

Abstract
Diabetes mellitus causes cardiac dysfunction and heart failure that is associated with metabolic abnormalities and autonomic impairment. Autonomic control of ventricular function occurs through regulation of cAMP-dependent protein kinase (PKA). The diabetic heart has suppressed β-adrenergic responsiveness, partly attributable to receptor changes, yet little is known about how PKA signaling is directly affected. Control and streptozotocin-induced diabetic mice were therefore administered 8-bromo-cAMP (8Br-cAMP) acutely to activate PKA in a receptor-independent manner, and cardiac hemodynamic function and PKA signaling were evaluated. In response to 8Br-cAMP treatment, diabetic mice had impaired inotropic and lusitropic responses, thus demonstrating postreceptor defects. This impaired signaling was mediated by reduced PKA activity and PKA catalytic subunit content in the cytoplasm and myofilaments. Compartment-specific loss of PKA was reflected by reduced phosphorylation of discrete substrates. In response to 8Br-cAMP treatment, the glycolytic activator PFK-2 was robustly phosphorylated in control animals but not diabetics. Control adult cardiomyocytes cultured in lipid-supplemented media developed similar changes in PKA signaling, suggesting that lipotoxicity is a contributor to diabetes-induced β-adrenergic signaling dysfunction. This work demonstrates that PKA signaling is impaired in diabetes and suggests that treating hyperlipidemia is vital for proper cardiac signaling and function.
Keywords: cardiac metabolism, cardiomyocyte, diabetes, heart, protein kinase A (PKA)
Introduction
Diabetes mellitus carries at least twice the risk of cardiovascular complications such as coronary artery disease, hypertension, and heart failure (1, 2). Diabetes can directly cause heart failure, in the case of diabetic cardiomyopathy, as well as exacerbate other comorbidities (3, 4). The reasons for this are multifaceted and involve both metabolic and mechanical cardiac stresses. Metabolic abnormalities include metabolic inflexibility caused by improper glucose uptake and oxidation and by lipotoxicity induced by lipid overload (5). Diabetes, in common with other forms of heart failure, has impairment of autonomic control and overstimulation of the sympathetic nervous system (6, 7). The etiology of chronic cardiac stimulation in diabetes is characterized by sympathetic enhancement and parasympathetic withdrawal (3, 8). Excess sympathetic stimulation over time causes extensive dysfunction to the β-adrenergic pathway, a process that is intimately tied with heart failure (9, 10).
Normally, ligand binding to β1-receptors activates Gs proteins, which stimulate adenylate cyclase to produce cAMP. The primary target of cAMP is cAMP-dependent protein kinase (PKA),2 a heterotetrameric protein comprised of two catalytic (PKA-C) and two regulatory subunits (either PKA-RI or PKA-RII isoforms). cAMP binds PKA-R subunits and induces the release of PKA-C, which then phosphorylates specific serines and threonines on protein targets in the cytoplasm, myofilaments, sarcoplasmic reticulum, sarcolemma, and nucleus to increase cardiac output, metabolism, and gene transcription. Subcellular localization of PKA is essential for proper signaling, and this is provided by protein kinase A-anchoring proteins (AKAPs) (11) and protein kinase inhibitors (PKIs) (12). AKAPs facilitate PKA interactions with protein substrates, whereas PKIs mediate shuttling of PKA-C from the nucleus to the cytoplasm.
Persistent activation of the β-adrenergic pathway ultimately leads to a blunted response to additional ligand stimulation (13). In diabetes, this is partly from a reduction in β1-adrenergic receptor content and function (14); however adrenergic impairment in the diabetic heart is likely more complex. For example, cardiomyocytes isolated from diabetic animals maintain a blunted response to adenylate cyclase and PKA agonists despite these drugs bypassing the receptor and cAMP production (15, 16). This suggests that there are post-receptor defects in the diabetic heart and implicates PKA signaling.
Several factors involving PKA signaling could contribute to this, including a decrease in PKA content or activity, alterations in its subcellular localization, loss of PKA substrates, or an increase in counteracting protein phosphatases. Previous studies have not evaluated such changes as contributors to impaired or altered β-adrenergic signaling in the diabetic heart. This is important to address because PKA dysfunction could constitute a driving factor in the progression of diabetic cardiomyopathy and development of heart failure.
In the present study, we used the streptozotocin (STZ) mouse model of type I diabetes and direct administration of a PKA agonist, 8-bromo-cAMP (8Br-cAMP) to discern the postreceptor changes in PKA signaling. Cardiac contractile function was measured, and PKA activation, content, substrate phosphorylation, and localization were evaluated. Adult cardiomyocytes isolated from control animals were used as a model system to define how metabolic stresses directly impair PKA signaling.
Experimental Procedures
Type I Diabetic Animal Model
STZ-induced diabetic male mice were obtained from the Jackson Laboratory on a C57BL/6J background. 3-month post-STZ diabetic animals were used for hemodynamic measurements, and 4-month post-STZ diabetic animals were used for biochemical analysis, all with age-matched controls. All mouse experiments were carried out in accordance with the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
Hemodynamic Measurements
Left ventricular hemodynamics were acquired using a pressure catheter (SPR-1000; Millar) placed in the left ventricle by the close chested approach via the right carotid artery (17). Anesthesia was maintained by 1.5% isoflurane, and temperature was maintained by homeothermic controller. Data were acquired by PowerLab 8/35 and analyzed by LabChart Pro 7.
Preparation of Cardiac Homogenates
The mice were euthanized by cervical dislocation. Hearts were quickly perfused, excised, and flash frozen. Frozen hearts were homogenized in sucrose-mannitol buffer with the addition of phosphatase, phosphodiesterase, and protease inhibitors. Protein content was standardized to 1 mg/ml via Bradford assay.
cAMP Assay
cAMP levels were assayed using a cAMP antibody-based kit (Cyclic AMP XP assay kit; Cell Signaling Technology).
SDS-PAGE and Western Blot Analysis
SDS-PAGE and Western blotting was performed as previously reported (18), using protein-standardized homogenates. All antibodies were from Cell Signaling Technology except PKA-RII, actin-HRP, and PKIα (Santa Cruz Biotechnology), and PKIγ (Proteintech). Actin was used as a loading control for all blots. Densitometry analysis was performed using ImageJ software (National Institutes of Health).
PKA Activity Assay
PKA activity was measured in the soluble fraction of cardiac homogenates using a nonradioactive assay (Promega). cAMP was not added to the reactions, and thus activities represent in vivo activities (19).
Native Gel Electrophoresis
Blue native gel electrophoresis was carried out using the soluble fraction of cardiac homogenates according to the manufacturer's protocol (NativePAGE Bis-Tris gel system; Life Technologies). Clear native electrophoresis was similarly performed, without the addition of Coomassie Blue.
Subcellular Fractionation
Heart homogenates were fractionated based upon their detergent solubility and centrifugation, as previously described, to produce enriched fractions (20, 21). The soluble fraction represents the cytosol. The fraction soluble in a 1% Triton X-100 buffer represents the membranes. The final fraction, soluble in 2% SDS (SDS-PAGE sample buffer), represents the myofilaments/nuclei. Fractionation was confirmed using compartment-specific antibodies, as shown.
Lactate Content
A fluorometric lactate assay was performed using heart homogenates according to the manufacturer's specifications (MAK064; Sigma).
RII-AKAP Blot Overlay
A complex of PKA-RII bound to HRP via primary and secondary antibody was overlaid on a blot of cardiac homogenates and visualized by enhanced chemiluminescence. The specificity of the RII-AKAP interaction was confirmed by the addition of an AKAP disrupting peptide, Ht31 (Promega), on a separate blot.
Cell Culture of Adult Mouse Ventricular Cardiomyocytes
Adult primary cardiomyocytes from C57BL/6J mice were isolated and cultured as previously described (22). Briefly, the heart was excised, and the aorta was cannulated within 1–2 min and digested with type II collagenase. Calcium was reintroduced to the single cell suspension, and cells were plated on laminin-coated plates. Media was switched to a serum-free culture media (minimum essential medium with Hanks' balanced salt solution; Gibco) supplemented with penicillin-G, 0.1% BSA, glutamine, 0.4 mg/ml NaHCO3, 10 mm butanedione monoxime, and insulin-transferrin-selenium or insulin as indicated. Metabolic conditions were varied as indicated. The cells were incubated overnight at 37 °C and 5% CO2 and used the subsequent day.
Statistical Analysis
The data are presented as means ± S.E. The data were analyzed using an unpaired, two-tailed Student's t test unless otherwise noted. p values of <0.05 were considered significant.
Results
8Br-cAMP Enhances Cardiac Inotropy, Lusitropy, and Chronotropy by Direct Activation of Cardiac PKA
Our goal was to bypass the receptor and signal amplification portions of the β-adrenergic pathway and activate cardiac PKA in vivo to examine the PKA signaling pathway directly. 8Br-cAMP was chosen because it is cell-permeable and resistant to phosphodiesterases (23). Initial experiments were undertaken to determine whether 8Br-cAMP acts as a direct inotropic agent in vivo. Control mice were treated with a saturating dose of metoprolol (50 mg/kg, intraperitoneally) to block potential confounding effects of global PKA activation on sympathetic output. Metoprolol significantly reduced dP/dt max, as expected, and this was largely reversed by subsequent administration of 8Br-cAMP with a peak at 8 min (Fig. 1). Tau followed a similar pattern, showing that diastolic relaxation is also enhanced. Longer time points were not used because confounding variables, such as vasodilation, become more prominent. This demonstrates that 8Br-cAMP acts as a positive inotrope by direct activation of cardiac PKA.
FIGURE 1.
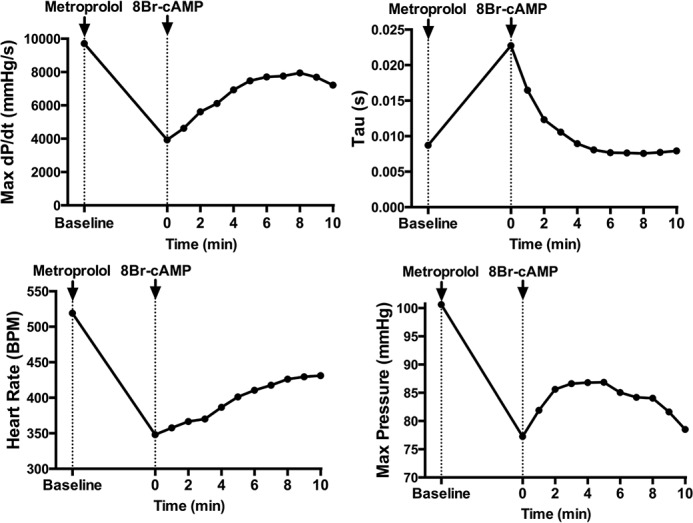
Acute 8Br-cAMP treatment directly activates cardiac PKA and increases cardiac function. Representative traces of left ventricular hemodynamics from 3-month-old C57BL/6J mice. The data were gathered continuously at 1 k/s and analyzed in LabChart Pro. Data points represent 1-min averages. The mice were pretreated with metoprolol (50 mg/kg, intraperitoneally) 10 min before treatment with 8Br-cAMP (330 mg/kg, intraperitoneally). The effect of metoprolol stabilized prior to 8Br-cAMP treatment.
Diabetic Mice Have Attenuated Hemodynamic Response to 8Br-cAMP Stimulation
We next tested the hypothesis that significant dysfunction occurs in PKA signaling in the STZ mouse heart. The ventricular hemodynamic responses of control and 3 month STZ-induced diabetic mice were evaluated following either isoproterenol or 8Br-cAMP administration. There was no pretreatment of animals with metoprolol in these experiments.
The ventricular hemodynamics of untreated diabetic mice had a reduced max pressure and dP/dt max, an extended systolic duration, and similar heart rates as compared with controls (Table 1). Diabetic mice, compared with age-matched controls, were desensitized to isoproterenol as illustrated by the change in dP/dt max, dP/dt min, and heart rate (Fig. 2, A, E, and F). Importantly, 8Br-cAMP treatment showed very similar desensitization for dP/dt max and to a lesser extent dP/dt min (Fig. 2, A and E). After 3 min of 8Br-cAMP treatment, the control mice exhibited an 89 ± 16% higher systolic response (dP/dt max) than the diabetic mice, similar to the maximum difference with isoproterenol (86 ± 11%). The peak increase in dP/dt max was 30% less in the diabetics than the controls, and the rate at which dP/dt max increased to these peak values was significantly decreased in the diabetics (Fig. 2, B–D).
TABLE 1.
Baseline left ventricular hemodynamics
The results for control and 3 months post-STZ diabetic mice are shown. Baseline left ventricular parameters were taken before treatments were given as shown in Fig. 2.
| Baseline left ventricular hemodynamics |
||
|---|---|---|
| Control | 3 months post-STZ | |
| n | 8 | 8 |
| Max pressure (mm Hg) | 108 ± 1.8 | 101 ± 3.1a |
| Min pressure (mm Hg) | −1.03 ± 1.8 | 0.283 ± 1.0 |
| EDP (mm Hg) | 3.54 ± 1.3 | 6.46 ± 1.4 |
| Mean pressure (mm Hg) | 46.2 ± 2.1 | 48.8 ± 2.2 |
| Maximum-minimum pressure (mm Hg) | 109 ± 2.1 | 97.1 ± 4.0a |
| Systolic duration (ms) | 53.4 ± 0.96 | 59.1 ± 1.0b |
| Diastolic duration (ms) | 57.0 ± 2.4 | 52.4 ± 0.78 |
| Heart rate (beats/min) | 545 ± 11 | 538 ± 5.7 |
| dP/dt max (mm Hg/s) | 9780 ± 530 | 7990 ± 480a |
| Contractility index (1/s) | 182 ± 5.9 | 144 ± 5.2b |
| dP/dt min (mm Hg/s) | −10700 ± 1100 | −9100 ± 370 |
| IRP average dP/dt (mm Hg/s) | −5770 ± 390 | −4990 ± 270 |
| Tau (ms) | 10.1 ± 1.4 | 10.6 ± 0.57 |
| Pressure time index (mm Hg*s) | 4.58 ± 0.12 | 4.74 ± 0.16 |
a p < 0.05 versus unpaired Student's t test.
b p < 0.005 versus unpaired Student's t test.
FIGURE 2.
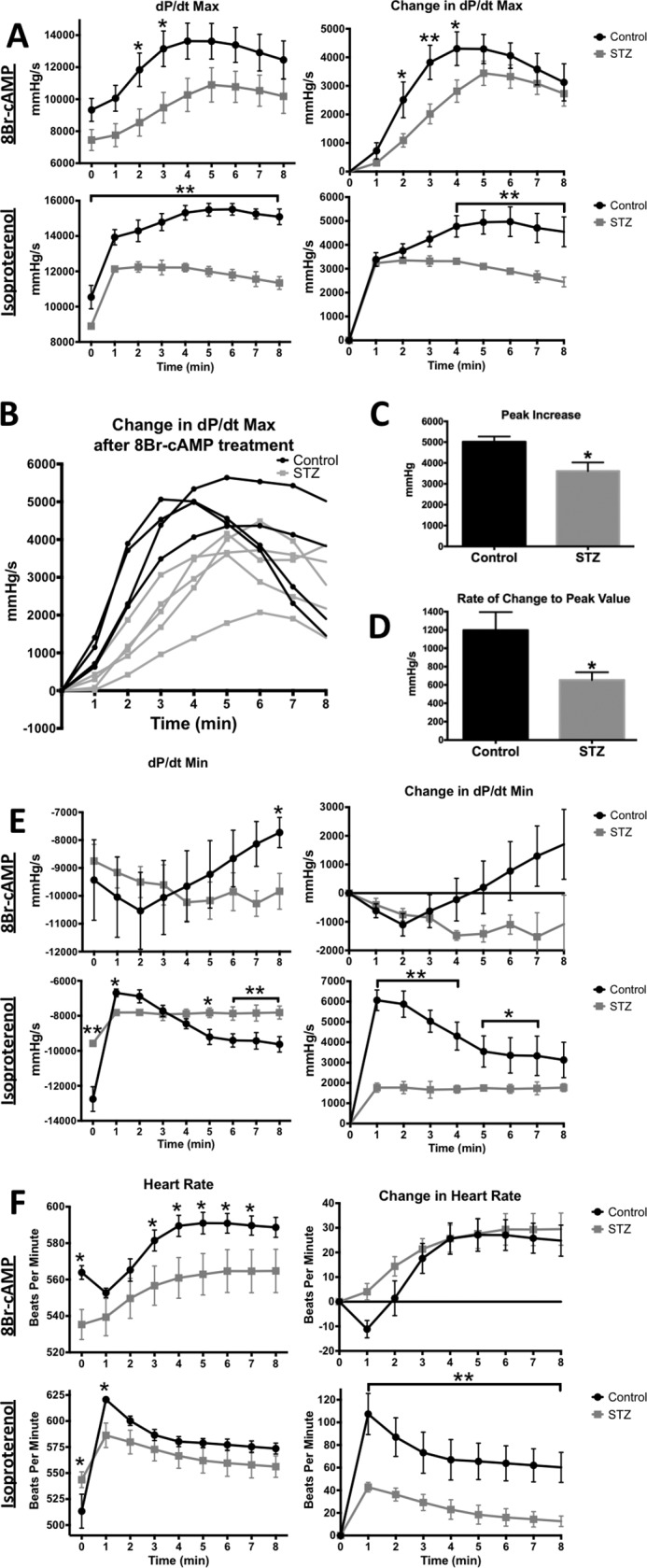
Diabetic mice show an early, blunted hemodynamic response to 8Br-cAMP and Isoproterenol. Left ventricular hemodynamics from control and diabetic (3 months post-STZ) mice in response to either a 330 mg/kg 8Br-cAMP or a 20 mg/kg isoproterenol intraperitoneal treatment. 1 min average data were obtained as in Fig. 1. Black traces, control; gray traces, STZ. A, systolic performance measured by dP/dt max. Average values (left) and the change with treatment (right). B, individual traces of change in dP/dt max after 8Br-cAMP treatment. C and D, peak increase and rate of change to peak refer to change in dP/dt max. B–D, one control data trace was removed from these figures for an experimental reason but was averaged into all other figures. E, diastolic performance measured by dP/dt min. F, heart rate. *, p < 0.05, unpaired Student's t test (n = 5 for 8Br-cAMP, n = 3 for isoproterenol).
In control mice, isoproterenol initially slowed dP/dt min followed by recovery, whereas 8Br-cAMP initially increased diastolic relaxation before slowing (Fig. 2E). In the diabetic, both isoproterenol and 8Br-cAMP treatments caused a similarly minimal initial response that did not change over time (Fig. 2E). These results demonstrate that attenuated systolic and diastolic responses occur independently of changes in adrenergic receptors and cAMP production.
Isoproterenol increased the heart rate in control significantly more than diabetic animals (Fig. 2F). In contrast, controls and diabetics heart rates both increased by a similar magnitude in response to 8Br-cAMP (30 and 25 beats/min, respectively), suggesting that the deficient heart rate response is from changes elsewhere in the adrenergic pathway.
Overall, these data show that the adrenergic effect on inotropy, and to a lesser extent lusitropy, is reduced in diabetic mice because of PKA signaling dysfunction. However, the defect in heart rate is likely upstream of PKA. This pattern of changes in response to 8Br-cAMP (deficient dP/dT max and systolic duration, for example) follows the ventricular hemodynamic deficiencies of the diabetic at baseline.
Diabetic Mice Have Attenuated PKA Substrate Phosphorylation and PKA Enzyme Activity
We next sought to determine the mechanism of the altered response to 8Br-cAMP. First, we determined whether 8Br-cAMP treatment was penetrating cardiac tissue by using an antibody-based assay that recognizes both cAMP and 8Br-cAMP. The cAMP levels at baseline were not significantly different between the two groups (Fig. 3A). With 8Br-cAMP treatment, the levels of total cAMP and 8Br-cAMP increased substantially in both groups, demonstrating similar absorption and distribution of the drug.
FIGURE 3.
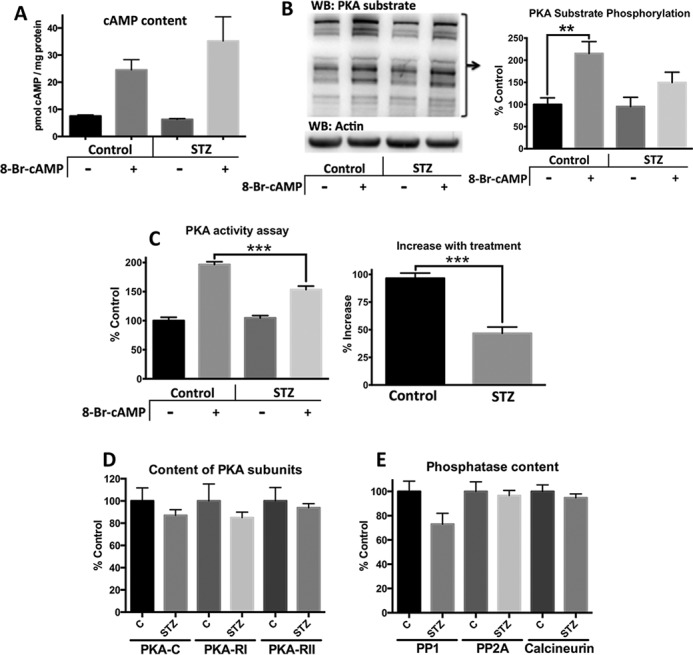
Diabetic mice have less PKA substrate phosphorylation and PKA activity than control mice following 8Br-cAMP treatment. Control and diabetic (4 months post-STZ) mice were treated for 8 min with saline or 330 mg/kg 8Br-cAMP and then euthanized. A, cAMP content was measured in the soluble fraction of cardiac homogenates. The antibody-based system detects both cAMP and 8Br-cAMP. B, phosphorylation of PKA substrates was measured by Western blot (WB) of cardiac homogenates and standardized to actin. Representative blot refers to bands larger than 60 kDa. Densitometric analysis used the summation of all bands >80 kDa (n = 5). C, PKA activity was measured in the soluble fraction of cardiac homogenates in the presence of a phosphatase inhibitor. D and E, PKA subunit content (D) and phosphatases (E) were measured by Western blot analysis. *, p < 0.05; **, p < 0.01 unpaired Student's t test (n = 5 for all four groups).
We next determined whether the attenuated contractile response to 8Br-cAMP was due to deficient PKA substrate phosphorylation by using a phospho-antibody that specifically recognizes the PKA substrate consensus sequence. Control mice showed a significant increase in PKA substrate phosphorylation that was twice as robust as in the diabetic mice (115 and 57%, respectively; Fig. 3B). This deficient substrate phosphorylation is reflected closely by PKA activity. 8Br-cAMP treatment increased PKA activity in the control and diabetic groups by 97 and 47%, respectively. This represents in vivo PKA activity because no additional cAMP was added to the assay. Furthermore, a phosphatase inhibitor is present in the assay and thus not affected by phosphatase activity. Collectively, these data support that reduced PKA activity contributes to decreased substrate phosphorylation.
The decrease in PKA activity was not accompanied by a significant loss of its protein content (Fig. 3D). For example, there was an average decrease of 13% for PKA-C and 15% for PKA-RI in the diabetic mice, but it failed to reach statistical significance. Nevertheless, a decrease in PKA may be a contributing factor to deficient PKA signaling. Likewise, the content of phosphatases that attenuate the β-adrenergic response were not significantly changed in diabetic animals (Fig. 3E), but there was a trend toward decreased PP1. However, overactivation of phosphatases cannot be completely ruled out as a contributor to suppressed PKA signaling.
Diabetic Mice Show Enhanced Activation of PKA-RII Holoenzyme Complexes with 8Br-cAMP Treatment
Normally, when activated by cAMP, PKA regulatory subunit dimers release two active catalytic subunits. Native gel electrophoresis, paired with Western blotting, was used to quantify this dissociation of subunits to test whether the release of active PKA was impaired in the diabetic mice. Clear native electrophoresis (Fig. 4A) and blue native electrophoresis (Fig. 4B) were found to resolve the RI and RII populations of PKA, respectively. The identities of the PKA holoenzymes and R subunit dimers were determined by adding 8Br-cAMP to a control heart homogenate prior to native PAGE. Furthermore, probing blots for PKA-C confirmed that the higher molecular weight species, but not lower, contained catalytic subunits (data not shown).
FIGURE 4.
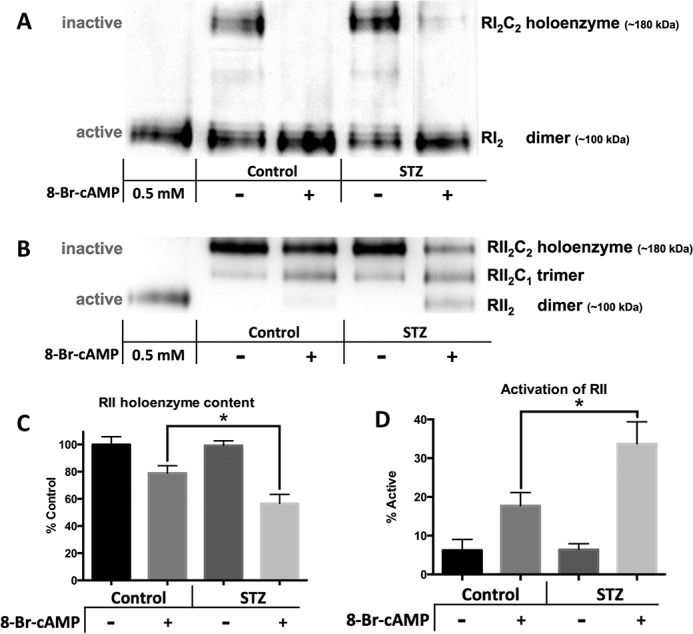
The RI population of PKA is fully activated by 8Br-cAMP in both control and diabetic mice. RII activation is enhanced in diabetic mice. The soluble fraction of cardiac samples from control and 4 months post-STZ diabetic mice treated with either saline or 330 mg/kg 8Br-cAMP were separated by native gel electrophoresis techniques indicated and then subjected to Western blot analysis. 0.5 mm 8Br-cAMP was added to control homogenates (lanes 1) to differentiate band identities. Representative blots are shown. A, clear native electrophoresis was used to separate the inactive RI holoenzyme from the active RI dimer. B, blue native gel electrophoresis was used to separate the three species of RII complexes. C and D, quantification of RII activation can be shown either by loss of the inactive holoenzyme complex (C) or by calculating the percent active from the percentage in the dimer (fully activated), trimer (half-activated), and holoenzyme (fully inactive) forms (where % active = %RII2C/2 + %RII2) (D). n = 5 for each figure. *, p < 0.05, unpaired Student's t test.
Control and diabetic hearts had similar amounts of RI holoenzyme and RI dimers under basal conditions, and 8Br-cAMP treatment resulted in complete activation in both groups (Fig. 4A). There were also similar amounts of RII populations under basal conditions in both groups. Upon 8Br-cAMP treatment, although, ∼20% of RII isoforms were activated in controls, and 40% were activated in diabetics. This increase was quantified both by the loss of holoenzyme (Fig. 4C) and by the calculation of % activation (Fig. 4D). This enhanced RII activation in the diabetic mice may compensate for deficient PKA signaling. Nevertheless, these results demonstrate that cAMP-induced release of the active catalytic subunit is not deficient in the diabetic mice. Thus, the loss of proper PKA substrate phosphorylation is likely from alterations to the PKA catalytic subunit.
PKA-C Content Is Altered in Diabetic Mice in a Location-specific Manner
Alterations in PKA signaling may be mediated by changes in localization. Subcellular fractionation was employed to separate the cytosol, membrane, and myofilament/nucleus fractions of cardiac homogenates. These fractions are aqueous soluble, 1% Triton X-100-soluble, and 2% SDS-soluble, respectively. GAPDH (cytosol), lamin B1 (nucleus), actin (myofilament), and SERCA2 (sarcoplasmic reticulum) confirm the fractional enrichments (Fig. 5B). PKA-C content was standardized to PKA-RII as a control, which was present in all fractions and did not change with treatment. Thus, subcellular PKA content can also be viewed as the C:RII ratio (Fig. 5A). In both groups, 8Br-cAMP treatment caused a reduction of PKA-C in the cytosol and an increase in the myofilament/nucleus fraction because of translocation (Fig. 5, A and B). However, relative to controls, diabetic animals exhibited reduced cytosolic PKA-C and a strong trend toward a reduction in the myofilament/nuclear fraction (Fig. 5A). The myofilament/nucleus fraction trend reached significance when evaluated in a paired manner to reduce day-to-day variability (p < 0.005). Interestingly, the majority of proteins phosphorylated by PKA reside in the myofilament/nucleus fraction (data not shown). Thus, reduction of PKA-C here may be contributing to contractile impairment.
FIGURE 5.
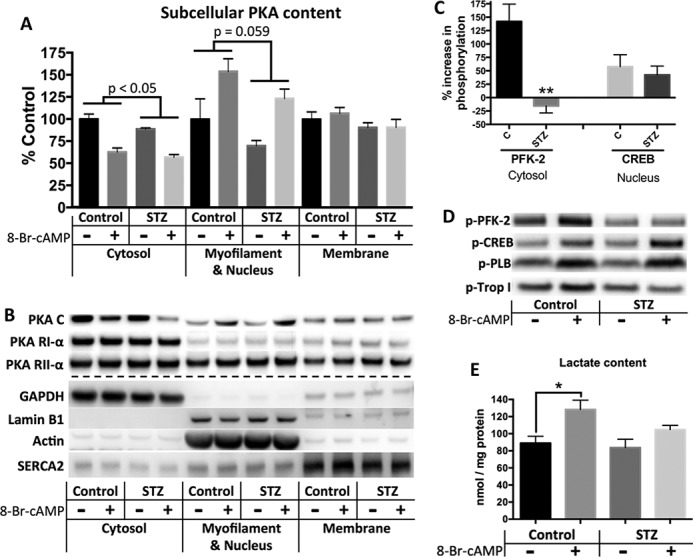
Improper subcellular PKA localization and content is accompanied by deficient PFK-2 phosphorylation and glycolytic flux in diabetic mice. A and B, subcellular fractionation of freshly homogenized cardiac tissue from control and 4 months post-STZ diabetic mice yields cytosol, membrane, and myofilament/nucleus enriched fractions. A, densitometric data showing PKA-catalytic fractional content standardized to PKA-RII as a control, because RII was present in all fractions and neither affected by diabetes status nor treatment. Thus, values can be viewed as the C:RII ratio. p values represent comparison of diabetes status by two-way analysis of variance. B, representative blots of PKA subunits in each fraction, as well as fraction markers. C and D, Western blots of cardiac homogenates using phospho-specific antibodies to PKA targets. C, percent increase in phosphorylation with treatment compared with basal level of phosphorylation. Phospho-antibodies were standardized to total content for each substrate with the ratio [phospho-substrate/actin]/[substrate/actin]. D, representative blots. Total levels of PFK-2, CREB, phospholamban (PLB), and troponin I (Trop I) were not affected by treatment. E, effect of 8Br-cAMP treatment on lactate content was determined using a fluorometric enzyme assay. n = 5 for each figure. *, p < 0.05; **, p < 0.005, unpaired Student's t test.
We next examined whether the changes in localized PKA content were reflected by altered phosphorylation of compartment specific substrates. 8Br-cAMP treatment of control animals caused robust phosphorylation of phosphofructokinase 2 (PFK-2), a cytosolic target of PKA yet was strikingly unresponsive in diabetic animals (Fig. 5, C and D). Phosphorylation of the nuclear cAMP response element-binding protein (CREB) was comparable in control and diabetic animals (Fig. 5, C and D). There was a trend toward decreased phosphorylation of the myofilament protein troponin I and no difference with the sarcoplasmic reticulum protein phospholamban (Fig. 5D). All phospho-substrate values were calculated as the ratios of phosphorylated to unphosphorylated intensities. The total content of each PKA substrate was not affected by 8Br-cAMP treatment (data not shown). Collectively, our results suggest that the magnitude of phosphorylation is dependent on the localized content of PKA-C and that changes in this content may be reflected by the phosphorylation of cytosolic and myofilaments substrates.
Phosphorylation of PFK-2 increases its production of fructose-2,6-bisphosphate, the most potent activator of PFK-1 and glycolytic flux (24–26). Improper phosphorylation of PFK-2 would therefore be expected to have metabolic consequences. Despite containing similar lactate content under basal conditions, treatment of control animals with 8Br-cAMP caused a significant increase in lactate in control mice (44 ± 12%) that was attenuated in diabetic mice (25 ± 5.9% increase; Fig. 5E). This suggests that decreased glycolytic flux is a potential consequence of deficient cytosolic PKA activity.
Mechanisms of Deficient PKA Signaling
The mechanisms of PKA signaling deficiency in diabetes may be multifold. One potential contributor is post-translational modifications to the PKA-C subunit. Thr-197 phosphorylation, required for activity (27), was not significantly different between groups (Fig. 6, A and B). Furthermore, post-translational analysis by isoelectric focusing found no evidence of PKA-C modifications (data not shown). Thus, our results do not support, but cannot exclude, a role for post-translational modification in impairing PKA-C activity and as a potential contributor to deficient substrate phosphorylation.
FIGURE 6.
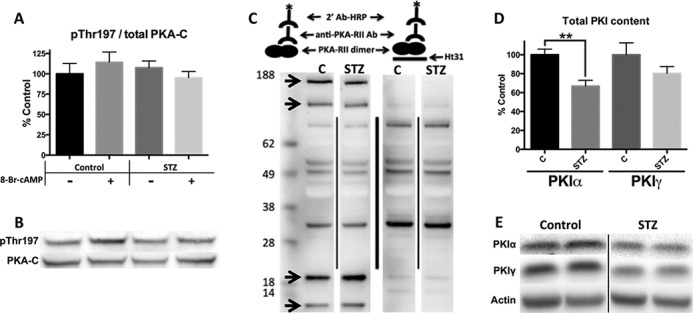
PKI is decreased in diabetic mice, but PKA phosphorylation and AKAP binding are unchanged. A and B, densitometry and Western blots of cardiac homogenates. The phosphorylated form of PKA standardized to total PKA. C, schematic and representative AKAP screen blot by PKA-RII overlay. PKA-RII bound to HRP via primary and secondary antibodies (left). Binding-deficient PKA-RII construct made by the addition of Ht31 (right) to show nonspecific bands. Arrows represent RII-AKAP interactions. D and E, PKI content shown by densitometry and representative Western blots. Standardized to actin. n = 5 for all data figures. **, p < 0.005, unpaired Student's t test.
AKAPs, which tether RII PKA isoforms to specific cell compartments, may be responsible for altered subcellular PKA content. We therefore adapted a PKA-RII overlay assay to quantify RII-AKAP interactions (28). Using the AKAP disruptor, Ht31, four RII interacting partners were identified (Fig. 6C). No differences in binding were found between control and diabetic samples, suggesting that AKAP content and RII-PKA binding capacity are maintained. This conclusion is also supported by the proportional distribution of RII in the cytosol versus noncytosolic fractions in control and diabetic animals (Fig. 5B).
Another group of proteins that regulate PKA localization are the PKIs. They are primarily nuclear proteins that inhibit and translocate PKA-C out of the nucleus (12), thus affecting the nuclear and nonnuclear concentration of PKA. PKIα was significantly reduced (33 ± 6%), and PKIγ trended toward a reduction (20 ± 7%) in diabetic animals (Fig. 6, D and E). This was not due to translational differences in the PKIs because mRNA transcripts were the same (data not shown), suggesting post-translational control of these proteins. Overall, our results support a potential role for PKIs as mediators of change in PKA localization in the diabetic heart.
Lipids Reduce PKA Content and Impair PKA Signaling
In the STZ diabetes model, three primary metabolic stresses are the lack of insulin signaling, hyperglycemia, and hyperlipidemia. Experiments were next undertaken to determine whether one or more of these factors are contributing to impaired PKA signaling. Primary adult mouse cardiomyocytes were isolated from control C57BL/6J mice and cultured with normal glucose, in the absence of insulin, with high glucose, or with fatty acids. This method allowed us to study each metabolic stress independently and isolated from the multifactorial pathologies of the diabetic cardiac environment. After 18 h under each culture condition, cells were treated with 100 μm 8Br-cAMP for 8 min. This treatment completely activated both isoforms of PKA. Cardiomyocytes tolerated all treatments and media supplements without any loss of viability (data not shown).
High glucose or the absence of insulin had no effect on PKIγ content. However, lipid supplementation decreased PKIγ content by 25% (Fig. 7A). Likewise, lipids blunted PKA substrate phosphorylation by 66%, whereas other metabolic stresses had no effect (Fig. 7, B–D). This decreased substrate phosphorylation was mirrored by a lipid-induced decrease in PKA activity. For example, addition of 8Br-cAMP increased PKA activity in control cells by 650% but only by 450% in cells cultured with lipid supplements (Fig. 7E). This suggests that hyperlipidemia may be the primary driving factor of deficient PKA substrate phosphorylation.
FIGURE 7.
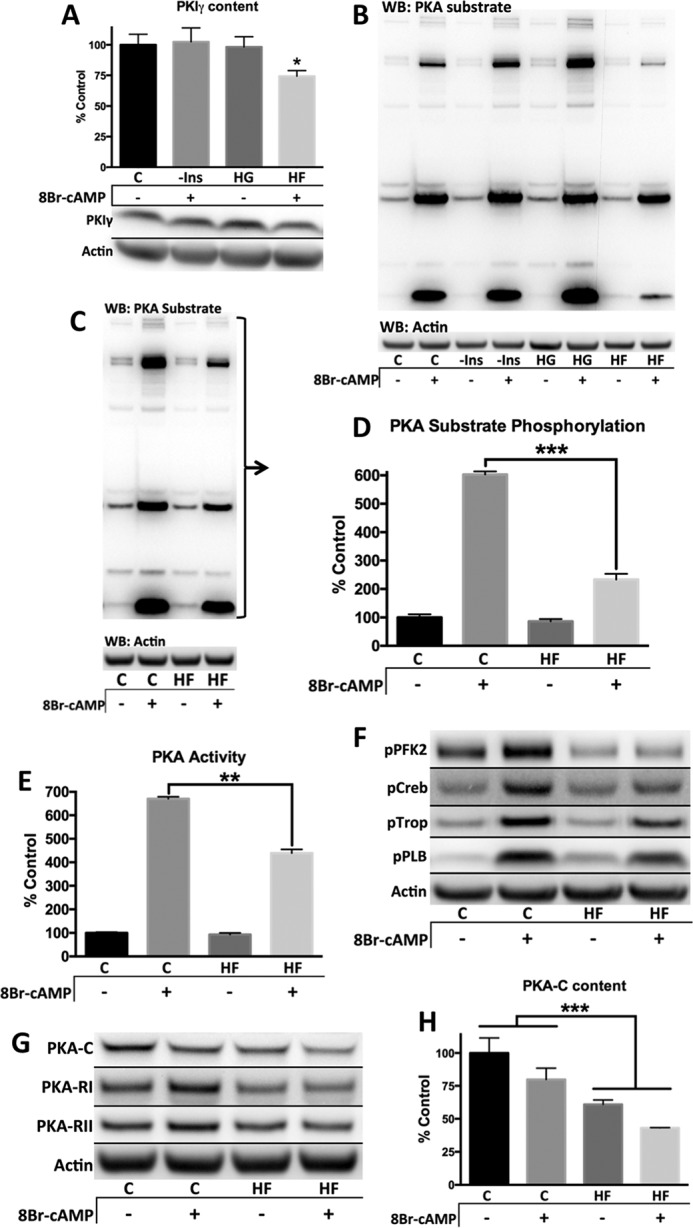
Lipid supplemented media causes blunted PKA signaling in primary adult mouse cardiomyocytes by decreasing PKA content. Primary adult mouse cardiomyocytes from control C57B6/J mice were cultured overnight with listed media modifications. All Western blots (WB, excludes E) represent cell pellets lysed in 1× SDS-PAGE sample buffer and were standardized to actin. A and B, primary adult cardiomyocyte culture media was supplemented with 10 mg/liter insulin, excluding the condition lacking insulin (−Ins). The glucose level in the high glucose condition (HG) was increased to 450 mg/dl from 100 mg/dl. The high fat medium (HF) was supplemented with 100 μm oleate/100 μm palmitate conjugated to 0.02% BSA, and all other conditions were supplemented with the appropriate vehicle. C, control. Where applicable, cells were treated with 100 μm 8Br-cAMP for 8 min. C–H, culture medium was supplemented with insulin-transferrin-selenium (10 mg/liter insulin, 5.5 mg/liter transferrin, 6.7 μg/liter sodium selenite) for both conditions, and either 100 μm oleate/100 μm palmitate bound BSA (HF) or vehicle (C). 8Br-cAMP treatment was 100 μm for 8 min. C and D, entire lane was quantified and standardized to actin. E, PKA activity measured in 1% Nonidet P-40 lysed cell fraction without additional cAMP. The values were standardized to protein concentration determined by Bradford protein assay. F–H, representative blots and quantification. n = 3 for all figures, except n = 5 for A. *, p < 0.05; **, p < 0.005; ***, p < 0.0005, unpaired Student's t test except for H, which was analyzed by two-way analysis of variance.
We have shown that PFK-2 is completely unresponsive to 8Br-cAMP-mediated phosphorylation in diabetic animals in vivo. Likewise, cells cultured with lipid supplements show a similar lack of PFK-2 phosphorylation upon 8Br-cAMP stimulation (Fig. 7F). The decrease in PKA substrate phosphorylation is likely due to decreased PKA content. Lipid supplementation caused a decrease in PKA-C and RI and, to a lesser extent, RII (Fig. 7, G and H). Interestingly, the diabetic mice also showed a trend toward decreased PKA-C and RI, but not RII subunits (Fig. 3). In cardiomyocytes, PKA-C decreased comparably in each subcellular fraction (data not shown), whereas in vivo there was a more prominent decrease in the nuclear/myofilament and cytosolic fractions.
These data support how changes in PKA-C content can have a proportionally larger effect on PKA substrate phosphorylation. Lipid-supplemented cardiomyocytes have 39% reduced PKA-C content, yet PKA substrate phosphorylation response to 8Br-cAMP is impaired by 66%. Similarly, PKA-C content is reduced on average by 13% in diabetic mice, and the phosphorylation response is reduced by 50%.
Our data support a model in which hyperlipidemia contributes to a loss of PKA-C and deficient substrate phosphorylation (Fig. 8). A distinction between our models is that in the diabetic heart, the decrease in PKA-C is more prominent in specific cell fractions, whereas in cardiomyocytes treated with lipids, there is a global decrease. PKI loss helps maintain nuclear phosphorylation as a compensatory response, yet the cytosol and myofilaments exhibit impaired PKA phosphorylation, most dynamically illustrated by the complete loss of PFK-2 phosphorylation. The blunted response to PKA activation results in contractile and metabolic deficiencies that affect the function of the diabetic heart.
FIGURE 8.
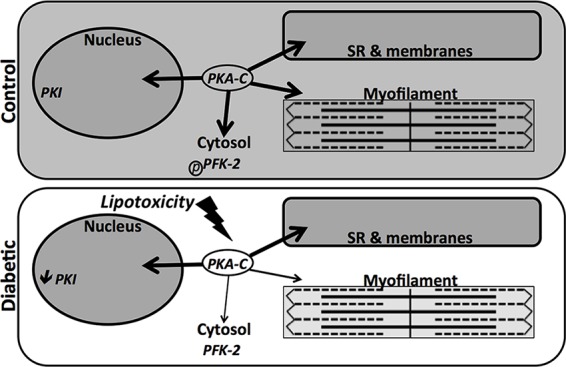
Decreased localized PKA content from lipotoxicity in the diabetic heart leads to deficient phosphorylation. The model represents how altered localized PKA content in diabetic mice may be influenced by lipids and the PKI deficit. Shading represents the extent of phosphorylation in the diabetic heart.
Discussion
This study shows that the diabetic heart has an impaired response to 8Br-cAMP that includes altered ventricular inotropy and lusitropy. These results support that postreceptor adrenergic deficiencies are present in the STZ rodent diabetic model. 8Br-cAMP-mediated phosphorylation of PKA substrates was attenuated in the diabetic heart and occurred concurrently with depressed PKA activity and altered localized content of the kinase. In addition to contractile consequences, the impact was also demonstrated metabolically by the lack of PFK-2 phosphorylation and attenuation of lactate production with 8Br-cAMP stimulation. The impaired PKA signaling present in vivo was replicated in primary adult mouse cardiomyocytes by the addition of lipids to the culture, suggesting that hyperlipidemia and lipotoxicity possess a role in the impairment of PKA signaling and thus the adrenergic pathway as a whole. Overall, we present a contributory mechanism to explain deficient ventricular performance and metabolism in the diabetic heart.
This work establishes the use of a cAMP analog as a tool for measuring the in vivo increase in cardiac performance induced by direct PKA activation, an inotropic effect that has not been previously measured in a mammalian model. cAMP analogs have been used in cell culture and ex vivo in a Langendorff perfusion system to study the response to PKA activation in disease states; however, those studies lacked the impact of the diabetic environment (15, 16, 29). The dose of 8Br-cAMP (330 mg/kg) administered was necessary for a robust inotropic response because this treatment bypasses the signal amplification portion of the β-adrenergic pathway. Further, the standard intravenous ramping protocol used with isoproterenol is not compatible with 8Br-cAMP treatment because any extended 8Br-cAMP treatment results in analysis confounding vasodilation. Thus, intraperitoneal treatments were used. The effective dosage of 8Br-cAMP was equivalent to 0.5–1 μm in primary adult mouse cardiomyocytes, as determined by comparable RII holoenzyme activation (data not shown).
Diabetic mouse models demonstrate that PKA is overactivated early on in the disease (10, 30), and this is accompanied by increased circulating norepinephrine (31). However, there is an eventual decrease in adrenergic sensitivity prior to cardiac functional decreases (13). This suggests that increased sympathetic activity compensates initially and follows other types of heart failure in which increased adrenergic signaling becomes impaired over time (10, 32, 33).
The β-adrenergic pathway is desensitized in diabetes, both in patients and animal models, and receptor changes are often considered a primary cause (14). However, cardiomyocytes isolated from STZ-induced diabetic animals show contractile impairment upon activation of β-adrenergic receptors, adenylate cyclase, or PKA (15, 16). The impaired hemodynamic response to 8Br-cAMP in diabetic mice supports the hypothesis that changes to PKA signaling are a predominant alteration and contribute to β-adrenergic dysfunction in the diabetic heart. For example, systolic function was diminished to a similar magnitude in diabetic mice whether stimulated by isoproterenol or 8Br-cAMP (control dP/dt max was 89 ± 16 and 86 ± 11% greater, respectively; Fig. 2A). The direct impact of PKA dysfunction is further supported by the findings that basal levels of cAMP and basal PKA holoenzyme activation were similar in control and diabetic animals (Figs. 3A and 4C).
Metabolic inflexibility, the rigid reliance on fatty acid oxidation for energy production, is an important component of diabetic cardiomyopathy (5, 18). This arises from both impaired mitochondrial pyruvate oxidation and glycolysis. β-Adrenergic activation primarily increases glycolysis to meet increased energetic demands. This occurs through phosphorylation and activation of PFK-2- and calcium-mediated activation of pyruvate dehydrogenase in the mitochondria (34). The complete unresponsiveness of PFK-2 to PKA phosphorylation in the diabetic heart may have a major impact on diabetic cardiac metabolism and would be especially notable under conditions of adrenergic activation and heart failure.
Lipid supplementation in culture media of primary adult mouse cardiomyocytes highlights the profound impact that elevated lipids have on PKA signaling. A reduction in PKA-C, with less loss of PKA-R, alters the ratio between the catalytic and regulatory subunits, exacerbating the effect on PKA signaling. Decreasing the ratio of PKA-C to its corresponding phosphatases exaggerates the impact on PKA signaling even further. This is proposed to explain how the reduced fractional PKA-C content in the diabetic heart significantly effects PKA substrate phosphorylation and cardiac function. Other contributing factors include the reduced PKI content, which would help maintain nuclear phosphorylation despite reduced PKA content (35).
Palmitate/oleate lipid supplementation of cardiomyocytes in culture was used to model in vivo hyperlipidemia. The concentration of lipids used for our experiments (200 μm) is similar to previous reports (36) and is in the recommended range to simulate in vivo conditions (37). Importantly, the effects of lipids on PKA signaling were dose-dependent (data not shown) and did not decrease cell viability (rod-shaped cells after 18-h incubations). Nevertheless, the impairment of PKA signaling induced by lipids in adult cardiomyocytes had some distinct differences from what was observed in diabetic hearts in vivo. For example, in cardiomyocytes the loss of PKA-C occurred in a cell wide manner, whereas in diabetic hearts, distinct pools of PKA-C were affected. These differences could be attributed to the relative short duration of lipid treatment and lack of chronic β-adrenergic stimulation in our culture model. What is clear is that in the short term, lipids impact PKA signaling, whereas other metabolic stresses, such as lack of insulin or hyperglycemia, do not.
This work shows that deficient PKA function in diabetes is a major contributing factor to adrenergic impairment and would likely decrease the efficacy of inotropes that bypass the adrenergic receptor such as phosphodiesterase inhibitors. Further, this process may be unique to diabetic cardiomyopathy compared with other forms of heart failure that do not involve hyperlipidemia. Although PKA itself is a poor therapeutic target because of its multitude of functions across many cell types, targeting the lipotoxicity-induced loss of PKA is feasible, and the specific mechanism needs further exploration. Considering how dynamically PKA regulates cardiac function and how commonly the adrenergic pathway is targeted in therapeutics, correcting the deficient PKA signaling present in the diabetic heart is a necessary direction.
Author Contributions
L. B. B. designed and performed the experiments, analyzed the data, and wrote the manuscript. K. M. H. conceived the idea for the project, assisted with experimental design and data interpretation, and reviewed/edited the manuscript.
This work was supported in part by an Institutional Development Award from the National Institute of General Medical Sciences of the National Institutes of Health through Grant P20GM104934, by Oklahoma Center for the Advancement of Science and Technology Grant HR13-183, and by American Heart Association Grant 14GRNT20510031. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PKA
- cAMP-dependent protein kinase
- 8Br-cAMP
- 8-bromoadenosine 3′,5′-cyclic monophosphate
- PKA-C
- protein kinase A catalytic subunit
- PKA-R
- protein kinase A regulatory subunit
- STZ
- streptozotocin
- PFK-2
- phosphofructokinase 2
- PKI
- protein kinase inhibitor
- AKAP
- A kinase anchoring-protein
- CREB
- cAMP response element-binding protein
- dP/dt max
- maximal rate of rise of left ventricular pressure
- dP/dt min
- maximal rate of decrease of left ventricular pressure.
References
- 1.Gilbert R. E., and Krum H. (2015) Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet 385, 2107–2117 [DOI] [PubMed] [Google Scholar]
- 2.Kannel W. B., Hjortland M., and Castelli W. P. (1974) Role of diabetes in congestive heart failure: the Framingham study. Am. J. Cardiol. 34, 29–34 [DOI] [PubMed] [Google Scholar]
- 3.Aneja A., Tang W. H., Bansilal S., Garcia M. J., and Farkouh M. E. (2008) Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757 [DOI] [PubMed] [Google Scholar]
- 4.McMurray J. J., Gerstein H. C., Holman R. R., and Pfeffer M. A. (2014) Heart failure: a cardiovascular outcome in diabetes that can no longer be ignored. Lancet Diabetes Endocrinol. 2, 843–851 [DOI] [PubMed] [Google Scholar]
- 5.Bugger H., and Abel E. D. (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57, 660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pappachan J. M., Varughese G. I., Sriraman R., and Arunagirinathan G. (2013) Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J. Diabetes 4, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pop-Busui R. (2010) Cardiac autonomic neuropathy in diabetes: a clinical perspective. Diabetes Care 33, 434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tillquist M. N., and Maddox T. M. (2012) Update on diabetic cardiomyopathy: inches forward, miles to go. Curr. Diab. Rep. 12, 305–313 [DOI] [PubMed] [Google Scholar]
- 9.Florea V. G., and Cohn J. N. (2014) The autonomic nervous system and heart failure. Circ. Res. 114, 1815–1826 [DOI] [PubMed] [Google Scholar]
- 10.Lymperopoulos A., Rengo G., and Koch W. J. (2013) Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res. 113, 739–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mauban J. R., O'Donnell M., Warrier S., Manni S., and Bond M. (2009) AKAP-scaffolding proteins and regulation of cardiac physiology. Physiology 24, 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalton G. D., and Dewey W. L. (2006) Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides 40, 23–34 [DOI] [PubMed] [Google Scholar]
- 13.Hoit B. D., Castro C., Bultron G., Knight S., and Matlib M. A. (1999) Noninvasive evaluation of cardiac dysfunction by echocardiography in streptozotocin-induced diabetic rats. J. Cardiac Failure 5, 324–333 [DOI] [PubMed] [Google Scholar]
- 14.Dinçer U. D., Bidasee K. R., Güner S., Tay A., Ozçelikay A. T., and Altan V. M. (2001) The effect of diabetes on expression of β1-, β2-, and β3-adrenoreceptors in rat hearts. Diabetes 50, 455–461 [DOI] [PubMed] [Google Scholar]
- 15.Tamada A., Hattori Y., Houzen H., Yamada Y., Sakuma I., Kitabatake A., and Kanno M. (1998) Effects of β-adrenoceptor stimulation on contractility, [Ca2+]i, and Ca2+ current in diabetic rat cardiomyocytes. Am. J. Physiol. 274, H1849–H1857 [DOI] [PubMed] [Google Scholar]
- 16.Yu Z., Quamme G. A., and McNeill J. H. (1994) Depressed [Ca2+]i responses to isoproterenol and cAMP in isolated cardiomyocytes from experimental diabetic rats. Am. J. Physiol. 266, H2334–H2342 [DOI] [PubMed] [Google Scholar]
- 17.Pacher P., Nagayama T., Mukhopadhyay P., Bátkai S., and Kass D. A. (2008) Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat. Protoc. 3, 1422–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vadvalkar S. S., Baily C. N., Matsuzaki S., West M., Tesiram Y. A., and Humphries K. M. (2013) Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem. J. 449, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphries K. M., Pennypacker J. K., and Taylor S. S. (2007) Redox regulation of cAMP-dependent protein kinase signaling: kinase versus phosphatase inactivation. J. Biol. Chem. 282, 22072–22079 [DOI] [PubMed] [Google Scholar]
- 20.Hwang S. I., and Han D. K. (2013) Subcellular fractionation for identification of biomarkers: serial detergent extraction by subcellular accessibility and solubility. Methods Mol. Biol. 1002, 25–35 [DOI] [PubMed] [Google Scholar]
- 21.McFarland B. H., and Inesi G. (1971) Solubilization of sarcoplasmic reticulum with Triton X-100. Arch. Biochem. Biophys. 145, 456–464 [DOI] [PubMed] [Google Scholar]
- 22.O'Connell T. D., Rodrigo M. C., and Simpson P. C. (2007) Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 357, 271–296 [DOI] [PubMed] [Google Scholar]
- 23.Sandberg M., Butt E., Nolte C., Fischer L., Halbrügge M., Beltman J., Jahnsen T., Genieser H. G., Jastorff B., and Walter U. (1991) Characterization of Sp-5,6-dichloro-1-β-d-ribofuranosylbenzimidazole-3′,5′-monophosphorothioate (Sp-5,6-DCl-cBiMPS) as a potent and specific activator of cyclic-AMP-dependent protein kinase in cell extracts and intact cells. Biochem. J. 279, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q., Donthi R. V., Wang J., Lange A. J., Watson L. J., Jones S. P., and Epstein P. N. (2008) Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am. J. Physiol. Heart Circ. Physiol. 294, H2889–H2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donthi R. V., Ye G., Wu C., McClain D. A., Lange A. J., and Epstein P. N. (2004) Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J. Biol. Chem. 279, 48085–48090 [DOI] [PubMed] [Google Scholar]
- 26.Ros S., and Schulze A. (2013) Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams J. A., McGlone M. L., Gibson R., and Taylor S. S. (1995) Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry 34, 2447–2454 [DOI] [PubMed] [Google Scholar]
- 28.Hausken Z. E., Coghlan V. M., and Scott J. D. (1998) Overlay, ligand blotting, and band-shift techniques to study kinase anchoring. Methods Mol. Biol. 88, 47–64 [DOI] [PubMed] [Google Scholar]
- 29.van den Ende R., Batink H. D., Michel M. C., and van Zwieten P. A. (1994) Influence of ischaemia and reperfusion on cardiac signal transduction: G protein content, adenylyl cyclase activity, cyclic AMP content, and forskolin and dibutyryl cyclic AMP-induced inotropy in the rat Langendorff heart. Fundam. Clin. Pharmacol. 8, 408–416 [DOI] [PubMed] [Google Scholar]
- 30.Hicks K. K., Seifen E., Stimers J. R., and Kennedy R. H. (1998) Effects of streptozotocin-induced diabetes on heart rate, blood pressure and cardiac autonomic nervous control. J. Auton. Nerv. Syst. 69, 21–30 [DOI] [PubMed] [Google Scholar]
- 31.Paulson D. J., and Light K. E. (1981) Elevation of serum and ventricular norepinephrine content in the diabetic rat. Res. Commun. Chem. Pathol. Pharmacol. 33, 559–562 [PubMed] [Google Scholar]
- 32.Antos C. L., Frey N., Marx S. O., Reiken S., Gaburjakova M., Richardson J. A., Marks A. R., and Olson E. N. (2001) Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ. Res. 89, 997–1004 [DOI] [PubMed] [Google Scholar]
- 33.Engelhardt S., Hein L., Wiesmann F., and Lohse M. J. (1999) Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 96, 7059–7064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Depre C., Ponchaut S., Deprez J., Maisin L., and Hue L. (1998) Cyclic AMP suppresses the inhibition of glycolysis by alternative oxidizable substrates in the heart. J. Clin. Invest. 101, 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J. H., Polanowska-Grabowska R. K., Smith J. S., Shields C. W. 4th, and Saucerman J. J. (2014) PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to beta-adrenergic signaling. J. Mol. Cell Cardiol. 66, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asrih M., Lerch R., Papageorgiou I., Pellieux C., and Montessuit C. (2012) Differential regulation of stimulated glucose transport by free fatty acids and PPARα or -δ agonists in cardiac myocytes. Am. J. Physiol. Endocrinol. Metab. 302, E872–E884 [DOI] [PubMed] [Google Scholar]
- 37.Watt M. J., Hoy A. J., Muoio D. M., and Coleman R. A. (2012) Distinct roles of specific fatty acids in cellular processes: implications for interpreting and reporting experiments. Am. J. Physiol. Endocrinol. Metab. 302, E1–E3 [DOI] [PMC free article] [PubMed] [Google Scholar]