

Background: Jak3 is a tyrosine kinase and its role in metabolic syndrome is not known.
Results: Jak3 was essential for intestinal mucosal tolerance through suppressed expression and activation of TLRs.
Conclusion: Jak3 expression prevented development of obesity and metabolic syndrome.
Significance: Understanding mucosal functions of Jak3 have important implications for patients with diabetes.
Keywords: diabetes, inflammation, innate immunity, Janus kinase (JAK), metabolic syndrome, Toll-like receptor 4 (TLR4)
Abstract
Obesity, a worldwide epidemic, is a major risk factor for the development of metabolic syndrome (MetS) including diabetes and associated health complications. Recent studies indicate that chronic low-grade inflammation (CLGI) plays a key role in metabolic deterioration in the obese population. Previously, we reported that Jak3 was essential for mucosal differentiation and enhanced colonic barrier functions and its loss in mice resulted in basal CLGI and predisposition to DSS induced colitis. Since CLGI is associated with diabetes, obesity, and metabolic syndrome, present studies determined the role of Jak3 in development of such conditions. Our data show that loss of Jak3 resulted in increased body weight, basal systemic CLGI, compromised glycemic homeostasis, hyperinsulinemia, and early symptoms of liver steatosis. Lack of Jak3 also resulted in exaggerated symptoms of metabolic syndrome by western high-fat diet. Mechanistically, Jak3 was essential for reduced expression and activation of Toll-like receptors (TLRs) in murine intestinal mucosa and human intestinal epithelial cells where Jak3 interacted with and activated p85, the regulatory subunit of the PI3K, through tyrosine phosphorylation of adapter protein insulin receptor substrate (IRS1). These interactions resulted in activation of PI3K-Akt axis, which was essential for reduced TLR expression and TLR associated NFκB activation. Collectively, these results demonstrate the essential role of Jak3 in promoting mucosal tolerance through suppressed expression and limiting activation of TLRs thereby preventing intestinal and systemic CLGI and associated obesity and MetS.
Introduction
Obesity is a major risk factor for the development of metabolic syndrome (MetS)3 and associated health complications. MetS is a group of interrelated metabolic disorders that include hyperinsulinemia, hyperglycemia, hyperlipidemia, and hepatic steatosis (1). Studies suggest that obesity associated systemic chronic low grade inflammation (CLGI) contributes to the development of MetS (1). Results from some recent studies indicate that CLGI particularly of liver and adipose tissues plays a key role in metabolic deterioration in the obese population (2–4) where adipose tissue is reported as the key source for increased pro-inflammatory cytokines during obesity (5–9). The roles of Toll-like receptors (TLRs) in the pathogenesis of inflammation-mediated insulin resistance and associated metabolic complications in context with adipocytes, hepatocytes, and immunocytes have been reported; however the role of enterocytes in such a setting is less understood (46). Though gastrointestinal (GI) tracts connects the outer environment with the inner body and several TLRs are expressed on mucosal epithelial cells of the GI tracts, the regulation of the hypo-responsiveness and tolerance of these receptors toward huge load of commensal bacteria and dietary components in the human intestine remains unclear (42, 43). Moreover, TLR-mediated activation of NF-κB plays pivotal role in the production of various inflammatory cytokines, such as IL-6, TNF-α, MCP-1, IL-1β, etc (10, 11). Results of various studies have provided evidence of increased expression of TLRs, especially TLR4 in obesity and nonalcoholic steatohepatitis (12–15) where CLGI-driven obesity and related metabolic syndrome appears to be key factors responsible for associated health complications (16–19). However, the specific roles of intestinal mucosa in general and mucosal Janus kinase-3 (Jak3) in particular in the development of CLGI and associated diabesity (diabetes and obesity) are not known.
Janus kinases (Jaks) are a family of non-receptor tyrosine kinase with four members; Jak1, Jak2, Jak3, and Tyk2 (20). Studies have shown that Jak3 is widely expressed in different organs including intestine of both humans and mice (20, 21). Like other members, Jak3 mediates signals initiated by different cytokines through interactions with the common γ (gamma) chain of several cytokine receptors (20, 22). Gastrointestinal (GI) tract is the first organ that interacts with dietary components and luminal microbiota both of which are known to regulate diabesity (16, 17, 23, 24). Our previous works show multiple roles of mucosal Jak3 in the regulation of intestinal functions including mucosal cytoskeletal remodeling (25), intestinal restitution (26), mucosal homeostasis and differentiation (27), mucosal barrier functions (28), trans-molecular regulation of Jak3 activation, and assessment of Jak3-directed therapies (29, 30). Recently, we reported that loss of colonic mucosal expression of Jak3 not only led to reduced mucin expression and development of intestinal CLGI in mice but they also had tendency of gaining body weight, a phenotype consistent with human metabolic syndrome (28). Though CLGI play a key role in the development of metabolic syndrome in the obese population (2–4), the specific roles of Jak3 in the development of CLGI and associated diabesity are not known. The present study demonstrates mechanisms through which Jak3 regulates mucosal tolerance, CLGI, and associated metabolic syndrome.
Experimental Procedures
Materials
Materials were obtained from the following sources: Caco2 (ATCC), HT-29 Cl-19A (a kind gift from Dr. A. P. Naren, CFRC, Cincinnati, OH); a permanently differentiated homogenous clone derived from human colonic epithelial cells HT-29 (31) was used as reported before (25, 28, 32); Hematoxylin, Eosin, Xylene, Alcian blue (VWR), Antibodies: pY20 (MP Biomedicals); Jak3, IgG, β-actin, TLR4, villin (Santa Cruz Biotechnology Inc.), TLR2 (Novous Biologicals), NF-κB, p NF-κB, IRS1, pIRS1, p85, p85(y463), pAkt, Akt, (Cell signaling), F4/80 (eBioscience); LPS, Akt inhibitor A6730, wortmannin (Sigma), LPS-RS (Invivogen), Rat/mouse Insulin ELISA-Kit (EMD-Millipore), Multianylate cytokine assay kit (Qiagen). Pierce® BCA Protein Assay Kit (Thermo Scientific). Recombinant human insulin was from Novo Nordisk, and glucose was from Baxter Clintec.
Animals
6–8-week old C57BL/6 mice (WT) or C57BL/6-background jak3−/− mice (KO): S form (33), with intact kinase domain (34) were from the Jackson Laboratory (USA). jak3−/− mice were back-crossed to C57BL/6 jak3+/+ to generate jak3−/+ mice which were then in-bred to generate jak3−/−, and jak3+/+ littermates, which were either co-housed or housed separately according to sex and genotype. For HFD studies, male C57/BL6 mice, weighing 22 ± 2 g, were fed a high-fat diet (65% cal from fat) or a normal diet (6.5% cal from fat) for 8 weeks. The animals were housed in a temperature- and light-controlled room. The mice had food and water ad libitum. Mouse chow was purchased from Research Diets (New Brunswick, NJ). This study adhered to the institutional guidelines of Texas A&M University Institutional Animal Care and Use Committee.
Assessment of Body Weight and MPO Activity
Mice were weighed on the day of weaning and regular fixed intervals thereafter. Body weights were expressed as percent (%) compared with initial body weight at the time of weaning (as 100%). For HFD studies, mice on western high fat diet or respective control diet were weighed every alternate day for 10 weeks and then the difference in the body weight between percentage changes from the baseline was calculated. Quantitative determination of serum MPO was done as reported before (28). Briefly, serum samples were diluted 1:500 with dilution buffer and measured in duplicates using micro plate reader (Awareness Technology Inc.).
Epidydamal Fat-pad and Liver Weight Determination
Twenty-month-old male C57Bl/6 mice were placed on western high fat diet, or the control diets and analyzed for changes in epidydamal fat pad weight and the gross liver weight.
Liver Histological Examination
For histological analysis, OCT mold embedded tissue sections of liver from WT and KO mice with or without were air-dried for 20 min at RT and fixed in 95% ethanol and tissue sections were stained with hematoxylin-eosin (H&E) and examined under light microscopy at 200× magnification as reported before (28). The mean degree of liver steatosis in liver was calculated from observation of 25 different fields of H&E-stained sections of liver from each animal (35).
Fasting Blood Glucose, Glucose Tolerance Test, and Insulin Tolerance Test
After mice were deprived of food for 4h (before the insulin tolerance test) or overnight (12 h; for fasting glucose), basal blood glucose levels were measured via tail bleeding using a glucometer (Free style). For glucose tolerance test the mice were intraperitoneally injected with a bolus of glucose (2 mg of glucose per g of body weight). For insulin tolerance tests, the mice were subject to a bolus injection of insulin (0.75 m-units of insulin/g of body weight). Blood glucose levels were subsequently measured at the times indicated.
Immunofluorescence Microscopy (IFM)
For IFM, OCT mold embedded tissue sections were air-dried for 20 min at RT, fixed using 4% paraformaldehyde, and blocked using 5% BSA in PBS for 30 min. Sections were than incubated with primary antibodies for Jak3, TLR4, TLR2, villin, F4/80, pNF-κB, NF-κB, pIRS1, and IRS1 followed by incubation with cy3 or Alexa-fluor 488 conjugated secondary antibodies. The sections were than rinsed twice with PBS and mounted using Vectashield (Vector Lab). For all negative controls primary antibodies were replaced with a control non-immune IgG at the same concentrations. IFM for Caco2 cells transduced with RFP-tagged Jak3 shRNA were done as reported previously. The immunostained slides were visualized using C1-plus Nikon laser scanning confocal microscope and the images were processed using NIS element software (NikonR). All experiments were conducted at least in triplicate and representative images were shown.
Serum Cytokine Assay
IL-6 and IL-17a were determined in the sera from WT and KO mice using Multi-Anylate ELISA Array Kit per the manufacturer's protocol. The absorbance at 450 nm was measured using micro plate reader (Awareness Technology Inc.).
Tissue Lysis, Immunoprecipitation (IP), Western Blot (IB)
The frozen colon tissue samples from WT and KO mice were resuspended (2 ml/g of tissue) in ice-cold lysis buffer (50 mmol/liter Tris, pH 7.4, containing 1% Nonidet P-40, 0.1 mmol/liter EGTA, 0.1 mmol/liter EDTA, 2 mmol/L β-mercaptoethanol, 5 μmol/liter leupeptin, and 4 μmol/liter pepstatin) for 20 min. The suspended tissues were cut into small pieces using a sterilized surgical scissor and homogenized using a homogenizer (Wheaton Overhead Stirrer, CT) with 20 brief pulses. The resulting homogenates were centrifuged at 14,000 rpm for 10 min at 4 °C. Protein concentrations in the post-nuclear supernatants (total protein extract) were determined using BCA protein assay kit. SDS-PAGE separated and electro blotted samples were analyzed for protein expression using indicated antibodies. IP and IB using tissue lysate or cell lysates from Caco2 cells grown under indicated experimental conditions were done as reported previously (27).
Cell Culture, LPS, CP-690550 (tofacitinib), and A6730 Treatment
Caco2 or HT-29 Cl-19A cells were grown to confluence followed by treatment with lentiviruses for doxycycline-regulated RFP-tagged jak3-shRNA expression in presence or absence of doxycycline to knock-down Jak3-expression as reported (27). For Jak3 and Akt inhibition studies cells were treated with or without Jak3 inhibitor CP-690550 (tofacitinib) or Akt inhibitor A6730 in presence or absence of LPS (100 ng/ml) as reported before (25–27). Expression of TLR4 and pNF-κB were determined using Western analysis (IB) lysates of these cells.
Data Analysis
All data are presented as mean (±S.E.) and analyzed using Microcal OriginR software version 9.2. Differences in the parametric data were evaluated by the Student's t test. Significance in all tests was set at a 95% or greater confidence level.
Results
Loss of Jak3 Leads to Increased Body-Weight
Previously we reported that loss of Jak3 not only resulted in predisposition to DSS-induced colitis but during the same period the control untreated Jak3-KO mice also tend to gain weight (28). To confirm that, these effects of weight gain were due to loss of Jak3-driven obesity and not an anomaly associated with familial transmission of microbiota, we back-crossed Jak3-KO mice to C57BL/6-wt in our facility to generate WT and Jak3-KO siblings and co-housed them by sex only right from birth so that both WT and Jak3-KO mice were exposed to the similar microbiota from the beginning of their life. In another set, mice were housed by sex and genotype only (non-co-housed) to determine if these have an effect. Determination of body-weight from co-housed or non-co-housed (Fig. 1, A–C) mice showed that irrespective of housing, loss of Jak3 led to approximately two (in male; Fig. 1A) to 3-fold (in female; Fig. 1B) increase in body-weight compared with their WT littermates. Since caging practices had little influence on body weight, rests of the experiments were performed using mice housed by sex and genotype (as indicated in Fig. 1C) unless otherwise mentioned.
FIGURE 1.

Loss of Jak3 leads to increased body weight and systemic chronic low-grade inflammation: A–B, jak3−/− mice were back-crossed to C57BL/6 jak3+/+ to generate jak3−/+ mice, which were then in-bred to generate jak3−/− and jak3+/+ littermates. Body weight of co-housed Jak3-KO and their WT siblings male (A) or female (B) or non-co-housed males Jak3-KO and WT (C) were plotted at the indicated intervals as a percentage of their body weight at the time of weaning. D–E, increased pro-inflammatory cytokines IL-6 and IL17a in serum of KO mice: cytokine level in the blood serum from WT and KO mice were measured using a mouse Multianylate cytokine assay kit (Qiagen) per the manufacturer's protocol, and mean values from each group (n = 6 mice per group) are shown. F, MPO in serum of WT and KO mice: MPO activity was determined as described in “Experimental Procedures” and plotted as mean values from each group (n = 6 mice per group). G–H, loss of Jak3 leads to increased colonic macrophages in mice: Colonic tissue sections from WT and Jak3-KO mouse were immunostained for macrophages using F4/80 antibody (G). Images were acquired using Nikon C1-plus laser confocal microscope and representative images are shown from each group (n = 6). Scale bar 100 μm. The number of macrophages in five visual fields (VF) chosen at random were counted for each animal at 200× magnification and the results are quantified as the average number of macrophage positive cells per visual field (H). I, modest increase in average food intake by the Jak3-KO mice. Age and sex matched WT or Jak3-KO mice were transferred to new cage (n = 5 mice per cage) with known amount of diet. After 24 h, food was reweighed and the amounts consumed were calculated by difference. Average (n = 10 cages per group) weight of food consumed per cage was plotted verses each group. A–F, H, values are mean ± S.E. Asterisks (*) indicate statistically significant differences between WT and KO groups (p < 0.05, n = three independent experiments). G, representative images are from n = 3 images from n = 6 mice per group.
Loss of Jak3 Leads to Increased Basal Chronic Low-grade Inflammation
Increased body weight is associated with IL17a- and IL6-mediated CLGI (36). Previously, we reported that Jak3 plays an essential role in several intestinal functions (26, 37, 38) where loss of Jak3 led to increased level of intestinal and colonic IL17a and IL6 (28). To determine if these basal CLGI was restricted only to GI tract (28), we determined the effects of loss of Jak3 on basal systemic CLGI in Jak3-KO mice. Fig. 1 shows that Jak3-KO mice not only had substantial increase in systemic level of pro-inflammatory cytokines IL6 (D) and IL17a (E) in their serum but also there was a 4-fold increase in systemic myeloperoxidase (MPO) activity (F). These results indicate that the basal chronic inflammation was not restricted to GI tract (28), but also extended to systemic circulation. Since increased colonic (28) and systemic MPO activity indicated increased tissue infiltration and activation of neutrophils, we also determined if colonic infiltration of macrophages were increased. Our data showed that loss of Jak3 resulted in increased colonic macrophages (G–H). Next, we determined if systemic CLGI-associated increased body weight was due to hyperphagia. Fig. 1I shows that there was modest but non-significant increase in average food intake by Jak3-KO mice.
Loss of Jak3 Leads to Increased Body Fat and Impaired Ability of Glucose Homeostasis
Since mice with loss of Jak3 had systemic CLGI and increased body-weight, a phenotype reminiscent of human metabolic syndrome (28), we determined if loss of Jak3 had an impact on other symptoms of metabolic syndrome. As shown in Fig. 2, similar to co-housed mice (A), in non-cohoused mice, loss of Jak3 resulted in statistically significant increase in fasting blood glucose level (B) and epididymal fat-pad (C). Since the KO mice had a higher fasting glucose level than WT, indicating loss of glycemic control, we sought to confirm this by administering a bolus of glucose in both groups of mice and determined their ability to restore blood glucose. Fig. 2D shows that, Jak3-KO mice had impaired ability to restore blood glucose to baseline levels. Moreover, in Jak3-KO mice the basal insulin levels were also elevated (Fig. 2E).
FIGURE 2.

Loss of Jak3 leads to increased body fat and impaired ability of glucose homeostasis. A–B, loss of Jak3 leads to elevated 12h fasting glucose in mice. 12 h fasting blood glucose concentration in either co-housed KO and WT siblings mice (A) or non-cohoused KO and WT mice (n = 6 per group) were determined using FreeStyleTM Glucometer. Note that KO mice have statistically significant high level of fasting blood glucose level compared with WT irrespective of housing. C, loss of Jak3 leads to increased body fat in mice: Epididymal fat was excised and weighed in both WT and KO mice (n = 6/group). The ratio of the weight of the fat tissue relative to body weight (on the day fat excised) is shown. D, loss of Jak3 leads to impaired ability to restore blood glucose: To determine glucose tolerance, blood glucose concentration in fasted mice was measured after intraperitoneal injection with 2g/kg body weight of d-glucose followed by measuring their level as in B at the indicated time intervals post-injection (n = 5/group). E, loss of Jak3 leads to increased fasting serum insulin level: mice were fasted for 12 h followed by collection of their blood for the analysis of serum insulin as described in “Experimental Procedures.” A–D, values are mean ± S.E. Asterisks (*) indicate statistically significant differences between WT and KO groups (p < 0.05 n = three independent experiments).
Loss of Jak3 Leads to Early Symptoms of Liver Steatosis
Since metabolic syndrome also affects liver, we determined the impact of loss of Jak3 on liver. Jak3-KO mice showed enlarged liver (Fig. 3A) where average liver to body weight ratio was 1.3 times higher in KO mice compared with the corresponding WT group (Fig. 3B). Assessment of liver sections by blinded reviewer indicated that loss of Jak3 resulted in less-dense hepatocytic nuclei (representative section in Fig. 3C, 100× and 400×), which were also comparatively bigger (400×, green arrow). Assessments also indicated more unstained regions (black arrow) that were scattered throughout the H&E sections with fewer endothelial cells surrounding the liver triad (yellow arrow) in Jak3-KO liver compared with corresponding WT. Together, these data were consistent with early symptoms of liver steatosis.
FIGURE 3.

Loss of Jak3 leads to early symptoms of liver steatosis: Enlarged liver in Jak3 KO mice. A, age and sex matched WT and Jak3-KO mice (n = 6 per group) were euthanized, livers were excised and representative (n = 6) macroscopic images of liver are shown. B, before euthanasia, the weight of mice were determined in A followed by measurement of liver weight after euthanasia. Average liver to body weight ratio was calculated for WT and Jak3-KO group. C, histological analysis of liver sections of mice from B: fresh frozen liver sections from WT and Jak3-KO mice were stained with H&E and representative sections are shown with indicated magnifications from each group (n = 6). Arrows; green hepatocyte, black-non-stained regions, yellow cells surrounding triad. A–B, representative images are from n = 3 images from n = 6 mice per group. B, values are mean ± S.E., asterisks (*) indicate statistically significant differences (p < 0.05) from at least (n = 3) independent experiments.
Loss of Jak3-mediated Symptoms of Metabolic Syndrome Is Exaggerated by Western HFD
Development of metabolic syndrome in humans is thought to be promoted by a diet high in saturated fats (39–41). To investigate the effect of such a diet on metabolic syndrome in Jak3 KO mice, we fed WT and Jak3-KO mice either normal diet (6.5% cal from fat) or a high-fat diet (65% cal from fat) for 8 weeks and the difference between percent change in body weight of high-fat and normal diet fed mice were plotted verses weeks of treatment (Fig. 4A). Though, both WT and Jak3-KO mice on high-fat diet showed increase in body mass, the difference in body mass was 2-fold higher in Jak3-KO mice compared with the corresponding WT group. Jak3-KO mice also showed increased fasting blood glucose level (Fig. 4B), increased liver to body-weight ratio (Fig. 4C), and severe hepatic steatosis as assessed by blinded reviewer (Fig. 4D). Together, these indicated an exaggerated manifestation of metabolic syndrome from high-fat diet as a result of loss of Jak3.
FIGURE 4.

Loss of Jak3-mediated symptoms of metabolic syndrome is exaggerated by western high-fat diet. A, time course of body weight change during high fat diet-induced obesity. WT and (KO) mice (n = 6, each group) were treated with high fat diet for 8 weeks, and the percent change in body weight was plotted versus weeks of feeding. B, during this “A” period, fasting blood glucose concentrations of both the WT and KO groups were measured weekly. C, at the end of 8 weeks mice were sacrificed and liver to body-weight ratio was determined for each mouse in both the group. D, histological analysis of liver sections from WT and KO mice treated with HF diet; a representative sections are shown with indicated magnifications from each group (n = 6). A-C, value represents mean ± S.E. (n = 6). Asterisks (*) indicate statistically significant differences (p < 0.05).
Loss of Jak3 Results in Increased Activation of Toll-like Receptor Signaling
Since activation of TLRs in general and TLR2 and TLR4 in particular has been associated with diabetes emanating from chronic inflammation (10) and our data showed that loss of Jak3 resulted in systemic and intestinal CLGI with early symptoms of liver steatosis, we determined if colonic and hepatic expression of TLR2 and TLR4 was altered under basal conditions in these tissues. As shown in Fig. 5A, expression of TLR4 was absent in the colonic epithelium of WT mice. However, as shown by co-immunostaining studies using epithelial specific marker villin, loss of Jak3 resulted not only in expression of TLR4 in colonic epithelium but these were also associated with increased expression and activation of NF-κB, the downstream target of TLR4 activation. These results were further confirmed by Western analysis using colonic (Fig. 5B) and hepatic (Fig. 5C) tissues of from WT and Jak3-KO mice. To determine if these increased expression was due to increased sensitivity toward TLR4 ligand LPS (or TLR2 ligand pam3CKS4) and if Jak3-expression influenced such sensitivity, using colonic epithelial model of Caco2 and HT-29, we investigated the effects of loss of Jak3 on the expression of TLR4 in presence of LPS. As shown in Fig. 5D, knock-down of Jak3 expression by lentivirus carrying doxycycline-regulated RFP-tagged jak3-shRNA (27) resulted in a substantial increase in TLR4 expression in both Caco2 and HT29 (data not shown) cells. Western analysis (Fig. 5E) of these cells further confirmed that LPS treatment led to increased expression of TLR4 and activated-NFκB, the downstream target of TLR4 activation which was significantly higher in the cells where the expression of Jak3 was knocked down (RFP: red). We also noticed an increased TLR2 expression in response to its ligand pam3CKS4 in Jak3 knocked down cells where doxycycline alone (control) didn't have any effects on either TLR4 (Fig. 5E, 6th and 7th panels) or TLR2 (data not shown) expression. These results indicated that expression of Jak3 was required for reduced surface expression and activation of TLR2 and TLR4. Since our data suggested that Jak3 expression was essential for reduced expression of TLRs and decreased TLR-mediated signaling, we determined if activation of Jak3 proteins was also required for suppressed expression of TLRs and associated decreased TLR-signaling. Fig. 5F shows that activation by LPS led to tyrosine phosphorylation of Jak3 (1st and 2nd panels) that was associated with suppressed expression of TLR4 (4th panel), and these effects were reversed during inhibition of Jak3 activation by CP-690505.
FIGURE 5.

Loss of Jak3 results in an increased expression and activation of toll-like receptor signaling. A, immunofluorescence staining of colonic mucosa of WT and Jak3-KO mice: Colonic tissue sections from each mouse were immunostained using indicated primary antibodies. Images were acquired using Nikon C1-plus laser confocal microscope and representative images are shown from each group (n = 6). Note the comparatively increased expression of both TLR-4 and pNF-κB in colonic mucosa of KO mice. Scale bar 550 μm. B, colonic tissue lysates of mice from “A” were analyzed using IB for indicated proteins and β-actin as control. C, liver tissue lysates of mice from “A” were analyzed using IB for indicated proteins and β-actin as control. Representative blots (n = 3) are shown. D, Jak3 regulates TLR-4 expression and activation in human IEC: Expression of TLR4 was determined in 2 weeks post-confluent Caco2 cells transduced with lentiviruses as described in “Experimental Procedures” followed by treatment with LPS for 24 h to activate TLR-4 in presence or absence of doxycycline to knock-down Jak3 expression. IFM were performed using antibody for TLR4. Representative images are shown where the green indicates TLR-4 and the red color confirms RFP-tagged jak3-shRNA expression mediated knock-down of Jak3 (27). Bar 14 μm. E, loss of Jak3 results in increased activation of TLR signaling; Cells from “D” were lysed using lysis buffer followed by Western analysis of the cell lysates using indicated antibodies. F, activation of Jak3 is required for suppression of TLR-4 expression: Cells were pretreated with CP-690505 for 30 min to inhibit Jak3 activation followed by treatment with LPS as in “D” to activate TLR-4. Western analysis of the cell lysates using indicated antibodies are shown. B-C, E-F, representative blots are shown (n = 3 from three independent experiments; B and E, lower bar graphs) Densitometric analysis was done using Image LabTM software (Bio-Rad), and ratio of average densities (n = 3 from three independent experiments) between pNF-κB and NF-κB is shown where asterisks (*) indicate statistically significant differences (p < 0.05).
Activated Jak3 Interacts With and Facilitates Tyrosine Phosphorylation of Insulin Receptor Substrate (IRS)1
Since loss of Jak3 resulted in compromised glucose regulatory phenotype with hyperinsulinemia in mice and tyrosine phosphorylation of IRS1 are necessary for insulin sensitivity of both colon and liver (42), we tested if Jak3 interacted with IRS1 in WT mice and if loss of Jak3 affected tyrosine phosphorylation of IRS1 in the colon of Jak3 KO mice. Fig. 6A (1st and 2nd panels) shows that Jak3 not only interacted with IRS1 in colonic tissues of WT-mice, but Jak3- associated IRS1 was also tyrosine phosphorylated. Fig. 6B (1st and 2nd panels) shows that expression of IRS1 was not affected in Jak3-KO mice; however, loss of Jak3 resulted in loss of tyrosine phosphorylation of IRS1.
FIGURE 6.

Jak3 interacts with and activates p85, the regulatory sub-unit of the PI3K through IRS1. A, Jak3 is required for IRS1 interactions with p85 and tyrosine phosphorylation of both IRS1 and p85 in mice colon: Western analysis (IB) of co-immunoprecipitates (IP) from tissue lysates of colon from WT and KO were done using indicated antibodies. For input controls, IB was done using β-actin, p85, and IRS1 antibodies. B–C, loss of Jak3 results in loss of tyrosine phosphorylation of both IRS1 and Akt, the downstream target for PI3K in colon and liver of mice: IP followed by IB of proteins from tissue lysates of colon (B) and liver (C) of WT and KO were done using indicated antibodies. D, inhibition of Jak3 decreases tyrosine phosphorylation of p85 and IRS1 in LPS-stimulated human IEC: Human IEC Caco2 were stimulated with TLR4 agonist LPS in presence or absence of Jak3-specific inhibitor CP-690505 (tofacitinib) as in Fig. 5 and cell lysates were subjected to Co-IP followed by IB using indicated antibodies. E, inhibition of Akt exerts similar effects as inhibition of Jak3 on the reversal of TLR4 expression in human IEC: Caco2 were pretreated with or without Akt-specific inhibitor A6730 for 30 min followed by stimulation with LPS as in D, and cell lysates were subjected to Co-IP followed by IB using indicated antibodies. A–E, representative blots are shown (n = 3) from (A–C; n = 6 mice) or from human IEC with (n = 3) independent experiments.
Jak3 Interacts With and Activates p85, the Regulatory Subunit of the PI3K through IRS1
Since phosphoinositide 3 kinase (PI3K) negatively regulates TLR4 signaling and Jak3-KO mice not only showed increased TLR4 signaling in both colon and liver but Jak3 also interacted with IRS1 in colon of wild-type mice, we tested if Jak3 facilitated the activation of PI3K through IRS1 in these tissues. As shown in Fig. 6A (3rd and 4th panels), loss of Jak3 not only resulted in disruption of the interactions between p85 and IRS1, but also tyrosine phosphorylation of p85 was lost in Jak3-KO mice. To determine the reason for the loss of tyrosine phosphorylation of p85, using co-immunoprecipitation, we tested if Jak3 interacted with p85 in these tissues. Fig. 6A, 5th panel, shows that Jak3 co-immunoprecipitated with p85, which was lost in KO mice. Together, these results indicate that Jak3 interacted with IRS1 and p85 and these interactions resulted in tyrosine phosphorylation of both IRS1 and p85. Next, we further confirmed if these interactions were essential for tyrosine phosphorylation of the downstream target of PI3K, the pAkt in these tissues. Fig. 6, B and C show that expression of Jak3 was essential for the activation of Akt in both colon (3rd and 4th) and liver (1st, 2nd, and 3rd panels) tissues of mice. To further confirm these, we tested if inhibition of Jak3 affects tyrosine phosphorylation of p85 and IRS1 in LPS-stimulated Caco2 cells. Fig. 6D shows that inhibition of Jak3 resulted in decreased phosphorylation of both p85 and IRS1 that correlated with increased activation of NF-κB in the absence of Jak3 (Fig. 5, B and C).
Jak3-mediated Activation of PI3K-Akt Axis Is Essential for Reduced TLR Expression
Since Jak3 activation was essential for the activation of p85, the regulatory subunit of PI3K, we determined if activations of the downstream target of activated PI3K, Akt was necessary for suppressed TLR signaling in IEC. Fig. 6E shows that inhibition of Akt in Jak3 activated IEC led to reversal of suppression of Jak3-mediated TLR signaling as reflected by increased expression of activated NFκB.
Modulation of PI3K and TLR4 Impact Glycemic Regulation in Mice
Since Jak3-mediated activation of PI3K was essential for reduced TLR expression, to confirm this we determined if PI3K inhibition increases TLR4 expression and NF-κB activation and if this leads to phenotypes associated with metabolic syndrome in WT mice, colonic PI3K was inhibited by in vivo administration of PI3K inhibitor wortmannin in mice, and its effects on fasting blood glucose, colonic expression of TLR4 and associated activation of NF-κB were evaluated. As shown in Fig. 7, intraperitoneal administration of PI3K inhibitor wortmannin led to significant increase in fasting blood glucose levels in these mice (A). Colonic tissue analysis showed that these effects were associated with corresponding increase in TLR4 expression and associated NF-κB activation (B). To further demonstrate whether colonic TLR4 activation was responsible for LGCI and associated glycemic dysregulation in Jak3-KO mice, intraperitoneal administration of TLR4 antagonist LPS-RS in Jak3-KO mice showed modest but significant reversal of not only the fasting blood glucose levels (C) but these effects were also associated with reduced activation of NF-κB in colonic tissue of Jak3-KO mice (D).
FIGURE 7.

Modulation of PI3K and TLR4 impact glycemic regulation in mice. A–B, colonic inhibition of PI3K promotes TLR4 expression associated NF-κB activation and elevation of fasting blood glucose in WT mice. WT mice (n = 5 per group) were injected intraperitoneally with either vehicle or PI3K inhibitor wortmannin (1 mg/kg body weight) once every 24 h for 5 days and 12 h fasting blood glucose were measured as in Fig. 1 on day 5 (A), followed by post sacrifice colonic tissue analysis by IB using indicated antibodies (B). C–D, TLR4 antagonist LPS-RS partially ameliorates glycemic dysregulation and associated colonic elevation of pNF-κB in Jak3-KO mice. Jak3-KO mice (n = 5 per group) were injected intraperitoneally with either vehicle or TLR4 antagonist LPS-RS (40 mg/Kg body weight) once every 24 h for 5 days, and 12 h fasting blood glucose were measured as above on day 5 (C), followed by postsacrifice colonic tissue analysis by IB using indicated antibodies (D). B and D; right bar graphs, densitometric analysis was done using Image LabTM software (Bio-Rad), and ratio of average densities (n = 3 from three independent experiments) between pNF-κB and NF-κB is shown where asterisks (*) indicate statistically significant differences (p < 0.05).
Discussion
Though obesity in most cases is associated with CLGI which contributes to the development of metabolic syndrome involving compromised glucose homeostasis, hyperlipidemia, hyperinsulinemia, and together culminating into type 2 diabetes, the mechanisms of origin and mode of perpetuation of such CLGI is not known. Gastrointestinal (GI) tract is the first organ that constantly interacts with dietary components and luminal microbiota and both these environmental factors are known to regulate CLGI-associated diseases such as obesity and diabetes (16, 17, 23, 24). To understand the mechanism of GI inflammation, previously we reported that absence of Jak3 expression in colon led to basal colonic CLGI, which were associated with reduced colonic mucin expression, and a tendency of gaining body weight (28). Since increased body weight is mostly considered a phenotype indicative of human metabolic syndrome (1, 28), in the present study we characterized how loss of Jak3 contributes to development of obesity and metabolic syndrome.
Caging practices by genotype upon weaning can sometime results in cages of mice developing familial transmitted distinct microbiota composition irrespective of genotype that may influence the symptoms of metabolic syndrome (43). Our data using age and sex matched WT and Jak3-KO co-housed siblings or age and genotype matched independently housed WT or Jak3-KO mice showed that obesity and metabolic syndrome in Jak3-KO mice were genotype driven where loss of Jak3 resulted in manifestation of these symptoms. As these symptoms were associated with basal colonic (28) and systemic CLGI, it is possible that colonic CLGI may be the origin for systemic CLGI. These were further corroborated by increased macrophages and neutrophils in colonic tissue of Jak3-KO mice, which could be the source of increased cytokine levels responsible for CLGI in these mice (Fig. 1). Previously, we reported that loss of Jak3 increases the susceptibility toward DSS induced colitis (28). We speculate that histopathologically evident basal low-grade gut inflammation as seen in Jak3-KO mice that predisposed to colitis (28) could be severe but relatively rare outcome of an altered host-microbiota relationship while a more common consequence of such disturbances could be “low-grade” chronic inflammation associated metabolic syndrome as seen in other mice model (44, 45). To demonstrate whether this was the case in Jak3-KO mice, we determined whether these mice had symptoms of metabolic syndrome. Our data showed that loss of Jak3 led to significant increase in body fat and loss of glycemic control (Fig. 2). In human, the loss of glycemic controls is mostly associated with insulin resistance resulting in hyperinsulinemia (46). Indeed, Jak3-KO mice not only had impaired ability of glucose homeostasis but these symptoms were also associated with hypeinsulinemia indicating type-2 diabetes.
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of the metabolic syndrome, which describes a spectrum of liver pathology ranging from simple steatosis to hepatic cirrhosis (47, 48). High-fat diets (HFD) are among the major environmental factor that induces pro-inflammatory signaling including through TLRs, which correlates with subsequent development of obesity, NAFLD, and insulin resistance (49, 50). Our data suggested that loss of Jak3 not only resulted in predisposition toward HFD-induced obesity as indicated by exaggerated effects of such diets in Jak3-KO mice but these effects were also associated with severe hepatic steatosis and steatohepatitis as indicated by comparatively increased accumulation of fats in the liver of Jak3-KO mice (Fig. 4). These effects were an extension of early symptoms of liver steatosis under basal conditions as indicated by relatively enlarged liver having lesser-dense hepatocytic nuclei and prominent unstained regions scattered throughout the H&E sections of liver with fewer endothelial cells surrounding the hepatic triad in Jak3-KO mice (Fig. 3).
To demonstrate the mechanism of CLGI associated predisposition to MetS in Jak3-KO mice, our data showed that loss of Jak3 results in increased expression of TLR2 and TLR4 in the colonic mucosa of mice, which were associated with increased activation of NFκβ in these tissues (Fig. 5). Previously, we reported that Jak3 facilitates intestinal barrier functions by two mechanisms; (a) facilitating the expression of colonic mucins and (b) facilitating adherens junction (AJ) formation through its interactions with AJ protein β-catenin (28). It is possible that loss of Jak3 led to relatively milder leaky gut barrier that resulted in compromised tolerance of gut epithelial mucosa toward residential microbiota through increased LPS sensitization led enhanced expression of colonic TLR2 and TLR4 and associated pNFκβ in colon. Increased sensitization of LPS by LPS-high responder mucosal epithelial cells results in posttranscriptional regulation of TLR4 mRNA where LPS is known to promote binding of human antigen R (HuR) proteins to the 3′-UTR of TLR4 mRNA. Through these interactions with cis-acting elements in TLR4 3′-UTR, HuR enhances TLR4 expression (51), where activated pNFκβ may further facilitate these interactions in Jak3 deficient IEC. Additionally, ZNF160 a transcription factor expressed more only in IEC less sensitized to LPS than a highly sensitive IES and could represses TLR4 transcription. It is known to that ZNF160 interact with scaffold proteins to recruit histone deacetylase and histone deacetylation at the 5′ region of the TLR4 gene is significantly higher in IEC less sensitive to LPS. These epigenetic regulation repressed TLR4 expression in less sensitive IEC (52). Since activation of NF-κβ is associated with expression of pro-inflammatory cytokines by mucosal epithelial cells (53), it is possible that leaky barrier mediated compromised mucosal tolerance acted as a key source of the initiation of inflammation resulting in increased tissue infiltration of neutrophils and macrophages ultimately culminating in colonic and systemic CLGI in Jak3-KO mice. These results were further corroborated by the findings that TLR4-KO mice fed with high-fat diet (HFD) showed significant reduction of circulating levels of pro-inflammatory cytokines (5, 11). Though CLGI play a key role in the development of metabolic syndrome (2–4), the specific roles of mucosal-Jak3 in development of CLGI and associated diabesity were not known. Our data showed that expression and activation of Jak3 was essential for reduced expression of both TLR4 and TLR2 in both human and mouse intestinal epithelium. In these cells, Jak3 interacted with and tyrosine phosphorylated p85, which is the regulatory sub-unit of PI3K, and these interactions took place through the mediation of tyrosine phosphorylated adapter protein IRS1 (Fig. 6). Previously it was reported that stimulation of human monocyte by LPS led to activation of Jak3 (11). Here we showed that activation of TLR4 was sufficient for the intracellular activation of Jak3 in the intestinal epithelial cells and Jak3 activations suppressed TLR4 expression. Together, these interactions resulted in activation of PI3K-Akt axis, which was essential for reduced TLR expression and TLR associated NFκB activation. Since activation of NFκB is responsible for the production of pro-inflammatory cytokines, these results show an essential role of Jak3 in prevention of pro-inflammatory cytokine production in intestinal mucosa. To investigate the contribution of colonic PI3K-Akt axis, and TLR signaling in the development of metabolic syndrome, our data showed that inhibition of PI3K led to significant glycemic dysregulation that was associated with increased colonic TLR4 expression and NFκB activation in WT mice and modest but significant reversal of these upon inhibition of TLR4 signaling in Jak3-KO mice (Fig. 7). These modest but significant effects indicate additional mechanisms could play a role in predisposition to metabolic syndrome including loss of Jak3 induced compromised intestinal barrier functions, aberrant feeding behavior, and increases infiltration of neutrophils and macrophages in colonic mucosa (Fig. 1). These infiltrated cells of innate immune system could produce additional pro-inflammatory cytokines resulting in colonic and systemic CLGI and associated metabolic syndrome.
To modulate the immune functions, Jak3-directed drugs are widely used in the treatment of different medical conditions including asthma, allergies (54), hematopoietic malignancies (9–11), organ transplant (55–59), and thromboembolitic complications (60). However, because of lack of knowledge about the roles of Jak3 in obesity and metabolic syndrome, the implications of functional inhibition of Jak3 on diabetes and MetS can not be assessed. Present study demonstrates the role of Jak3 in innate immunity through preventing development of diabetes and MetS through multiple mechanisms including those through promoting mucosal tolerance. It would be worthwhile to develop drugs for colonic modulators of Jak3 functions that cannot only facilitate mucosal tolerance under normal condition but may also be useful for the prevention of diabesity in high risk population who are predisposed toward and development of obesity and associated metabolic syndrome. Additionally, this will also be useful for preventing chronic inflammation in patients undergoing Jak3-directed therapy. Though we have shown the predisposition of diabesity in Jak3-KO mice, it remains to be tested if under the same conditions either the transgenic mice that overexpress Jak3 or pharmacological manipulator that facilitates Jak3 expression/activation could protect from mucosal inflammation and associated diabetic symptoms. Since Jak3-SH2 domain acts as a regulator for Jak3 activation (25, 61), SH2-directed drugs could be developed as potential manipulator for Jak3 activation.
In summary we showed that Jak3 played a critical role in the pathogenesis of obesity and associated metabolic syndrome under basal conditions and during those induced by high-fat diets where colonic expression of Jak3 was essential for the maintenance of a healthy mucosal barrier, reduced infiltration of macrophages and neutrophils in colonic tissue, and overall mucosal tolerance (Fig. 8). Mechanistically, we demonstrated that Jak3 facilitated epithelial tolerance toward LPS-induced increased expression of TLR4 and associated activation of NF-κβ through interactions with and tyrosine phosphorylation of p85, the regulatory subunit of PI3K (Fig. 8). Thus, these results showed for the first time the essential role of Jak3 in prevention of obesity and associated MetS where Jak3 facilitated prevention of CLGI through several mechanisms including those through mucosal tolerance toward commensal gut-microbiota by suppressed expression and limited activation of TLRs.
FIGURE 8.
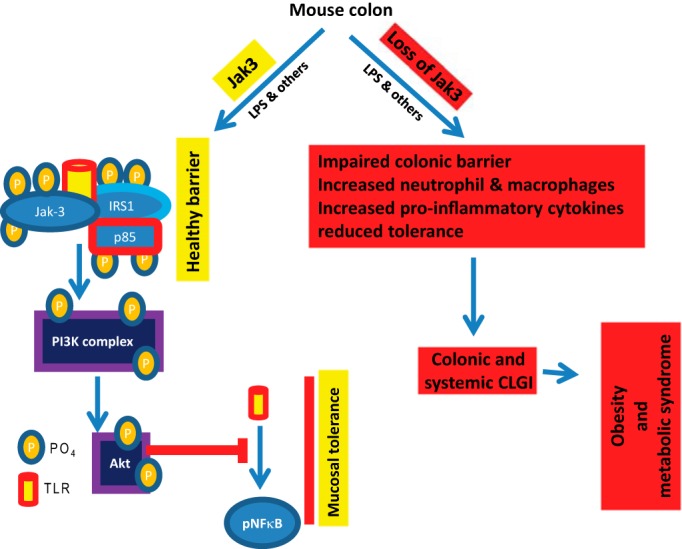
Proposed models for Jak3-mediated mucosal tolerance and predisposition to obesity and MetS.
Author Contributions
J. M.: conception, drafting, design, acquisition, analysis, and interpretation of data. R. K. V.: acquisition, analysis, and interpretation of data. G. A.: analysis and interpretation of data, critical revision for important intellectual content. F. M.: analysis, and interpretation of data. N. K.: conception, design, analysis, and interpretation of data, drafting of article, critical revision for intellectual content, and final approval of the version published.
The work was supported by grants from Crohn's & Colitis Foundation of America (CCFA Ref. 2188) and NIH (Grant DK081661) (to N. K.), NIH (sub-awarded) Grant 1R43GM109528-01 (to N. K. and J. M.), and the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology endowment from Baylor Scott & White and VA Merit award (5I01BX000574) (to G. A.). The authors declare that they have no conflicts of interest with the content of this article.
- MetS
- metabolic syndrome
- CLGI
- chronic low grade inflammation
- HFD
- high-fat diet
- TLR
- Toll-like receptor
- KO
- knock-out
- LPS
- lipopolysaccharides
- MPO
- myeloperoxidase
- IEC
- intestinal epithelial cells
- IP
- immunoprecipitation
- IB
- immunoblotting
- Jak3
- Janus kinase 3.
References
- 1.Bleau C., Karelis A. D., St-Pierre D. H., and Lamontagne L. (2014) Crosstalk between intestinal microbiota, adipose tissue and skeletal muscle as an early event in systemic low-grade inflammation and the development of obesity and diabetes. Diabetes Metab. Res. Rev. 31, 545–561 [DOI] [PubMed] [Google Scholar]
- 2.Kanneganti T. D., and Dixit V. D. (2012) Immunological complications of obesity. Nat. Immunol. 13, 707–712 [DOI] [PubMed] [Google Scholar]
- 3.Nunemaker C. S., Chen M., Pei H., Kimble S. D., Keller S. R., Carter J. D., Yang Z., Smith K. M., Wu R., Bevard M. H., Garmey J. C., and Nadler J. L. (2008) 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am. J. Physiol. Endocrinol. Metab. 295, E1065–E1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reilly S. M., Chiang S. H., Decker S. J., Chang L., Uhm M., Larsen M. J., Rubin J. R., Mowers J., White N. M., Hochberg I., Downes M., Yu R. T., Liddle C., Evans R. M., Oh D., Li P., Olefsky J. M., and Saltiel A. R. (2013) An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat. Med. 19, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crunkhorn S. (2013) Metabolic disorders: Breaking the links between inflammation and diabetes. Nat. Rev. Drug Discov. 12, 261. [DOI] [PubMed] [Google Scholar]
- 6.Onat A., Can G., Hergenç G., Yazici M., Karabulut A., and Albayrak S. (2007) Serum apolipoprotein B predicts dyslipidemia, metabolic syndrome and, in women, hypertension and diabetes, independent of markers of central obesity and inflammation. Int. J. Obes. 31, 1119–1125 [DOI] [PubMed] [Google Scholar]
- 7.Ding S., and Lund P. K. (2011) Role of intestinal inflammation as an early event in obesity and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 14, 328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips C. M., and Perry I. J. (2013) Does inflammation determine metabolic health status in obese and nonobese adults? J. Clin. Endocrinol. Metab. 98, E1610–E1619 [DOI] [PubMed] [Google Scholar]
- 9.Wildman R. P., Muntner P., Reynolds K., McGinn A. P., Rajpathak S., Wylie-Rosett J., and Sowers M. R. (2008) The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 168, 1617–1624 [DOI] [PubMed] [Google Scholar]
- 10.Lucas K., and Maes M. (2013) Role of the toll like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 48, 190–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Brown J., Gao S., Liang S., Jotwani R., Zhou H., Suttles J., Scott D. A., and Lamont R. J. (2013) The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells. J. Immunol. 191, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Könner A. C., and Brüning J. C. (2011) Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol. Metab. 22, 16–23 [DOI] [PubMed] [Google Scholar]
- 13.Fresno M., Alvarez R., and Cuesta N. (2011) Toll-like receptors, inflammation, metabolism and obesity. Arch. Physiol. Biochem. 117, 151–164 [DOI] [PubMed] [Google Scholar]
- 14.Kawai T., and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 15.Jialal I., and Kaur H. (2012) The Role of toll-like receptors in diabetes-induced inflammation: implications for vascular complications. Curr. Diab. Rep. 12, 172–179 [DOI] [PubMed] [Google Scholar]
- 16.Park J. S., Seo J. H., and Youn H. S. (2013) Gut microbiota and clinical disease: obesity and nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 16, 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everard A., and Cani P. D. (2013) Diabetes, obesity and gut microbiota. Best Pract. Res. Clin. Gastroenterol. 27, 73–83 [DOI] [PubMed] [Google Scholar]
- 18.Després J. P., Moorjani S., Lupien P. J., Tremblay A., Nadeau A., and Bouchard C. (1992) Genetic aspects of susceptibility to obesity and related dyslipidemias. Mol. Cell. Biochem. 113, 151–169 [DOI] [PubMed] [Google Scholar]
- 19.Bertrand R. L., Senadheera S., Markus I., Liu L., Howitt L., Chen H., Murphy T. V., Sandow S. L., and Bertrand P. P. (2011) A Western diet increases serotonin availability in rat small intestine. Endocrinology 152, 36–47 [DOI] [PubMed] [Google Scholar]
- 20.Safford M. G., Levenstein M., Tsifrina E., Amin S., Hawkins A. L., Griffin C. A., Civin C. I., and Small D. (1997) JAK3: expression and mapping to chromosome 19p12–13.1. Exp. Hematol. 25, 374–386 [PubMed] [Google Scholar]
- 21.Takahashi T., and Shirasawa T. (1994) Molecular cloning of rat JAK3, a novel member of the JAK family of protein tyrosine kinases. FEBS Lett. 342, 124–128 [DOI] [PubMed] [Google Scholar]
- 22.Murata Y., Yamashita A., Saito T., Sugamura K., and Hamuro J. (2002) The conversion of redox status of peritoneal macrophages during pathological progression of spontaneous inflammatory bowel disease in Janus family tyrosine kinase 3(-/-) and IL-2 receptor γ(-/-) mice. Int. Immunol. 14, 627–636 [DOI] [PubMed] [Google Scholar]
- 23.Ding S., Chi M. M., Scull B. P., Rigby R., Schwerbrock N. M., Magness S., Jobin C., and Lund P. K. (2010) High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 5, e12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flint H. J., Scott K. P., Louis P., and Duncan S. H. (2012) The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589 [DOI] [PubMed] [Google Scholar]
- 25.Mishra J., Karanki S. S., and Kumar N. (2012) Identification of molecular switch regulating interactions of Janus kinase 3 with cytoskeletal proteins. J. Biol. Chem. 287, 41386–41391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar N., Mishra J., Narang V. S., and Waters C. M. (2007) Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin. J. Biol. Chem. 282, 30341–30345 [DOI] [PubMed] [Google Scholar]
- 27.Mishra J., Waters C. M., and Kumar N. (2012) Molecular mechanism of interleukin-2-induced mucosal homeostasis. Am. J. Physiol. Cell Physiol. 302, C735–C747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra J., Verma R. K., Alpini G., Meng F., and Kumar N. (2013) Role of Janus kinase 3 in mucosal differentiation and predisposition to colitis. J. Biol. Chem. 288, 31795–31806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mishra J., Drummond J., Quazi S. H., Karanki S. S., Shaw J. J., Chen B., and Kumar N. (2013) Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit. Rev. Oncol. Hematol. 86, 232–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar N., Mishra J., and Quazi S. H. (2012) Training the defense system for modern-day warfare: the horizons for immunotherapy and vaccines for cancer. J. Immunodefic. Disord. 1, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilly B. C., Kansen M., van Gageldonk P. G., van den Berghe N., Galjaard H., Bijman J., and de Jonge H. R. (1991) G-proteins mediate intestinal chloride channel activation. J. Biol. Chem. 266, 2036–2040 [PubMed] [Google Scholar]
- 32.Hotamisligil G. S., Peraldi P., Budavari A., Ellis R., White M. F., and Spiegelman B. M. (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271, 665–668 [DOI] [PubMed] [Google Scholar]
- 33.Lai K. S., Jin Y., Graham D. K., Witthuhn B. A., Ihle J. N., and Liu E. T. (1995) A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J. Biol. Chem. 270, 25028–25036 [DOI] [PubMed] [Google Scholar]
- 34.Thomis D. C., Gurniak C. B., Tivol E., Sharpe A. H., and Berg L. J. (1995) Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 270, 794–797 [DOI] [PubMed] [Google Scholar]
- 35.Grima P., Guido M., Chiavaroli R., Stano F., Tundo P., Tana M., de Donno A., and Zizza A. (2012) Altered phosphate metabolism in HIV-1-infected patients with metabolic syndrome. Scand. J. Infect. Dis. 44, 133–137 [DOI] [PubMed] [Google Scholar]
- 36.Plagemann A., Harder T., Brunn M., Harder A., Roepke K., Wittrock-Staar M., Ziska T., Schellong K., Rodekamp E., Melchior K., and Dudenhausen J. W. (2009) Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587, 4963–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maziak W. (2005) The asthma epidemic and our artificial habitats. BMC Pulm. Med. 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stempel D. A. (2005) September epidemic of asthma exacerbations in children: a manifestation of persistent or episodic disease? J. Allergy Clin. Immunol. 115, 230–232 [DOI] [PubMed] [Google Scholar]
- 39.Phillips C. M., Goumidi L., Bertrais S., Field M. R., McManus R., Hercberg S., Lairon D., Planells R., and Roche H. M. (2012) Dietary saturated fat, gender and genetic variation at the TCF7L2 locus predict the development of metabolic syndrome. J. Nutr. Biochem. 23, 239–244 [DOI] [PubMed] [Google Scholar]
- 40.Cuda C., Badawi A., Karmali M., and El-Sohemy A. (2011) Polymorphisms in Toll-like receptor 4 are associated with factors of the metabolic syndrome and modify the association between dietary saturated fat and fasting high-density lipoprotein cholesterol. Metabolism 60, 1131–1135 [DOI] [PubMed] [Google Scholar]
- 41.Tulk H. M., and Robinson L. E. (2009) Modifying the n-6/n-3 polyunsaturated fatty acid ratio of a high-saturated fat challenge does not acutely attenuate postprandial changes in inflammatory markers in men with metabolic syndrome. Metabolism 58, 1709–1716 [DOI] [PubMed] [Google Scholar]
- 42.Kim J. J., and Sears D. D. (2010) TLR4 and insulin resistance. Gastroenterol. Res. Pract. 2010, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ubeda C., Lipuma L., Gobourne A., Viale A., Leiner I., Equinda M., Khanin R., and Pamer E. G. (2012) Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J. Exp. Med. 209, 1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chassaing B., and Gewirtz A. T. (2014) Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol. Pathol. 42, 49–53 [DOI] [PubMed] [Google Scholar]
- 45.Chassaing B., Ley R. E., and Gewirtz A. T. (2014) Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology 147, 1363–1377.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esteve E., Ricart W., and Fernández-Real J. M. (2011) Gut microbiota interactions with obesity, insulin resistance and type 2 diabetes: did gut microbiote co-evolve with insulin resistance? Curr. Opin. Clin. Nutr. Metab. Care 14, 483–490 [DOI] [PubMed] [Google Scholar]
- 47.Arslan N. (2014) Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 20, 16452–16463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guarner V., and Rubio-Ruiz M. E. (2015) Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 40, 99–106 [DOI] [PubMed] [Google Scholar]
- 49.Kim F., Pham M., Maloney E., Rizzo N. O., Morton G. J., Wisse B. E., Kirk E. A., Chait A., and Schwartz M. W. (2008) Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler. Thromb. Vasc. Biol. 28, 1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Wit N. J., Bosch-Vermeulen H., de Groot P. J., Hooiveld G. J., Bromhaar M. M., Jansen J., Müller M., and van der Meer R. (2008) The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med. Genomics 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan Z. Q. (2006) Regulation of TLR4 expression is a tale about tail. Arterioscler. Thromb. Vasc. Biol. 26, 2582–2584 [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K., Sugi Y., Hosono A., and Kaminogawa S. (2009) Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J. Immunol. 183, 6522–6529 [DOI] [PubMed] [Google Scholar]
- 53.Stadnyk A. W. (2002) Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can. J. Gastroenterol. 16, 241–246 [DOI] [PubMed] [Google Scholar]
- 54.Malaviya R., Zhu D., Dibirdik I., and Uckun F. M. (1999) Targeting Janus kinase 3 in mast cells prevents immediate hypersensitivity reactions and anaphylaxis. J. Biol. Chem. 274, 27028–27038 [DOI] [PubMed] [Google Scholar]
- 55.Tortolani P. J., Lal B. K., Riva A., Johnston J. A., Chen Y. Q., Reaman G. H., Beckwith M., Longo D., Ortaldo J. R., Bhatia K., McGrath I., Kehrl J., Tuscano J., McVicar D. W., and O'Shea J. J. (1995) Regulation of JAK3 expression and activation in human B cells and B cell malignancies. J. Immunol. 155, 5220–5226 [PubMed] [Google Scholar]
- 56.Ward A. C., Touw I., and Yoshimura A. (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 95, 19–29 [PubMed] [Google Scholar]
- 57.Hall B. M. (1991) Cells mediating allograft rejection. Transplantation 51, 1141–1151 [DOI] [PubMed] [Google Scholar]
- 58.Stepkowski S. M., Erwin-Cohen R. A., Behbod F., Wang M. E., Qu X., Tejpal N., Nagy Z. S., Kahan B. D., and Kirken R. A. (2002) Selective inhibitor of Janus tyrosine kinase 3, PNU156804, prolongs allograft survival and acts synergistically with cyclosporine but additively with rapamycin. Blood 99, 680–689 [DOI] [PubMed] [Google Scholar]
- 59.Säemann M. D., Diakos C., Kelemen P., Kriehuber E., Zeyda M., Böhmig G. A., Hörl W. H., Baumruker T., and Zlabinger G. J. (2003) Prevention of CD40-triggered dendritic cell maturation and induction of T-cell hyporeactivity by targeting of Janus kinase 3. Am. J. Transplant. 3, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 60.Cetkovic-Cvrlje M., and Tibbles H. E. (2004) Therapeutic potential of Janus kinase 3 (JAK3) inhibitors. Curr. Pharm. Des. 10, 1767–1784 [DOI] [PubMed] [Google Scholar]
- 61.Mishra J., and Kumar N. (2014) Adapter protein Shc regulates Janus kinase 3 phosphorylation. J. Biol. Chem. 289, 15951–15956 [DOI] [PMC free article] [PubMed] [Google Scholar]