 2: p.D226A; Stretch 3: p.L259P). The recombinant mutant proteins behaved similarly to wild type in terms of expression level, protein solubility (not shown), and thermal stability (Fig. 5C), where only p.D226A led to slightly unstable protein. We found interaction of HsMMACHCFL (incubated with MeCbl and GSH) with HsMMADHCΔ123-T182N and HsMMADHCΔ123-W189A to be severely decreased (Fig. 5D, lanes 3 and 5), but retainedfor HsMMADHCΔ123-M186A, HsMMADHCΔ123-D226A, and HsMMADHCΔ123-L259P (Fig. 5D, lanes 4, 6, and 7). These findings suggest that residues within Stretch 1 (Thr-182, Trp-189) occur at the binding interface with MMACHC.
FIGURE 5.

Structural and biochemical analysis of MMADHC missense mutations. A, domain diagram of MMADHC, with missense mutations color-coded according to their cellular defects (10–12), namely MMA (green), combined HC+MMA (stretch 1, red), or HC (stretches 2 and 3, blue) phenotypes. Purple line indicates aa region observed in the crystal structure. B, graphic (left) and surface (right) representations of MmMMADHCΔ128 mapped with stretches 1–3 and individual mutations found within these regions. Dotted lines indicate disordered regions. C, analysis of thermal unfolding curves of various protein constructs reveals a melting temperature (Tm) of 62 °C (right) for HsMMADHCΔ123, which is relatively unchanged for all mutants with the exception of HsMMADHCΔ123-D226A where the Tm is slightly decreased as compared with wild type. Error bars indicate means ± S.E. D, interaction of HsMMACHCFL with wild-type or mutant HsMMADHCΔ123 studied by BN-PAGE. Depicted are: HsMMACHCFL with MeCbl and GSH alone (lane 1), and in combination with HsMMADHCΔ123 (lane 2), HsMMADHCΔ123-T182N (lane 3), HsMMADHCΔ123-M186A (lane 4), HsMMADHCΔ123-W189A (lane 5), HsMMADHCΔ123-D226A (lane 6), and HsMMADHCΔ123-L259P (lane 7). White asterisks indicate the MMACHC-MMADHC heterodimer.
A Structural Model of the MMACHC-MMADHC Complex
We next utilized SAXS, a low-resolution structural characterization method in solution (34), to construct a model of the MMACHC-MMADHC interaction. To ensure that the SAXS data most closely resembled the ordered aa regions revealed from the MMACHC and MMADHC structures, we reconstituted a complex from untagged MmMMADHCΔ128 and untagged HsMMACHCΔC proteins in the presence of MeCbl and GSH. The Guinier approximation and pair distribution function P(r) calculated from the scattering data (Fig. 6, A and B) suggest a radius of gyration (Rg) of 24.7 Å and maximal intraparticle dimension (Dmax) of 80 Å for the complex, both parameters larger than those from either protein component alone (Fig. 6C). Further, the elution time of the protein complex from the in-line HPLC column was earlier than either protein alone, giving rise to a calculated molecular weight consistent with a 1:1 stoichiometry (Fig. 6C). The ab initio envelope of the MmMMADHCΔ128-alone sample fits well to one monomer (Fig. 6D). MMACHC is known to exist in a monomer:dimer equilibrium (Fig. 1) (16), and the monomeric species selected for SAXS analysis is consistent with one protomer in the ab initio envelope (Fig. 6E). However, for the MMACHC-MMADHC complex, the ab initio envelope (Fig. 6F) gives an elongated dimension of 80, 40, and 40 Å. One protomer from each of HsMMACHCΔC and MmMMADHCΔ128 would account for all the mass of the envelope, further indicating a 1:1 heterodimeric stoichiometry that agrees with our BN-PAGE, size-exclusion chromatography, and native mass spectrometry data (Fig. 1). Although the relative orientation of both proteins cannot be ascertained by SAXS, we applied our mutagenesis and interaction data (Fig. 5) to direct protein docking of the complex using the ZDOCK server (35), given that residue Thr-182 of MmMMADHC should be involved in the interaction. With this constraint, the top seven models generated from ZDOCK (Fig. 6F) all yield a good fit (χ2 ∼0.03–0.04) to the measured scattering. These models are consistent with an MMACHC-MMADHC interface that involves the Cbl binding region of MMACHC and aa 182–189 (Fig. 5, stretch 1) of MMADHC (Fig. 6G). A similar docking run was performed with HADDOCK (36), yielding consistent models (data not shown).
FIGURE 6.

SAXS analysis of the MMACHC-MMADHC interaction. A, raw scattering curves for MmMMADHCΔ128 (red), HsMMACHCΔC (blue), and their complex (black). B, P(r) plots, calculated with Scatter (27) and normalized to peak height, show differences between the single proteins and the complex. Guinier plots indicate aggregation-free data. C, table of radius of gyration (Rg), maximal intraparticle dimension (Dmax) as calculated from the Guinier plot, apparent molecular weight (MW), and elution time from the in-line HPLC column. D and E, ab initio bead models calculated with 13 runs of DAMMIF (39) for MmMMADHCΔ128 fitted with its x-ray structure (this study) (D) and HsMMACHCΔC fitted with its x-ray structure (PDB: 3SOM) (E). F, top seven models of the HsMMACHCΔC-MmMMADHCΔ128 complex produced by ZDOCK with fitted χ2 values shown. The SAXS envelope is overlaid with modeled orientations of HsMMACHCΔC (blue) and MmMMADHCΔ128 (red) structures. MmMMADHCΔ128 is presented in the same orientation on all models for better comparison. G, HsMMACHCΔC-MmMMADHCΔ128 complex fitted with a representative example of the ZDOCK (35) complex model. In E and G, Cbl is shown as purple sticks.
Discussion
The role of MMACHC within the early stages of the Cbl processing pathway has previously been clarified from the available structural and biochemical evidence of its Cbl binding and processing activities (16–20). By contrast, the role of MMADHC, tasked with directing Cbl to the destination enzymes yet unable to bind Cbl itself, is much less clear. This study presents the first structural characterization of MMADHC to provide novel insights into its unknown function, with the key findings that: (i) MMADHC functions by binding Cbl-laden MMACHC, but only post-Cbl processing, i.e. after the upper axial ligand of Cbl has been removed; (ii) the MMACHC interaction module of MMADHC contains an ingeniously modified NTR fold that abolishes homodimerization, favors heterodimerization with another modified NTR fold from MMACHC, and accounts for the reported lack of Cbl binding (13); and (iii) missense mutations of MMADHC that reduced both AdoCbl and MeCbl production in cells can be explained at least in part by an abrogated interaction with MMACHC.
Our data illustrate the application of protein-protein interaction in the early Cbl-targeting steps of the pathway for both the mitochondrial (AdoCbl to MUT) and the cytosolic (MeCbl to MS) targeting routes. Such protein-protein interaction is essential to the targeting process, and is disrupted by a known disease mutation of MMACHC that precludes B12 processing and by mutations of MMADHC that alter the binding interface. We propose a mechanistic model for the MMACHC-MMADHC complex whereby the adaptation of the NTR fold by both proteins favors heterodimerization to form the “Cbl trafficking chaperone.” In this model, MMACHC in the Cbl-free form could exist in a monomer-homodimer equilibrium (Fig. 7A). The self-association of MMACHC into homodimer, likely with a high dissociation rate that precludes its isolation in vitro (13, 16, 17), is enriched in the presence of Cbl ligand (Fig. 7B) and may serve to close the enzymatic active site for proper Cbl processing (16) (Fig. 7C). Cbl-laden MMACHC (monomer/homodimer), with the upper axial ligand now removed, preferentially binds monomeric MMADHC as a 1:1 heterodimer rather than binding to itself, resulting in the “trafficking chaperone” that delivers processed Cbl to its target destinations (Fig. 7D).
FIGURE 7.

Proposed role of the MMACHC-MMADHC complex in Cbl targeting. A, MMACHC (blue sector) in the unbound state exists primarily as a monomer. B, upon binding unprocessed Cbl with an intact upper axial ligand (triangle), it can shift toward the homodimeric state. C, following processing of Cbl (green to red triangle) via GSH-mediated removal of its upper axial ligand (green stick and square), MMACHC is available for interaction with MMADHC (orange sector). D, MMADHC binds MMACHC in a 1:1 heterodimer and escorts Cbl-laden MMACHC either toward the mitochondria (star) for use by methylmalonyl-CoA MUT or to MS in the cytosol.
Interestingly, only the C-terminal 154 aa of MMADHC is required for the interaction with MMACHC. As such, the exact function of the MMADHC N terminus remains to be determined. Predicted to be largely disordered, the N-terminal ∼115 aa harbors a mitochondrial targeting sequence (aa 1–12) and has been shown to be required for cellular AdoCbl production only (11). Thus, this protein region is likely only required for function within, or direction to, the mitochondria.
Because MMADHC mutations that abolished direct interaction with MMACHC result in the combined HC+MMA phenotype (11, 12), it is conceivable that disruption of the MMACHC-MMADHC complex will result in an inability to deliver Cbl to both destination enzymes MS and MUT. A possible role of MMACHC in the mitochondria, where it has not been detected either on its own or in complex (37), remains undetermined. The dysfunction caused by MMADHC missense mutations, which result in a loss of MS activity only, i.e. the so-called cblD-HC mutations (10), also remains unexplained. Because MMACHC has recently been shown to bind various isoforms of MS (38), the cblD-HC mutations could affect a larger, yet unidentified multi-protein complex, likely including at least MMACHC and MS, but possibly also methionine synthase reductase.
In conclusion, we have structurally characterized the MMACHC-interacting module of MMADHC and demonstrated that specific MMADHC mutations that cause combined loss of AdoCbl and MeCbl production in cells can interfere with MMACHC interaction. Further studies will clarify the affinity of these proteins for each other, and how this protein-protein interaction results in Cbl delivery in the cellular context.
Author Contributions
B. F., D. S. F., M. R. B., and W. W. Y. conceived of the study. D. S. F., T. P., and V. F. cloned the MMADHC constructs. D. S. F., J. K., and F. F. purified, crystallized and determined structure of MMADHC. F. F. and J. K. reconstituted the complex and carried out SAXS analysis. M. S. and T. J. M. performed BN-PAGE. M. S. and R. C. performed and analyzed native mass spectrometry. D. S. F. and W. W. Y. wrote the manuscript with editing and proofreading support from all other co-authors.
Acknowledgments
We thank the Diamond Light Source for access to the beamlines I04 and B21 (MX10619), and James Doutch for help with SAXS data collection and processing.
Addendum
During the revision of this manuscript, Yamada et al. (J. Biol. Chem., 10.1074/jbc.M115.682435 (40)) reported the human MMADHC structure of a region corresponding to our mouse structure, and consistent with our structural observations described here.
This work was supported by the Swiss National Science Foundation (Grants SNSF 31003A_156907 and SNSF 31003A_138521) (to D. S. F., M. R. B., B. F. and T. P.). This work was also supported by the Rare Disease Initiative Zurich, Clinical Research Priority Program, University of Zurich (to D. S. F. and M. R. B.). The Structural Genomics Consortium is a registered charity (Number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, Glaxo-SmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust (092809/Z/10/Z). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5A4R) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Cbl
- cobalamin
- MeCbl
- methyl-Cbl
- AdoCbl
- adenosyl-Cbl
- CNCbl
- cyanocobalamin
- MS
- methionine synthase
- MUT
- methylmalonyl-CoA mutase
- HC
- homocystinuria
- MMA
- methylmalonic aciduria
- aa
- amino acid(s)
- SAXS
- small-angle x-ray scattering
- NTR
- nitroreductase
- SeMet
- selenomethionine
- BN-PAGE
- blue native-PAGE
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- r.m.s.d
- root mean square deviation
- FL
- full-length
- Hs
- Homo sapiens
- Mm
- Mus musculus.
References
- 1.Watkins D., and Rosenblatt D. S. (2014) Inherited disorders of folate and cobalamin transport and metabolism. in The Online Metabolic and Molecular Bases of Inherited Disease (Valle D., Beaudet A. L., Vogelstein B., Kinzler K. W., Antonarakis S. E., Ballabio A., Gibson M., and Mitchell G., eds), McGraw-Hill, New York [Google Scholar]
- 2.Froese D. S., and Gravel R. A. (2010) Genetic disorders of vitamin B metabolism: eight complementation groups–eight genes. Expert Rev. Mol. Med. 12, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gherasim C., Lofgren M., and Banerjee R. (2013) Navigating the B12 road: assimilation, delivery, and disorders of cobalamin. J. Biol. Chem. 288, 13186–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumgartner M. R. (2013) Vitamin-responsive disorders: cobalamin, folate, biotin, vitamins B1 and E. Handb. Clin. Neurol. 113, 1799–1810 [DOI] [PubMed] [Google Scholar]
- 5.Harding C. O., Arnold G., Barness L. A., Wolff J. A., and Rosenblatt D. S. (1997) Functional methionine synthase deficiency due to cblG disorder: a report of two patients and a review. Am. J. Med. Genet. 71, 384–390 [DOI] [PubMed] [Google Scholar]
- 6.Hörster F., Baumgartner M. R., Viardot C., Suormala T., Burgard P., Fowler B., Hoffmann G. F., Garbade S. F., Kölker S., and Baumgartner E. R. (2007) Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut−, cblA, cblB). Pediatr. Res. 62, 225–230 [DOI] [PubMed] [Google Scholar]
- 7.Fowler B., Leonard J. V., and Baumgartner M. R. (2008) Causes of and diagnostic approach to methylmalonic acidurias. J. Inherit. Metab. Dis. 31, 350–360 [DOI] [PubMed] [Google Scholar]
- 8.Lerner-Ellis J. P., Anastasio N., Liu J., Coelho D., Suormala T., Stucki M., Loewy A. D., Gurd S., Grundberg E., Morel C. F., Watkins D., Baumgartner M. R., Pastinen T., Rosenblatt D. S., and Fowler B. (2009) Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum. Mutat. 30, 1072–1081 [DOI] [PubMed] [Google Scholar]
- 9.Suormala T., Baumgartner M. R., Coelho D., Zavadakova P., Kozich V., Koch H. G., Berghaüser M., Wraith J. E., Burlina A., Sewell A., Herwig J., and Fowler B. (2004) The cblD defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J. Biol. Chem. 279, 42742–42749 [DOI] [PubMed] [Google Scholar]
- 10.Coelho D., Suormala T., Stucki M., Lerner-Ellis J. P., Rosenblatt D. S., Newbold R. F., Baumgartner M. R., and Fowler B. (2008) Gene identification for the cblD defect of vitamin B12 metabolism. N. Engl. J. Med. 358, 1454–1464 [DOI] [PubMed] [Google Scholar]
- 11.Stucki M., Coelho D., Suormala T., Burda P., Fowler B., and Baumgartner M. R. (2012) Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Hum. Mol. Genet. 21, 1410–1418 [DOI] [PubMed] [Google Scholar]
- 12.Jusufi J., Suormala T., Burda P., Fowler B., Froese D. S., and Baumgartner M. R. (2014) Characterization of functional domains of the cblD (MMADHC) gene product. J. Inherit. Metab. Dis. 37, 841–849 [DOI] [PubMed] [Google Scholar]
- 13.Deme J. C., Miousse I. R., Plesa M., Kim J. C., Hancock M. A., Mah W., Rosenblatt D. S., and Coulton J. W. (2012) Structural features of recombinant MMADHC isoforms and their interactions with MMACHC, proteins of mammalian vitamin B12 metabolism. Mol. Genet. Metab. 107, 352–362 [DOI] [PubMed] [Google Scholar]
- 14.Gherasim C., Hannibal L., Rajagopalan D., Jacobsen D. W., and Banerjee R. (2013) The C-terminal domain of CblD interacts with CblC and influences intracellular cobalamin partitioning. Biochimie 95, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plesa M., Kim J., Paquette S. G., Gagnon H., Ng-Thow-Hing C., Gibbs B. F., Hancock M. A., Rosenblatt D. S., and Coulton J. W. (2011) Interaction between MMACHC and MMADHC, two human proteins participating in intracellular vitamin B12 metabolism. Mol. Genet. Metab. 102, 139–148 [DOI] [PubMed] [Google Scholar]
- 16.Froese D. S., Krojer T., Wu X., Shrestha R., Kiyani W., von Delft F., Gravel R. A., Oppermann U., and Yue W. W. (2012) Structure of MMACHC reveals an arginine-rich pocket and a domain-swapped dimer for its B12 processing function. Biochemistry 51, 5083–5090 [DOI] [PubMed] [Google Scholar]
- 17.Koutmos M., Gherasim C., Smith J. L., and Banerjee R. (2011) Structural basis of multifunctionality in a vitamin B12-processing enzyme. J. Biol. Chem. 286, 29780–29787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Froese D. S., Zhang J., Healy S., and Gravel R. A. (2009) Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol. Genet. Metab. 98, 338–343 [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Gherasim C., and Banerjee R. (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. U.S.A. 105, 14551–14554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannibal L., Kim J., Brasch N. E., Wang S., Rosenblatt D. S., Banerjee R., and Jacobsen D. W. (2009) Processing of alkylcobalamins in mammalian cells: A role for the MMACHC (cblC) gene product. Mol. Genet. Metab. 97, 260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Froese D. S., Healy S., McDonald M., Kochan G., Oppermann U., Niesen F. H., and Gravel R. A. (2010) Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol. Genet. Metab. 100, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 24.Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., and Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foerster S., Apostol L., and Bras W. (2010) Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Crystallogr. 43, 639–646 [Google Scholar]
- 28.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 30.Kim J., Hannibal L., Gherasim C., Jacobsen D. W., and Banerjee R. (2009) A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J. Biol. Chem. 284, 33418–33424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z., Gherasim C., Lesniak N. A., and Banerjee R. (2014) Glutathione-dependent one-electron transfer reactions catalyzed by a B12 trafficking protein. J. Biol. Chem. 289, 16487–16497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gherasim C., Ruetz M., Li Z., Hudolin S., and Banerjee R. (2015) Pathogenic mutations differentially affect the catalytic activities of the human B12-processing chaperone CblC and increase futile redox cycling. J. Biol. Chem. 290, 11393–11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McWilliam H., Li W., Uludag M., Squizzato S., Park Y. M., Buso N., Cowley A. P., and Lopez R. (2013) Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 41, W597–W600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mertens H. D., and Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141 [DOI] [PubMed] [Google Scholar]
- 35.Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wassenaar T., van Dijk M., Loureiro-Ferreira N., van der Schot G., de Vries S., Schmitz C., van der Zwan J., Boelens R., Giachetti A., Ferella L., Rosato A., Bertini I., Herrmann T., Jonker H. A., Bagaria A., Jaravine V., Güntert P., Schwalbe H., Vranken W., Doreleijers J., Vriend G., Vuister G., Franke D., Kikhney A., Svergun D., Fogh R., Ionides J., Laue E., Spronk C., Jurkša S., Verlato M., Badoer S., Dal Pra S., Mazzucato M., Frizziero E., and Bonvin A. J. J. (2012) WeNMR: structural biology on the grid. J. Grid Computing 10, 743–767 [Google Scholar]
- 37.Mah W., Deme J. C., Watkins D., Fung S., Janer A., Shoubridge E. A., Rosenblatt D. S., and Coulton J. W. (2013) Subcellular location of MMACHC and MMADHC, two human proteins central to intracellular vitamin B12 metabolism. Mol. Genet. Metab. 108, 112–118 [DOI] [PubMed] [Google Scholar]
- 38.Fofou-Caillierez M. B., Mrabet N. T., Chéry C., Dreumont N., Flayac J., Pupavac M., Paoli J., Alberto J. M., Coelho D., Camadro J. M., Feillet F., Watkins D., Fowler B., Rosenblatt D. S., and Guéant J. L. (2013) Interaction between methionine synthase isoforms and MMACHC: characterization in cblG-variant, cblG and cblC inherited causes of megaloblastic anaemia. Hum. Mol. Genet. 22, 4591–4601 [DOI] [PubMed] [Google Scholar]
- 39.Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada K., Gherasim C., Banerjee R., and Koutmos M. (2015) Structure of human B12 trafficking protein Cb1D reveals molecular mimicry and identifies a new subfamily of nitro-FMN reductases. J. Biol. Chem. 290, 29155–29166 [DOI] [PMC free article] [PubMed] [Google Scholar]