

Background: Kv2.1 channels are components of plasma membrane:endoplasmic reticulum membrane contact sites (PM:ER MCS).
Results: Kv2.1 exhibits clustering and enhanced phosphorylation in M phase versus interphase COS-1 cells, resulting in enhanced PM:ER MCS.
Conclusion: Cell cycle-dependent regulation of Kv2.1 localization and phosphorylation impacts PM:ER MCS.
Significance: PM:ER MCS are regulated by changes in PM protein localization and phosphorylation during mitosis.
Keywords: endoplasmic reticulum (ER), membrane structure, mitosis, plasma membrane, potassium channel, protein phosphorylation, subcellular organelle
Abstract
The plasma membrane (PM) comprises distinct subcellular domains with diverse functions that need to be dynamically coordinated with intracellular events, one of the most impactful being mitosis. The Kv2.1 voltage-gated potassium channel is conditionally localized to large PM clusters that represent specialized PM:endoplasmic reticulum membrane contact sites (PM:ER MCS), and overexpression of Kv2.1 induces more exuberant PM:ER MCS in neurons and in certain heterologous cell types. Localization of Kv2.1 at these contact sites is dynamically regulated by changes in phosphorylation at one or more sites located on its large cytoplasmic C terminus. Here, we show that Kv2.1 expressed in COS-1 cells undergoes dramatic cell cycle-dependent changes in its PM localization, having diffuse localization in interphase cells, and robust clustering during M phase. The mitosis-specific clusters of Kv2.1 are localized to PM:ER MCS, and M phase clustering of Kv2.1 induces more extensive PM:ER MCS. These cell cycle-dependent changes in Kv2.1 localization and the induction of PM:ER MCS are accompanied by increased mitotic Kv2.1 phosphorylation at several C-terminal phosphorylation sites. Phosphorylation of exogenously expressed Kv2.1 is significantly increased upon metaphase arrest in COS-1 and CHO cells, and in a pancreatic β cell line that express endogenous Kv2.1. The M phase clustering of Kv2.1 at PM:ER MCS in COS-1 cells requires the same C-terminal targeting motif needed for conditional Kv2.1 clustering in neurons. The cell cycle-dependent changes in localization and phosphorylation of Kv2.1 were not accompanied by changes in the electrophysiological properties of Kv2.1 expressed in CHO cells. Together, these results provide novel insights into the cell cycle-dependent changes in PM protein localization and phosphorylation.
Introduction
The plasma membrane (PM)2 of mammalian cells is composed of numerous subcompartments whose structure and function needs to be coordinated with diverse cellular events. Although much is known of signaling pathways linking stimulation of PM receptors by extracellular signals to intracellular signaling events, much less is known of how events triggered within cells influence the PM. The PM voltage-gated K+ channel Kv2.1 is expressed in a wide variety of excitable and non-excitable cells, and is notable for its high level of expression in brain neurons (1, 2). Kv2.1 is also expressed in cardiac and skeletal muscle (3), and in numerous proliferating cell types, including smooth muscle (4), pancreatic β cells (5), mesenchymal stem cells (6), and various tumor cell lines (7). In brain neurons, Kv2.1 is present in large PM clusters (2, 8–10), which are located at sites overlaying subsurface cisternae (11), which in certain neurons contain high levels of ryanodine receptor intracellular Ca2+ release channels (12–14). These sites represent specialized PM:endoplasmic reticulum (ER) membrane contact sites (i.e. PM:ER MCS (15)). Recombinant Kv2.1 is also present in large clusters in certain heterologous cell lines, such as Madin-Darby canine kidney (8) and HEK293 (16) cells, but not in others, one example being COS-1 cells (16, 17). Clustering of Kv2.1 endogenously expressed in neurons (18) and exogenously expressed in heterologous HEK293 cells (16) is dynamically regulated by changes in the phosphorylation state. Kv2.1 clustering is impacted by the activity of a variety of protein kinases and phosphatases, including CDK5 (19), calcineurin (18, 20, 21), and PP1 (19), with enhanced Kv2.1 phosphorylation correlating with enhanced clustering, and Kv2.1 dephosphorylation with dispersion of Kv2.1 and its uniform PM localization. Stimulation of phosphatase activity leading to dispersion of Kv2.1 clusters in neurons causes Kv2.1 to move away from PM:ER MCS (22, 23), suggesting that localization of Kv2.1 with these specialized membrane domains is conditional. In addition to regulating clustering, changes in the Kv2.1 phosphorylation state leads to complex effects on Kv2.1 voltage-dependent gating (18, 20, 21, 24–26) and expression level (27, 28). Consistent with its complex phosphorylation-dependent regulation, a large number (>35) of in vivo phosphorylation sites (phosphosites) have been identified on Kv2.1, most of which are on the large (400 amino acid) cytoplasmic C terminus (reviewed in Ref. 29). Among these is a single site (Ser(P)-586) that when mutated results in loss of Kv2.1 clustering (9), although a direct mechanistic requirement for phosphorylation at this site in regulating Kv2.1 clustering has not been definitively established.
Overexpression of Kv2.1 in brain neurons (12, 23) and in heterologous HEK293 cells (23) enhances PM:ER MCS, suggesting a role for this PM channel in induction or stabilization of these specialized membrane contact sites. The conditional localization of Kv2.1 at these sites, and the impact of Kv2.1 on their structure, suggests a possible role for Kv2.1 phosphorylation in conditionally regulating association of the ER with the PM. However, the clustering, phosphorylation state, and association with PM:ER MCS of Kv2.1 during mitosis, when robust changes in membrane structure throughout the cell are driven by cell cycle-dependent changes in protein kinase and phosphatase activity (30) leading to widespread changes in cellular protein phosphorylation (31), has not been investigated. During mitosis, the ER becomes relocalized to the cell periphery, and is excluded from the mitotic spindle (32). It has been suggested that relocalization of the ER to the cell periphery during mitosis facilitates its even distribution into the daughter cells (32). Much is known of the cell cycle-dependent changes in the structure of the nuclear envelope (33), the Golgi apparatus (34), and ER (35) during mitosis, and the signaling pathways that couple mitotic machinery to changes in phosphorylation of components of these membrane organelles. A prominent example is the ER resident protein STIM1, which is a substrate for mitotic phosphorylation that alters its interaction with the microtubule plus tip binding protein EB1 and mediates loss of ER binding to the mitotic spindle (36). Interestingly, STIM1 phosphorylation at mitosis also leads to a loss of binding to its PM binding partner Orai1 (37), resulting in both the functional loss of store-operated calcium entry and the structural loss of this class of PM:ER MCS at mitosis. However, PM substrates that may be involved in phosphorylation-dependent changes in the structure of PM:ER MCS during mitosis have not been identified.
The activity and expression of multiple types of ion channels vary in a cell cycle-dependent manner, often leading to changes in membrane potential throughout the cell cycle (38). The mechanisms by which ion channels are regulated during the cell cycle is not fully understood, although changes in mRNA expression (39–41), phosphorylation (37, 42), and localization (43) have been observed. K+ channels, including Kv2.1, are important regulators of membrane potential and proliferation, and act via both ion conduction-dependent and -independent mechanisms (38). Given the fundamental role of cell cycle-dependent changes in protein phosphorylation during mitosis, and the dynamic regulation of Kv2.1 by changes in multisite phosphorylation in neurons, we investigated whether the cell cycle impacts phosphorylation of Kv2.1 in COS-1 cells, which unlike many other cells types do not typically cluster Kv2.1. We also determined the impact of M phase clustering of Kv2.1 on PM:ER MCS, and defined the cell cycle-dependent changes in the Kv2.1 phosphorylation state, domains of Kv2.1 mediating cell cycle-dependent clustering, and the functional characteristics of Kv2.1 in M phase versus interphase cells. Our findings on cell cycle-dependent phosphorylation and clustering of Kv2.1 suggest that changes in the subcellular compartmentalization of PM proteins, including those associated with PM:ER MCS, may be a more common feature of cell cycle-dependent changes in cell structure than previously recognized.
Experimental Procedures
Antibodies
The rabbit polyclonal antibodies used were anti-Kv2.1e (RRID:AB_2531887), generated against a Kv2.1 S1-S2 extracellular region peptide CLPELQSLDEFGQSRDNPQL (44); anti-phospho-Histone H3 Ser-10 (06-570;, EMD Millipore, Temecula, CA RRID:AB_310177), and phosphospecific antibodies specific against Kv2.1 phosphosites Ser(P)-453 (S453P; RRID:AB_2315784), Ser(P)-563 (S563P; AB_2315785), Ser(P)-603 (S603P; RRID:AB_2531883), and Ser(P)-715 (S715P; RRID:AB_2531884) generated as described previously (21, 45). The mouse mAbs used were anti-mortalin mAb N52A/42 (RRID:AB_10674108) and anti-Kv1.4 mAb K13/31 (RRID:AB_10673576), directed against the intracellular N terminus of rat Kv1.4 (46); anti-Kv2.1 mAb L83/11 (RRID:AB_2315864), a non-phosphospecific mAb generated against the synthetic peptide SKPPEELEM(pS)SMPSPVAPLPA; anti-Kv2.1 mAb K89/34 (RRID:AB_10672253), generated against the cytoplasmic distal C-terminal end of Kv2.1; anti-Kv2.1 recombinant mAb K89/34 IgG2a (RRID:AB_2315768), which is the recombinant version of K89/34 with the IgG1 heavy chain constant region replaced with that of the IgG2a subclass (14); and anti-Kv2.1 Ser(P)-603 phosphospecific mAb L61/14 (RRID:AB_2315769), generated against the p603 phosphopeptide (14, 18, 21, 45). The anti-Kv2.1 Ser(P)-586 phosphospecific mAb L100/1 (RRID:AB_2531886), directed against the Ser(P)-586 phosphopeptide CMSSID(pS)FISSA, was generated for this study. Of the top 96 L100 candidate mAbs originally selected by their binding to this phosphopeptide by ELISA, L100/1 was distinguished by a lack of binding to the corresponding dephosphopeptide, and that in immunocytochemistry experiments in transfected HEK293 cells it recognized cells expressing wild-type Kv2.1 but not those expressing the S586A mutant. Moreover, in immunohistochemistry analyses of brain sections, L100/1 immunolabeling overlaps precisely with that of Kv2.1 in control brains, but is eliminated in brains from animals subjected to hypoxia, which triggers dephosphorylation of Kv2.1. L100/1 immunolabeling is also eliminated in brain sections from Kv2.1 knock-out mice. The mouse mAbs N52A/42, K13/31, K89/34, and L83/11 are available from the University of California, Davis/NIH NeuroMab Facility, a not-for profit supplier of mAbs administered through the University of California. The secondary antibodies used were from ThermoFisher (Waltham, MA) and were goat anti-rabbit IgG (H+L)-Alexa Fluor 488 (RRID:AB_10563748), goat anti-mouse IgG1-Alexa Fluor 488 (RRID:AB_10053811), goat anti-mouse IgG2a-Alexa Fluor 488 (RRID:AB_10562578), goat anti-rabbit IgG (H+L)-Alexa Fluor 555 (AB_10561552), goat anti-mouse IgG1-Alexa Fluor 555 (RRID:AB_10562375), goat anti-mouse IgG2a-Alexa Fluor 555 (RRID:AB_1500824), goat anti-rabbit IgG (H+L)-Alexa Fluor 647 (RRID:AB_10562581), and goat anti-mouse IgG1-Alexa Fluor 647 (RRID:AB_10565021).
Cell Culture and Transfection
COS-1 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% bovine calf serum (HyClone), 1% penicillin/streptomycin, and 1× GlutaMAX (ThermoFisher). COS-1 cells were split to 15–20% confluence, then immediately transiently transfected with rat Kv2.1, Kv1.4, or Kv2.1 mutants in the mammalian expression vectors pRBG4 (47) or pCGN (48) using Lipofectamine 2000 (ThermoFisher) transfection reagent following the manufacturer's protocol. COS-1 cells were used 40–48 h post-transfection and splitting. Generation of plasmids was described previously (9, 17, 45, 49). CHO cells stably transfected with rat Kv2.1 under a tetracycline (Tet) inducible promoter (CHO-Kv2.1) were generated as previously described (50). CHO-Kv2.1 cells were maintained in Ham's F-12 medium containing 10% fetal bovine serum, 1% penicillin/streptomycin, 1 μg/ml of blasticidin, and 25 μg/ml of Zeocin. T-REx-CHO cells, which were used to generate the CHO-Kv2.1 stable cell line but do not express Kv2.1, were used as control cells, and were maintained in identical conditions as CHO-Kv2.1 cells except without the addition of Zeocin in the maintenance media (50). To initiate expression of Kv2.1, tetracycline was added to the maintenance media at a concentration of 1 μg/ml. CHO cells were used 24 h after addition of tetracycline. INS-1 cells were a generous gift from Dr. David Keller, California State University, Chico. INS-1 cells were maintained in RPMI 1640 with 11.1 mm d-glucose supplemented with 10% fetal clone III, 1% penicillin/streptomycin, 25 mm HEPES, 2 mm l-glutamine, 1 mm sodium pyruvate, and 50 μm 2-mercaptoethanol. All cell lines were grown in a humidified incubator at 37 °C and 5% CO2. For metaphase arrest, 1 μg/ml of colchicine was added to maintenance media 16–17 h before use.
Immunofluorescence Labeling of Mammalian Cell Lines Expressing Kv2.1
COS-1 and CHO cells expressing Kv channels were grown on poly-l-lysine-coated glass coverslips and fixed for 15 min in ice-cold, 4% formaldehyde prepared fresh from paraformaldehyde, 4% sucrose, 0.1% Triton X-100 in DPBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.76 mm KH2PO4, pH 7.4) with 1 mm CaCl2 and 1 mm MgCl2. Cells were blocked with 3% (w/v) bovine serum albumin, 0.1% Triton X-100 in TBS (20 mm Tris, 150 mm NaCl), pH 7.4, then incubated with primary antibodies. Primary antibodies were detected with mouse IgG subclass-specific or rabbit goat secondary antibodies conjugated to Alexa Fluor (1:2000; ThermoFisher). Cells were labeled with dyes (Hoechst 33258; Phalloidin, ThermoFisher) during incubation with secondary antibodies. Images were acquired with an AxioCam MRm digital camera installed on a Zeiss AxioImager M2 microscope or with an AxioCam HRm digital camera installed on a Zeiss AxioObserver Z1 microscope with a ×63/1.40 NA plan-Apochromat oil immersion objective or a ×20/0.8 NA plan-Apochromat objective and an ApoTome coupled to Axiovision software (Zeiss, Oberkochen, Germany).
Live Cell Imaging
For live cell imaging of GxTX-labeled cells, COS-1 cells transfected with GFP-Kv2.1 were imaged in a modified Ringer's solution (3.5 mm KCl, 155 mm NaCl, 1.5 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, pH 7.4) containing 0.1% BSA and 10 mm glucose. Cells were incubated in 10 nm GxTX-550 (51). DNA was labeled with a formulation of Hoechst 33342, a live cell compatible nuclear dye (ThermoFisher R37605). Images were acquired with an EMCCD camera (Photometrics QuantEM 512SC) mounted on an Zeiss AxioObserver Z1 microscope with a ×40/1.3 NA plan Neofluar oil-immersion objective, and run by the Micromanager software suite 1.4 (51). To select M phase cells for electrophysiology, DNA was labeled with Hoechst 33342, and images were acquired with a ×40/0.95 NA plan Apochromat air objective.
For total internal reflection fluorescence (TIRF) imaging, live COS-1 cells transfected with pDsRed2-ER with or without GFP-Kv2.1 were imaged in a physiological saline solution (4.7 mm KCl, 146 mm NaCl, 2.5 mm CaCl2, 0.6 mm MgSO4, 1.6 mm NaHCO3, 0.15 mm NaH2PO4, 20 mm HEPES, pH 7.4) containing 8 mm glucose and 0.1 mm ascorbic acid. DNA was labeled with Hoechst 33342. Cells were maintained at 37 °C during the course of imaging with a heated stage and objective heater. Images were obtained with an Andor iXon EMCCD camera installed on a TIRF/widefield equipped Nikon Eclipse Ti microscope using a Nikon LUA4 laser launch with 405, 488, 561, and 647 nm lasers with a ×100 plan Apo TIRF, 1.49 NA objective run with NIS Elements software (Nikon).
Image Analysis
All of the images were transferred to Photoshop software (Adobe Systems, San Jose, CA) or ImageJ (National Institutes of Health) as linear 8-bit TIF files. For analyses of clustering, three independent samples of at least 20 cells each were scored for having clustered or dispersed localization. For analysis of clustering by coefficient of variation, ImageJ was used to measure the mean and S.D. of the pixel intensity values of regions of interest drawn around each cell, excluding the edge of the cell and perinuclear labeling (which is often Golgi labeling), from an n of three independent samples of at least 30 cells each, imaged with a ×63 objective. For measuring the levels of phospho-Kv2.1 immunolabeling, the intensities of phosphospecific Kv2.1 immunolabeling and total Kv2.1 immunolabeling were measured in regions of interests drawn around each cell, excluding the edge of the cell in the ImageJ. To account for variations in the overall level of total Kv2.1 and slight differences in background labeling between M phase and interphase cells, the average intensity of phosphospecific immunolabeling in control M phase cells was subtracted from the average intensity of phosphospecific immunolabeling in Kv2.1 expressing M phase cells, and the average intensity of phosphospecific immunolabeling in control interphase cells was subtracted from the average intensity of Kv2.1 expressing interphase cells. The same calculation was performed with total Kv2.1 immunolabeling. The difference in the ratio of phosphospecific immunolabeling to the total Kv2.1 immunolabeling between M phase and interphase COS-1 cells was calculated based on the background-subtracted values. This analysis was performed in an n of three independent samples of at least 28 cells each, imaged with a ×20 objective. All images were measured from z-stacks of optical sections taken at equal exposure across all images within an experiment. Representative images were taken with optimal exposure time.
For cortical endoplasmic reticulum (cER) morphology analysis, images were taken at optimal exposure to adjust for the inherent variability in intensity observed with transient transfection. To measure cluster size, regions of interests were drawn around individual clusters in ImageJ, and the surface area of regions was measured. Up to 15 of the largest clusters on each cell were chosen for analysis. Analysis was performed blind. This analysis was performed on three independent samples of at least 5 cells each, imaged with a ×100 objective using TIRF microscopy.
SDS-PAGE and Immunoblotting
Analyses of COS-1 and CHO cell lysates prepared from transfected cells were performed as described (17, 52). In brief, cells were lysed for 5 min on ice in an ice-cold lysis buffer solution containing TBS (20 mm Tris, 150 mm NaCl, pH 8.0), 10 mm EDTA, 2% Triton X-100, 10 mm iodoacetamide, 10 mm NaF, and a protease inhibitor mixture (2 μg/ml of aprotinin, 1 μg/ml of leupeptin, 2 μg/ml of antipain, 10 μg/ml of benzamidine, and 1 mm phenylmethylsulfonyl fluoride). The detergent lysate was centrifuged in a microcentrifuge for 5 min at 16,100 × g to pellet nuclei and debris, and the resulting supernatant (cleared lysate) was saved for analysis. For INS-1 cells, crude membrane preparations were performed as described (53). In brief, cells were pelleted by centrifugation at 400 × g, and then homogenized in phosphate buffer (50 mm K2HPO4, pH 7.4, 0.1 m NaCl) containing 10 mm iodoacetamide, 10 mm NaF, and a protease inhibitor mixture (described above). The homogenate was centrifuged at 400 × g for 5 min to remove nuclei and unbroken cells, and then centrifuged at 38,900 × g for 90 min to pellet membranes. Membranes were resuspended in homogenization buffer (320 mm sucrose, 5 mm NaPO4, pH 7.4) to prepare a membrane fraction. For immunoblots, the COS-1 or CHO cell-cleared lysate or INS-1 cell membrane fraction was added to one-third volume of 4× reducing SDS sample buffer and fractionated on 9% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with various antibodies, as described above. Note that “lauryl sulfate” (Sigma L-5750) was used in SDS gel recipes to accentuate electrophoretic mobility differences between different phosphorylation states of Kv2.1, as described previously for other proteins (17, 54). For rabbit polyclonal antibodies specific against Kv2.1 phosphosites Ser(P)-603 (S603P) and Ser(P)-715 (S715P), peptide absorption was performed to block non-phosphospecific antibodies; antisera were incubated at 4 °C overnight with 123 μg/ml of nonphosphorylated Kv2.1 fusion protein (containing the Kv2.1 C-terminal amino acids 529–775) tagged with glutathione S-transferase (GST), immediately before being used for immunoblotting. Primary antibodies were detected with mouse IgG subclass-specific or rabbit goat secondary antibodies conjugated to Alexa Fluor (1:1500; ThermoFisher). Quantitation of immunoreactivity was performed by densitometry analysis using AlphaView software (ProteinSimple, Santa Clara, CA).
Phosphatase digestion was performed on COS-1 cell lysates with 0.1 unit/μl of calf intestinal alkaline phosphatase (Roche Life Science, Indianapolis, IN) at 37 °C for 2 h in 0.1 mm EDTA, 50 mm Tris, pH 8.5, in the presence of 0.1% pure SDS (161–0301, Bio-Rad) and a protease inhibitor mixture (described above) with the addition of 100 mm NaF in control samples not treated with alkaline phosphatase. The reaction was stopped by the addition of one-third volume of 4× reducing SDS sample buffer.
Electrophysiology
Whole cell voltage clamp recordings were used to measure currents from Kv2.1 channels. CHO-Kv2.1 cells stably transfected with rat Kv2.1 under control of a tetracycline repressor (50) were maintained as described above. For patch clamp recordings, cells were grown for 1–2 days on uncoated glass bottom 35-mm dishes (D35-20-1.5-N, Cellvis, Mountain View, CA) in 2 ml of Ham's F-12 medium with 10% FBS and 100 units/ml of penicillin-streptomycin, without the selection agents described above. To induce channel expression, minocycline (1 μg/ml) was added 1–1.5 h before removal from the incubator. To label chromosomes, 2 drops of Hoechst 33342 solution was added 20 min before removal from the incubator. Upon removal from the incubator, cells were rinsed with an external solution containing (in mm): 3.5 KCl, 155 NaCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, adjusted to pH 7.4, with NaOH. The internal (pipette) solution contained (in mm): 155 KCl, 15 NaCl, 2 MgCl2, 10 HEPES, 0.1 EGTA adjusted to pH 7.2 with KOH. Pipettes were pulled from thin-wall borosilicate glass (Sutter BF150-110-10HP), coated with Sylgard (SYLG184, Dow Corning, Midland, MI), heat cured, and fire polished. Pipette tip resistances were less than 4 mΩ. Spatially isolated cells with Hoechst fluorescence indicating either M-phase or interphase chromatin structure were selected for patch clamp. Recordings were at room temperature (21–23 °C). Voltage clamp was achieved with an Axon Axopatch 200B amplifier (MDS) run by Patchmaster software (HEKA Instruments, Holliston, MA). Holding potential was −100 mV. Liquid junction potential offset was calculated to be less than 4 mV and was not corrected. Series resistance compensation was used when necessary, to constrain voltage error to less than 10 mV. Capacitance and Ohmic leak were subtracted using a P/5 protocol. Recordings were low pass filtered by the amplifier at 10 kHz and digitized at 100 kHz. Current traces were digitally filtered at 1 kHz for display in figures. Data were analyzed and plotted with IgorPro software (Version 6, Wavemetrics, Lake Oswego, OR). Conductance, G, was determined from the current level at the end of a 100-ms step to the indicated voltage, normalized by the driving force from a calculated reversal potential of −97 mV. Conductance change with voltage was fit from −80 to +40 mV with the fourth power of a Boltzmann distribution described previously (51). Vmid is the voltage when the fit reaches half-maximal amplitude, z is the number of electronic charges determining slope, A is maximal amplitude. To measure τ activation, kinetics during a voltage step from −100 to +40 mV were fit from 10 to 90% current rise by the power of an exponential function as described previously (55). Steady state conductance, GSS, was measured from current amplitude at the end of a 100-ms step to 0 mV following a 10-s pre-pulse to the indicated voltage and fit by a Boltzmann distribution of the form,
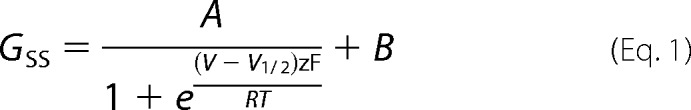 |
where V is pre-pulse voltage, V½ is the voltage when the fit reaches half-maximal amplitude, z is the number of electronic charges determining slope, B is the baseline of the fit, A is maximal amplitude above baseline. To reduce the number of free parameters, fits were truncated at 0 mV to avoid complications due to U-type inactivation at more positive voltages (56). To measure τ inactivation, kinetics were fit by a monoexponential function.
Results
The Plasma Membrane Ion Channel Kv2.1 Is Preferentially Clustered at M Phase
To investigate cell cycle-dependent regulation of Kv2.1, we first expressed recombinant Kv2.1 in proliferating heterologous COS-1 cells. We initially used nuclear morphology as assessed by staining of DNA with Hoechst dye to identify M phase cells based on their condensed chromosomes. We found that in cultures of transiently transfected COS-1 cells that, as previously shown (8, 16, 17, 22), the vast majority of transfected cells express exogenous Kv2.1 uniformly throughout the plasma membrane (see representative cell in the lower right of panels in Fig. 1A). However, a subpopulation of cells exhibited robust Kv2.1 clustering (see representative cell in the upper left of panels in Fig. 1A). The cells with clustered Kv2.1 typically had condensed chromosomes as revealed with the Hoechst dye (Fig. 1A). We confirmed that these robust differences in Kv2.1 localization in different subpopulations within the same cultures were related to the cell cycle stage by immunolabeling for histone H3 phosphorylated at Ser-10 (H3pS10; Fig. 1A), an M phase marker (57). In contrast to Kv2.1, the related Kv1.4 Kv channel, which also has a uniform expression in COS-1 cells (e.g. Ref. 52), retains this uniform expression in both interphase and M phase COS-1 cells (Fig. 1B). Surface plots (58) of selected regions of the COS-1 cell plasma membrane (Fig. 1C) demonstrate that Kv2.1 immunolabeling of the M phase cell at the upper left of panel A has a greater difference between the fluorescence intensity local minima and maxima relative to the interphase cell at the lower right, consistent with the non-uniform (i.e. clustered) subcellular localization of Kv2.1 in M phase cells. Labeling for Kv1.4 is similar in interphase and M phase cells (Fig. 1C). A significantly increased incidence of cells with clustered Kv2.1 was observed in M phase cells regardless of whether the cell cycle stage was determined by Hoechst staining and the presence or absence of condensed chromosomes, or by H3pS10 immunolabeling (Fig. 1D). Moreover, analysis of the intensity of Kv2.1 immunolabeling across the M phase and interphase cells revealed that the coefficient of variation of labeling intensity is significantly greater in M phase cells, further supporting increased Kv2.1 clustering in M phase cells, and more diffuse, uniform expression in interphase cells (Fig. 1E). As such, Kv2.1 clustering in COS-1 is distinct from that seen for recombinant Kv2.1 expressed in heterologous Madin-Darby canine kidney (8) and HEK293 (16) cells, and endogenous and exogenous Kv2.1 in neurons (2, 8–10), in that Kv2.1 clustering in COS-1 cells is not seen during all stages of the cell cycle but is limited to M phase cells.
FIGURE 1.
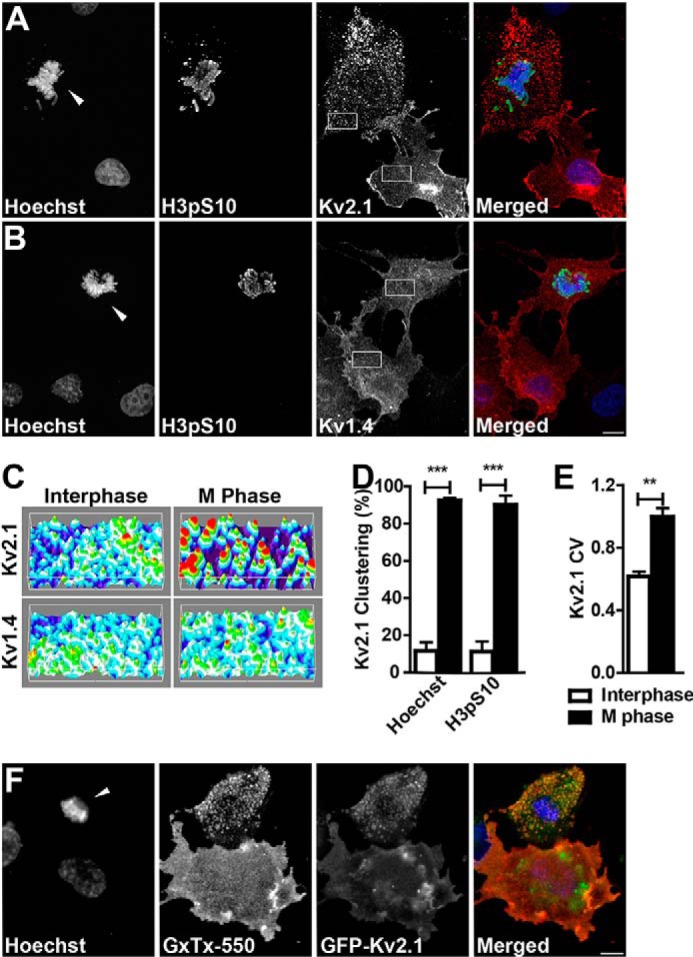
M phase COS-1 cells cluster Kv2.1. A, M phase and interphase Kv2.1 expressing COS-1 cells stained with Hoechst (blue) and immunolabeled for H3pS10 (green) and Kv2.1 (red). B, M phase and interphase Kv1.4 expressing COS-1 cells stained with Hoechst (blue) and immunolabeled for H3pS10 (green) and Kv1.4 (red). Arrowheads indicate M phase nuclei. Scale bar, 10 μm. C, three-dimensional surface plots of the regions indicated in A and B showing relative Kv immunolabeling intensity plotted along the cell surface area. D, cells categorized as interphase (white bars) or M phase (black bars) were scored as either clustered or unclustered. Cells with over 25% of their membrane covered in clusters were considered clustered. Cell cycle stage was determined by either nuclear morphology as revealed by Hoechst staining (left two bars) or the presence of H3pS10 immunolabeling (right two bars). Data are from at least 20 cells in an n of three independent samples. E, coefficient of variation (CV) of Kv2.1 fluorescence intensity in interphase (white bar) or M phase (black bar) cells. Cell cycle stage was determined by nuclear morphology as revealed by Hoechst staining. Data are from at least 30 cells in an n of three independent samples. Data in D and E are the mean ± S.E. **, p < 0.01; ***, p < 0.001 (two-tailed unpaired t test). F, Live COS-1 cells expressing GFP-Kv2.1 (green) labeled with GxTX-550 (red) and Hoechst nuclear dye (blue).
We next assessed whether the M phase Kv2.1 clusters are found on the cell surface. If Kv2.1 clusters are plasma membrane localized, they are targeted by guangxitoxin-1E, a tarantula toxin that selectively binds to Kv2 channels (51, 59). We applied guangxitoxin-1E conjugated to a DyLight 550 fluorophore (GxTX-550) to live COS-1 cells expressing GFP-Kv2.1. GxTX-550 labeled the Kv2.1 clusters present in M phase cells as well as the diffuse Kv2.1 in interphase cells (Fig. 1F), indicating cell surface localization of Kv2.1 during both M phase and interphase. The clustered GFP-Kv2.1 present in M phase cells appears to have a somewhat decreased GxTX-550 labeling intensity relative to the diffuse GFP-Kv2.1 in interphase cells. The cause of this decreased intensity is unclear, but could result from self-quenching of clustered fluorophores, different conformational states of Kv2.1 impacting the binding of this state-specific toxin (51), or a subpopulation of subsurface GFP-Kv2.1 (60).
Multisite Phosphorylation of Kv2.1 Increases at M Phase
Clustering of Kv2.1 in brain neurons is reversibly regulated by stimuli that cause changes in the Kv2.1 phosphorylation state (18). The large (440 amino acid) carboxyl-terminal cytoplasmic tail of Kv2.1 contains both the “proximal restriction and clustering” (PRC) domain (9), which is necessary for clustering of Kv2.1 in neurons, and the bulk (33/37) of the known in vivo phosphosites (reviewed in Ref. 29), a subset of which independently regulate phosphorylation-dependent modulation of Kv2.1 expression, localization, and function. Studies in neurons (18) and heterologous HEK293 cells (16) show that increased Kv2.1 clustering is correlated with increased phosphorylation, and vice versa. Because changes in phosphorylation state regulate many cell cycle-dependent changes in protein function, we next addressed whether the changes in Kv2.1 localization depicted in Fig. 1 occur in concert with cell cycle-dependent changes in Kv2.1 phosphorylation state.
We employed previously described phosphorylation-specific (phosphospecific) antibodies against four Kv2.1 phosphosites (Ser(P)-453, Ser(P)-563, Ser(P)-603, and Ser(P)-715) that LC-MS/MS analyses identified as phosphosites found on Kv2.1 purified from HEK293 cells and brain, and that stable isotope labeling by amino acids in cell culture-based quantitative LC-MS/MS analyses of HEK293 cells had shown are regulated by calcineurin-dependent dephosphorylation (45). Studies employing these phosphospecific antibodies showed that these sites exhibit activity-dependent changes in the phosphorylation state in brain neurons (21). We also employed a novel phosphospecific monoclonal antibody (mAb) “L100/1” against the Ser(P)-586 phosphosite, which is located within the PRC domain and critical for Kv2.1 clustering (9). Although we identified Ser(P)-586 as an in vivo phosphosite in analyses of Kv2.1 purified from brain and HEK293 cells (45), in our subsequent stable isotope labeling by amino acids in cell culture analyses of HEK293 cells (45, 61), we could not detect either the phosphorylated or unphosphorylated tryptic peptides containing Ser-586, perhaps due to the lower amounts of starting material used in these experiments involving metabolic labeling of cultured cells. We used these phosphospecific anti-Kv2.1 antibodies in conjunction with antibodies against total Kv2.1 and Hoechst dye in multiplex immunofluorescence labeling experiments on COS-1 cells. We found that the intensity of phosphorylation at all five phosphosites (Ser(P)-453, Ser(P)-563, Ser(P)-586, Ser(P)-603, and Ser(P)-715) was higher in M phase cells than in interphase cells (Fig. 2, A and C). In each case phosphospecific labeling was more prominent in cells with clustered Kv2.1 than with diffuse Kv2.1. These results show that similar to brain neurons, clustering of Kv2.1 channels in COS-1 cells is associated with increased Kv2.1 phosphorylation.
FIGURE 2.
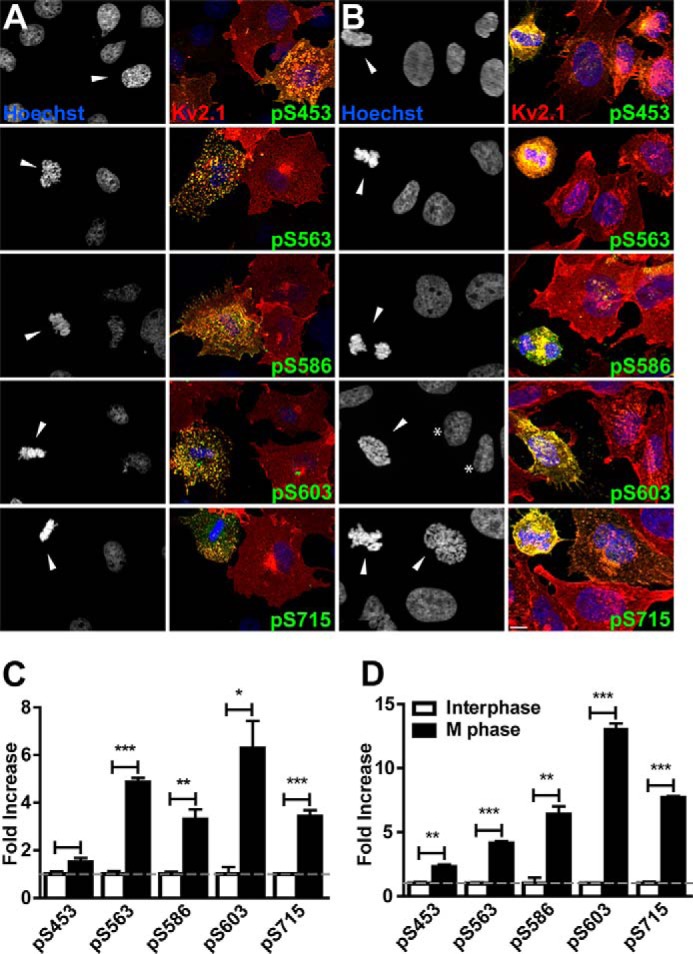
Kv2.1 is phosphorylated at M phase. Kv2.1 expressing COS-1 (A) and CHO (B) cells stained with Hoechst (blue) and immunolabeled for total Kv2.1 (red), and the indicated Kv2.1 phosphosite (green). Arrowheads indicate M phase nuclei. Asterisks in B denote interphase CHO cells with Kv2.1 clustering. Scale bar, 10 μm. The ratio of the fluorescence intensity of each Kv2.1 phosphosite to total Kv2.1 was measured in COS-1 (C) and CHO (D) cells in interphase (white bars) and M phase (black bars). Data are from at least 28 cells in an n of three independent samples. Values are normalized to interphase cells. Data are the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (two-tailed unpaired t test). Cell cycle stage was determined by nuclear morphology as revealed by Hoechst staining.
To determine whether the cell cycle-dependent changes in Kv2.1 phosphorylation seen in non-human primate COS-1 cells occur in other cell types, we next examined Kv2.1 stably expressed in rodent CHO cells. We found that similarly to transiently transfected COS-1 cells, the stable CHO cell line exhibited increased levels of phosphospecific Kv2.1 antibody labeling at this same set of five phosphosites in M phase cells (Fig. 2, B and D). Interestingly, distinct from COS-1 cells, Kv2.1 expressed in CHO cells exhibited some degree of clustering in a subpopulation of interphase cells (asterisks in Fig. 2B). Together, these results support that diverse cells types (African green monkey COS-1 kidney fibroblasts and Chinese hamster ovary CHO cells) exhibit cell cycle-dependent regulation of Kv2.1 phosphorylation.
M Phase Kv2.1 Clusters Are Located at PM:ER MCS and Alter Cortical ER Morphology
Kv2.1 clusters are localized to PM:ER MCS in neurons (11–14) and when heterologously expressed in HEK293 cells (23), where they induce PM:ER MCS formation (23). The ability of Kv2.1 to induce and/or stabilize PM:ER MCS in neurons and HEK293 cells depends on the localization of Kv2.1 to clusters, as acutely disrupting Kv2.1 clusters pharmacologically or with mutations blocks the effect of Kv2.1 on PM:ER MCS (23). To determine whether M phase-specific Kv2.1 clusters promote PM:ER MCS formation, we expressed the ER marker pDsRed2-ER in COS-1 cells with or without GFP-Kv2.1. A live cell compatible nuclear dye was used to distinguish M phase and interphase COS-1 cells. Live COS-1 cells were imaged using TIRF microscopy to selectively illuminate the basal plasma membrane and the juxtaposed cER. The cER in interphase cells is primarily composed of small puncta and occasional tubules, regardless of coexpression with Kv2.1 (Fig. 3, A and C). A similar pattern was observed in M phase cells in the absence of Kv2.1 (Fig. 3B). In the presence of Kv2.1, cER puncta show robust colocalization with Kv2.1 clusters during M phase, but not during interphase when Kv2.1 is diffuse (Fig. 3, C–E). M phase Kv2.1 clusters alter cER morphology in a manner dependent on the size of the Kv2.1 clusters (Fig. 3G). M phase cells with small Kv2.1 clusters have small cER puncta, similar in appearance to M phase cER in the absence of Kv2.1 (Fig. 3, B, D, F, and G). On the other hand, large M phase Kv2.1 clusters induce the formation of large cER clusters, a morphology that was never observed in the absence of large Kv2.1 clusters (Fig. 3, E–G). These results demonstrate a role for M phase Kv2.1 clusters in regulating cER morphology specifically in regards to its association with PM:ER MCS.
FIGURE 3.
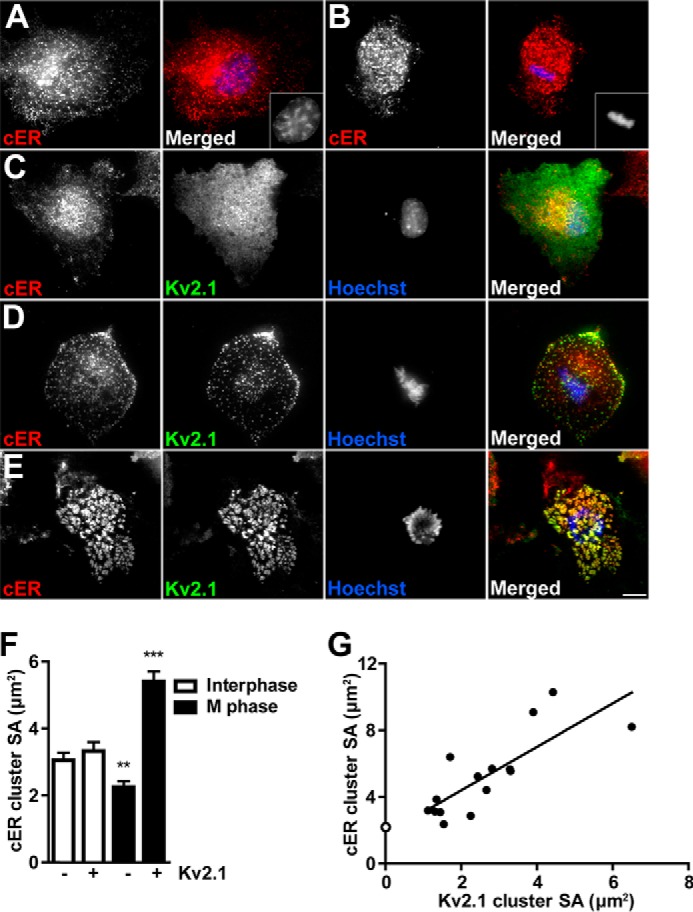
M phase clustering of Kv2.1 recruits cER to PM:ER MCS. Control COS-1 cells in interphase (A) and M phase (B) expressing pDsRed2-ER (red) and stained with Hoechst nuclear dye (blue). Insets show nuclei. Kv2.1 expressing COS-1 cells in interphase (C) and M phase (D and E) expressing pDsRed2-ER (red) and GFP-Kv2.1 (green) stained with Hoechst nuclear dye (blue). Red and green images were acquired with TIRF microscopy, blue images were acquired with widefield fluorescence microscopy. Scale bar, 10 μm. F, average cER surface area (SA) was measured in M phase (black bars) and interphase (white bars) cells in the presence or absence of Kv2.1 (indicated below). Data are from an n of at least 180 clusters from at least 5 different cells in three independent samples. Data are the mean ± S.E. **, p < 0.01; ***, p < 0.001 (two-tailed unpaired t test). G, average cER cluster SA was plotted against the average Kv2.1 cluster SA. Filled circles are average surface areas per cell. Linear regression analysis was performed, r2 = 0.6326. Open circle is average cER cluster size from M phase cell lacking Kv2.1 from F.
Analysis of Kv2.1 Clustering Mutants Suggests Similar Mechanisms Underlie Kv2.1 Clustering in Neurons and in M Phase Heterologous Cells
Previous analyses of Kv2.1 clustering determinants used a series of Kv2.1 internal deletion, truncation, and point mutants to define a specific segment on the Kv2.1 C terminus (amino acids 573–598; the so-called PRC domain, Fig. 4A), and four critical residues therein (Ser-583, Ser-586, Phe-587, and Ser-589) that are necessary for Kv2.1 clustering in cultured hippocampal neurons (9). We used a subset of these mutants to determine whether the same determinants are involved in M phase clustering of Kv2.1. We expressed this panel of Kv2.1 mutants in COS-1 cells, and examined their subcellular localization in M phase and interphase cells. COS-1 cells transfected with the Kv2.1 C-terminal truncation mutants ΔC187, ΔC237, and ΔC247 retain M phase-specific Kv2.1 clustering (Fig. 4B). However, eliminating larger portions of the C terminus of Kv2.1 including the PRC domain, in the ΔC263, ΔC273, and ΔC318 truncation mutants, abolished M phase clustering (Fig. 4B). These results are broadly consistent with the patterns of clustering of these mutants in cultured hippocampal neurons (9), as summarized in the “neuronal clustering” column in Fig. 4B. This may be expected given the similar phosphorylation-dependent regulation of Kv2.1 clustering in neurons (18) and in heterologous HEK293 cells (16).
FIGURE 4.
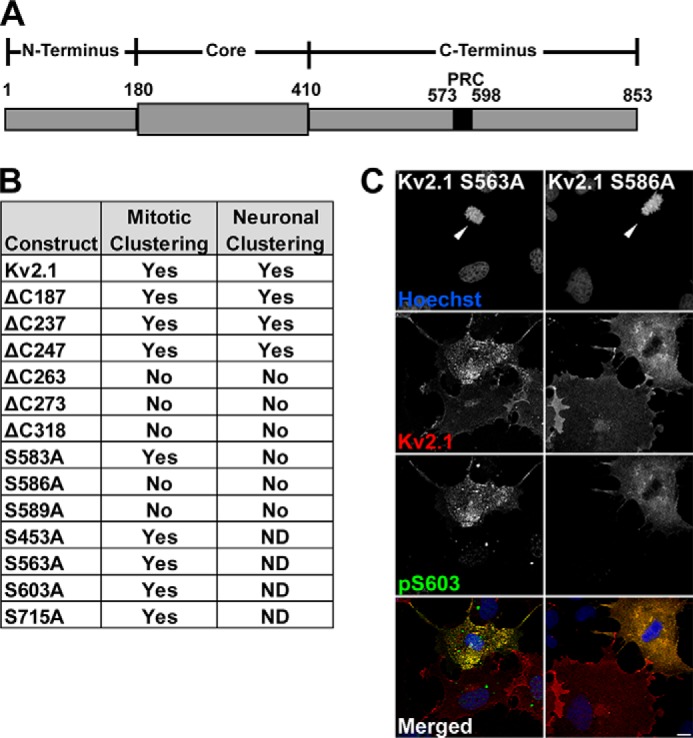
Determinants of M phase clustering of Kv2.1. A, schematic diagram of Kv2.1, indicating the location of the PRC domain within the C terminus. B, comparison of Kv2.1 clustering in M phase COS-1 cells and neurons (neuron results from Ref. 9). C, COS-1 cells expressing mutant Kv2.1 constructs S563A, left, and S586A, right, stained with Hoechst (blue) and immunolabeled for total Kv2.1 (red), and Ser(P)-603 (green). Arrowheads indicate M phase nuclei. Scale bar, 10 μm.
M phase Kv2.1 clustering was also eliminated in the point mutants S586A and S589A (Fig. 4, B and C), also consistent with their diffuse localization in neurons (9). However, the S583A mutant retained the cell cycle-dependent clustering typical of wild-type Kv2.1 (Fig. 4B), inconsistent with its diffuse localization in neurons (9). This suggests that although clustering of Kv2.1 in neurons and its cell cycle-dependent clustering in proliferating cells require the same overall domain of Kv2.1, exhibit phosphorylation dependence, and share the bulk of the same specific residues, the two types of clustering are distinct in their requirement for an intact serine residue at Ser-583.
Although Kv2.1 is phosphorylated at Ser(P)-453, Ser(P)-563, Ser(P)-586, Ser(P)-603, and Ser(P)-715 in M phase cells (Fig. 2), the truncation analyses presented in Ref. 9 would suggest that except for Ser(P)-586, these other phosphosites are not critical for clustering, as these residues are deleted in truncation and/or internal deletion mutants that retain clustering (9). Consistent with this prediction, we found that Kv2.1 phosphosite mutants S453A, S563A, S603A, and S715A were similar to WT Kv2.1 in their cell cycle-dependent clustering (Fig. 4, B and C). Conversely, the S586A mutant, which lacks clustering, still shows an overall increase in cell cycle-dependent phosphorylation, for example, at Ser(P)-603 (Fig. 4, B and C). This suggests a widespread increase in Kv2.1 phosphorylation at numerous sites during M phase, although of the sites that exhibit enhanced M phase labeling with phosphospecific antibodies, only the alanine mutation at Ser-586 disrupted cell cycle-dependent clustering. These results also show that cell cycle-dependent phosphorylation of Kv2.1 can occur in the absence of Kv2.1 clustering.
Metaphase Arrest Causes Increased Kv2.1 Phosphorylation
We next determined whether experimentally arresting cells in metaphase would impact phosphorylation of Kv2.1. Control cultures of COS-1 and CHO cells have very low levels of M phase cells, as judged by immunolabeling for the M phase marker H3pS10 (Fig. 5A). In this experiment, H3pS10 labels only 5.6 ± 0.6% of COS-1 cells, and 3.7 ± 1.0% of CHO cells (Fig. 5A). We used colchicine treatment to induce metaphase arrest and increase steady-state levels of M phase cells. Colchicine treatment increased the percentage of H3pS10-positive COS-1 and CHO cells to 42.9 ± 3.9 and 32.8 ± 0.5%, respectively (Fig. 5A). Immunoblots of cell lysates prepared from such cultures using phosphospecific antibodies showed Kv2.1 phosphorylation at Ser(P)-563, Ser(P)-603, and Ser(P)-715 were significantly increased in lysates prepared from both COS-1 (Fig. 5, B and C) and CHO (Fig. 5, B and D) cells subjected to colchicine treatment. Although phosphorylation at these sites could be analyzed by immunofluorescence immunocytochemistry, phosphorylation at Ser(P)-453 and Ser(P)-586 could not be studied by immunoblot, as our phosphospecific Ser(P)-453 antibody had an interfering nonspecific band on immunoblots that was present in samples of the S453A mutant (not shown) and the phosphospecific Ser(P)-586 L100/1 mAb does not work on immunoblots. Immunoblots probed for total Kv2.1 showed the addition of a higher Mr band in colchicine-treated COS-1 and CHO cells (Fig. 5, B and E). Treatment with alkaline phosphatase eliminated this higher Mr Kv2.1 band (Fig. 5E), indicating an increase in the overall phosphorylation of a subpopulation of Kv2.1 with colchicine treatment. Together, these results show that experimentally inducing metaphase arrest leads to increased Kv2.1 phosphorylation at three sites, further strengthening the link between cell cycle and changes in the Kv2.1 phosphorylation state.
FIGURE 5.
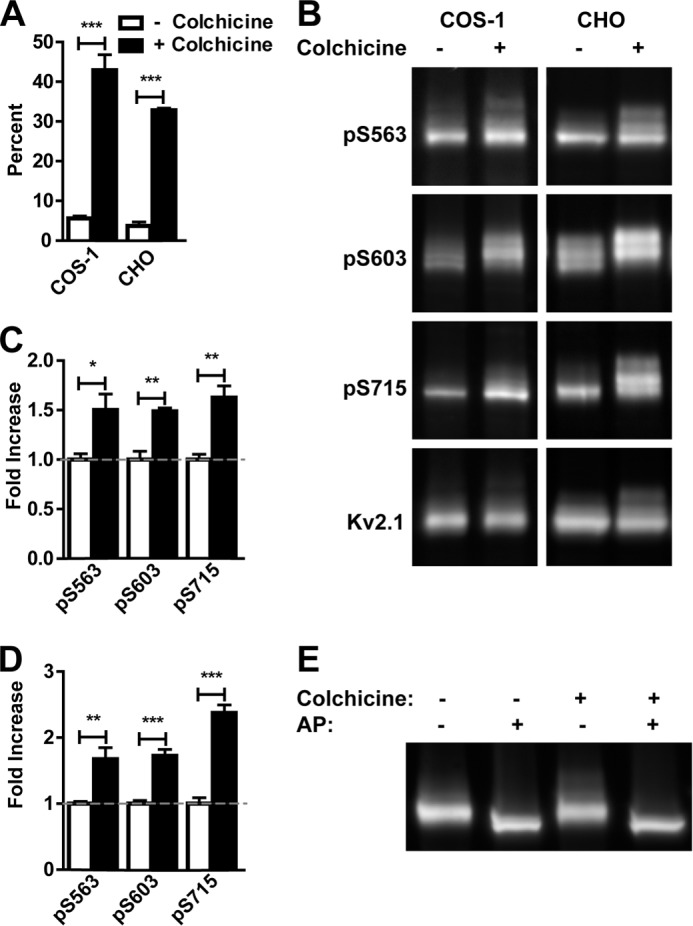
M phase arrest leads to enhanced Kv2.1 phosphorylation. A, percent of H3pS10 positive nuclei in control (white bars) or colchicine-treated (black bars) COS-1 and CHO cells. B, lysates of Kv2.1 expressing COS-1 and CHO cells treated with or without colchicine probed for the indicated Kv2.1 phosphosites and total Kv2.1. The ratio of the intensity of each Kv2.1 phosphosite to total Kv2.1 was measured in control (white bars) and colchicine-treated (black bars) COS-1 (C) and CHO (D) cell lysates. Values are normalized to control lysates. Data are the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (two-tailed unpaired t test). E, COS-1 cells were treated with or without colchicine, then lysates were incubated with or without alkaline phosphatase (AP).
Kv2.1 Phosphorylation Is Increased at M Phase in Pancreatic β Cells
Pancreatic β cells express endogenous Kv2.1, where it plays a critical role in regulating glucose-dependent insulin secretion (5, 62). The immortalized INS-1 cell line is derived from rat pancreatic β cells, expresses Kv2.1 (63), and has been used as a cell culture model of β cell function (e.g. Refs. 64 and 65). To evaluate cell cycle-dependent regulation of Kv2.1 phosphorylation in a proliferative cell type that endogenously expresses Kv2.1, we prepared samples from INS-1 cells that had been treated with colchicine to arrest cells in metaphase. Immunoblots with anti-Kv2.1 phosphospecific antibodies revealed increased levels of Ser(P)-563 and Ser(P)-603 phosphorylation in membrane fractions from colchicine-treated (metaphase arrested) INS-1 cells compared to untreated INS-1 cells (Fig. 6, A and B), consistent with results from COS-1 and CHO cells exogenously expressing recombinant Kv2.1. This suggests that M phase Kv2.1 phosphorylation is not an effect seen only upon exogenous expression of Kv2.1 in heterologous cell lines, but also occurs in cells endogenously expressing Kv2.1.
FIGURE 6.
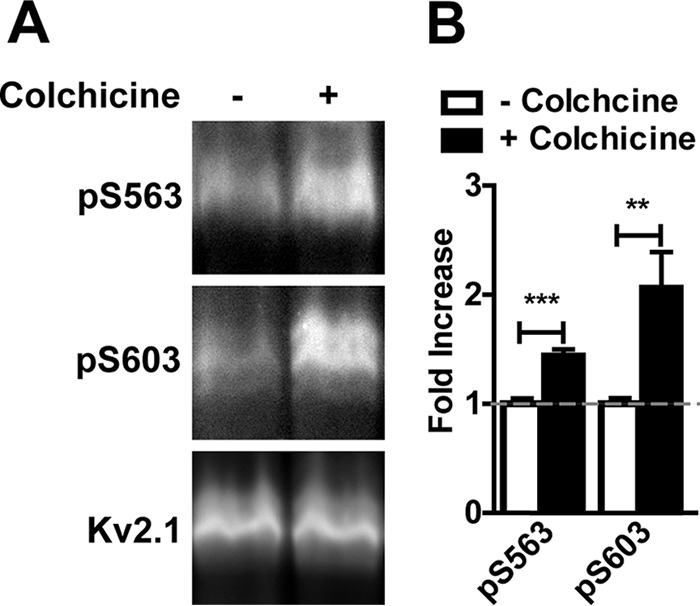
Enhanced phosphorylation of endogenous Kv2.1 in a pancreatic β cell line. A, membrane fractions of INS1 cells treated with or without colchicine probed with the indicated Kv2.1 phosphospecific antibodies and total Kv2.1. B, the ratio of the intensity of each Kv2.1 phosphosite to total Kv2.1 was measured in control (white bars) and colchicine-treated (black bars) INS1 cells. Values are normalized to control cells. Data are the mean ± S.E. **, p < 0.01; ***, p < 0.001 (two-tailed unpaired t test).
The Electrophysiological Properties of Kv2.1 Expressed in CHO Cells Appear Unchanged during M Phase
Changes in Kv2.1 phosphorylation are associated with diverse changes in the amplitude and/or gating characteristics of Kv2.1 currents in neurons and heterologous cells (18, 24–27, 66, 67). To determine whether Kv2.1 amplitude and/or gating are impacted by the altered phosphorylation state observed in M phase cells, we performed whole cell patch clamp in CHO cells stably expressing Kv2.1. A live cell compatible nuclear dye was used to distinguish M phase and interphase CHO cells (Fig. 7, A and B). Voltage step protocols elicited outward currents typical of Kv2.1 in this cell line (Fig. 7C). Within the limits of error in our dataset, the macroscopic Kv2.1 current amplitude, voltage-dependent activation, steady state inactivation, as well as kinetics of activation and inactivation were not significantly different between interphase and M phase cells (Fig. 7, D–H). These data suggest that the cell cycle-dependent changes in Kv2.1 phosphorylation and localization (Figs. 1–3) do not result in alteration of the ionic currents of Kv2.1.
FIGURE 7.

Electrophysiological characterization of Kv2.1. A, differential interference contrast image of live Kv2.1-expressing CHO cells plated for electrophysiology. B, Hoechst fluorescence from cells in A. Arrowheads indicate M phase nuclei, arrows indicate interphase nuclei. C, Kv2.1 activation in an interphase (left) or M phase cell (right). Representative whole cell currents in response to 100-ms steps to −20, 0, 20, and 40 mV were from a holding potential of −100 mV. Scale bars: 1 nA, 50 ms. D, amplitude of Kv2.1 currents at +40 mV from interphase (I) or M phase (M) cells. Circles are measurements from individual cells. Bars throughout the figure are mean ± S.E. E, mean conductance-voltage profile of Kv2.1 during M phase (solid circles, n = 4) or interphase (hollow circles, n = 5). Lines are fits of a fourth-power Boltzmann function. Parameters from fits ± S.D.: interphase (dotted line), Vmid = 7 ± 2 mV, z = 1.9 ± 0.2 e+; M phase (solid line), Vmid = 9 ± 1 mV, z = 1.9 ± 0.1 e+. F, individual cell parameters from fits to conductance (left and center) or activation kinetics at +40 mV (right). G, mean steady state inactivation-voltage profile during M phase (solid circles, n = 7) or interphase (hollow circles, n = 4). Lines are fits of a Boltzmann function. Parameters from fits ± S.D.: interphase (dotted line), Vmid = −35 ± 1 mV, z = 3.2 ± 0.5 e+; M phase (solid line), Vmid = −38.1 ± 0.5 mV, z = 3.6 ± 0.2 e+. H, individual cell parameters from fits to steady-state conductance (left and center) or inactivation kinetics at 0 mV (right).
Discussion
Our findings, together with those previously published, show that clustered Kv2.1 is associated with PM:ER MCS, and that the association is conditional and regulated by the Kv2.1 phosphorylation state. This conditional association of Kv2.1 with PM:ER MCS in COS-1 cells is distinct in that it is specific to M phase cells and is not present in interphase cells, as it is in other proliferating cells (e.g. HEK293 cells). Kv2.1 is not expressed in all cell types, and has been most thoroughly characterized in cells that in adults are post-mitotic, such as neurons, and cardiac and skeletal muscle (3). As such, whereas Kv2.1 localizes to and modulates the structure of PM:ER MCS in all cells in which it has been examined, it is not a fundamental component of the PM:ER MCS in all cells. Kv2.1 is expressed in mitotic cells such as smooth muscle (4), pancreatic β cells (68), insulinoma cells such as INS-1 cells (63), rat mesenchymal stem cells (6), and certain types of uterine cancer cells (7). In these latter cell types blockade or knockdown of Kv2.1 suppresses proliferation (6, 7), suggesting a possible reciprocal relationship between cell cycle-dependent regulation of Kv2.1 and cell cycle progression. The lack of robust cell cycle regulation of the functional properties of Kv2.1, at least when expressed in heterologous CHO cells, argues against a model where enhanced M phase phosphorylation impacts gating of Kv2.1 channels and this altered channel activity contributes to the cycle-dependent changes in membrane potential that have been observed in many cell types (69). Future studies may define whether cycle-dependent changes in localization and the phosphorylation state of Kv2.1 impact cell cycle progression in the diverse proliferating cell types that express Kv2.1.
A number of K+ channels play a critical role in cell cycle progression (38). This expands the initially established role of K+ channels as mediators of rapid changes in membrane potential in neurons and other excitable cells (70), to that of important effectors of proliferation in non-excitable cells, and as potential targets and biomarkers for cancer (71–73). In addition to their canonical role in conducting K+ ions across the plasma membrane to regulate membrane potential, K+ concentration, and cell volume, K+ channels play non-canonical roles independent of their ionic conductance (38). The role of Kv2.1 at PM:ER MCS is still unclear, but may lie outside of its function as a voltage-gated K+ channel. Previous studies in HEK293 cells have suggested that most if not all of the Kv2.1 channels clustered at PM:ER MCS may be non-functional (74). Taken together with the finding that Kv2.1 stabilizes and/or enhances PM:ER MCS, as shown here in M phase COS-1 cells, and previously in HEK293 cells and neurons (12, 23) suggests a structural, rather than ion conducting, role for Kv2.1 at these sites. As tools to study functions of ion channel proteins outside of their ionic conductance advance, a theme whereby ion channels have functions supplementary to transmembrane ion flux has begun to emerge, for example, serving as PM scaffolding proteins for intracellular signaling complexes (75). There are well established roles for other PM ion channels that localize to PM:ER MCS, most involving the specialized Ca2+ signaling that occurs at these sites (76). For example, voltage-gated Ca2+ channels are localized to junctional triads in skeletal and cardiac muscle (77), and Ca2+-activated BK K+ channels to sites overlaying subsurface cisternae in neurons (78), and SR in smooth muscle (79). Kv2.1 function is not intrinsically Ca2+-sensitive (1), although Kv2.1 is regulated through changes in phosphorylation state as mediated by Ca2+-dependent protein kinases and phosphatases (18–21). Another distinct aspect of the PM:ER MCS of many cells is that they represent specialized lipid environments (77). Activity of a number of ion channels is regulated through binding to specialized lipids that are highly enriched at PM:ER MCS, such as PIP2 (80). While a comprehensive analysis of Kv2.1 regulation by PIP2 and its molecular determinants has not been performed, PIP2 does prevent the rundown of Kv2.1 seen during excised patch clamp recordings (81), although depletion of PIP2 in intact cells does not affect Kv2.1 function (82); see Ref. 83 for a discussion of conflicting reports. In certain cell types Kv2.1 is associated with noncaveolar lipid rafts, and this association impacts Kv2.1 function (84–87). The presence of Kv2.1 at PM:ER MCS, which have been purported to be enriched in raft-like lipid domains (76), may have a further impact on Kv2.1 function. Our data show that cell cycle-dependent Kv2.1 clustering is mediated by the same determinants (with the exception of Ser-583) that mediate clustering in neurons, suggesting common mechanisms. The overall mechanisms of Kv2.1 clustering are not fully elucidated, although compelling data have been presented for a perimeter fence mechanism (88). Future studies may reveal how changes in the phosphorylation state of Kv2.1 are mechanistically connected to changes in Kv2.1 clustering and association with PM:ER MCS.
Our results reveal novel cell cycle-dependent phosphorylation of a PM protein associated with PM:ER MCS. To date, no specific examples of cell cycle-dependent changes in phosphorylation of PM components of PM:ER MCS, such as demonstrated here for Kv2.1, have been reported. Moreover, PM proteins that exhibit altered localization within the PM during mitosis, as we observe for Kv2.1, have not been reported in mammalian cells. As such Kv2.1 is emerging as having an intriguing role, not only as one of the few known PM components of PM:ER MCS, but also as a unique example of a PM protein whose phosphorylation state, and association with these sites, changes in a cell cycle-dependent manner. A well established PM component of one class of PM:ER MCS is the Orai subunit of the STIM1·Orai complex (89). The STIM1 component of this complex is localized to the ER membrane, and reversibly interacts with PM Orai to form the ion channel that underlies store-operated Ca2+ entry (90). STIM1 also becomes phosphorylated at mitosis, resulting in a loss of interaction with Orai, providing the mechanism for mitotic inhibition of store-operated Ca2+ entry (37). Our data suggest a conditional association of Kv2.1 with PM:ER MCS in COS-1 cells that is regulated by phosphorylation, such that its presence at PM:ER MCS is induced by M phase phosphorylation. As such, cell cycle-dependent phosphorylation events can lead to coordinated dissociation of STIM1 from Orai and disruption of this class of PM:ER MCS (37) and formation/stabilization of Kv2.1-containing PM:ER MCS.
The protein kinases and phosphatases that regulate cell cycle-dependent clustering are not known. Moreover, it is possible that other forms of post-translational modifications of Kv2.1 during M phase could also contribute to the cell cycle-dependent changes in Kv2.1 clustering and association with PM:ER MCS. None of the potential phosphoacceptor amino acids whose mutation leads to loss of Kv2.1 clustering (9), including Ser(P)-586, are predicted CDK phosphosites, as are three of the other sites (Ser(P)-563, Ser(P)-603, and Ser(P)-715; but not Ser(P)-453) that we found are phosphorylated at mitosis. One of these sites (Ser(P)-603) is phosphorylated in neurons and HEK293 cells, as well as in vitro, by the neuronal CDK family member CDK5 (19), an atypical CDK that is not regulated by mitosis. It is not known whether canonical cyclin-dependent CDKs phosphorylate PM substrates, as does CDK5, whose activity is regulated by both cytoplasmic cyclins such as cyclin E (involved in CDK5-mediated Kv2.1 phosphorylation (58)), and by the PM-associated non-cyclin proteins Cdk5R1 (p35) or Cdk5R2 (p39) (91). Roscovitine, an inhibitor of CDK1, CDK2, CDK5, and CDK7, eliminates Kv2.1 phosphorylation at Ser(P)-603, and Kv2.1 clustering, in neurons and HEK293 cells (19). We found that Roscovitine also eliminated the subpopulation of COS-1 cells exhibiting enhanced phosphorylation and clustering of Kv2.1 (data not shown), but as Roscovitine inhibits cell cycle progression (92), and eliminated the subpopulation of M phase COS-1 cells from our cultures, we could not use this pharmacological approach to determine whether these CDK kinases are required for M phase Kv2.1 phosphorylation.
That each of the five Kv2.1 phosphosites analyzed is subjected to cell cycle-dependent phosphorylation suggests widespread changes in phosphorylation of PM proteins may occur at M phase. The cell cycle-dependent phosphorylation and localization of Kv2.1 at PM:ER MCS provides a valuable reporter for characterizing intracellular signaling pathways regulating the phosphorylation state of PM proteins during mitosis, and that serve to couple nuclear events to those at the PM. That the PM Kv2.1 channel is subject to M phase-specific phosphorylation raises questions as to whether other PM proteins may be targets for phosphorylation events that impact PM:ER MCS. It is possible that our findings on cell cycle-dependent phosphorylation and clustering of Kv2.1 may represent an event more common for PM proteins than generally appreciated, as the cell-cycle dependence of Kv2.1 modulation was apparent due to the dramatic phosphorylation-dependent clustering characteristics of Kv2.1, and the availability of anti-Kv2.1 phosphospecific antibodies. The identification of Kv2.1 as a component of PM:ER MCS provides an opportunity to use this PM ion channel as a starting point for studies defining the molecular machinery at these sites and their dynamic regulation during the cell cycle.
Author Contributions
M. M. C. conducted most of the experiments, analyzed the results, and wrote most of the paper. D. C. A. conducted and analyzed electrophysiology experiments, under the supervision of J. T. S. J. S. T. supervised the project, and wrote the paper with M. M. C. and J. T. S.
Acknowledgments
We thank Drs. John Albeck and Michael Paddy at the University of California Davis for help with live cell imaging, which was performed at the University of California Davis MCB Imaging Facility. We thank Dr. David Keller, California State University, Chico, for the INS-1 cells. We thank Shu Ti, Michael Kirmiz, and Naoki Hirasawa for their contributions to the study.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS042225 (to J. S. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- PM
- plasma membrane
- TIRF
- total internal reflection fluorescence
- PRC
- proximal restriction and clustering
- cER
- cortical endoplasmic reticulum
- PIP2
- phosphatidylinositol 4,5-biphosphate
- MCS
- membrane contact sites.
References
- 1.Frech G. C., VanDongen A. M., Schuster G., Brown A. M., and Joho R. H. (1989) A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature 340, 642–645 [DOI] [PubMed] [Google Scholar]
- 2.Trimmer J. S. (1991) Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 10764–10768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drewe J. A., Verma S., Frech G., and Joho R. H. (1992) Distinct spatial and temporal expression patterns of K+ channel mRNAs from different subfamilies. J. Neurosci. 12, 538–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel A. J., Lazdunski M., and Honoré E. (1997) Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 16, 6615–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson D. A., Kuznetsov A., Lopez J. P., Kash S., Ammälä C. E., and Philipson L. H. (2007) Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 6, 229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng X. L., Lau C. P., Lai K., Cheung K. F., Lau G. K., and Li G. R. (2007) Cell cycle-dependent expression of potassium channels and cell proliferation in rat mesenchymal stem cells from bone marrow. Cell Prolif. 40, 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T., and Takimoto K. (2004) Selective expression of HERG and Kv2 channels influences proliferation of uterine cancer cells. Int. J. Oncol. 25, 153–159 [PubMed] [Google Scholar]
- 8.Scannevin R. H., Murakoshi H., Rhodes K. J., and Trimmer J. S. (1996) Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J. Cell Biol. 135, 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim S. T., Antonucci D. E., Scannevin R. H., and Trimmer J. S. (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25, 385–397 [DOI] [PubMed] [Google Scholar]
- 10.Trimmer J. S. (2015) Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85, 238–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du J., Tao-Cheng J. H., Zerfas P., and McBain C. J. (1998) The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84, 37–48 [DOI] [PubMed] [Google Scholar]
- 12.Antonucci D. E., Lim S. T., Vassanelli S., and Trimmer J. S. (2001) Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108, 69–81 [DOI] [PubMed] [Google Scholar]
- 13.King A. N., Manning C. F., and Trimmer J. S. (2014) A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J. Comp. Neurol. 522, 2594–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandikian D., Bocksteins E., Parajuli L. K., Bishop H. I., Cerda O., Shigemoto R., and Trimmer J. S. (2014) Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol. 522, 3555–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prinz W. A. (2014) Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 205, 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohapatra D. P., and Trimmer J. S. (2006) The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J. Neurosci. 26, 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi G., Kleinklaus A. K., Marrion N. V., and Trimmer J. S. (1994) Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J. Biol. Chem. 269, 23204–23211 [PubMed] [Google Scholar]
- 18.Misonou H., Mohapatra D. P., Park E. W., Leung V., Zhen D., Misonou K., Anderson A. E., and Trimmer J. S. (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718 [DOI] [PubMed] [Google Scholar]
- 19.Cerda O., and Trimmer J. S. (2011) Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J. Biol. Chem. 286, 28738–28748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misonou H., Mohapatra D. P., Menegola M., and Trimmer J. S. (2005) Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J. Neurosci. 25, 11184–11193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misonou H., Menegola M., Mohapatra D. P., Guy L. K., Park K. S., and Trimmer J. S. (2006) Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J. Neurosci. 26, 13505–13514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misonou H., Mohapatra D. P., and Trimmer J. S. (2005) Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26, 743–752 [DOI] [PubMed] [Google Scholar]
- 23.Fox P. D., Haberkorn C. J., Akin E. J., Seel P. J., Krapf D., and Tamkun M. M. (2015) Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 128, 2096–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murakoshi H., Shi G., Scannevin R. H., and Trimmer J. S. (1997) Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol. Pharmacol. 52, 821–828 [DOI] [PubMed] [Google Scholar]
- 25.Ikematsu N., Dallas M. L., Ross F. A., Lewis R. W., Rafferty J. N., David J. A., Suman R., Peers C., Hardie D. G., and Evans A. M. (2011) Phosphorylation of the voltage-gated potassium channel Kv2.1 by AMP-activated protein kinase regulates membrane excitability. Proc. Natl. Acad. Sci. U.S.A. 108, 18132–18137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baver S. B., and O'Connell K. M. (2012) The C-terminus of neuronal Kv2.1 channels is required for channel localization and targeting but not for NMDA-receptor-mediated regulation of channel function. Neuroscience 217, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redman P. T., He K., Hartnett K. A., Jefferson B. S., Hu L., Rosenberg P. A., Levitan E. S., and Aizenman E. (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. U.S.A. 104, 3568–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song M. Y., Hong C., Bae S. H., So I., and Park K. S. (2012) Dynamic modulation of the Kv2.1 channel by SRC-dependent tyrosine phosphorylation. J. Proteome Res. 11, 1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trimmer J. S., and Misonou H. (2015) Phosphorylation of voltage-gated ion channels. in Handbook of Ion Channels (Zheng J., and Trudeau M. C., eds) pp. 531–544, CRC Press, Boca Raton, FL [Google Scholar]
- 30.Medema R. H., and Lindqvist A. (2011) Boosting and suppressing mitotic phosphorylation. Trends Biochem. Sci. 36, 578–584 [DOI] [PubMed] [Google Scholar]
- 31.Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 32.McCullough S., and Lucocq J. (2005) Endoplasmic reticulum positioning and partitioning in mitotic HeLa cells. J. Anat. 206, 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Güttinger S., Laurell E., and Kutay U. (2009) Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 10, 178–191 [DOI] [PubMed] [Google Scholar]
- 34.Preisinger C., and Barr F. A. (2001) Signaling pathways regulating Golgi structure and function. Sci STKE 2001, pe38. [DOI] [PubMed] [Google Scholar]
- 35.English A. R., and Voeltz G. K. (2013) Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 5, a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smyth J. T., Beg A. M., Wu S., Putney J. W. Jr., and Rusan N. M. (2012) Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr. Biol. 22, 1487–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smyth J. T., Petranka J. G., Boyles R. R., DeHaven W. I., Fukushima M., Johnson K. L., Williams J. G., and Putney J. W. Jr. (2009) Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat. Cell Biol. 11, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urrego D., Tomczak A. P., Zahed F., Stühmer W., and Pardo L. A. (2014) Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouadid-Ahidouch H., Le Bourhis X., Roudbaraki M., Toillon R. A., Delcourt P., and Prevarskaya N. (2001) Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K+ channel. Receptors Channels 7, 345–356 [PubMed] [Google Scholar]
- 40.Ouadid-Ahidouch H., Roudbaraki M., Ahidouch A., Delcourt P., and Prevarskaya N. (2004) Cell-cycle-dependent expression of the large Ca2+-activated K+ channels in breast cancer cells. Biochem. Biophys. Res. Commun. 316, 244–251 [DOI] [PubMed] [Google Scholar]
- 41.Ouadid-Ahidouch H., Roudbaraki M., Delcourt P., Ahidouch A., Joury N., and Prevarskaya N. (2004) Functional and molecular identification of intermediate-conductance Ca(2+)-activated K+ channels in breast cancer cells: association with cell cycle progression. Am. J. Physiol. Cell Physiol. 287, C125–134 [DOI] [PubMed] [Google Scholar]
- 42.Zheng Y. J., Furukawa T., Ogura T., Tajimi K., and Inagaki N. (2002) M phase-specific expression and phosphorylation-dependent ubiquitination of the ClC-2 channel. J. Biol. Chem. 277, 32268–32273 [DOI] [PubMed] [Google Scholar]
- 43.Huang X., Dubuc A. M., Hashizume R., Berg J., He Y., Wang J., Chiang C., Cooper M. K., Northcott P. A., Taylor M. D., Barnes M. J., Tihan T., Chen J., Hackett C. S., Weiss W. A., James C. D., Rowitch D. H., Shuman M. A., Jan Y. N., and Jan L. Y. (2012) Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 26, 1780–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohapatra D. P., Siino D. F., and Trimmer J. S. (2008) Interdomain cytoplasmic interactions govern the intracellular trafficking, gating, and modulation of the Kv2.1 channel. J. Neurosci. 28, 4982–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park K. S., Mohapatra D. P., Misonou H., and Trimmer J. S. (2006) Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313, 976–979 [DOI] [PubMed] [Google Scholar]
- 46.Bekele-Arcuri Z., Matos M. F., Manganas L., Strassle B. W., Monaghan M. M., Rhodes K. J., and Trimmer J. S. (1996) Generation and characterization of subtype-specific monoclonal antibodies to K+ channel α- and β-subunit polypeptides. Neuropharmacology 35, 851–865 [DOI] [PubMed] [Google Scholar]
- 47.Lee B. S., Gunn R. B., and Kopito R. R. (1991) Functional differences among nonerythroid anion exchangers expressed in a transfected human cell line. J. Biol. Chem. 266, 11448–11454 [PubMed] [Google Scholar]
- 48.Tanaka M., and Herr W. (1990) Differential transcriptional activation by Oct-1 and Oct-2: interdependent activation domains induce Oct-2 phosphorylation. Cell 60, 375–386 [DOI] [PubMed] [Google Scholar]
- 49.Nakahira K., Shi G., Rhodes K. J., and Trimmer J. S. (1996) Selective interaction of voltage-gated K+ channel β-subunits with α-subunits. J. Biol. Chem. 271, 7084–7089 [DOI] [PubMed] [Google Scholar]
- 50.Trapani J. G., and Korn S. J. (2003) Control of ion channel expression for patch clamp recordings using an inducible expression system in mammalian cell lines. BMC Neurosci. 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tilley D. C., Eum K. S., Fletcher-Taylor S., Austin D. C., Dupré C., Patrón L. A., Garcia R. L., Lam K., Yarov-Yarovoy V., Cohen B. E., and Sack J. T. (2014) Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc. Natl. Acad. Sci. U.S.A. 111, E4789–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manganas L. N., and Trimmer J. S. (2000) Subunit composition determines Kv1 potassium channel surface expression. J. Biol. Chem. 275, 29685–29693 [DOI] [PubMed] [Google Scholar]
- 53.Ukomadu C., Zhou J., Sigworth F. J., and Agnew W. S. (1992) muI Na+ channels expressed transiently in human embryonic kidney cells: biochemical and biophysical properties. Neuron 8, 663–676 [DOI] [PubMed] [Google Scholar]
- 54.Ward G. E., Garbers D. L., and Vacquier V. D. (1985) Effects of extracellular egg factors on sperm guanylate cyclase. Science 227, 768–770 [DOI] [PubMed] [Google Scholar]
- 55.Sack J. T., and Aldrich R. W. (2006) Binding of a gating modifier toxin induces intersubunit cooperativity early in the Shaker K channel's activation pathway. J. Gen. Physiol. 128, 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klemic K. G., Shieh C. C., Kirsch G. E., and Jones S. W. (1998) Inactivation of Kv2.1 potassium channels. Biophys. J. 74, 1779–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hendzel M. J., Wei Y., Mancini M. A., Van Hooser A., Ranalli T., Brinkley B. R., Bazett-Jones D. P., and Allis C. D. (1997) Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106, 348–360 [DOI] [PubMed] [Google Scholar]
- 58.Shah N. H., Schulien A. J., Clemens K., Aizenman T. D., Hageman T. M., Wills Z. P., and Aizenman E. (2014) Cyclin e1 regulates Kv2.1 channel phosphorylation and localization in neuronal ischemia. J. Neurosci. 34, 4326–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrington J., Zhou Y. P., Bugianesi R. M., Dulski P. M., Feng Y., Warren V. A., Smith M. M., Kohler M. G., Garsky V. M., Sanchez M., Wagner M., Raphaelli K., Banerjee P., Ahaghotu C., Wunderler D., Priest B. T., Mehl J. T., Garcia M. L., McManus O. B., Kaczorowski G. J., and Slaughter R. S. (2006) Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes 55, 1034–1042 [DOI] [PubMed] [Google Scholar]
- 60.Deutsch E., Weigel A. V., Akin E. J., Fox P., Hansen G., Haberkorn C. J., Loftus R., Krapf D., and Tamkun M. M. (2012) Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol. Biol. Cell 23, 2917–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park K. S., Mohapatra D. P., and Trimmer J. S. (2007) Proteomic analyses of Kv2.1 channel phosphorylation sites determining cell background specific differences in function. Channels 1, 59–61 [DOI] [PubMed] [Google Scholar]
- 62.Li X. N., Herrington J., Petrov A., Ge L., Eiermann G., Xiong Y., Jensen M. V., Hohmeier H. E., Newgard C. B., Garcia M. L., Wagner M., Zhang B. B., Thornberry N. A., Howard A. D., Kaczorowski G. J., and Zhou Y. P. (2013) The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J. Pharmacol. Exp. Ther. 344, 407–416 [DOI] [PubMed] [Google Scholar]
- 63.Su J., Yu H., Lenka N., Hescheler J., and Ullrich S. (2001) The expression and regulation of depolarization-activated K+ channels in the insulin-secreting cell line INS-1. Pflugers Arch. 442, 49–56 [DOI] [PubMed] [Google Scholar]
- 64.Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., and Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 [DOI] [PubMed] [Google Scholar]
- 65.Skelin M., Rupnik M., and Cencic A. (2010) Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX 27, 105–113 [DOI] [PubMed] [Google Scholar]
- 66.Bocksteins E., Raes A. L., Van de Vijver G., Bruyns T., Van Bogaert P. P., and Snyders D. J. (2009) Kv2.1 and silent Kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am. J. Physiol. Cell Physiol. 296, C1271–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Misonou H., Thompson S. M., and Cai X. (2008) Dynamic regulation of the Kv2.1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J. Neurosci. 28, 8529–8538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roe M. W., Worley J. F. 3rd, Mittal A. A., Kuznetsov A., DasGupta S., Mertz R. J., Witherspoon S. M. 3rd, Blair N., Lancaster M. E., McIntyre M. S., Shehee W. R., Dukes I. D., and Philipson L. H. (1996) Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J. Biol. Chem. 271, 32241–32246 [DOI] [PubMed] [Google Scholar]
- 69.Levin M. (2014) Molecular bioelectricity: how endogenous voltage potentials control cell behavior and instruct pattern regulation in vivo. Mol. Biol. Cell 25, 3835–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hille B. (2001) Ionic channels of excitable membranes, 3 Ed., Sinauer, Sunderland, MA [Google Scholar]
- 71.Huang X., and Jan L. Y. (2014) Targeting potassium channels in cancer. J. Cell Biol. 206, 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hervé J. C. (2015) Membrane channels and transporters in cancers. Biochim. Biophys. Acta 1848, 2473–2476 [DOI] [PubMed] [Google Scholar]
- 73.Lastraioli E., Iorio J., and Arcangeli A. (2015) Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 1848, 2685–2702 [DOI] [PubMed] [Google Scholar]
- 74.O'Connell K. M., Loftus R., and Tamkun M. M. (2010) Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl. Acad. Sci. U.S.A. 107, 12351–12356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee A., Fakler B., Kaczmarek L. K., and Isom L. L. (2014) More than a pore: ion channel signaling complexes. J. Neurosci. 34, 15159–15169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Helle S. C., Kanfer G., Kolar K., Lang A., Michel A. H., and Kornmann B. (2013) Organization and function of membrane contact sites. Biochim. Biophys. Acta 1833, 2526–2541 [DOI] [PubMed] [Google Scholar]
- 77.Henne W. M., Liou J., and Emr S. D. (2015) Molecular mechanisms of inter-organelle ER-PM contact sites. Curr. Opin. Cell Biol. 35, 123–130 [DOI] [PubMed] [Google Scholar]
- 78.Kaufmann W. A., Ferraguti F., Fukazawa Y., Kasugai Y., Shigemoto R., Laake P., Sexton J. A., Ruth P., Wietzorrek G., Knaus H. G., Storm J. F., and Ottersen O. P. (2009) Large-conductance calcium-activated potassium channels in purkinje cell plasma membranes are clustered at sites of hypolemmal microdomains. J. Comp. Neurol. 515, 215–230 [DOI] [PubMed] [Google Scholar]
- 79.Wellman G. C., and Nelson M. T. (2003) Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium 34, 211–229 [DOI] [PubMed] [Google Scholar]
- 80.Hille B., Dickson E. J., Kruse M., Vivas O., and Suh B. C. (2015) Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 1851, 844–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hilgemann D. W., Feng S., and Nasuhoglu C. (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, re19. [DOI] [PubMed] [Google Scholar]
- 82.Kruse M., Hammond G. R., and Hille B. (2012) Regulation of voltage-gated potassium channels by PI(4,5)P2. J. Gen. Physiol. 140, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hilgemann D. W. (2012) Fitting K(V) potassium channels into the PIP(2) puzzle: Hille group connects dots between illustrious HH groups. J. Gen. Physiol. 140, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martens J. R., Navarro-Polanco R., Coppock E. A., Nishiyama A., Parshley L., Grobaski T. D., and Tamkun M. M. (2000) Differential targeting of Shaker-like potassium channels to lipid rafts. J. Biol. Chem. 275, 7443–7446 [DOI] [PubMed] [Google Scholar]
- 85.Martens J. R., Sakamoto N., Sullivan S. A., Grobaski T. D., and Tamkun M. M. (2001) Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J. Biol. Chem. 276, 8409–8414 [DOI] [PubMed] [Google Scholar]
- 86.Xia F., Gao X., Kwan E., Lam P. P., Chan L., Sy K., Sheu L., Wheeler M. B., Gaisano H. Y., and Tsushima R. G. (2004) Disruption of pancreatic beta-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J. Biol. Chem. 279, 24685–24691 [DOI] [PubMed] [Google Scholar]
- 87.Wu X., Hernandez-Enriquez B., Banas M., Xu R., and Sesti F. (2013) Molecular mechanisms underlying the apoptotic effect of KCNB1 K+ channel oxidation. J. Biol. Chem. 288, 4128–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O'Connell K. M., Rolig A. S., Whitesell J. D., and Tamkun M. M. (2006) Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J. Neurosci. 26, 9609–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prakriya M. (2013) Store-operated Orai channels: structure and function. Curr. Top. Membr. 71, 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soboloff J., Rothberg B. S., Madesh M., and Gill D. L. (2012) STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Malumbres M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cicenas J., Kalyan K., Sorokinas A., Stankunas E., Levy J., Meskinyte I., Stankevicius V., Kaupinis A., and Valius M. (2015) Roscovitine in cancer and other diseases. Ann. Transl. Med. 3, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]