

Background: Activation of the immune response following pathogen infection appears to play a critical role in the elicitation of neurodegeneration.
Results: P. aeruginosa infection induced neurodegeneration, and mutation of the maternal effect sterile gene mes-1 resulted in resistance to P. aeruginosa-induced neurodegeneration in C. elegans.
Conclusion: Germ line loss results in resistance to P. aeruginosa-induced neurodegeneration.
Significance: Our results reveal that pathogen infection results in neurodegeneration in C. elegans.
Keywords: Caenorhabditis elegans (C. elegans), host-pathogen interaction, infection, innate immunity, neurodegeneration, pathogen-induced neurodegeneration, P. aeruginosa infection, neurodegeneration, germ line, mes-1
Abstract
Increasing evidence indicates that immune responses to microbial infections may contribute to neurodegenerative diseases. Here, we show that Pseudomonas aeruginosa infection of Caenorhabditis elegans causes a number of neural changes that are hallmarks of neurodegeneration. Using an unbiased genetic screen to identify genes involved in the control of P. aeruginosa-induced neurodegeneration, we identified mes-1, which encodes a receptor tyrosine kinase-like protein that is required for unequal cell divisions in the early embryonic germ line. We showed that sterile but not fertile mes-1 animals were resistant to neurodegeneration induced by P. aeruginosa infection. Similar results were observed using animals carrying a mutation in the maternal effect gene pgl-1, which is required for postembryonic germ line development, and the germ line-deficient strains glp-1 and glp-4. Additional studies indicated that the FOXO transcription factor DAF-16 is required for resistance to P. aeruginosa-induced neurodegeneration in germ line-deficient strains. Thus, our results demonstrate that P. aeruginosa infection results in neurodegeneration phenotypes in C. elegans that are controlled by the germ line in a cell-nonautonomous manner.
Introduction
Neurodegenerative diseases represent a serious health problem in modern society. Increasing evidence indicates that immune responses in humans may play a critical role in the elicitation of neurodegeneration (1–3), but the mechanisms underlying the process remain obscure. Here, we used the nematode Caenorhabditis elegans to investigate the potential relationship between bacterial infection and neurodegeneration. C. elegans was chosen as a model genetic organism because of a number of traits that facilitate the study of neurodegeneration, including a hermaphroditic lifestyle, a short 2–3-week life span, and nervous system simplicity. Indeed, the C. elegans nervous system consists of only 302 neurons, which represent most types of neurons in the mammalian brain, including sensory neurons, motor neurons, and interneurons (4). Because development is reproducible among individuals, each neuron can be individually identified (5). Moreover, the entire connectivity of the C. elegans nervous system has been reconstructed by serial electron micrographs (5).
It is well known that aging can induce neurodegeneration in C. elegans (6–9). For example, C. elegans touch receptor neurons exhibit morphological changes that are associated with neurodegeneration during aging, including the accumulation of novel neuronal outgrowths and decline in synaptic integrity (9). In addition, many environmental contaminants, including metals, insecticides, pesticides, and fungicides, also cause morphological changes in the nervous system of C. elegans that result in neurodegeneration (10–17). Previous studies have shown that at least seven sensory neurons play an important role in controlling innate immunity against infections by Pseudomonas aeruginosa in C. elegans (18–20); however, whether the pathogen affects the rate of neurodegeneration remains unknown.
Here, we first investigated whether P. aeruginosa infection causes morphological changes in the sensory neurons of C. elegans that are associated with neurodegeneration. We found that infected animals exhibited a number of neural changes that are hallmarks of neurodegeneration. To identify genes that when mutated may enhance resistance to P. aeruginosa-induced neurodegeneration, we conducted a forward genetic analysis of nematodes exhibiting enhanced resistance to killing by P. aeruginosa. We isolated a strain carrying a deletion in mes-1, which was resistant to both P. aeruginosa-mediated killing and P. aeruginosa-induced neurodegeneration. The mes-1 gene encodes a receptor tyrosine kinase-like protein that is required for unequal cell divisions in the early embryonic germ line. Thus, our findings indicate that the survival of neuronal cells during exposure to bacterial pathogens is controlled by the C. elegans germ line in a cell-nonautonomous manner.
Experimental Procedures
Nematode and Bacterial Strains
The C. elegans strains used in this study were wild-type N2 Bristol, OH4168 otIs151 (Pceh-36::RFP + rol-6(su1006));otEx2419 (Pgcy-1::GFP + unc-122::GFP), CX5974 kyIs262 (unc-86::myr GFP + odr-1::RFP) IV, OH3192 ntIs1(Pgcy-5::gfp + lin-15(+)), CZ1200 juIs76 (Punc-25::gfp + lin-15(+)), SS149 mes-1(bn7), and SS579 pgl-1(bn101), which were obtained from the Caenorhabditis Genetics Center (supported by the National Institutes of Health National Center for Research Resources). Strains AY109 mes-1(bn7);ntIs1(Pgcy-5::gfp + lin-15(+)), AY110 pgl-1(bn101);ntIs1(Pgcy-5::gfp + lin-15(+)), AY111 glp-1(e2141);ntIs1(Pgcy-5::gfp + lin-15(+)), and AY112 glp-4(bn2);ntIs1(Pgcy-5::gfp + lin-15(+)) were constructed using standard genetic manipulation techniques. The rescue strain AY113 was constructed by injecting Pmes-1::mes-1 and co-injecting the marker Punc-122::gfp into ac319 animals. C. elegans were grown and maintained at 20 °C on plates containing nematode growth medium (NGM)2 that were seeded with Escherichia coli OP50 as a food source as described previously (21) unless otherwise mentioned. The bacterial strains used were E. coli OP50 and P. aeruginosa PA14. E. coli and P. aeruginosa cultures were grown in Luria-Bertani (LB) broth overnight at 37 °C.
C. elegans Killing Assay
Gravid adult nematodes were allowed to lay eggs on NGM plates for 3 h at 25 °C to obtain synchronized animals. The bacterial lawns used for the C. elegans killing assays were prepared by placing a 30-μl drop of an overnight culture of P. aeruginosa on modified NGM agar medium (0.35% instead of 0.25% peptone) in 3.5-cm diameter plates. The plates were incubated overnight at 37 °C and cooled to room temperature for at least 1 h before seeding with synchronized young adult animals. The killing assay was performed at 25 °C, and live animals were transferred daily to fresh plates. Young adult animals were used in all cases unless otherwise indicated. The scored animals were considered dead if they failed to respond to the gentle touch of a platinum wire. Three replicates were analyzed in each experiment.
Genetic Screen of Mutants Resistant to P. aeruginosa
Ethylmethanesulfonate mutagenesis was performed using the OH3192 strain, which expresses GFP in the ASER neuron. Mutant isolation was performed as described previously (22). Briefly, early L4 larvae of OH3192 were treated with 50 mm ethylmethanesulfonate for 4 h and recovered for 2 h. Approximately 10 late L4 larvae (P0 generation) were transferred to large Petri dishes and allowed to lay ∼100 eggs of F1 progeny. The P0s were then removed, and the F1s were allowed to grow to adulthood. After 1 day of egg laying, the F1s and any hatched F2 progeny were removed by washing. The eggs of the F2s, which stuck to the plates, were allowed to develop to L4 larvae on E. coli OP50, transferred to plates containing P. aeruginosa PA14, and incubated for 72 h at 25 °C. Live worms were collected and cultured singly on NGM plates seeded with E. coli OP50. These worms were allowed to lay eggs overnight. Mixtures of the eggs and hatched L1 larvae were transferred to fresh NGM plates seeded with E. coli OP50. After incubation for 4–5 days, the worms were lysed using a solution of sodium hydroxide and bleach (5:2 ratio) and washed, and the eggs were synchronized for 22 h in S basal liquid medium at room temperature. The synchronized L1 larvae were placed on NGM plates seeded with E. coli OP50 and grown at 20 °C until the animals had reached the L4 larvae or young adult stage. The animals were collected and used for the survival and neurodegeneration assays on P. aeruginosa PA14.
C. elegans Neurodegeneration Assay
The bacterial lawns used for the neurodegeneration assay were prepared as indicated below.
Partial Lawn
The bacterial lawns used for the neurodegeneration assay were prepared by placing 50 μl of an overnight culture of PA14 on modified NGM agar medium in 3.5-cm diameter plates.
Full Lawn
The full lawn plates used for the C. elegans neurodegeneration assay were prepared by spreading 50 μl of an overnight culture of PA14 over the entire surface of modified NGM agar medium in 3.5-cm diameter plates.
P. aeruginosa PA14 was incubated overnight at 37 °C and then incubated at 25 °C for 24 h before seeding with synchronized animals. L4 larvae or young adult nematodes were transferred to PA14 plates and grown for 30 h at 25 °C.
The animals were mounted onto agar pads and visualized using a Zeiss Axioscope microscope or Leica M165 FC microscope. The worms were analyzed for a number of morphological changes that are hallmarks of neurodegeneration, including beaded dendrites, wavy dendrites, branching dendrites, and changes in the soma described as branching. A worm was scored as exhibiting neurodegeneration when the neurons displayed the aforementioned changes. Three independent experiments were performed, and ∼60 animals were used for each experiment.
Chemotaxis Assay
Chemotaxis was studied as described previously (23, 24). Briefly, 9-cm assay plates (5 mm potassium phosphate (pH 6.0), 1 mm CaCl2, 1 mm MgSO4, and 2% agar) with a gradient of NaCl were used. The gradient was created by placing overnight an agar plug excised from a plate containing 100 mmol/liter NaCl. Shortly before the chemotaxis assay, the NaCl plug was removed, and 1 μl of 0.5 mol/liter sodium azide was spotted onto the same position to anesthetize the animals. As a control, sodium azide was spotted at a position ∼4 cm away from the center of the NaCl gradient. Synchronized young adult nematodes were transferred to plates seeded with E. coli or P. aeruginosa for 30 h at 25 °C. After the incubation, ∼100 animals/condition were placed equidistant (∼3 cm) from the two spots of the aforementioned 9-cm assay plates. The animals were allowed to move freely for 30 min at 25 °C. The assay plates were then chilled to 4 °C, and the number of animals around each spot was counted. The chemotaxis index was calculated as (A − B)/(N − C), where A is the number of nematodes within 1 cm of the peak of the salt gradient, B is the number of nematodes within 1 cm of the control spot, N is the number of all nematodes on the assay plate, and C is the number of animals that did not move from the original region. All of the assays were conducted independently in triplicate.
RNA Interference
RNA interference was used to generate loss-of-function RNAi phenotypes by feeding nematodes E. coli strain HT115 expressing double-stranded RNA homologous to the target gene daf-16. E. coli HT115 strain was grown in LB broth containing ampicillin (100 μg/ml) at 37 °C overnight and plated onto NGM containing 100 μg/ml ampicillin and 5 mm isopropyl 1-thio-β-d-galactopyranoside. The dsRNA-expressing bacteria were allowed to grow overnight at 37 °C. L2 or L3 larval animals were placed on RNAi or vector control plates for 3 days at 15 °C until the nematodes became gravid. Gravid adults were then transferred to fresh RNAi-expressing bacterial lawns and allowed to lay eggs at 25 °C for 2 h to synchronize a second-generation RNAi population. After removing the adults, the hatched eggs and nematodes were grown at 25 °C for 60 h. unc-22 RNAi served as a control.
Statistical Analysis
Animal survival was plotted as a nonlinear regression curve using Prism (version 4.00) (GraphPad Software, La Jolla, CA). The survival curves were considered significantly different from the control when the p values were <0.05. Prism uses the product limit or Kaplan-Meier method to calculate survival fractions and the log-rank test, which is equivalent to the Mantel-Haenszel test, to compare survival curves. A two-sample Student's t test for independent samples was used to analyze the neurodegeneration rate, and p < 0.05 was considered significant. All experiments were repeated at least three times unless otherwise indicated.
Results
P. aeruginosa Infection Induces Neurodegeneration in C. elegans
To investigate the effect of bacterial infection on sensory neurons, we infected animals expressing RFP or GFP in amphid neurons with P. aeruginosa strain PA14. We found that the sensory neurons of animals infected with P. aeruginosa exhibited a number of morphological changes that can be considered hallmarks of neurodegeneration. These morphological changes included beaded dendrites, wavy dendrites, branching dendrites, and changes in the soma described as branching (Fig. 1A). Strain OH4168, which expresses a ceh-36::rfp transgene that allows visualization of ASE neurons (25), exhibited a number of morphological changes associated with neurodegeneration 30 h after infection with P. aeruginosa (Fig. 1B). These changes were confirmed using strain OH3192, which expresses GFP only in the ASER neuron driven by the promoter of the gcy-5 gene (26, 27) (Fig. 1B). Similar changes were induced by P. aeruginosa infection in strain CX5974 (Fig. 1C). This strain uses the odr-1 promoter to express RFP in AWC, AWB, and I1 neurons (28), but only AWC can be observed in the confocal plane. None of these changes were observed when the animals were exposed for 30 h to E. coli OP50 (Fig. 1, B and C), which is the normal food source of C. elegans in the laboratory. These results indicated that exposure to pathogenic bacteria resulted in neurodegeneration.
FIGURE 1.

Neurodegeneration phenotypes induced by P. aeruginosa infection. A, summary of the morphological changes observed in animals infected with P. aeruginosa. B, images of ASE neurons of strain OH4168 and the ASER neuron of strain OH3192 exposed to control E. coli or P. aeruginosa. C, images of the AWC neuron of strain CX5974 exposed to control E. coli or P. aeruginosa. Different morphological changes in the neurons are indicated by white arrows. A–C, synchronized L4 nematodes were cultured on plates containing full lawns of P. aeruginosa for 30 h at 25 °C and then visualized using a Leica M165 FC stereomicroscope. Scale bars, 50 μm. D, chemotaxis of L4 nematodes exposed to E. coli or P. aeruginosa. Asterisks indicate statistical significance (**, p < 0.01). The data represent the results of three independent experiments. Bars, mean ± S.E. (error bars).
Although Fig. 1, B and C, shows important changes related to neurodegeneration, we performed a functional assay to address the function of ASE neurons after infection with P. aeruginosa. We took advantage of chemotaxis toward NaCl, which is uniquely controlled by ASE sensory neurons (23). Thus, we examined the chemotaxis toward NaCl of strain OH3192 cultured on E. coli or P. aeruginosa plates. As shown in Fig. 1D, P. aeruginosa infection caused a significant decrease in chemotaxis toward NaCl, which indicated that the infection not only affected neural morphology but also impaired ASE function.
Chemosensation is used by C. elegans to search for food or to avoid noxious conditions (29). Indeed, pathogen avoidance is part of the defense response of C. elegans against P. aeruginosa infection (20, 30–32). To assess whether pathogen avoidance protects C. elegans against neurodegeneration induced by P. aeruginosa infection, assays were performed in plates containing partial or full lawns of P. aeruginosa. By infecting the animals on full lawns of P. aeruginosa, the ability to reduce pathogen exposure by leaving the lawn is eliminated. Consistent with the idea that pathogen avoidance protects against P. aeruginosa infection, animals grown on full lawns of P. aeruginosa exhibited significantly increased neurodegeneration compared with those grown on partial lawns (p < 0.01) (Fig. 2A). The exposure of animals on partial lawns of P. aeruginosa resulted in only moderate neurodegeneration (Fig. 2A).
FIGURE 2.
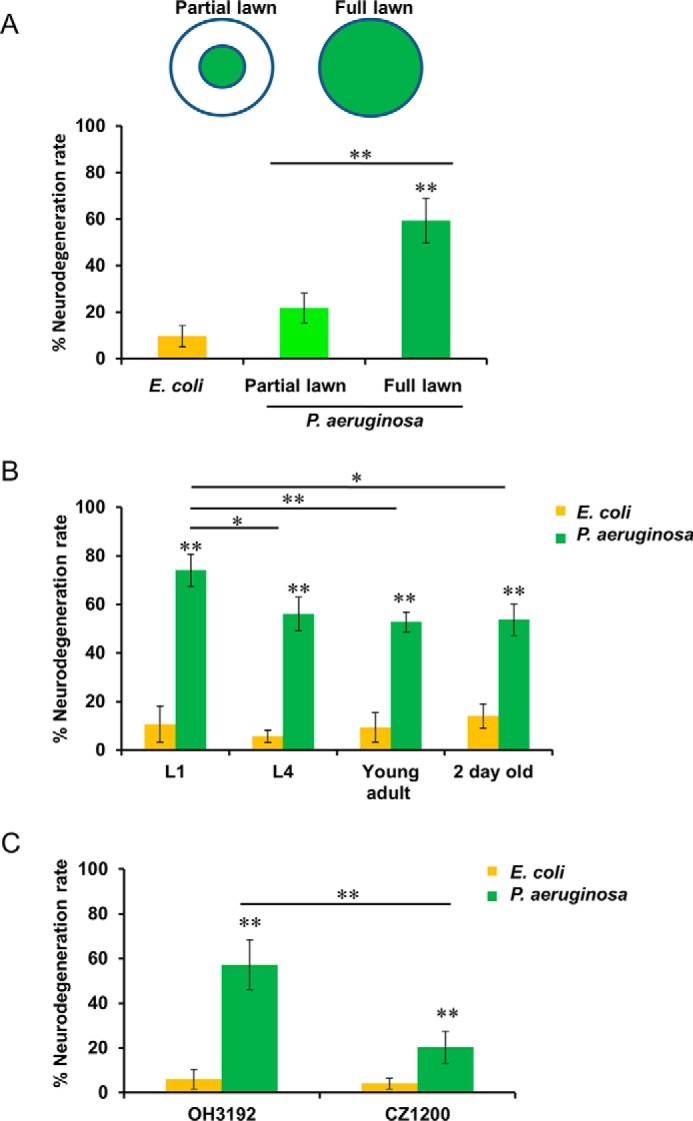
Effect of pathogen avoidance and development on neurodegeneration caused by P. aeruginosa infection. A, neurodegeneration rates in OH3192 L4 nematodes cultured on full lawns or partial lawns or P. aeruginosa for 30 h at 25 °C. B, neurodegeneration rates in OH3192 nematodes placed on full lawn plates of P. aeruginosa or E. coli for 30 h at 25 °C, starting at different developmental stages. C, neurodegeneration rates in OH3192 or CZ1200 young adult nematodes cultured on full lawn plates of P. aeruginosa or E. coli for 30 h at 25 °C. Asterisks indicate significant differences (*, p < 0.05; **, p < 0.01). The data represent the results of three independent experiments (n = 60 ± 5 animals). Bars, mean ± S.E. (error bars).
To examine the susceptibility of animals at different developmental stages to P. aeruginosa-induced neurodegeneration, L1 larvae, L4 larvae, young adults, or 2-day-old nematodes were infected. P. aeruginosa-induced neurodegeneration was increased in L1 larvae compared with L4 larvae, young adults, or 2-day-old animals (Fig. 2B), suggesting that larval neurons may be more susceptible to P. aeruginosa-mediated neurodegeneration than adult neurons. We also examined the susceptibility of motor neurons to pathogen-induced neurodegeneration. P. aeruginosa-induced neurodegeneration in strain OH3192 was compared with that induced in CZ1200 animals, which express GFP in GABAergic motor neurons (Fig. 2C). The results indicated that sensory neurons were more sensitive to pathogen-induced neurodegeneration than motor neurons.
We also assessed whether osmotic stress could induce neurodegeneration. Similarly to P. aeruginosa exposure, osmotic stress caused by exposure to 450 or 500 mm NaCl killed 50–60% of the animals after 48 h of exposure (Fig. 3A). Although osmotic stress initially killed C. elegans more rapidly than P. aeruginosa (Fig. 3A), it did not induce significant neurodegeneration (Fig. 3B). These results indicated that neurodegeneration induced by P. aeruginosa infection was not due to the premature death of the animals.
FIGURE 3.

Effect of osmotic stress on survival and neurodegeneration. A, survival curves of OH3192 young adults grown on standard 50 mm NaCl or increasing concentrations of NaCl. B, neurodegeneration rates in OH3192 young adults cultured on plates containing 50 mm NaCl or increasing concentrations of NaCl for 30 h at 25 °C followed by visualization using a Leica M165 FC stereomicroscope. Asterisks indicate significant differences (**, p < 0.01; NS, not significant). The data represent the results of three independent experiments (n = 60 ± 5 animals). Bars, mean ± S.E. (error bars).
Isolation of C. elegans Mutants That Are Resistant to P. aeruginosa-induced Neurodegeneration
To identify C. elegans mutants that were resistant to P. aeruginosa-induced neurodegeneration, we used a forward genetic screen. First, 340 mutagenized OH3192 nematodes were used to isolate mutants with enhanced resistance to pathogen infection. Of the five initial mutants isolated, only three exhibited significant resistance to P. aeruginosa-mediated killing after four backcrosses (Fig. 4, A–E). Mutant 319 was named ac319 and chosen for further analysis based on its strong resistance to pathogen infection. In addition, compared with other mutants, the ac319 mutant exhibited the most highly significant enhanced resistance to P. aeruginosa-induced neurodegeneration (Fig. 4F). Interestingly, among all of the isolated mutants, the ac319 mutant segregated both fertile animals and sterile animals that did not exhibit a germ line (Fig. 5A). Because a number of germ line-deficient mutants are temperature-sensitive (33, 34), we decided to assess the sterility of ac319 animals at 15, 20, or 25 °C individually. We found that the percentage of sterile nematodes increased with the rising temperature (Fig. 5B).
FIGURE 4.

Isolation of mutants that are resistant to P. aeruginosa-mediated killing and P. aeruginosa-induced neurodegeneration. A–E, survival curves of mutagenized OH3192 strains exposed to P. aeruginosa. Young adult animals were used, and three replicates were analyzed in each experiment. F, neurodegeneration rates in young adult mutants cultured on full lawn plates of P. aeruginosa for 30 h at 25 °C. Asterisks indicate significant differences (*, p < 0.05; **, p < 0.01). The data represent the results of three independent experiments (n = 60 ± 5 animals). Bars, mean ± S.E. (error bars).
FIGURE 5.

Mutant ac319 exhibits a maternal effect sterile phenotype. A, images of 1-day-old fertile and sterile ac319 animals. The normal germ line in ac319 fertile animals is indicated by an arrow. Scale bar, 50 μm. B, eggs were cultured at 15, 20, or 25 °C until the animals reached the gravid adult stage and were scored for sterility. The graph represents the combined results of three independent experiments. Bars, mean ± S.E. (error bars). C, 12 fertile ac319 young adult animals were individually cultured for 24 h, and their progeny were scored for sterility after growth for 3 days at 20 °C.
To confirm that the aforementioned sterile phenotype of the ac319 mutants was not due to heterozygosity, we analyzed the percentage of germ line-deficient animals among the progeny of fertile ac319 mutants. We found that 40–50% of the progeny of the ac319 animals were sterile (Fig. 5C), indicating that a maternal effect sterile mutation caused the offspring of homozygous mutant mothers to develop into sterile adults. Further analysis revealed that the ac319 mutants with normal germ lines survived as long as the wild-type animals following exposure to P. aeruginosa, whereas the germ line-deficient ac319 mutants exhibited greater resistance to P. aeruginosa infection (p < 0.01) (Fig. 6A). The germ line-deficient animals also displayed a strong resistance to P. aeruginosa-induced neurodegeneration, whereas the fertile animals demonstrated neurodegeneration comparable with that of control animals (Fig. 6, B and C). These results suggested that germ line-deficient animals might be more resistant to P. aeruginosa-induced neurodegeneration.
FIGURE 6.
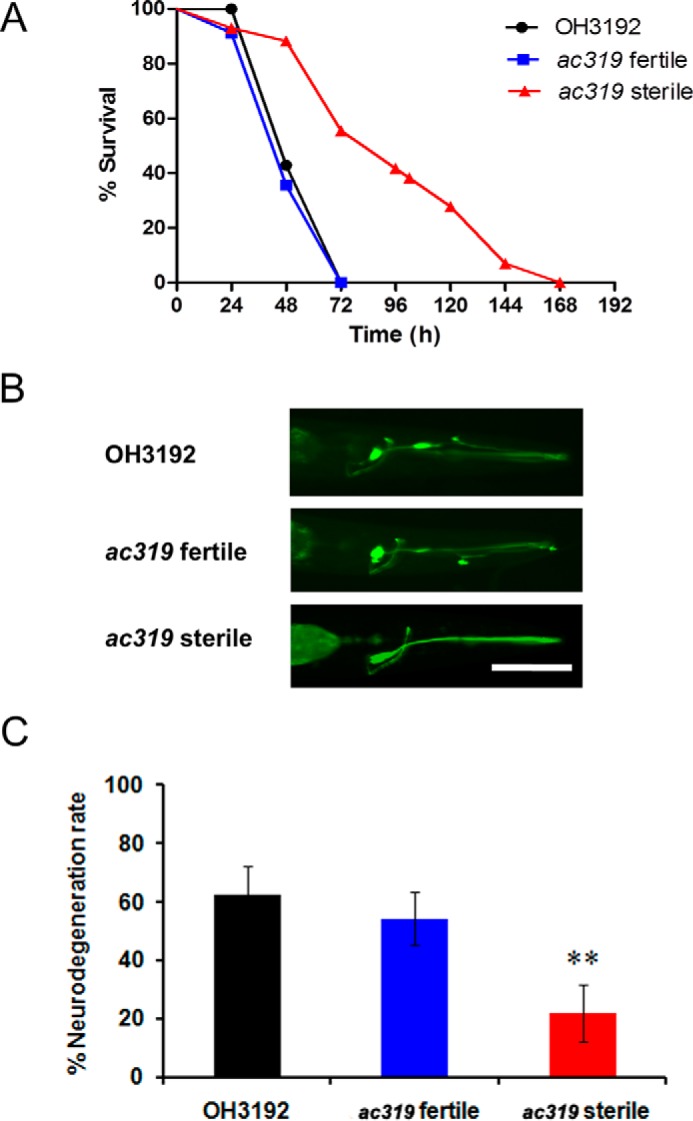
ac319 sterile animals are resistant to P. aeruginosa infection and P. aeruginosa-induced neurodegeneration. A, survival curves of OH3192, ac319 fertile, and ac319 sterile young adults exposed to P. aeruginosa. p < 0.0001, OH3192 versus ac319 sterile animals. B, images of the ASER neuron of OH3192, ac319 fertile, and ac319 sterile young adults exposed to P. aeruginosa. Scale bar, 50 μm. C, neurodegeneration rates of OH3192, ac319 fertile, and ac319 sterile young animals exposed to P. aeruginosa. Synchronized young adult animals were cultured on full lawn plates of P. aeruginosa for 30 h at 25 °C. The neurodegeneration rate of OH3192, ac319 fertile, and ac319 sterile animals was scored using a Leica M165 FC stereomicroscope. Asterisks indicate significant differences (*, p < 0.05; **, p < 0.01). The graph represents the combined results of three independent experiments (n = 60 ± 5 animals). Bars, mean ± S.E. (error bars).
Germ Line-deficient Mutants Are Resistant to P. aeruginosa-induced Neurodegeneration
As a first step in the identification of the ac319 mutation, we sequenced genes that are known to cause germ line deficiency and enhanced resistance to pathogen killing, including glp-1, mes-1, and pgl-1 (35–37). We did not identify mutations in the glp-1 or pgl-1 genes in the ac319 mutant animals. However, using flanking primers, we initially failed to amplify and sequence the mes-1 gene in ac319 animals. Further analysis showed that ac319 animals carry an 11,490-bp deletion on chromosome X that ranges from 18,338 to 29,827 (Fig. 7A). This deletion encompasses three genes, including xtr-1, F54F7.8, and mes-1. The mes-1 gene encodes a receptor tyrosine kinase-like protein that is required for unequal cell division in the early embryonic germ line (38). In addition, animals carrying mutations in mes-1 exhibit a maternal effect sterility phenotype similar to that observed in ac319 mutants (39). Thus, we decided to study the susceptibility of mes-1(bn7) animals to P. aeruginosa-induced neurodegeneration. To observe neurodegeneration, mes-1(bn7) was crossed with strain OH3192. As shown in Fig. 7B, sterile but not fertile mes-1(bn7) animals were resistant to neurodegeneration induced by P. aeruginosa infection. Similar results were observed using OH3192 animals carrying a mutation in the maternal effect gene pgl-1(bn101), which is required for postembryonic germ line development (Fig. 7B).
FIGURE 7.

Germ line-deficient animals are resistant to neurodegeneration induced by P. aeruginosa. A, location of the mutation in both fertile and sterile ac319 animals. A deletion of 11,490 bp on chromosome X from 18,338 to 29,827 was identified. B, neurodegeneration rates in pgl-1(bn101), mes-1(bn7), ac319, ac319;Pmes-1::mes-1 young animals crossed with strain OH3192. C, neurodegeneration rate in glp-1(e2141) and glp-4(bn2) animals crossed with strain OH3192. Young adult nematodes were cultured on full lawn plates of P. aeruginosa for 30 h at 25 °C. Asterisks indicate significant differences (*, p < 0.05; **, p < 0.01). The graph represents the combined results of three independent experiments (n = 30–60 animals). Bars, mean ± S.E. (error bars).
To confirm that the absence of mes-1 caused the ac319 animals to become resistant to pathogen-induced neurodegeneration, we created ac319 animals expressing mes-1 under its own promoter. As shown in Fig. 7B, the enhanced resistance to neurodegeneration of ac319 animals was completely abolished in these animals. The expression of mes-1 also rescued the sterility of the ac319 animals (data not shown). We used two additional germ line-deficient mutants, glp-1(e2141) and glp-4(bn2), to further investigate the role of sterility in resistance to pathogen-induced neurodegeneration. Both glp-1(e2141) and glp-4(bn2) animals displayed reduced levels of neurodegeneration following P. aeruginosa infection (Fig. 7C), confirming that sterility may confer protective effects to neurons in response to pathogen infection.
Taken together, these results suggest that the deletion of mes-1 in ac319 animals may be responsible for the maternal effect sterility and enhanced resistance to pathogen-induced neurodegeneration and that sterile animals more efficiently deal with the deleterious consequences of bacterial infections compared with wild-type animals.
DAF-16 Is Required for the Resistance to Pathogen-induced Neurodegeneration of Germ Line-deficient Animals
The FOXO transcription factor DAF-16 is known to control a variety of physiological processes in C. elegans, including longevity, the stress response, and immunity (40–44). Thus, we investigated whether the enhanced resistance to pathogen-induced neurodegeneration of mes-1(bn7) animals also required DAF-16 activity. RNA interference (RNAi) was used to knock down DAF-16 activity in mes-1(bn7) animals. The resistance of mes-1(bn7) animals to P. aeruginosa-induced neurodegeneration was significantly reduced by daf-16 RNAi (Fig. 8). DAF-16 was also required for resistance to the pathogen-induced neurodegeneration of glp-1(e2141) animals (Fig. 8). However, daf-16 RNAi did not fully suppress the enhanced resistance to P. aeruginosa-induced neurodegeneration of mes-1(bn7) or glp-1(e2141) animals, indicating that some other factors might be involved in this process.
FIGURE 8.

DAF-16 is required for the resistance to P. aeruginosa-induced neurodegeneration of germ line-deficient animals. Shown are neurodegeneration rates in glp-1(e2141) and mes-1(bn7) animals crossed with strain OH3192. The animals were grown on control or daf-16 RNAi plates until the young adult stage and then cultured on full lawn plates of P. aeruginosa for 30 h at 25 °C. The graph represents the combined results of three independent experiments (n = 30 animals). Bars, mean ± S.E. (error bars).
Discussion
Increasing evidence indicate that microbial infections and neuroinflammation can potentially trigger neurodegenerative diseases. Given the molecular conservation of neuronal signaling pathways across vertebrates and invertebrates, a number of investigators have utilized the nematode C. elegans to study the mechanisms underlying neurodegenerative disease pathology. Here, we sought to investigate the potential relationship between bacterial infection and neurodegeneration using C. elegans. Our findings indicate that host changes induced by P. aeruginosa infection result in axonal degeneration and aberrant neuronal sprouting similar to that observed in aged animals and patients with neurodegenerative disorders.
To study the neurodegeneration induced by P. aeruginosa infection, we used three C. elegans strains that express fluorescent proteins in sensory neurons and discovered four types of morphological changes, including beaded dendrites, branched somas, wavy dendrites, and branched dendrites (Fig. 1). In C. elegans, novel outgrowth phenotypes, including branching from the main dendrite or new growth from the soma, appear at a high frequency in some aging touch neurons, but the incidence of beads and protrusions declines in older PLM neurons (9). We found that P. aeruginosa-induced neurodegeneration results in both new dendrite growth and the generation of beads and protrusions, suggesting that different mechanisms may be involved in neurodegeneration caused by aging or pathogen infection.
To provide insight regarding the molecular mechanisms that regulate P. aeruginosa-induced neurodegeneration, we employed an unbiased forward genetics approach that resulted in the isolation of a mutant carrying a deletion that includes the mes-1 gene. In subsequent studies, we demonstrated that animals carrying mutations in the maternal effect sterility genes mes-1 or pgl-1, which are required for postembryonic germ line development, were resistant to P. aeruginosa-induced neurodegeneration. These findings indicate that germ line loss results in resistance to pathogen-induced neurodegeneration. We also demonstrated that the DAF-16 transcription factor is required for resistance to neurodegeneration in mes-1 and glp-1 animals. DAF-16 plays an important role in the control of longevity and immunity (40–44). In addition, it is known to control the extended life span of glp-1 animals and the enhanced resistance to pathogens of glp-4 animals (37, 45, 46). Our results strengthen the notion that DAF-16 functions at the intersection of pathways that control immunity, longevity, and stress responses. The enhanced resistance of germ line-deficient animals to P. aeruginosa-mediated killing and P. aeruginosa-induced neurodegeneration may be due to general stress resistance. It is also possible that neurodegeneration induced by P. aeruginosa requires cell-nonautonomous signals that originate in the germ line.
Author Contributions
Q. W. and A. A. conceived, designed, and interpreted the main body of experiments. Q. W. performed the majority of the experiments. X. C. performed experiments. D. Y. and D. W. contributed to the data interpretation. The manuscript was primarily written by Q. W. and A. A., with contributions from the other authors.
Acknowledgment
We thank the Caenorhabditis Genetics Center (University of Minnesota) for supplying the strains used in this study.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NGM
- nematode growth medium.
References
- 1.Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., and Kummer M. P. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heneka M. T., Golenbock D. T., and Latz E. (2015) Innate immunity in Alzheimer's disease. Nat. Immunol. 16, 229–236 [DOI] [PubMed] [Google Scholar]
- 3.Amor S., Puentes F., Baker D., and van der Valk P. (2010) Inflammation in neurodegenerative diseases. Immunology 129, 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hobert O. (2010) Neurogenesis in the nematode Caenorhabditis elegans. WormBook 10.1895/wormbook.1.12.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White J. G., Southgate E., Thomson J. N., and Brenner S. (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340 [DOI] [PubMed] [Google Scholar]
- 6.Pan C. L., Peng C. Y., Chen C. H., and McIntire S. (2011) Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 9274–9279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chew Y. L., Fan X., Gẗz J., and Nicholas H. R. (2013) Aging in the nervous system of Caenorhabditis elegans. Commun. Integr. Biol. 6, e25288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng C. Y., Chen C. H., Hsu J. M., and Pan C. L. (2011) C. elegans model of neuronal aging. Commun. Integr. Biol. 4, 696–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toth M. L., Melentijevic I., Shah L., Bhatia A., Lu K., Talwar A., Naji H., Ibanez-Ventoso C., Ghose P., Jevince A., Xue J., Herndon L. A., Bhanot G., Rongo C., Hall D. H., and Driscoll M. (2012) Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du M., and Wang D. (2009) The neurotoxic effects of heavy metal exposure on GABAergic nervous system in nematode Caenorhabditis elegans. Environ. Toxicol. Pharmacol. 27, 314–320 [DOI] [PubMed] [Google Scholar]
- 11.Mocko J. B., Kern A., Moosmann B., Behl C., and Hajieva P. (2010) Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson's disease. Neurobiol. Dis. 40, 120–129 [DOI] [PubMed] [Google Scholar]
- 12.Negga R., Stuart J. A., Machen M. L., Salva J., Lizek A. J., Richardson S. J., Osborne A. S., Mirallas O., McVey K. A., and Fitsanakis V. A. (2012) Exposure to glyphosate- and/or Mn/Zn-ethylene-bis-dithiocarbamate-containing pesticides leads to degeneration of γ-aminobutyric acid and dopamine neurons in Caenorhabditis elegans. Neurotox. Res. 21, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanduyn N., Settivari R., Wong G., and Nass R. (2010) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol. Sci. 118, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benedetto A., Au C., Avila D. S., Milatovic D., and Aschner M. (2010) Extracellular dopamine potentiates Mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 10.1371/journal.pgen.1001084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanDuyn N., Settivari R., LeVora J., Zhou S., Unrine J., and Nass R. (2013) The metal transporter SMF-3/DMT-1 mediates aluminum-induced dopamine neuron degeneration. J. Neurochem. 124, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison Brody A., Chou E., Gray J. M., Pokyrwka N. J., and Raley-Susman K. M. (2013) Mancozeb-induced behavioral deficits precede structural neural degeneration. Neurotoxicology 34, 74–81 [DOI] [PubMed] [Google Scholar]
- 17.Chen P., Martinez-Finley E. J., Bornhorst J., Chakraborty S., and Aschner M. (2013) Metal-induced neurodegeneration in C. elegans. Front. Aging Neurosci. 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J., Singh V., Kajino-Sakamoto R., and Aballay A. (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332, 729–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun J., Liu Y., and Aballay A. (2012) Organismal regulation of XBP-1-mediated unfolded protein response during development and immune activation. EMBO Rep. 13, 855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh V., and Aballay A. (2012) Endoplasmic reticulum stress pathway required for immune homeostasis is neurally controlled by arrestin-1. J. Biol. Chem. 287, 33191–33197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen E. M., and Mango S. E. (2002) The art and design of genetic screens: Caenorhabditis elegans. Nat. Rev. Genet. 3, 356–369 [DOI] [PubMed] [Google Scholar]
- 23.Bargmann C. I., and Horvitz H. R. (1991) Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7, 729–742 [DOI] [PubMed] [Google Scholar]
- 24.Saeki S., Yamamoto M., and Iino Y. (2001) Plasticity of chemotaxis revealed by paired presentation of a chemoattractant and starvation in the nematode Caenorhabditis elegans. J. Exp. Biol. 204, 1757–1764 [DOI] [PubMed] [Google Scholar]
- 25.O'Meara M. M., Bigelow H., Flibotte S., Etchberger J. F., Moerman D. G., and Hobert O. (2009) Cis-regulatory mutations in the Caenorhabditis elegans homeobox gene locus cog-1 affect neuronal development. Genetics 181, 1679–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng G., Cochella L., Liu J., Hobert O., and Li W. H. (2011) Temporal and spatial regulation of microRNA activity with photoactivatable cantimirs. ACS Chem. Biol. 6, 1332–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doerks T., Copley R. R., Schultz J., Ponting C. P., and Bork P. (2002) Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 12, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L., Fu Y., Ren M., Xiao B., and Rubin C. S. (2011) A RasGRP, C. elegans RGEF-1b, couples external stimuli to behavior by activating LET-60 (Ras) in sensory neurons. Neuron 70, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bargmann C. I. (2006) Chemosensation in C. elegans. WormBook 10.1895/wormbook.1.123.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Styer K. L., Singh V., Macosko E., Steele S. E., Bargmann C. I., and Aballay A. (2008) Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322, 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meisel J. D., Panda O., Mahanti P., Schroeder F. C., and Kim D. H. (2014) Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell 159, 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shivers R. P., Youngman M. J., and Kim D. H. (2008) Transcriptional responses to pathogens in Caenorhabditis elegans. Curr. Opin. Microbiol. 11, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pepper A. S. R., Killian D. J., and Hubbard E. J. A. (2003) Genetic analysis of Caenorhabditis elegans glp-1 mutants suggests receptor interaction or competition. Genetics 163, 115–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beanan M. J., and Strome S. (1992) Characterization of a germ-line proliferation mutation in C. elegans. Development 116, 755–766 [DOI] [PubMed] [Google Scholar]
- 35.TeKippe M., and Aballay A. (2010) C. elegans germline-deficient mutants respond to pathogen infection using shared and distinct mechanisms. PloS One 5, e11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha A., and Rae R. (2014) A functional genomic screen for evolutionarily conserved genes required for lifespan and immunity in germline-deficient C. elegans. PLoS One 9, e101970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alper S., McElwee M. K., Apfeld J., Lackford B., Freedman J. H., and Schwartz D. A. (2010) The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J. Biol. Chem. 285, 1822–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkowitz L. A., and Strome S. (2000) MES-1, a protein required for unequal divisions of the germline in early C. elegans embryos, resembles receptor tyrosine kinases and is localized to the boundary between the germline and gut cells. Development 127, 4419–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arantes-Oliveira N., Apfeld J., Dillin A., and Kenyon C. (2002) Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295, 502–505 [DOI] [PubMed] [Google Scholar]
- 40.Lin K., Dorman J. B., Rodan A., and Kenyon C. (1997) daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319–1322 [DOI] [PubMed] [Google Scholar]
- 41.Ogg S., Paradis S., Gottlieb S., Patterson G. I., Lee L., Tissenbaum H. A., and Ruvkun G. (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999 [DOI] [PubMed] [Google Scholar]
- 42.Chávez V., Mohri-Shiomi A., Maadani A., Vega L. A., and Garsin D. A. (2007) Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics 176, 1567–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh V., and Aballay A. (2006) Heat shock and genetic activation of HSF-1 enhance immunity to bacteria. Cell Cycle 5, 2443–2446 [DOI] [PubMed] [Google Scholar]
- 44.Singh V., and Aballay A. (2009) Regulation of DAF-16-mediated innate immunity in C. elegans. J. Biol. Chem. 284, 35580–35587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berman J. R., and Kenyon C. (2006) Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124, 1055–1068 [DOI] [PubMed] [Google Scholar]
- 46.TeKippe M., and Aballay A. (2010) C. elegans germ line-deficient mutants respond to pathogen infection using shared and distinct mechanisms. PLoS One 5, e11777. [DOI] [PMC free article] [PubMed] [Google Scholar]