

Background: Myosin motor function in muscle is dependent upon inter-domain interactions.
Results: Charge reversal for either of two amino acids in the interacting relay or converter domains disables myosin function in vitro and in vivo, whereas the double mutation largely restores it.
Conclusion: These residues link myosin relay and converter domains via a salt bridge.
Significance: Disrupting this communication may cause human hypertrophic cardiomyopathy.
Keywords: ATPase, cardiomyopathy, Drosophila, muscle, myosin, electron microscopy, myofibril, suppressor mutation
Abstract
Our molecular modeling studies suggest a charge-dependent interaction between residues Glu-497 in the relay domain and Arg-712 in the converter domain of human β-cardiac myosin. To test the significance of this putative interaction, we generated transgenic Drosophila expressing indirect flight muscle myosin with charge reversal mutations in the relay (E496R) or converter (R713E). Each mutation yielded dramatic reductions in myosin Ca-ATPase activity (∼80%) as well as in basal (∼67%) and actin-activated (∼84%) Mg-ATPase activity. E496R myosin-induced in vitro actin-sliding velocity was reduced by 71% and R713E myosin permitted no actin motility. Indirect flight muscles of late pupae from each mutant displayed disrupted myofibril assembly, with adults having severely abnormal myofibrils and no flight ability. To understand the molecular basis of these defects, we constructed a putative compensatory mutant that expresses myosin with both E496R and R713E. Intriguingly, ATPase values were restored to ∼73% of wild-type and actin-sliding velocity increased to 40%. The double mutation suppresses myofibril assembly defects in pupal indirect flight muscles and dramatically reduces myofibril disruption in young adults. Although sarcomere organization is not sustained in older flies and flight ability is not restored in homozygotes, young heterozygotes fly well. Our results indicate that this charge-dependent interaction between the myosin relay and converter domains is essential to the mechanochemical cycle and sarcomere assembly. Furthermore, the same inter-domain interaction is disrupted when modeling human β-cardiac myosin heavy chain cardiomyopathy mutations E497D or R712L, implying that abolishing this salt bridge is one cause of the human disease.
Introduction
Muscle myosin heavy chain is composed of an N-terminal motor domain that binds myosin light chains at its lever arm. This connects to a rod domain that dimerizes with another heavy chain molecule and assembles into multimeric thick filaments. Thick filaments interdigitate with actin containing thin filaments within sarcomeres. Myosin powers muscle contraction in an ATP-dependent manner through its interaction with actin, allowing thin filaments to slide past thick filaments. The mechanochemical cycle of the myosin molecular motor is dependent upon inter-domain interactions that communicate changes in nucleotide state, actin binding, and lever arm position (1). In this regard, the myosin relay domain serves as a critical communication center, interacting indirectly with the nucleotide and actin binding sites and directly with the converter domain, which is a key element in driving lever arm movement during the recovery and power strokes (1, 2). In this communication we use the Drosophila melanogaster transgenic system to demonstrate that a salt bridge between an amino acid residue in the relay and a residue in the converter domain is critical to myosin function and that disruption of this salt bridge is likely the root cause of two forms of hypertrophic cardiomyopathy (HCM).2
HCM entails myocardial disarray and ventricular thickening that may obstruct blood flow from the heart (3). This disease affects ∼0.2% of the population and is typically caused by dominant mutations in contractile protein genes. Mutations in the MYH7 gene, which encodes the slow skeletal muscle/β-cardiac (ventricular) isoform of human myosin heavy chain, can cause HCM as well as dilated or restrictive cardiomyopathies (4). We found that two MYH7 HCM mutations affect regions of the myosin protein that we predict to interact during the contractile cycle. The E497D mutation, which results in left ventricular hypertrophy with non-obstructive apical hypertrophic cardiomyopathy that is greatly exacerbated by coronary artery disease (5), modifies an amino acid residue in the myosin relay domain. The R712L allele, which is the cause of an age-dependent moderate to severe HCM in an Indian family (6), affects a residue in the converter domain. By using charge reversal and suppressor mutations, we demonstrate here that disruption of the 497–712 relay-converter interaction results in both mechanochemical and myofibrillar defects in the Drosophila model system.
D. melanogaster serves as an excellent model for testing myosin function in that organisms lacking endogenous myosin in their indirect flight muscles are available to act as transgene recipients (7). This facilitates isolation of pure mutant myosin for in vitro studies (8). Because Drosophila has a single Mhc gene, which encodes multiple myosin heavy chain isoforms via alternative RNA splicing (9, 10), there is no concern that isoforms produced by other muscle myosin genes will complicate the analysis. Furthermore, because indirect flight muscle function is not required for viability, the effects of expressing transgenic myosin on muscle structure and function can be assessed, no matter how severe the mutant phenotype.
For the current study, we employed an integrative approach to assess the roles of Drosophila residues Glu-496 and Arg-713 (corresponding to residues Glu-497 and Arg-712 of human MYH7) in myosin and muscle function. Molecular modeling predicted that relay domain residue Glu-496 interacts with converter residue Arg-713 via a salt bridge in both the pre-power stroke and post-rigor states. We tested the importance of this interaction by employing mutagenesis to create transgenic lines expressing E496R, R713E, or a putative compensatory mutant (E496R/R713E) designed to restore function to the single-site mutants. Biochemical, ultrastructural, locomotory, and molecular modeling data reveal that this interaction is critical for proper myosin and muscle structure and function and is expected to alter communication of the nucleotide binding state to the power stroke-generating lever arm (11, 12). The Drosophila model sheds light on the abnormality inherent in E497D and R712L human HCM mutations, suggesting that aberrations in relay-converter interaction are mechanistically linked to altered myosin function and abnormal muscle morphology that lead to induction of the HCM phenotype.
Experimental Procedures
DNA Constructs and P Element Transformation
Using the pwMhc2 genomic DNA construct (7) and site-directed mutagenesis, we constructed three transgenes: pwMhcE496R, pwMhcR713E, and pwMhcE496R-R713E. To produce pwMhcE496R, the wild-type genomic Mhc construct pwMhc2 containing P element ends and a white+ eye color marker was digested with EagI. A resulting 12.5-kb fragment was gel isolated and then subcloned into the pBluescriptKS EagI site (Stratagene, La Jolla, CA) to yield pMhc-3′. The remainder of the pwMhc2 clone from the EagI digest was re-ligated to yield pwMhc-5′. The pwMhc-5′ subclone was further digested with EcoRV. A 2.9-kb EcoRV-digested fragment from pwMhc-5′ was gel isolated and ligated into an EcoRV site in pBluescriptKS, to produce pMhc-5′-RV. The pMhc-5′-RV subclone was digested with SalI and the 0.8-kb SalI fragment was gel isolated and ligated into the SalI site of pBluescriptKS to produce pMhc-5′-Sal. The pMhc-5′-Sal subclone contains exon 9a of the relay domain. The pMhc-5′-Sal subclone was subjected to the QuikChange II kit (Stratagene) for in vitro mutagenesis. For this purpose, exon-specific primer 5′-GTTCGTCATGAGGCAAGAGGAATA-3′ was used to create the pE496R-Sal subclone (E496R nucleotide change shown as bold). The mutagenized subclone was sequenced to ensure presence of the appropriate mutated nucleotides with no other changes. Then this subclone was digested with SalI. The 0.8-kb SalI fragment was gel isolated and ligated back into the SalI site of the pMhc-5′-RV subclone to yield a pMhcE496R-5′-RV subclone. This was digested with AvrII and SphI, which produced a 2.4-kb fragment. This fragment was gel isolated and ligated into pwMhc-5′, which had been digested with AvrII and SphI to produce pwMhcE496R-5′.
To produce pwMhcR713E, the pwMhc-5′ subclone (see above) was digested with PstI and EagI. A 2.5-kb PstI/EagI fragment was gel isolated and ligated into pBluescriptKS to yield pMhc-5′-Pst-Eag. A BamHI/EagI 0.6-kb fragment from pMhc-5′-Pst-Eag was then subcloned into the BamHI/EagI sites of pBluescriptKS to yield pMhc-5′-Bam-Eag. This subclone was subjected to in vitro mutagenesis using exon-specific primer 5′-CTTCCCTAACGAGATGATGTACCC-3′ to create the pR713E-Bam subclone (R713E nucleotide change shown as bold). This was sequenced to ensure the presence of the appropriate mutated nucleotides with no other changes. pR713E-Bam was digested with BamHI and EagI. The BamHI/EagI 0.6-kb fragment was then ligated back into pMhc-5′-Pst-Eag subclone, which had been digested with BamHI and EagI, to produce the pR713E-5′-Pst subclone. This subclone was digested with SphI and EagI, yielding a 2.1-kb SphI/EagI fragment that was gel isolated and ligated back into pwMhc-5′ at its SphI/EagI sites to produce the pwMhcR713E-5′ subclone. To construct pwMhcE496R-R713E a 2.1-kb SphI/EagI fragment from pR713E-5′-Pst was subcloned into the SphI/EagI site of pwMhcE496R-5′ to produce pwMhcE496R-R713E-5′.
The resulting subclones (pwMhcE496R-5′, pwMhcR713E-5′, and pwMhcE496R-R713E-5′) were separately digested with EagI. The wild-type 3′ end subclone pMhc-3′ was digested with EagI and the 12.5-kb fragment was gel isolated and ligated into each of the three EagI-digested subclones, to yield transgenes pwMhcE496R, pwMhcR713E, and pwMhcE496R-R713E.
We sequenced the entire coding region, splice junctions, and cloning sites of the final plasmids and detected no differences compared with wild-type, except for the expected amino acid changes arising from site-directed mutagenesis. Transgenic lines containing each of these plasmids were generated by BestGene, Inc. (Chino Hills, CA), using P element-mediated transformation (13). Transgenic lines were identified by the presence of pigmented eyes and transgenes were mapped to their chromosomal locations using balancer chromosomes and standard genetic crosses. Transgenes were crossed into an Mhc10 background that is null for myosin heavy chain in the indirect flight muscles (14), resulting in expression of two copies of each transgene and no endogenous copies of Mhc in these muscles. Two such homozygous lines for each construct were analyzed in detail (Table 1).
TABLE 1.
Transgenic lines, myosin expression, and flight ability
Each transgenic line was crossed into an Mhc10 (indirect flight and jump muscle myosin null) background to determine myosin protein accumulation relative to actin. Two of the three independent lines from each transgene are shown. Values are compared to pwMhc2 (wild-type transgenic). Protein amounts from 2-day-old adult female fly upper thoraces are mean ± S.E. for at least five protein preparations and gels. Homozygous or heterozygous transgenic flies (4 h or 2 day-old) were assayed for the ability to fly up (U), horizontal (H), down (D), or not at all (N). Flight index equals 6U/T + 4H/T + 2D/T + 0N/T; T is the total number of flies tested (18). Flight abilities (up, horizontal, down, not at all) are expressed as percentages mean ± S.E.
| Line name | Chromosome location | Protein accumulation ± S.E. | No. flight tested (4 h/2 day) | Up (4 h/2 day) | Horizontal (4 h/2 day) | Down (4 h/2 day) | Not at all (4 h/2 day) | Flight index ± S.E. (4 h/2 day) |
|---|---|---|---|---|---|---|---|---|
| % | % | % | % | |||||
| pwMhc2 | X | 1.00 ±0.03 | 116/116 | 37.1/57.8 | 38.8/14.6 | 13.8/15.5 | 10.3/12.1 | 4.1 ±0.1a/4.4 ±0.1 |
| pwMhc2/+ | 101/138 | 46.5/58.0 | 35.6/20.3 | 8.9/16.7 | 8.9/5.0 | 4.4 ±0.2/4.6 ±0.1 | ||
| E496R-3 | 3 | 0.98 ±0.03 | 97/112 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| E496R-3/+ | 106/99 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 | ||
| E496R-9 | 3 | 0.96 ±0.04 | 100/117 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| E496R-9/+ | 103/107 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 | ||
| R713E-8 | X | 0.88 ±0.02 | 96/123 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| R713E-8/+ | 95/102 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 | ||
| R713E-9 | 4 | 0.98 ±0.05 | 94/117 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| R713E-9/+ | 102/103 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 | ||
| E496R/R713E-6 | X | 0.96 ±0.07 | 98/113 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| (E496R/R713E-6)/+ | 97/110 | 27.8/0 | 35.1/0 | 20.6/9 | 16.5/91 | 3.5 ± 0.1/0.2 ± 0.1 | ||
| E496R/R713E-12 | 4 | 0.98 ±0.06 | 100/132 | 0/0 | 0/0 | 0/0 | 100/100 | 0/0 |
| (E496R/R713E-12)/+ | 93/114 | 27.9/0 | 30.1/0 | 23.7/11.4 | 18.3/88.6 | 3.3 ±0.2/0.2 ±0.1 |
a Flight index data for 2 day pwMhc2 are from Ref. 35.
Transgene Expression Verification
We confirmed that each transgene expressed RNA containing the appropriate mutation and that transcripts were correctly alternatively spliced by using reverse transcription and the polymerase chain reaction (RT-PCR) and DNA sequencing. RNA was extracted from upper thoraces of 2-day-old adult female flies from each transgenic line using a LiCl2 method (15). The New England Biolabs Protoscript cDNA synthesis RT-PCR kit was used with 0.5 μg of total RNA from each transgenic line and 3 μmol of specific reverse primer (see below). PCR was performed using 1 μl of cDNA and 3 μmol of each specific primer pair (as noted below) using the following conditions: 60 s at 94 °C, then 30 cycles of: 30 s at 94 °C, 30 s at 55 °C, and 2 min at 68 °C. RT-PCR products were subcloned using the GeneJet PCR Cloning Kit from Fermentas and sequenced. At least 10 independent clones for each RT-PCR were analyzed. Alternative exons 3 and 7 were examined using a reverse primer specific to exon 8 (5′-GTTCGTCACCCAGGGCCGTA-3′) and a forward primer specific to exon 2 (5′-TGGATCCCCGACGAGAAGGA-3′). Alternative exons 9 and 11 were examined using a reverse primer specific to exon 12 (5′-GAGCTTCTTGAAGCCCTTACGG-3′) and a forward primer specific to exon 8 (5′-TCTGGATACCCAGCAGAAGCGT-3′). Alternative exon 15 was examined using a reverse primer specific to exon 16 (5′-GGGTGACAGACGCTGCTTGGT-3′) and a forward specific primer to exon 14 (5′-CTCAAGCTCACCCAGGAGGCT-3′).
Levels of myosin expression were determined by SDS-PAGE of protein from upper thoraces, followed by digital scanning. One-dimensional SDS-PAGE gels were used to determine myosin expression levels of each transgenic line, as previously described (16). Briefly, each transgenic line was crossed into an Mhc10 background to eliminate wild-type myosin accumulation in indirect flight and jump muscles. Six dissected upper thoraces from 2-day-old adult female flies for each transgenic line were homogenized in 60 μl of SDS gel sample-loading buffer (Bio-Rad, Hercules, CA). Samples were boiled for 5 min prior to loading 6 μl on a 9% polyacrylamide gel. Each transgenic line was examined at least five times, each time with a newly prepared sample. Coomassie Blue-stained polyacrylamide gels were digitally scanned using an Epson Expression 636 flatbed scanner. Myosin heavy chain and actin levels were determined by NIH image software and the myosin to actin ratio was compared with the ratio for wild-type flies. All lines examined expressed the expected mutations, splicing patterns, and wild-type levels of myosin in adult upper thoraces (Table 1).
Flight Assay
At least 100 4-h-old or 2-day-old adult flies from each transgenic line (homozygotes in an Mhc10 background or heterozygotes in an Mhc10/+ background) were assayed for flight ability at 22 °C in a Plexiglas box with a light at its top (17). Upward flight (U) was given a value of 6, horizontal (H) a value of 4, downward (D) a value of 2, and no flight (N) a value of 0. Flight index was calculated as 6U/T + 4H/T + 2D/T + 0N/T, where T is the total number of flies tested (18).
Actin and Myosin Preparation, ATPase Activity, and in vitro Motility
Actin was prepared from chicken skeletal muscle after multiple steps of polymerization-depolymerization. Filamentous actin concentration was determined by spectrophotometry. Myosin was isolated from dissected dorsolongitudinal indirect flight muscles of ∼250 transgenic flies and its concentration was determined by spectrophotometry. A typical yield was 150–250 μg. Steady-state ATPase activities of myosin were obtained using [γ-32P]ATP and increasing concentrations of filamentous actin. Basal Mg-ATPase activities obtained in the absence of actin were subtracted from all data points, which were fit with the Michaelis-Menten equation to determine actin-stimulated ATPase (Vmax) and actin affinity relative to ATPase (Km). Ca-ATPase levels, although non-physiological, were measured to assess maximal ATPase activities in the absence of actin. In vitro motility assays were carried out by adding ATP and filamentous actin labeled with fluorescent phalloidin to coverslips coated with wild-type or mutant myosin. Video sequences captured under fluorescence optics were analyzed computationally to determine actin sliding velocity. A final concentration of 2.0 μg/μl of myosin was used for ATPase assays and 0.5 μg/μl was used for the in vitro motility assays. We recently described each of these procedures in detail (19). For all ATPase and motility assays (minimum of three independent experiments), statistical significance was determined by one-way analysis of variance followed by Tukey's post hoc test using KaleidaGraph software.
Analysis of Indirect Flight Muscle Ultrastructure
Myofibril assembly and stability were examined for lines expressing each transgene in an Mhc10 background using transmission electron microscopy as previously described (20). Transverse and longitudinal thin sections were taken from fixed indirect flight muscles of each transgenic line and viewed on a Tecnai 12 transmission electron microscope. Three stages of development were assessed: pupa, 2-h-old adult, and 2-day-old adult. At least three different samples were examined from each stage of development for two transgenic lines expressing each construct.
To quantitate myofibril integrity at each developmental stage, sarcomere structure was rated on a 1–10 scale, with 10 signifying wild-type structure and 1 indicating complete disorganization. Intermediate levels of disorganization were rated based upon degree of disruption in M- and Z-lines and number and size of gaps within a sarcomere. Four sarcomeres from different myofibrils were examined for each of 25 longitudinal images taken from four flies expressing each construct, yielding a total of 100 sarcomeres rated per stage for each type of transgene. Mean scores ± S.E. are reported.
Protein Structure Analysis
Scallop muscle myosin II crystal structures were utilized as templates to predict protein structures of the Drosophila myosin mutants in the pre-power stroke state (Protein Data Bank (PDB) code 1QVI) (21) and the actin-detached post-rigor state (PDB code 1KK8) (22). The coordinates of scallop myosin II along with the amino acid sequence of Drosophila indirect flight muscle myosin S1 (with or without mutations) was submitted to the ESyPred3D 1.0 homology modeling server (23). Structures were viewed and interaction distances were measured using PyMOL. Human β-cardiac myosin (PDB code 4DB1) was also used for modeling in this manner.
Results
Molecular Modeling Predicts Interaction between Relay Domain Residue Glu-496 and Converter Domain Residue Arg-713
We modeled the Drosophila indirect flight muscle myosin heavy chain sequence onto scallop muscle myosin II crystal structures to study the location and orientation of the amino acid residues corresponding to human HCM mutations E497D and R712L (Fig. 1A). Drosophila Glu-496 is embedded in the helix of the relay domain (black), whereas Arg-713 is near the N terminus of the converter domain (green) and is adjacent to the SH1 helix (orange). The residues are in close apposition in both the pre-power stroke state (Fig. 1B) and the actin-detached post-rigor state (Fig. 1C). The charged residues form a salt bridge of 3–4 Å at both stages despite the re-orientation of the relay and converter domains relative to each other during the contractile cycle.
FIGURE 1.
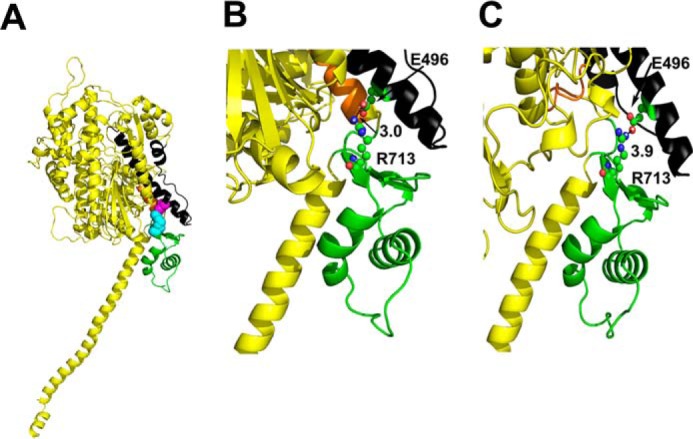
Location of interacting residues Glu-496 and Arg-713 in the myosin head. A, predicted structure of Drosophila myosin modeled using the pre-power stroke state of scallop myosin S1 (PDB code 1QVI) as a template. The relay domain is shown in black and the converter domain in green. Relay domain residue Glu-496 (magenta) is negatively charged and interacts with positively charged converter residue Arg-713 (cyan), which is just adjacent to the SH1-SH2 helix (orange). This interaction results in the formation of an inter-domain salt bridge. B, in the modeled pre-power stroke state (Proteiin Data Bank code 1QVI), relay domain residue Glu-496 shows a strong contact potential with a distance of 3.0 Å from residue Arg-713. C, in the modeled post-power stroke/near rigor state (PDB code 1KK8), relay domain residue Glu-496 shows a slightly weaker contact potential of 3.9 Å with Arg-713. Drosophila muscle myosin numbering is used here.
E496R, R713E, and E496R/R713E Transgenic Lines Lack Flight Ability, but Flight Is Restored in (E496R/R713E)/+ Heterozygotes
We generated three transgenes using in vitro mutagenesis techniques and inserted them into an indirect flight muscle myosin null background (Mhc10) by P element-mediated germline transformation and standard genetic crosses. Two of the three transgenes encode single amino acid changes (E496R and R713E) and the third putative compensatory transgene contains both mutations (E496R/R713E). RT-PCR confirmed that the alternative splicing pattern for each transgenic line is the same as in wild-type and that each transgenic line contains the appropriate site-directed mutation (E496R, R713E, or E496R/R713E) (see ”Experimental Procedures“). Myosin heavy chain accumulation levels were normal (Fig. 2), as determined by scanning of stained protein gels (Table 1). In contrast to control transgenics, where the flight index (18) is 4.1 at 4 h and 4.4 at 2 days (0 is no flight, 2 is downward, 4 is horizontal, 6 is upward), each of the transgenic lines examined (two for each construct) had absolutely no flight ability at either stage (Table 1). This was true for heterozygotes for each single mutation, E496R/+ or R713E/+ (Table 1). In contrast, the flight ability of (E496R/R713E)/+ approached normal levels (∼3.4 versus 4.6, respectively), but only at the earlier time point. Because one functional copy of the Mhc gene is not sufficient for flight (20), we conclude that the double mutation was capable of rescuing the flightless phenotype in the less restrictive heterozygous genetic background.
FIGURE 2.
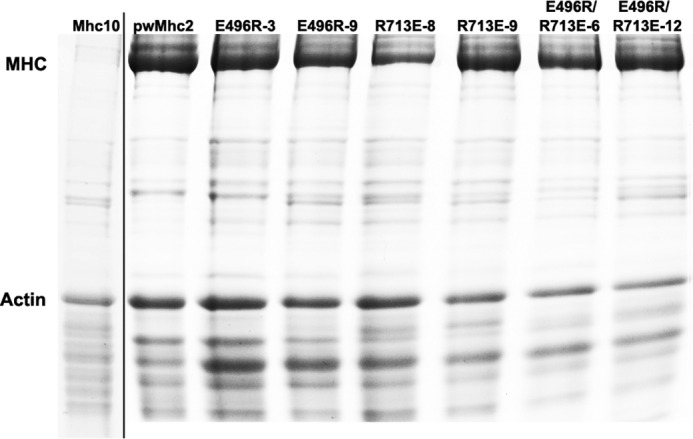
Expression of myosin heavy chain in homozygous transgenic lines. SDS-polyacrylamide gel electrophoresis of upper thoraces shows myosin heavy chain and actin levels. Quantitative results for the median relative levels of five such gels are presented in Table 1. On the left of the figure is a lane from a different gel that demonstrates the nearly complete absence of myosin in the Mhc10 line, which was the recipient genetic background for the transgenes.
E496R or R713E Mutations Dramatically Reduce Myosin ATPase Activities, but Activities Are Largely Restored by the E496R/R713E Double Mutation
Hydrolysis of ATP by myosin is an actin-stimulated process that is essential for progress through the mechanochemical cycle. As shown in Fig. 3A, myosins prepared from the E496R and R713E mutants display ∼5-fold reduction of Ca-ATPase (2.23 ± 0.32 and 1.67 ± 0.42 s−1, respectively) compared with wild-type transgenic control myosin (9.55 ± 0.92 s−1) (p < 0.001). A similar trend was obtained for basal Mg-ATPase (Fig. 3B). Compared with control, E496R and R713E mutants display ∼3-fold reduction in basal Mg-ATPase activity (0.23 ± 0.03 s−1 versus 0.09 ± 0.02 and 0.06 ± 0.02 s−1, respectively). Additionally, the actin-stimulated Mg-ATPase activities (Vmax) of E496R and R713E mutants were suppressed by ∼7-fold (0.29 ± 0.07 and 0.20 ± 0.04 s−1, respectively) compared with wild-type transgenic control myosin (1.63 ± 0.37 s−1) (Fig. 3, C and G). Interestingly, the double mutant (E496R/R713E) showed significant enhancement (p < 0.001) in Ca- (∼3.6-fold), basal Mg- (∼2.3-fold), and actin-stimulated (∼4.7-fold) ATPase activities (6.96 ± 1.23, 0.17 ± 0.04, and 1.15 ± 0.27 s−1, respectively) compared with both mutants (Fig. 3, A–C and G). However, the ATPase activities of the double mutant remain lower than those of the wild-type control (p < 0.05).
FIGURE 3.

Effects of E496R, R713E, and E496R/R713E mutations on myosin enzymatic activity and in vitro actin sliding velocity. Myosins isolated from indirect flight muscles of wild-type transgenic control, E496R, R713E, and E496R/R713E lines were assessed for (A) Ca-ATPase activity, (B) basal Mg-ATPase activity, (C) actin-stimulated Mg-ATPase activity (Vmax), and (D) actin affinity relative to ATPase (Km). To calculate the latter two parameters, Mg-ATPase activities (see panel G) were fit with the Michaelis-Menten equation. E, catalytic efficiency (determined by dividing Vmax by Km). For the ATPase activity parameters, the histograms show calculated values, standard deviations and statistical significances from multiple experiments (n = 4). F, actin sliding velocity and standard deviations as assessed by video microscopy from multiple biological replicates (n = 3; at least 40 filaments tracked per replicate). G, comparison of actin-stimulated Mg-ATPase activity for myosins isolated from indirect flight muscles of wild-type transgenic control, E496R, R713E, and E496R/R713E lines. Basal Mg-ATPase activities were subtracted from all data points. For all parameters, statistical significance is denoted as: * = p < 0.05; ** = p < 0.01; *** = p < 0.001; ns = not significant.
Based upon Mg-ATPase activity, additional ATPase parameters were compared among control, mutants, and the potential suppressor. The Km values, which measure actin affinity relative to ATPase stimulation, showed no substantial differences for both single mutants and the double mutant (0.39 ± 0.12 μm for E496R, 0.43 ± 0.15 μm for R713E, 0.35 ± 0.09 μm for E496R/R713E) as compared with control (0.31 ± 0.12 μm) (Fig. 3, D and G), suggesting that the mutant proteins remain appropriately folded to interact with actin. The significant reductions in the actin-stimulated Mg-ATPase (Vmax) (Fig. 3, D and G) result in ∼9-fold reduced catalytic efficiency (defined as Vmax/Km) in both single mutants (0.74 ± 0.18 μm−1 s−1 for E496R and 0.46 ± 0.17 μm−1 s−1 for R713E) compared with the wild-type transgenic control (5.26 ± 1.15 μm−1 s−1; p < 0.001) (Fig. 3E). The suppressor mutant (E496R/R713E) showed more than 5-fold enhancement of catalytic efficiency at 3.29 ± 1.18 μm−1 s−1 (p < 0.001) compared with both single mutants, but remains significantly lower than that for control myosin (p < 0.05). Thus although mutation of either residue associated with HCM dramatically reduces myosin ATPase activities, the double mutation (a putative suppressor) substantially rescues activities, suggesting that the interaction of the two residues is essential for myosin ATPase function.
E496R and R713E Mutations Decrease in vitro Motility, whereas the E496R/R713E Double Mutation Improves this Parameter
The in vitro actin-sliding assay essentially measures the unloaded velocity of actin filaments driven by myosin molecules and is dependent upon ATPase kinetics, myosin duty ratio, and working stroke size. We found that in vitro actin sliding velocity is greatly perturbed in both the E496R and R713E mutant myosins (Fig. 3F). E496R actin velocity was reduced more than 3-fold (p < 0.001) compared with wild-type transgenic control myosin (2.04 ± 0.13 versus 6.60 ± 0.21 μm s−1). Mutant R713E, which possesses even lower ATPase activity than E496R, did not support actin movement. The actin sliding velocity of double mutant myosin (E496R/R713E) rescues the lack of motility of the R713E myosin and is significantly higher (1.3-fold; p < 0.05) than the E496R mutant myosin (2.62 ± 0.06 versus 2.04 ± 0.13 μm s−1). However, it remains significantly (∼2.5-fold) lower (p < 0.001) than the control. The data suggest that interaction of relay domain residue 496 with converter residue 713 is important for driving in vitro actin motility.
E496R and R713E Mutations Disrupt Myofibril Assembly, whereas the E496R/R713E Double Mutation Rescues Assembly and Improves Myofibril Stability
Deficiencies in myosin function can be mirrored in defective assembly or stability of the myofibrillar apparatus (19, 24). Therefore, we used transmission electron microscopy to assay the ultrastructure of the indirect flight muscles from wild-type (pwMhc2), E496R, R713E, and E496R/R713E transgenics at the late pupal stage, in 2-h-old and 2-day-old adults (Fig. 4). Late pupal stage organisms expressing E496R (Fig. 4B) and R713E (Fig. 4C) show assembly defects, with disrupted myofibril morphology and sarcomeres with gaps and peripheral fraying compared with controls (Fig. 4A). Myofibrils containing R713E appear more severely affected. In contrast, the putative compensatory mutant E496R/R713E shows normal hexagonal packing of thick and thin filaments that is essentially identical to wild-type muscle structure (Fig. 4D). Two-h-old adults of both single mutants (Fig. 4, F and G) show continued disruption in myofibril morphology, with thick and thin filaments poorly organized and scattered throughout the cytoplasm compared with controls (Fig. 4E). In contrast, the double mutant shows only minor myofibrillar gaps, with subtle disruption in the double hexagonal packing of thick and thin filaments (Fig. 4H). In 2-day-old adults, all three mutant lines show severe myofibrillar disruption (Fig. 4, J–L). Myofibril boundaries are not defined, thick and thin filaments are scattered throughout the cell and sarcomere structure is disrupted compared with controls that retain normal structure (Fig. 4I).
FIGURE 4.

Effects of E496R, R713E, and E496R/R713E mutations on myofibril assembly and stability in dorsal longitudinal indirect flight muscles. Transverse and longitudinal sections are shown from homozygous late stage pupae, 2-h-old and 2-day-old adults. A, wild-type transgenic myosin yields normal myofibril assembly in late stage pupa. z, Z-line; m, M-line; mc, mitochondrion. B, myofibril morphology and assembly defects are present in E496R late stage pupa, with abnormal myofibril shapes as well as Z-line and M-line disruption. C, R713E late stage pupa myofibrils show poor myofilament packing. Sarcomeres display gaps and fraying. D, E496R/R713E late stage pupa myofibrils show essentially wild-type hexagonal packing of thick and thin filaments and normal arrangement of Z- and M-lines. E, wild-type transgenic retains normal myofibril structure in 2-h-old adult. F, E496R 2-h-old adult myofibril morphology has degenerated relative to the pupal stage, with scattered thick and thin filaments. Sarcomere M-line structure is lost, with large gaps and fraying of filaments from the sarcomere. G, R713E 2-h-old adult myofibril morphology shows myofibril fusion and disarray of myofilaments. Sarcomeric M-lines are barely visible. H, E496R/R713E 2-h-old adults display only minor disruption in myofibril morphology and myofilament packing compared with E496R and R713E young adults. M- and Z-lines remain intact. I, 2-day-old wild-type transgenic adult displays normal myofibril morphology, whereas 2-day-old adults of E496R (J), R713E (K), and E496R/R713E (L) lines all show severely disrupted myofibril morphology with thick and thin filaments scattered throughout the cytoplasm. Z-line structures are aberrant and M-lines are disrupted or absent. In general, mitochondrial morphology showed degeneration in older mutants, e.g. panel L, lower right. Scale bars, 0.5 μm.
We visually rated the integrity of sarcomeres for the transgenic lines at each developmental stage (Table 2). The quantitative results are in concordance with the images described above. Overall, our data indicate that defective myosin function induced by the single mutations in the relay or converter domains inhibits normal myofibril assembly, resulting in myofibril degeneration. In contrast, mutation E496R interacts with mutation R713E to re-establish proper myosin-driven myofibril assembly and to a lesser extent, muscle structure stability.
TABLE 2.
Assessment of sarcomere structure for transgenic lines
Electron micrographs of indirect flight muscles for each transgenic homozygote at multiple developmental stages were scored as to sarcomere integrity. Twenty-five longitudinal images collected from four organisms were examined at each stage of development for flies expressing each transgenic construct. For each image examined, one sarcomere was assessed from four different myofibrils. Sarcomeres were assigned values ranging from 1 to 10, with 1 indicating complete sarcomere disorganization and 10 resembling wild-type sarcomere structure. Disorganization was defined by degree of disruption in M- or Z-lines and/or gaps within a sarcomere. The mean determined from the assigned scores is reported along with the mean ± S.E.
| Stage | pwMhc2 sarcomere integrity score | E496R sarcomere integrity score | R713E sarcomere integrity score | E496R/R713E sarcomere integrity score |
|---|---|---|---|---|
| Pupal | 10 | 5.85 ±0.01 | 4.10 ±0.02 | 9.35 ±0.01 |
| Two hour | 10 | 4.40 ±0.01 | 2.90 ±0.01 | 6.70 ±0.01 |
| Two day | 10 | 2.30 ±0.01 | 2.20 ±0.02 | 2.50 ±0.01 |
Modeling Predicted Structures of E496R, R713E, and E496R/R713E Myosin Mutants Indicates Disrupted, Re-established and New Ionic Interactions
To define the predicted molecular effects of the single and double mutations, we modeled the Drosophila myosin sequence onto the scallop muscle myosin II crystal structures (Fig. 5) in the pre-power stroke state (left) and in the actin-detached post-rigor state (right). In wild-type (Fig. 5A), the closest distances between negatively charged Glu-496 and the positively charged Arg-713 residue is 3.0 Å (pre-power stroke) and 3.9 Å (post-rigor), allowing formation of a salt bridge as previously discussed (Fig. 1). In E496R (Fig. 5B), introduction of a positive charge creates repulsion with Arg-713 and increases inter-atomic distances to 4.0 Å (pre-power stroke) and 7.0 Å (post-rigor). In addition, the negatively charged Glu-499 residue forms a more stable salt-bridge with Arg-713 (2.9 Å) compared with wild-type (4.5 Å) at the post-rigor state (Fig. 5B). For mutant R713E, molecular modeling indicates that disruption of the charge attraction interaction between Glu-496 and Arg-713 increases the closest interatomic distances to 4.2 and 5.7 Å in the pre-power stroke and post-rigor configurations, respectively (Fig. 5C).
FIGURE 5.
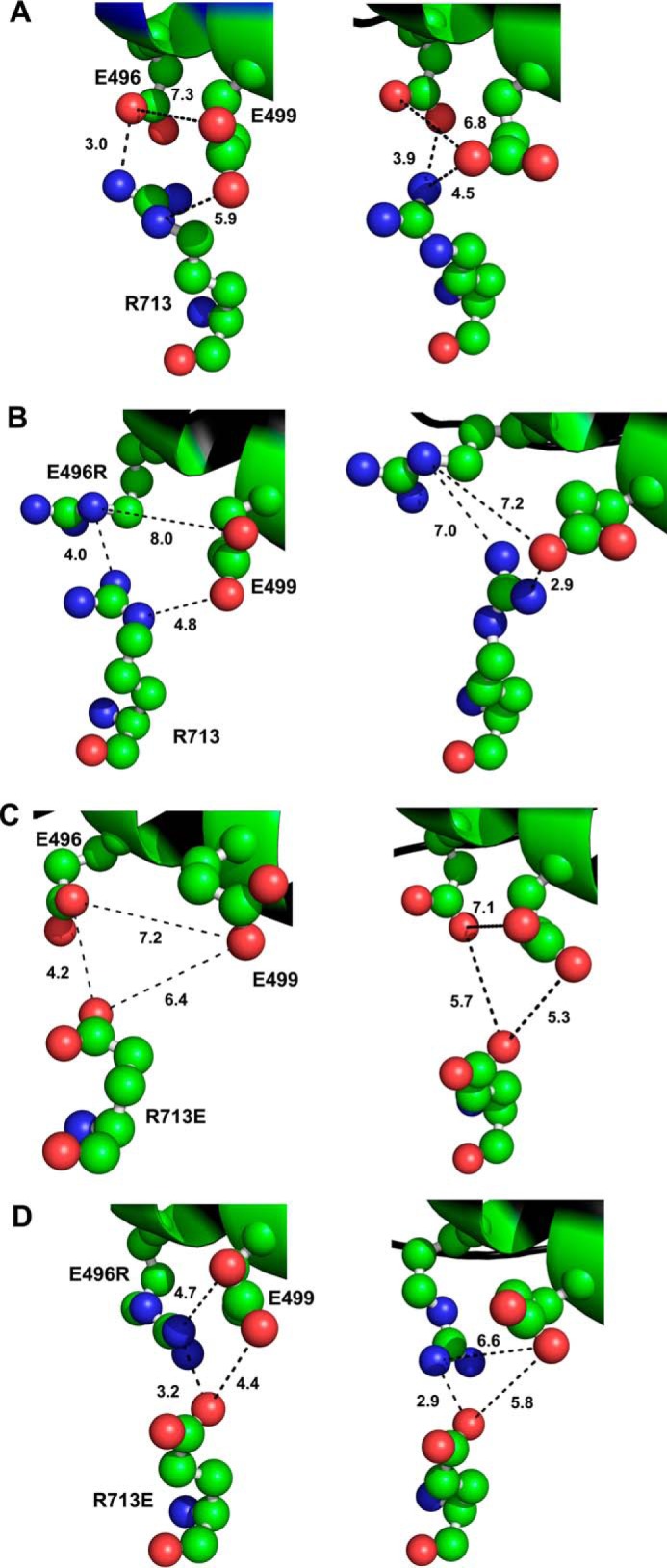
Potential interactions of amino acid residues in mutants E496R, R713E, and in putative compensatory mutant E496R/R713E. Location of residues 496, 499, and 713 in Drosophila indirect flight muscle myosin were mapped onto the crystal structure of scallop myosin S1 in the pre-power stroke state (PDB code 1QVI; left panels) and the post-power stroke, actin-detached state (PDB code 1KK8; right panels). A, in wild-type myosin, negatively charged Glu-496 is predicted to form a salt bridge with positively charged Arg-713 in the pre- (3.0 Å) and post- (3.9 Å) power stroke states. Residue Glu-499 is distant from both Arg-713 (5.9 Å) and Glu-496 (7.3 Å) in the pre-power stroke state, but shows propensity for a weak interaction with oppositely charged Arg-713 (4.5 Å) in the post-power stroke state. B, for mutant E496R, the salt bridge with Arg-713 is disrupted, although the residues remain in close proximity in the pre-power stroke state only (4.0 Å). In addition, negatively charged Glu-499 has a reduced contact distance from positively charged Arg-713 compared with wild-type at both the pre- (4.8 versus 5.9 Å) and post- (2.9 versus 4.5 Å) power stroke stages. Other distances measured between the residues would not allow significant interactions. C, for mutant R713E, the salt bridge with Glu-496 is disrupted, although the residues remain in relatively close proximity in the pre-power stroke state (4.2 Å). All other interactions previously mapped are neither stabilized via salt bridges, nor within close enough proximity to be significant. D, putative compensatory mutant E496R/R713E, restores the salt bridge of wild-type in both the pre- (3.2 Å versus 3.0 Å) and post- (2.9 Å versus 3.9 Å) power stroke states. The double mutation disrupts any propensity for a salt bridge to form between glutamic acid residues 713 and 499 (4.4 Å), but allows a unique weak interaction between E496R and Glu-499 at the pre-power stroke state (4.7 Å). Drosophila muscle myosin numbering is used throughout.
In the case of the putative compensatory mutant E496R/R713E (Fig. 5D), the charge interaction is predicted to be restored, but with some re-orientation of the side chains compared with wild type (Fig. 5A). The contact distance between E496R and R713E in the pre-power stroke state (Fig. 5D) is essentially the same as in wild-type (3.2 compared with 3.0 Å, respectively). In the post-rigor conformation, the contact distance of 2.9 Å (Fig. 5D) is less than in wild-type (3.94 Å). This reduced distance might hinder conformational changes that take place during transition states, which could account for the failure of the double mutation to fully rescue the wild-type phenotypes. In addition, absence of a weak salt bridge between residue 713 and Glu-499 that might be present in the wild-type post-rigor state (Fig. 5A, 4.5 Å) could have deleterious effects upon communication between the relay and converter domains. Furthermore, a new weak interaction between E496R and negatively charged residue Glu-499 (Fig. 5D, 4.7 Å) in the pre-power stroke state could dampen the flexibility of the relay helix.
Discussion
Mapping of myosin mutations that induce hypertrophic cardiomyopathy onto myosin crystal structures has identified hot spots that include loop 1 near the site of nucleotide exchange, the HCM loop near the actin binding site, the relay domain, and the converter domain (reviewed in Ref. 4). Several investigations have examined the effects of myosin HCM mutations on ATPase and in vitro motility properties after expressing these mutations in Dictyostelium myosin (25), smooth muscle myosin (26), transgenic mouse cardiac myosin (27), or human β-cardiac myosin from cultured mammalian muscle cells (28). Results have varied among the studies, but a consensus appears to be developing that increases in ATPase, in vitro motility, and/or force development are typical properties associated with HCM myosins (29). It thus appears that increased power (force X velocity) or alteration in the range of forces over which peak power occurs may be the basis for generating HCM (reviewed in Ref. 30).
Our novel approach demonstrated that interactions between residues analogous to human β-cardiac myosin (MYH7) Glu-497 and Arg-712 are required for normal motor function and myofibril assembly. We find that a salt bridge of 2.7 Å links these residues in human β-cardiac myosin (Fig. 6A). Furthermore, the Drosophila myosin sequence modeled onto the human β-cardiac myosin backbone yields an identical inter-residue distance, supporting the use of the Drosophila protein to predict human myosin domain interactions (Fig. 6B). Although the Drosophila mutations did not duplicate the human mutations affiliated with HCM, and thus would not necessarily mirror the changes in biochemical and physiological properties that arise in the human HCM patients, they were designed to test the concept that the mutated residues are critical for communication between the relay helix and converter domain of the molecule. Based on our results, it appears likely that disruption of the relay-converter communication pathway in human cardiomyopathy patients results in myosin dysfunction that leads to HCM in the human mutant alleles.
FIGURE 6.
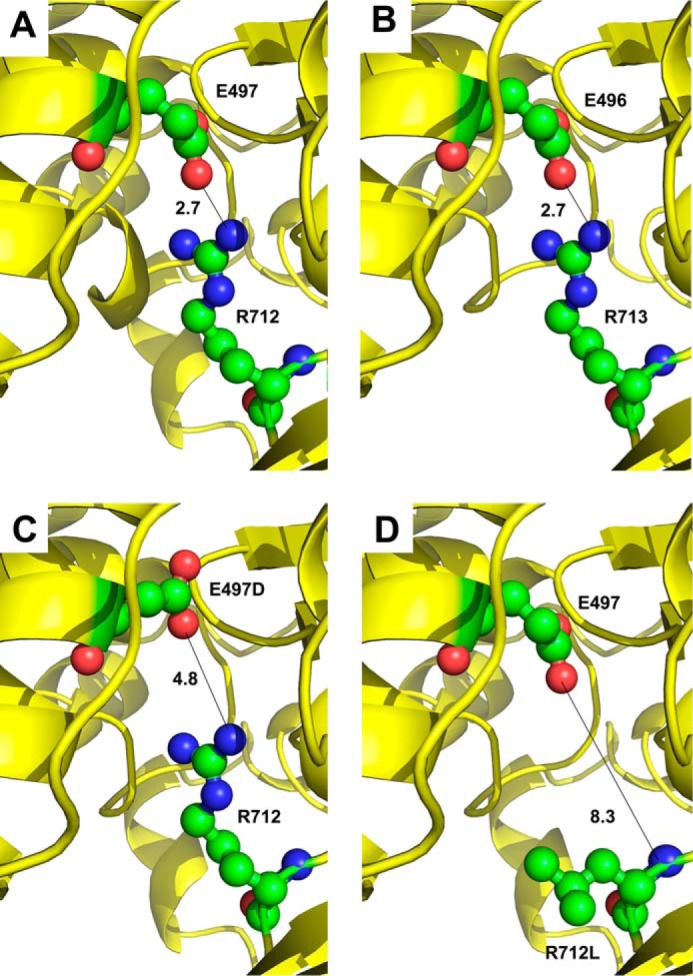
Mapping amino acid residues on the human β-cardiac myosin backbone. A, crystal structure of human β-cardiac myosin (PDB code 4DB1) in an intermediate conformation between the pre-power stroke and actin-detached post-power stroke states displays a salt bridge of 2.7 Å between human residues Glu-497 and Arg-712. B, mapping the Drosophila myosin sequence onto human β-cardiac myosin also yields a salt bridge of 2.7 Å between modeled Drosophila residues Glu-496 and Arg-713. A short N-terminal helix is absent in this model compared with panel A. C, HCM mutation E497D disrupts salt bridge formation with Arg-712 in human β-cardiac myosin, resulting in a distance of 4.8 Å between the charged atoms. D, HCM mutation R712L disrupts salt bridge formation with Glu-497 in human β-cardiac myosin, resulting in a distance of 8.3 Å between the nearest residues.
In patients, the E497D mutation results in apical hypertrophic cardiomyopathy, which is limited to the left ventricular apex (5). The effects of this mutation can be compounded by coronary artery disease, in that a relative of the proband with both coronary artery disease and the HCM mutation displayed massive concentric hypertrophy (5). Although the mutant E497D residue retains its negative charge, its reduced bulkiness results in a greater distance between residues 497 and 712, obviating a strong charge interaction (Fig. 6C). For the R712L mutation, the proband displayed electrocardiogram and echocardiograph evidence diagnostic of HCM, and six patients with the mutation showed a moderate to severe clinical course that was age dependent (6). This mutation disrupts the salt bridge to Glu-497 (Fig. 6D) as did the Drosophila E496R mutation. Unlike the Drosophila allele, it would not create a repulsive force. For the human E497D and R712L mutations, it remains to be tested whether mutations in the human myosin backbones yield enhanced or reduced power production.
Another human mutation that affects the residue 497 equivalent, E498G in the MYH3 gene, causes Freeman-Sheldon syndrome, a severe form of distal arthrogryposis (31). MYH3 is primarily expressed in fetal skeletal muscle and Freeman-Sheldon syndrome causes multiple contractures in feet and hands of the affected children. Based upon molecular modeling, Toydemir et al. (31) predicted that the E498G mutation would disrupt the salt bridge interaction with Arg-712. Our study verifies this thesis and shows that this salt bridge is indispensable for normal motor function, myofibril stability, and locomotion.
Several computational studies have provided evidence for the relay functioning as a communication pathway between the nucleotide-binding pocket and the converter domain. This pathway links the state of the nucleotide pocket to the ability of the lever arm to be cocked in the pre-power stroke state or for its release to produce the power stroke. Fischer et al. (32) used the conjugate peak refinement method in conjunction with crystal structures to build a model that transitions myosin from the post-power stroke to the pre-power stroke state via the ATP-induced recovery stroke. By using multiple myosin sequence alignments and physical proximity, Tang et al. (12) calculated the shortest paths between the catalytic site and the lever arm to identify conserved interacting residues in pre- and post-power stroke structures. Some of these residues had been shown previously to be functionally important via mutagenesis experiments. Interestingly, all of the rigor and post-rigor structures have the equivalent of human residue Glu-497 identified as a pathway member, although not by interaction with Arg-712. This is likely because the authors chose an alternative pathway termination point, even though Arg-712 was identified as a part of a residue cluster near the beginning of the lever arm (12). In another study, Yu et al. (33) used targeted molecular dynamics to define amino acid residues important to the recovery stroke. They indicated that breaking of a hydrogen bond between Glu-497 and Met-493 occurs when the relay helix unwinds (also see Ref. 32), but that Glu-497 subsequently interacts with Arg-712 and then Arg-712 interacts with Glu-500 to stabilize a kink in the relay helix. Clearly the interacting Glu-497 and Arg-712 residues we focused on in this study are key elements in relaying information from the active site to the converter domain/lever arm. Additional mutagenesis studies have shown that other residues at this interface serve a critical role in relay-converter interaction (19, 34).
A remarkable aspect of our analysis is that ultrastructural defects in sarcomere assembly can arise from mutations in the relay or converter domains. This is consistent with our previous studies on mutants and isoform domain switches (19, 35, 36), and suggests that a functional relay-converter interface permits necessary interactions required for myofibrillogenesis. Structural defects may be exacerbated by altered actin-myosin interactions during attempted muscle use. Our results raise the question as to whether such abnormalities in sarcomere assembly, even if they are subtle, might arise due to HCM mutations in human cardiac muscle. Perhaps these sorts of structural defects, in conjunction with myosin biochemical abnormalities, trigger the compensatory program that eventually results in hypertrophy. It is possible that structural defects in the myofibril in the absence of motor abnormalities can lead to HCM, which could explain the ability of rod domain mutations to induce HCM (4), even though they are unlikely to affect myosin motor activity.
In summary, we found that two myosin residues implicated in HCM interact via a salt bridge and that disruption of this bridge by charge reversal mutations results in severe defects in the myosin ATPase, actin sliding velocity, myofibril assembly/stability, and muscle function. Using a unique genetic suppression approach, we showed that compensatory mutations could rescue many of these defects, thereby verifying that this specific interaction between the relay helix and the converter domain is absolutely critical to myosin function and that its disruption is the likely primary cause of two varieties of HCM.
Author Contributions
W. A. K. and S. I. B. designed the study. S. I. B. coordinated it. W. A. K., S. I. B., and G. C. M. performed overall data interpretation and wrote the paper. W. A. K. made DNA constructs, performed RT-PCR, sequence analysis, genetic crosses, protein electrophoresis, flight testing, electron microscopy and molecular modeling. A. M. purified proteins and analyzed in vitro motility. G. C. M. performed and analyzed ATPase data. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgment
We appreciate comments provided on the manuscript by Dr. Douglas Swank (Rensselaer Polytechnic Institute).
Note Added in Proof
Following submission of this article, a human β-cardiac myosin structure (PDB code 4P7H) was published, detailing the interaction between Glu-497 and Arg-712 (37). An additional structure described in this study (PDB code 4PA0) shows binding of the force-enhancing drug omecamtiv mecarbil to a pocket that includes these residues, likely stabilizing their interaction.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM32443 (to S. I. B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HCM
- hypertrophic cardiomyopathy
- PDB
- Protein Data Bank.
References
- 1.Geeves M. A., and Holmes K. C. (2005) The molecular mechanism of muscle contraction. Adv. Protein Chem. 71, 161–193 [DOI] [PubMed] [Google Scholar]
- 2.Bloemink M. J., Dambacher C. M., Knowles A. F., Melkani G. C., Geeves M. A., and Bernstein S. I. (2009) Alternative exon 9-encoded relay domains affect more than one communication pathway in the Drosophila myosin head. J. Mol. Biol. 389, 707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gersh B. J., Maron B. J., Bonow R. O., Dearani J. A., Fifer M. A., Link M. S., Naidu S. S., Nishimura R. A., Ommen S. R., Rakowski H., Seidman C. E., Towbin J. A., Udelson J. E., Yancy C. W., and American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. (2011) 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 58, e212–260 [DOI] [PubMed] [Google Scholar]
- 4.Colegrave M., and Peckham M. (2014) Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat. Rec. (Hoboken) 297, 1670–1680 [DOI] [PubMed] [Google Scholar]
- 5.Arad M., Penas-Lado M., Monserrat L., Maron B. J., Sherrid M., Ho C. Y., Barr S., Karim A., Olson T. M., Kamisago M., Seidman J. G., and Seidman C. E. (2005) Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112, 2805–2811 [DOI] [PubMed] [Google Scholar]
- 6.Sakthivel S., Joseph P. K., Tharakan J. M., Vosberg H. P., and Rajamanickam C. (2000) A novel missense mutation (R712L) adjacent to the “active thiol” region of the cardiac beta-myosin heavy chain gene causing hypertrophic cardiomyopathy in an Indian family. Hum. Mutat. 15, 298–299 [DOI] [PubMed] [Google Scholar]
- 7.Swank D. M., Wells L., Kronert W. A., Morrill G. E., and Bernstein S. I. (2000) Determining structure/function relationships for sarcomeric myosin heavy chain by genetic and transgenic manipulation of Drosophila. Microsc. Res. Tech. 50, 430–442 [DOI] [PubMed] [Google Scholar]
- 8.Swank D. M., Bartoo M. L., Knowles A. F., Iliffe C., Bernstein S. I., Molloy J. E., and Sparrow J. C. (2001) Alternative exon-encoded regions of Drosophila myosin heavy chain modulate ATPase rates and actin sliding velocity. J. Biol. Chem. 276, 15117–15124 [DOI] [PubMed] [Google Scholar]
- 9.Bernstein S. I., Mogami K., Donady J. J., and Emerson C. P. Jr. (1983) Drosophila muscle myosin heavy chain encoded by a single gene in a cluster of muscle mutations. Nature 302, 393–397 [DOI] [PubMed] [Google Scholar]
- 10.Rozek C. E., and Davidson N. (1983) Drosophila has one myosin heavy-chain gene with three developmentally regulated transcripts. Cell 32, 23–34 [DOI] [PubMed] [Google Scholar]
- 11.Holmes K. C., Schröder R. R., Sweeney H. L., and Houdusse A. (2004) The structure of the rigor complex and its implications for the power stroke. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang S., Liao J. C., Dunn A. R., Altman R. B., Spudich J. A., and Schmidt J. P. (2007) Predicting allosteric communication in myosin via a pathway of conserved residues. J. Mol. Biol. 373, 1361–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubin G. M., and Spradling A. C. (1982) Genetic transformation of Drosophila with transposable element vectors. Science 218, 348–353 [DOI] [PubMed] [Google Scholar]
- 14.Collier V. L., Kronert W. A., O'Donnell P. T., Edwards K. A., and Bernstein S. I. (1990) Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 4, 885–895 [DOI] [PubMed] [Google Scholar]
- 15.Becker K. D., O'Donnell P. T., Heitz J. M., Vito M., and Bernstein S. I. (1992) Analysis of Drosophila paramyosin: identification of a novel isoform which is restricted to a subset of adult muscles. J. Cell Biol. 116, 669–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Donnell P. T., Collier V. L., Mogami K., and Bernstein S. I. (1989) Ultrastructural and molecular analyses of homozygous-viable Drosophila melanogaster muscle mutants indicate there is a complex pattern of myosin heavy-chain isoform distribution. Genes Dev. 3, 1233–1246 [DOI] [PubMed] [Google Scholar]
- 17.Drummond D. R., Hennessey E. S., and Sparrow J. C. (1991) Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. Mol. Gen. Genet. 226, 70–80 [DOI] [PubMed] [Google Scholar]
- 18.Tohtong R., Yamashita H., Graham M., Haeberle J., Simcox A., and Maughan D. (1995) Impairment of muscle function caused by mutations of phosphorylation sites in myosin regulatory light chain. Nature 374, 650–653 [DOI] [PubMed] [Google Scholar]
- 19.Kronert W. A., Melkani G. C., Melkani A., and Bernstein S. I. (2014) Mapping interactions between myosin relay and converter domains that power muscle function. J. Biol. Chem. 289, 12779–12790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Donnell P. T., and Bernstein S. I. (1988) Molecular and ultrastructural defects in a Drosophila myosin heavy chain mutant: differential effects on muscle function produced by similar thick filament abnormalities. J. Cell Biol. 107, 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gourinath S., Himmel D. M., Brown J. H., Reshetnikova L., Szent-Györgyi A. G., and Cohen C. (2003) Crystal structure of scallop myosin S1 in the pre-power stroke state to 2.6-Å resolution: flexibility and function in the head. Structure 11, 1621–1627 [DOI] [PubMed] [Google Scholar]
- 22.Himmel D. M., Gourinath S., Reshetnikova L., Shen Y., Szent-Györgyi A. G., and Cohen C. (2002) Crystallographic findings on the internally uncoupled and near-rigor states of myosin: further insights into the mechanics of the motor. Proc. Natl. Acad. Sci. U.S.A. 99, 12645–12650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert C., Léonard N., De Bolle X., and Depiereux E. (2002) ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 24.Bejsovec A., and Anderson P. (1990) Functions of the myosin ATP and actin binding sites are required for C. elegans thick filament assembly. Cell 60, 133–140 [DOI] [PubMed] [Google Scholar]
- 25.Liu X., Shu S., Kovács M., and Korn E. D. (2005) Biological, biochemical, and kinetic effects of mutations of the cardiomyopathy loop of Dictyostelium myosin II: importance of ALA400. J. Biol. Chem. 280, 26974–26983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita H., Tyska M. J., Warshaw D. M., Lowey S., and Trybus K. M. (2000) Functional consequences of mutations in the smooth muscle myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. J. Biol. Chem. 275, 28045–28052 [DOI] [PubMed] [Google Scholar]
- 27.Lowey S., Lesko L. M., Rovner A. S., Hodges A. R., White S. L., Low R. B., Rincon M., Gulick J., and Robbins J. (2008) Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α- or β-myosin heavy chain backbone. J. Biol. Chem. 283, 20579–20589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K., and Spudich J. A. (2013) Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. U.S.A. 110, 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore J. R., Leinwand L., and Warshaw D. M. (2012) Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spudich J. A. (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toydemir R. M., Rutherford A., Whitby F. G., Jorde L. B., Carey J. C., and Bamshad M. J. (2006) Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 38, 561–565 [DOI] [PubMed] [Google Scholar]
- 32.Fischer S., Windshügel B., Horak D., Holmes K. C., and Smith J. C. (2005) Structural mechanism of the recovery stroke in the myosin molecular motor. Proc. Natl. Acad. Sci. U.S.A. 102, 6873–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H., Ma L., Yang Y., and Cui Q. (2007) Mechanochemical coupling in the myosin motor domain: II. analysis of critical residues. PLoS Comput. Biol. 3, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shih W. M., and Spudich J. A. (2001) The myosin relay helix to converter interface remains intact throughout the actomyosin ATPase cycle. J. Biol. Chem. 276, 19491–19494 [DOI] [PubMed] [Google Scholar]
- 35.Kronert W. A., Dambacher C. M., Knowles A. F., Swank D. M., and Bernstein S. I. (2008) Alternative relay domains of Drosophila melanogaster myosin differentially affect ATPase activity, in vitro motility, myofibril structure and muscle function. J. Mol. Biol. 379, 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kronert W. A., Melkani G. C., Melkani A., and Bernstein S. I. (2012) Alternative relay and converter domains tune native muscle myosin isoform function in Drosophila. J. Mol. Biol. 416, 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkelmann D. A., Forgacs E., Miller M. T., and Stock A. M. (2015) Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat. Commun. 6, 7974. [DOI] [PMC free article] [PubMed] [Google Scholar]