

Abstract
Eukaryotic structural maintenance of chromosome proteins (SMC) are major components of cohesin and condensins that regulate chromosome structure and dynamics during cell cycle. We here determine the crystal structure of human condensin SMC hinge heterodimer with ∼30 residues of coiled coils. The structure, in conjunction with the hydrogen exchange mass spectrometry analyses, revealed the structural basis for the specific heterodimer formation of eukaryotic SMC and that the coiled coils from two different hinges protrude in the same direction, providing a unique binding surface conducive for binding to single-stranded DNA. The characteristic hydrogen exchange profiles of peptides constituted regions especially across the hinge-hinge dimerization interface, further suggesting the structural alterations upon single-stranded DNA binding and the presence of a half-opened state of hinge heterodimer. This structural change potentially relates to the DNA loading mechanism of SMC, in which the hinge domain functions as an entrance gate as previously proposed for cohesin. Our results, however, indicated that this is not the case for condensins based on the fact that the coiled coils are still interacting with each other, even when DNA binding induces structural changes in the hinge region, suggesting the functional differences of SMC hinge domain between condensins and cohesin in DNA recognition.
Keywords: analytical ultracentrifugation, chromatin structure, chromosomes, crystal structure, hydrogen exchange mass spectrometry, isothermal titration calorimetry (ITC), protein-DNA interaction
Introduction
Eukaryotic structural maintenance of chromosome proteins (SMC; in capital letters, used as a generic term for protein family)3 (1) plays a major role in multiple aspects of chromosomal behavior, such as the faithful duplication, condensation, segregation, and repair of chromosomes during cell cycle (2). Prokaryotes express a single type of SMC, either the SMC or a SMC-like core (MukB), which forms a homodimer via a specific interaction at the hinge region. On the other hand, eukaryotes have six different types of SMC (Smc1–Smc6). The SMC subunits form pairs at the hinge region to form three different heterodimers that bind three non-SMC subunits; these in turn lead to the establishment of the large, five-subunit condensin, cohesin, and Smc5–6 protein complexes. The Smc1–3 heterodimer is a component of cohesin, which is essential for chromosome cohesion; the Smc2–4 heterodimer is a component of condensin I and condensin II, which are localized to the axis of metaphase chromosomes in a double-stranded manner and contribute to the condensed chromosome structure (3–5). In addition, as functional studies of condensin, introduction of supercoiling to DNA and reannealing activity that convert single-stranded DNA (ssDNA) into double strand DNA (dsDNA) have been investigated (6–8), which might be responsible for diverse DNA metabolism mediated by condensin. The Smc5–6 heterodimer plays a role in DNA repair.
The overall architecture of the SMC complex is outstandingly conserved through organisms, as has been revealed by EM; this study showed that all of the SMC subunits are arranged as long (∼50 nm), rod-like, antiparallel coiled coils, with a globular ATPase “head” domain at one end and a “hinge” domain at the other (9, 10). The two subunits are dimerized via a hinge-hinge interaction, forming a cherry-shaped homo- or heterodimer that usually interacts with the heads of non-SMC subunits, to form a closed ring structure. This similarity in the ring-like architecture raises the basic question as to how this protein family functions multimodally. EM observation has revealed that the Smc heterodimers present in the condensin and cohesin complexes form similar cherry-like overall structures. The two coiled coil arms of the cohesin complex, however, appear to be more widely open compared with those of the condensin complex (10). In contrast, the prokaryotic Smc homodimer is known to display a wide variety of conformations; therefore, the arrangement of the two arms could be either open or closed. Based on these observations, it has been speculated that the difference in the overall configurations might be a possible factor affecting their machinery and that the hinge region of the protein plays an important role in this difference in configurations.
The crystal structures of the prokaryotic Smc hinge and the Smc hinge with the short coiled coil from Thermotoga maritima Smc (TmSmcH and TmSmcH-sCC, respectively), and two hinge structures from the SMC-like protein of Escherichia coli, MukB with a short coiled coil (MukBH-sCC) and MukB with a relatively long coiled coil (MukBH-lCC), have been reported so far (9, 11–13). These structures revealed that the hinge domain is primarily dimerized by the formation of β-sheets at the two binding surfaces of monomers with a donut-shaped, ring-like overall structure. The crystal structures of mouse condensin hinge (mSmc2–4H) and cohesin hinge (mSmc1–3H) have been independently reported, among eukaryotes (14, 15). These structures revealed that the eukaryotic Smc hinges form similar folds as the prokaryotic and bacterial Smc hinge domains, demonstrating that the overall structure of the Smc hinge domain is universal for a wide variety of SMC and SMC-like proteins. Under these circumstances, very recently Soh et al. (1) reported a SMC rod formation based on the crystal structures of the Pyrococcus furiosus Smc hinge (Pf SmcH) with long coiled coils and the yeast Smc2–4 hinge (ScSmc2–4H) with long coiled coils. This study paves the way for understanding structural difference of condensin and cohesin hinge region and for clarifying precise molecular machinery of chromosome structural alternations by bacterial Smc and cohesin.
Another important function of the SMC hinge, beyond its role in dimerization, is to regulate the interactions between the SMC and DNA, as revealed by a series of biochemical analyses conducted on Bacillus subtilis Smc (BsSmc). A number of previous studies have demonstrated the functional importance of the SMC hinge in the binding of DNA to the SMC (6–8, 16–18). The cohesin Smc1–3 complex has been extensively investigated among the eukaryotic SMC. The bovine cohesin Smc1–3 hinge with the transition region (TR) between the hinge domain and the coiled coil binds dsDNA, but not ssDNA, showing the specificity of the hinge region in recognizing specific DNA molecules (17). The SMC-DNA interaction has been emphasized in another recent study, where the artificially linked hinge domains of fission yeast cohesin Smc1–3 complex completely blocked the association of cohesin SMC complex with dsDNA and the subsequent engagement with chromosomes (19). These results surprisingly contradicted the previous idea that the cohesins are loaded into chromosome through their head domain, which undergoes conformational changes via ATP hydrolysis (19). The head region of the cohesin complex is currently believed to play an important role as an exit gate for SMC-DNA associations, which is distinct from the entrance gate (20, 21). Although the exact biological role of the SMC hinge in the SMC-DNA binding mechanics remains to be addressed, a theory has been proposed that the DNA is loaded into the ring of the cohesin SMC complex via transient binding to the SMC hinge domains, followed by dissociation of the hinge-hinge interaction between the SMC heterodimer. Therefore, although the information regarding this is currently limited, it has been hypothesized that the hinge domain plays a critical role in providing functional varieties of SMC, in addition to acting as a dimerization domain.
Recent evidence has shown that the condensin SMC hinge region also recognizes DNA; however, its preference is believed to be sharply different from that of cohesin. For example, biochemical analyses of the mouse Smc2–4 hinge with short stretch of coiled coils (mSmc2–4H-sCC) demonstrate its strong binding preference for ssDNA over dsDNA (14). The study using atomic force microscopy (18) clearly demonstrated that the fission yeast condensin Smc2–4 complex (cut3-cut14 heterodimer) bound to ssDNA via its hinge domain. In terms of the functional relevance of condensin, SMC interaction with ssDNA has been suggested by Sutani and Yanagida (6). They reported that reannealing of ssDNA to dsDNA was promoted by the cut3-cut14 heterodimer; together, these studies reveal the functional differences of the condensin and cohesin hinge domains in DNA recognition and manipulation (22).
To elucidate this further, we have focused on the hinge domains of human condensin SMC (hSmc2H and hSmc4H), and attempted to clarify their mechanistic function by analyzing their three-dimensional structures and physicochemical properties. For this purpose, hSmc2H and hSmc4H were expressed with middle coiled coils (30 residues each, hSmc2H-CC30 and hSmc4H-CC30, respectively), to stabilize the hinge fold. Surprisingly, hSmc2H-CC30 forms homodimer via β-sheet interactions, similar to prokaryotic SMC; on the other hand, one component of the dimer is easily replaced by a monomeric hSmc4H-CC30. The crystal structure of the hSmc2–4 with middle coiled coils was elucidated at a resolution of 1.89 Å. The structure clearly demonstrated the atomistic details of the mechanism of dimer selectivity and the unique coiled coil arrangement of condensin, where the arrangement of the coiled coils from two hinge domains is parallel and is similar to that reported in the quite recent study on the ScSmc2–4H with long coiled coils, even though the sequence identity between human and yeast condensin SMC hinge region is moderate (∼50%) (1). To probe coiled coil arrangement of condensin in solution, hydrogen exchange mass spectrometry (HDX-MS) of the hSmc2–4H with middle coiled coils was performed. The interactions between the hSmc2–4H with middle coiled coils and ssDNA were then investigated. The primary dissociation constant for hSmc2–4H with middle coiled coils and ssDNA interaction was determined to be 22.3 nm using isothermal titration calorimetry (ITC). Finally, the DNA binding regions of the hSmc2–4H with middle coiled coils in solution were determined using HDX-MS; the results suggested the existence of an encounter complex, and the conformational alternations in the heterodimer during ssDNA binding. Based on the present results together with previous reports, implications of condensin SMC hinge binding to ssDNA are discussed in terms of mitotic chromosome condensation.
Experimental Procedures
Cloning, Expression, and Purification of hSmc2H-CC30, hSmc2H(GD)-CC30, and hSmc4H-CC30
The human condensin Smc2 hinge domain with middle coiled coil sequence (hSmc2H-CC30) was cloned, expressed, and purified as previously described (23). The site-directed mutagenesis of hSmc2H-CC30 was performed using the QuikChange method, wherein the Gly residues at positions 661, 665, and 666 were substituted with Asp. The primers used were as follows: forward primer, 5′-GGTGATGTGTTTGATCCTCATGACACATTGAGTGACGATGCTCGATCCCAGGCAGCT-3′; and reverse primer, 5′-AGCTGCCTGGGATCGAGCATCGTCACTCAATGTGTCATGAGGATCAAACACATCACC-3′. Themutant protein was expressed and purified using the same procedure as for the wild-type protein. The protein purity was determined by SDS-PAGE analysis.
Cloning, Expression, and Purification of hSmc2H-CC30/hSmc4H-CC30
A pETDuet-1 vector was used for the coexpression of hSmc2H-CC30 and hSmc4H-CC30. The cells expressing the recombinant protein were cultured further for 24 h at 283 K and eventually harvested by centrifugation at 4,000 × g for 15 min at 277 K. The pellet was resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 300 mm sodium chloride) and disrupted by ultrasonication on ice. The lysate was centrifuged at 23,708 × g for 45 min to remove all insoluble debris. All subsequent procedures were carried out at 283 K unless otherwise stated. The supernatant was filtered and loaded onto a HisTrap HP column (GE Healthcare) previously equilibrated with buffer A. The protein was eluted via an imidazole gradient. The vector-encoded N-terminal His tag of the eluted protein was cleaved using HRV3C protease (Takara Bio Inc., Tokyo, Japan) in buffer B (50 mm Tris-HCl, pH 8.0, 150 mm sodium chloride) for 48 h at 277 K. The protein was completely cleaved using a 1:1000 (w/w) ratio of protease to target protein. The His tag-containing protein was eluted using a HisTrap HP column and further purified by gel filtration, using a HiLoad 26/60 Superdex 75 column (GE Healthcare). The purity of the protein was determined by SDS-PAGE analysis. The final product is a heterodimer composed of hSmc2H-CC30 (residues 476–707) and hSmc4H-CC30 (residues 566–809). The purified proteins were used in the crystallization, analytical ultracentrifugation (AUC), ITC, and HDX-MS experiments.
AUC
The sedimentation velocity AUC experiments were carried out using a 30 mm Tris-HCl (pH 8.0) and 150 mm sodium chloride buffer solution, in a Proteomelab XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA). 400-μl aliquots of all samples were sedimented at 42,000 rpm at 293 K, using 12-mm aluminum double sector centerpieces with quartz windows. The sedimentation behavior was monitored through the UV-visible absorbance optics. All sedimentation velocity data were analyzed using the continuous C(s) distribution model in the software program SEDFIT, version 11.8 (24). The parameters used for the analysis, i.e. partial specific volume of the protein, calculated from the amino acid composition, buffer density (ρ = 1.00314 g/cm3), and buffer viscosity (η = 1.0204 cP), were estimated using the program SEDNTERP, version 1.09.
Sedimentation equilibrium AUC experiments were performed on a Beckman Optima XL-A analytical ultracentrifuge (Beckman Coulter) at 293 K, using UV-visible absorbance optics. Samples of all proteins at multiple concentrations were loaded into 12-mm double sector charcoal-filled epon centerpieces. The rotor speeds were set to 12,000 or 16,000 rpm for hSmc2H-CC30/hSmc4H-CC30 and hSmc2H-CC30 and 16,000 or 20,000 rpm for hSmc4H-CC30. The data were collected as the averages of 16 scans at 0.001-cm intervals and were subjected to nonlinear least squares analysis using the software package (Microcal Origin) supplied by Beckman Coulter. The goodness of fit was evaluated on the basis of the magnitude and randomness of the residuals and expressed as the difference between the experimental data and the theoretical curve; in addition, each of the fit parameters were analyzed for physical reasonability.
Crystallization and Structure Determination
The crystallization conditions for the purified hSmc2H-CC30/hSmc4H-CC30 were initially screened by the sitting drop, vapor diffusion method (at 293 K), using a commercially available screening kit (Hampton Research, Aliso Viejo, CA). Each crystallization drop was prepared by mixing 2 μl each of the protein and reservoir solutions; this was equilibrated against 400 μl of the reservoir solution. Optimal crystals for x-ray diffraction studies were grown over a week from drops containing 10 mg/ml heterodimer in 19% PEG 3350, 50 mm sodium chloride, and 100 mm Tris-HCl buffer (pH 8.5). The crystals used to collect diffraction data displayed an ellipsoid-like shape, with typical dimensions of 0.4 × 0.1 × 0.1 mm. Prior to data collection, the droplet that contained single crystals of the hSmc2H-CC30/hSmc4H-CC30 was equilibrated against air without sealing for several hours. The dehydrated crystals were placed directly in a cold stream of nitrogen gas at 100 K. X-ray diffraction data were collected at Beamline BL-17A of the Photon Factory (Tsukuba, Japan), using an ADSC Quantum 315r CCD detector. The data were processed and scaled using iMosflm, version 1.0.7. and AIMLESS, from the CCP4 program suite (25). The hSmc2H-CC30/hSmc4H-CC30 heterodimer crystal belonged to the space group P212121, with one heterodimer molecule per asymmetric unit (unit-cell parameters, a = 50.40 Å, b = 93.98 Å, and c = 102.07 Å). During attempts to solve the structure with a molecular replacement approach, using the Phaser program of the PHENIX suite (26), utilizing the mouse condensin hinge domain heterodimer (Protein Data Bank code 3L51) as the search model. A molecular replacement solution was observed when each subunit of the mouse condensin hinge domain heterodimer was inputted separately as the initial coordinate for the Phaser calculation, yielding an interpretable electron density map. The data were subjected to several structural refinement rounds using Phenix.refine. This was followed by manual model building with the Coot software (27), which allowed the building of ∼89.7% of the heterodimer model. Further refinements, coupled with the addition of 303 water molecules, resulted in a final structure, with Rwork and Rfree values of 18.8 and 23.5%, respectively. The data collection, processing, and refinements statistics are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics of hSmc2H-CC30/hSmc4H-CC30
| Data collection | |
| Beamline | Photon Factory BL-17A |
| Wavelength (Å) | 0.9800 |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 50.40, 93.98, 102.07 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 46.99–1.89 (1.93–1.89)a |
| Rmergeb | 0.066 (0.476) |
| <I/σΙ> | 10.8 (2.6) |
| Completeness (%) | 98.4 (99.5) |
| No. of unique reflections | 47,639 |
| Redundancy | 4.40 (4.3) |
| Refinement | |
| Resolution range (Å) | 45.19–1.89 |
| No. of reflections | 38,707 |
| Rwork/free | 0.188/0.235 |
| No.atoms | |
| Protein | 3372 |
| Water | 303 |
| Average B-factors (Å2) | 28.0 |
| Wilson B-factors (Å2) | 23.4 |
| Root mean square deviation | |
| Bond length (Å) | 0.007 |
| Bond angles (°) | 0.974 |
| Ramachandran plot statistics | |
| Most favored (%) | 98.6 |
| Additional allowed (%) | 1.4 |
| Disallowed (%) | 0.0 |
a The values in parentheses are for the highest resolution shell.
b Rmerge = Σ|Ih − <Ih>|/ΣIh, where <Ih> is the average intensity of reflection h and symmetry-related reflections.
ITC
The 30-mer poly(T) ssDNA sample was employed in this experiment based on the observations of previous studies that condensin had no sequence specificity. The purified hSmc2H-CC30/hSmc4H-CC30 was dialyzed against 30 mm Tris-HCl (pH 8.0) and 10 mm sodium chloride buffer at 277 K overnight. The dialyzed protein solution (54 μm) was loaded into the cell of iTC200 (GE Healthcare). The titration was performed by injecting 2 μl of the syringe solution (0.3 mm DNA) at an interval of 120 s at 298 K. The three repeated thermograms were globally analyzed using the NITPIC (version 1.0.4b, April 2014) program (28). After fitting trials using several interacting models, the thermograms were well fit with the A + B + B ⇄ AB + B⇄ ABB interacting model (two symmetric sites, macroscopic K) using the SEDPHAT (version 12.01beta, May 2014) program (29). The 68.3% confidential intervals for fitting parameters were calculated by using the SEDPHAT program.
Hydrogen Deuterium Exchange Mass Spectrometry
DNA with the same sequence as those used in the ITC experiments were used in this study. Four microliter of the hSmc2H-CC30/hSmc4H-CC30 (50 μm in 30 mm Tris-HCl, 10 mm sodium chloride H2O, pH 8.0) was mixed with 4 μl of DNA solution (100 μm in same buffer), and incubated at 300 K for 30 min. Following the binding step, the complex solution was diluted 9-fold with 30 mm Tris-HCl and 10 mm sodium chloride D2O buffer (pH 8.0) and incubated at 277 K for different times (20 s and 1, 10, 60, and 240 min). The exchange reaction was quenched by reducing the pH to 2.5 using 5 μl of 8.45% formic acid (Merck-Millipore). Each mixing procedure was automated using the LEAP system (Waters Corporation, Milford, MA). The quenched samples (85 μl) were injected, desalted, and trapped using the mentioned above with minor modifications. 200 pmol of the exchanged and quenched heterodimer was injected into an immobilized pepsin column. The online digestion was performed over 5 min in water containing 0.05% formic acid at 277 K, at a flow rate of 24 μl/min. The peptides were separated using two types of linear acetonitrile gradients (8–40%, 7 min; and 40–85%, 5 min) with 0.1% formic acid. Each peptide was identified, and the deuterium incorporation was evaluated using the same software used in analyses of dimerization, supplied by Waters Corporation. All systems and lines were washed manually and via automation between each sample measurement, using 2 m guanidine hydrochloride in 4% acetonitrile and 0.8% formic acid. Engen et al. (30) had recently reported the certainty of the HDX-MS analysis and proposed the criteria to evaluate the obtained results. The reported criteria was applied throughout our study. When the sum of difference is over 1.1 Da between two states for a peptide, the result reflects differing amide proton environments of the region covered by the peptide in solution, with 98% confidence. In addition, a difference of over 0.5 Da (at a specific exposure time) in a peptide indicated the differing amide proton environments of the region. The two amino acids located at the N terminus of peptic peptides have a rapid exchange rate for their amide protons; consequently, the exchange of amide hydrogen to deuterium cannot be monitored via HDX-MS analysis. Therefore, 2 amino acids at the N terminus excluded from the interacting region were also included in the peptide, showing a difference in deuterium uptake rate.
Results
Dimerization Selectivity of Human Condensin SMC Hinge Domain
In contrast to prokaryotes, which express only one SMC, higher eukaryotes are known to express at least six paralogs of SMC, each of which specifically forms three types of heterodimers through the interaction of their hinge domains. Previous structural studies of the SMC hinges have clearly revealed the highly conserved nature of the SMC hinge fold from prokaryotes to eukaryotes; however, the mechanism of their dimerization selectivity remains to be addressed.
In this study, the hinge of human condensin SMC subunits (Smc2 and Smc4) comprising various lengths of coiled coils, were independently expressed. The inclusion of the coiled coils is expected to bind the N- and C-terminal segments of the hinge domain and could stabilize the dimerization interface (see “Experimental Procedures” and Fig. 1). Because the correct match of the segment length at the N and C termini is apparently important for stable coiled coil formation, several constructs were tested based on the results of secondary structure prediction, obtained using the programs COILS and PSIPRED (31, 32). Consequently, the constructs, which encode residues 476–707 of the hSmc2 and 566–809 of the hSmc4, successfully produced soluble hSmc2 and hSmc4 hinge domains, with a portion (∼30 residues) of predicted coiled coil sequences (referred as hSmc2H-CC30 and hSmc4H-CC30, respectively). It is of note that the AUC analyses indicated that hSmc2H-CC30 unexpectedly takes a homodimeric form in solution (Fig. 2 and Table 1), whereas hSmc4H-CC30 existed as a monomer (Fig. 2, b and d, and Table 1).
FIGURE 1.

Amino acid sequence alignment of human, mouse, yeast, Pyrococcus, and Bacillus Smc. Red amino acids are basic residues (9, 10), which play a major role in the interaction with DNA and are located at the TR (11). Highlighted residues are conserved among all aligned Smc. The secondary structures of hSmc4H-CC30 (top) and hSmc2H-CC30 (bottom) observed in the crystal structure are also indicated, along with their sequence. Disordered regions of hSmc2H-CC30/hSmc4H-CC30 in crystal are designated as dotted lines. Arrows indicated the basic residues mutated in the previous study of mouse condensin SMC hinges (8).
FIGURE 2.

AUC results of the SMC hinge domains. a–c, equilibrium concentration gradients in sedimentation equilibrium AUC were indicated for hSmc2H-CC30 (a), hSmc4H-CC30 (b), and hSmc2H-CC30/hSmc4H-CC30 (c). d, concentration dependences of 1/Mapp (where Mapp indicates apparent molecular weight) for each sample. Molecular weight estimates and deduced oligomeric states were summarized in Table 1. e, sedimentation velocity AUC results of hSmc2H-CC30 (solid line), hSmc2H(G/D)-CC30 (dashed line), and a mixture of hSmc2H-CC30 and hSmc4H-CC30 (dotted line) were indicated. Oligomeric states of each protein, which were identified based on the estimated molecular weights, were also shown.
TABLE 1.
Molecular weight estimates obtained by sedimentation equilibrium AUC
| Protein | Mcala | Mappb | Oligomeric state |
|---|---|---|---|
| kDa | kDa | ||
| hSmc2H/hSmc4H | 21.4 (hSmc2H) | 39.2 ± 2 | Heterodimer |
| 20.3 (hSmc4H) | |||
| hSmc2H-CC30 | 26.3 | 56.5 ± 3 | Homodimer |
| hSmc4H-CC30 | 28.1 | 30.7 ± 2 | Monomer |
| hSmc2H-CC30/hSmc4H-CC30 | 26.3 (hSmc2H-CC30) | 55.9 ± 3 | Heterodimer |
| 28.1 (hSmc4H-CC30) |
a Mcal indicates molecular weight calculated from amino acid sequences.
b Mapp indicates molecular weight estimates obtained by sedimentation equilibrium AUC experiments.
Previous biochemical analyses of the prokaryotic SMC hinge homodimer obtained from B. subtilis clearly revealed that the substitution of the three conserved glycine residues (Gly-658, Gly-662, and Gly-663), corresponded to, for example, Gly-661, Gly-665, and Gly-666 of hSmc2, found at the C-terminal region of the hinge domain, which are involved in the dimerization in a wide variety of organisms (Fig. 1), to the corresponding aspartate residues, disrupting the dimerization of the SMC (1, 8). Taking this into consideration, Gly → Asp mutations were introduced into the corresponding regions of hSmc2H-CC30 (hSmc2H(G/D)-CC30); they were analyzed by sedimentation velocity AUC experiments (Fig. 2e). The results clearly showed the conversion of the dimeric wild-type hSmc2H-CC30 into the monomeric hSmc2H(G/D)-CC30, indicating that hSmc2H-CC30 undergoes homodimerization through the same binding interface as the other SMC hinges reported so far. Interestingly, the sedimentation equilibrium AUC and sedimentation velocity AUC profiles of equimolar mixtures of hSmc2H-CC30 and hSmc4H-CC30 showed the formation of single species that corresponds to the heterodimer (Fig. 2, c–e, and Table 1). The heterodimer complex formation is consistent with the in vivo observation of the human condensin holo complex and clearly demonstrated that human SMC hinge is involved in specific recognition of its partner.
The hSmc2–4H with Middle Coiled Coils Crystal Structure Revealed Its Eukaryotic Type SMC Dimerization and Parallel Arm Configuration
Our results, showing that hSmc2H forms a homodimer in solution, appear to be inconsistent with the general theory that the eukaryotic Smc specifically forms a heterodimer in vivo. The homodimeric interaction of eukaryotic Smc has also been observed in the protein crystals of the yeast Smc1 head domain (33); however, the existence of eukaryotic SMC hinge homodimers, both in vitro and in vivo, has not yet been proved experimentally. Although the crystal structure of mSmc2–4H heterodimer has been reported with atomic resolution, this structure showed a disruption in one of the expected dimerization interfaces, which hampered the elucidation of the dimerization mechanism and specificity. Therefore, in this study, the dimeric interaction at the condensin SMC hinge was investigated by crystallizing the hSmc2–4H with middle coiled coils (hSmc2H-CC30/hSmc4H-CC30).
The crystal of hSmc2H-CC30/hSmc4H-CC30 belongs to the orthorhombic system, P212121, with one heterodimer molecule per asymmetric unit, and diffracted to a resolution of 1.89 Å (Table 2). The initial attempts to solve the structure using a molecular replacement approach with a previously reported mSmc2–4H structure (Protein Data Bank code 3L51) were unsuccessful, possibly because of the absence of coiled coils in the trial model and/or differences in their subunit interactions. Therefore, each subunit in the mSmc2–4H was then separately subjected to molecular replacement, which led to successful determination of the initial phase.
The subsequent structural refinement and model building procedures provided a final model of one hSmc2–4H, including several portions of coiled coils (Fig. 3a). The electron density was visible for all hSmc2H-CC30 residues, with the exception of Asn-707 at the C-terminal end. On the other hand, the N terminus (residues 566–571) and a majority of the C terminus (residues 768–809) of hSmc4H-CC30 was disordered, showing a void-like space with scarce electron density in the crystal lattice and was therefore not included in the final model.
FIGURE 3.

Structural characterization of human condensin Smc hinge-hinge interactions. a, overall structure of hSmc2–4 hinge with middle coiled coils (hSmc2H-CC30/hSmc4H-CC30). The hSmc2H-CC30 and hSmc4H-CC30 are colored light green and light blue, respectively. b, two dimerization interfaces (interface 1 and interface 2) between the hSmc2H-CC30 and hSmc2H-CC30 designated by a black arrow in the right panel of a. The hydrophobic interaction at two dimerization interfaces; interface 1 (left panel) and interface 2 (right panel) is shown. Amino acid residues involved in the interactions are indicated as stick and sphere models. The anti-parallel β-sheet interactions, with typically 4–6 intermolecular hydrogen bonds, are also indicated. c, three key intermolecular hydrogen bonding interactions at the top, middle, and bottom regions of the outer surface at each of the dimerization interfaces. d, dimerization interface observed in a mouse Smc2/Smc4 hinge heterodimer crystal (Protein Data Bank code 3L51), corresponding to interface 2 of the hSmc2H-CC30/hSmc4H-CC30 crystal structure. e, stereo view of the structural superposition of two subunit-subunit antiparallel β-sheets of the hSmc2H-CC30/hSmc4H-CC30. The β-sheets formed at interface 1 and 2 are colored red and light blue, respectively. Dimerization interface of hypothetical homodimer models of hSmc2H-CC30/hSmc2H-CC30 (middle panel) and hSmc4H-CC30/hSmc4H-CC30 (right panel) is also shown. The key interacting residues in b–e are presented as a stick model.
The crystal structure of hSmc2H-CC30/hSmc4H-CC30 (Fig. 3a) displayed a typical hinge fold, which was similar to previously determined SMC hinge structures (14, 15). As expected from the high sequence identity of 95% (91%) between mouse and human Smc2 (Smc4) hinges, each subunit is well superposed on each other, with root mean square deviation values of 0.673 and 0.803 Å for Smc2 and Smc4, respectively, when backbone Cα atoms were used. Contrary to the half-opened dimer structure of mSmc2H/mSmc4H (Fig. 3d), in which the N-terminal segment of mSmc4H (predicted to take the β-strand (β7)) is disordered, the constructs designed in this study include a middle length of coiled coil and consequently have two separated, complete interacting interfaces (interfaces 1 and 2; Fig. 3, b and c). This resulted in a 2-fold symmetric donut-shaped hSmc2H-CC30/hSmc4H-CC30 with minimum and maximum diameters of 1 and 3 nm for the cavity, respectively. The dimerization interactions were principally mediated by anti-parallel β-sheet interactions, with typically 4–6 intermolecular hydrogen bonds formed between the β8 of hSmc2H-CC30 and the β3 of hSmc4H-CC30 for interface 1 and between the β3 of hSmc2H-CC30 and β7 of hSmc4H-CC30 for interface 2 (Fig. 3b). These are further supported by the hydrophobic interactions formed among the residues located mainly on the β-strand, which are outstandingly conserved among the SMC protein family (Fig. 3b).
In addition to these conserved interactions at the inner region of the donut-shaped dimer, three key intermolecular hydrogen bonding interactions are formed at the top, middle, and bottom regions of the outer surface at each of the dimerization interfaces, where two interacting helices are closely aligned as designated in Fig. 3c. These characteristic interactions utilizing the outer helices are also formed at one of the dimer interfaces of the mSmc2H/mSmc4H heterodimer crystal (Fig. 3d), in a manner similar to the corresponding interface (interface 2) of hSmc2H-CC30/hSmc4H-CC30. At the middle region, however, the Lys-561 residue, in association with Glu-557 of mSmc2H, interacts with the Thr-723 residue of mSmc4H, utilizing a water-mediated hydrogen-bond network, whereas the Lys-561 of hSmc2H-CC30 directly interacts with Asp-722 of hSmc4H-CC30. At the bottom, the mSmc2H/mSmc4H heterodimer has another water-mediated hydrogen bond interaction between Glu-565 of mSmc2H and Leu-719 of mSmc4H; this interaction is not observed in our structure. Nevertheless, the direct interaction between the Arg-572 residue of hSmc2H-CC30 and Gly-742 of hSmc4H-CC30 (corresponding to Gly-740 of mSmc4H) is strictly conserved at the bottom region. These hydrogen bond interactions apparently reinforce the dimerization of the heterodimer and are absent in the bacterial SMC hinge, as judged from the reported crystal structure such as MukB. Therefore, it is believed that these types of interactions are one of the characteristics of eukaryotic SMC hinges, which assists in identifying their partners upon dimerization.
To better understand the dimerization specificity, the subunit interfaces of hSmc2H-CC30/hSmc4H-CC30 were further compared with those of a hypothetical homodimer model of each of the subunit, constructed by molecular modeling (Fig. 3e). The outstanding conservation of the anti-parallel β-sheet interactions demonstrated by the superposition of the structures enables the reasonable construction of a hypothetical model of the hSmc2H and the hSmc4H homodimers by simple superimposition (Fig. 3e, left panel). Surprisingly, tight interactions at two of the dimerization interfaces closely resembling those of hSmc2H-CC30/hSmc4H-CC30 were found in the hSmc2H homodimer model (Fig. 3e, middle panel). This observation is consistent with the results of the AUC analyses, proving the stable formation of the hSmc2H-CC30 dimer in solution (Fig. 2). Although no promising interactions were formed at the top region of the outer interaction surface in the hSmc4H homodimer model (Fig. 3e, right panel), a hydrogen bond interaction was observed between the Gln-652 and Thr-725, and the Lys-659 and Gln-743 side chains, at the middle and bottom regions, respectively, in addition to the β-sheet interaction. Specifically, we detected no apparent steric clash between any of the residues at the dimerization interfaces from our modeling analysis, suggesting the possibility of self-dimerization of the human Smc2 and Smc4. Therefore, the previously observed dimerization specificity of the condensin Smc hinges in vitro could not be delineated by simple examination of the hinge binding interfaces.
Therefore, as an alternative, we analyzed the unique structural characteristics of hSmc4H-CC30, where the N-terminal helix (α1) of the hinge domain was directly linked to the coiled coil region to form a 37-residue-long extended helix without the use of a transition loop and/or the kinks usually found in the SMC hinge region (Fig. 4a). The extended helix of hSmc4H-CC30 extensively interacted via a hydrogen bond network with the bottom region of interface 2 of hSmc2H-CC30 (Fig. 4a). This further stabilizes the dimerization interface and is believed to be a possible factor explaining the dimerization selectivity. It must be noted that this protruding nature of the helix prevents the self-dimerization of hSmc4H-CC30 because of steric clash, which is also explained by the results obtained in solution. Moreover, the newly observed subunit contacts allowed the bundling of the coiled coil regions of the subunits through the hydrogen bonds, for example between Glu-497 of hSmc2H-CC30 and His-577 of hSmc4H-CC30, resulting in an asymmetric heterodimer conformation with three coils protruding in the same direction. This feature markedly differs from other hinge structures (Fig. 4a, top left panel), such as MukBh-sCC and TmSMCh-sCC, both of which adopt a V-shaped dimer with an ∼120° angle between the coiled coils.
FIGURE 4.

Characteristic coiled coil arrangements in the human condensin Smc hinge with middle coiled coils. a, relative orientation of coiled coils in the hSmc2H-CC30/hSmc4H-CC30 heterodimer. Each subunit in the heterodimer is colored as in Fig. 1a. Crystal structure of T. maritima SMC hinge homodimer is also shown in the top left panel. An enlarged view of the interactions between hSmc2H-CC30 and one of the coils of hSmc4H-CC30 is presented in the bottom panel. Key interacting residues involved in the extensive hydrogen bond network are presented as a stick model. Water molecules involved in this network are colored in red. b, superimposition of hSmc4H-CC30 with mouse cohesin Smc3 hinge structure with a short stretch of coiled coil (mSmc3h-sCC) shows the characteristic structure of the N-terminal coil region of hSmc4H-CC30. Black arrows indicate the relative orientation of coils between hSmc4H-CC30 and mSmc3H-sCC. c, enlarged view of the kink region found in the N-terminal coil region of mSmc3H-sCC, in which Gly-501 plays a pivotal role; no such kink structure is observed in hSmc4H-CC30.
Structural comparisons with the recently published mouse Smc3 hinge with a short coiled coil (mSmc3H-sCC; Protein Data Bank code 2WD5) provides us with valuable information concerning the structural requirements for the formation of this elongated helix in hSmc4H-CC30 (Fig. 4, b and c). A glycine residue (Gly-501), which is highly conserved in cohesin SMC at the middle of the N-terminal helix (α1) in mSmc3H-sCC, produces a kink in the backbone to bend the helix to form coiled coil interactions with a C-terminal segment. Superimposition of the two structures showed that Asn-593 occupies the same position in hSmc4H-sCC; however, it is unlikely to form a kink structure similar to Gly-501 of mSmc3H-sCC because of the steric clash between the side chain of Asn-593 and the backbone N-H of Arg-594.
Importantly, sequence alignment results show that Gly and Pro residues that potentially promote the development of the kink and turn structure are often found at the corresponding region in human Smc1, Smc2, and Smc3. For example, a turn structure utilizing Pro-504 is formed at the corresponding region in hSmc2H-CC30 (Fig. 1). On the other hand, no such residues are found in the corresponding region in Smc4; this could promote the formation of an extended helix. Very recently, the existence of an extended helix of Smc4 results in a formation of aligned coiled coils was also confirmed experimentally by the crystal structural analyses of yeast condensin Smc2–4 hinge complex with long coiled coils and cross-linking analyses of chicken condensin Smc2–4 complex, showing that this characteristic coiled coil arrangement is universal feature of eukaryotic condensin SMC subunit (1, 34).
Although the N-terminal region of hSmc4H-CC30 interacts with one of the coils of hSmc2H-CC30, the C-terminal region (Val-768–His-809), which is expected to be involved in the coiled coil interaction with the N-terminal segment of hSmc4H-CC30 is, however, disordered. This is possibly because the predicted coiled coil segment of hSmc4H-CC30 is too short to have any stable interactions with each other in this highly asymmetric configuration and is consequently disordered in the crystal. The corresponding region is also found to be disordered in the crystal of yeast Smc2–4 hinge with long coiled coils showing potential flexibility of this region (1).
Interaction Analysis between the hSmc2–4H with Middle Coiled Coils and DNA by ITC
One of the major functions of condensin is the maintenance of the higher order structure of chromatin fiber, which is a repeat of nucleosomes composed of DNA and the histone octamer. Condensin is also involved in DNA repair. SMC proteins therefore should be capable of binding to the DNA. Previous studies have shown that bacterial SMC proteins preferentially interact with ssDNA rather than dsDNA, through their hinge and/or translational coiled coil regions (8). The higher affinity of eukaryotic Smc hinge dimer to 30-mer ssDNA compared with 30-mer dsDNA has also been confirmed for mSmc2–4H with short coiled coils (14). These studies concluded that the interaction of the condensin Smc hinge with dsDNA is weak and nonspecific, whereas that with ssDNA is stable. Then we investigated the interactions between hSmc2H-CC30/hSmc4H-CC30 and 30-mer ssDNA. As shown in Fig. 5a, the titration of 30-mer ssDNA into hSmc2H-CC30/hSmc4H-CC30 initially generated exothermic heat and then endothermic heat. Apparently the reaction represents other than 1:1 interaction because the first flexion point was confirmed at around molar ratio of 0.4. In fact, 1:1 binding model failed to fit the thermogram. The three repeated thermograms were well fitted globally with the A + B + B ⇄ AB + B ⇄ ABB interacting model, where two heterodimers (B) sequentially bind to one ssDNA (A) with two different dissociation constants. Statistical evaluation of fitting results employing different models excluded other models (supplemental Table S1 and Fig. S1). As summarized in Table 3 and Fig. 5b, the primary and secondary dissociation constants were determined to be 22.3 nm and 2.5 μm, respectively. The enthalpy changes accompanying the interaction were −1.26 kcal/mol for the primary binding and −4.78 kcal/mol for the secondary binding. The large entropic contribution (TΔS, 9.18 kcal/mol) to the total free energy change in the primary binding process was frequently observed in previous studies of ssDNA and protein interactions (35).
FIGURE 5.

Interaction analysis of hSmc2H-CC30/hSmc4H-CC30 with ssDNA observed by ITC. a, the representative titration profile of the triplicate experiments is shown. The thermogram of the titration of the ssDNA into hSmc2H-CC30/hSmc4H-CC30 is shown in the top panel. The normalized binding enthalpy data (black circles) with error bars provided by NITPIC are shown in the middle panel. The line in this panel represents the fitting result with the best parameters obtained from the global analysis based on A + B + B ⇄ AB + B ⇄ ABB interacting model, where A is ssDNA, and B is the heterodimer. The residuals are shown in the bottom panel. b, thermodynamic parameters obtained by nonlinear fitting of the thermogram using A + B + B ⇄ AB + B ⇄ ABB interacting model. c, dependences of fractions for the three species (B, AB, and ABB) on ssDNA (total A) concentration using the fitted thermodynamic parameters (primary Kd = 22.3 nm; secondary Kd = 2.5 μm) at the initial concentration of the heterodimer, 54 μm.
TABLE 3.
Thermodynamic parameters of ssDNA binding to hSmc2H-CC30/hSMC4H-CC30 heterodimer obtained from the ABB model
The values of Kd and ΔH were calculated by using the best fit value obtained from the global analysis of the triplicate data (supplemental Table S1). The values of ΔG and −TΔS were calculated by using the thermodynamic relationships. A was defined as ssDNA, and B was defined as hSmc2H-CC30/hSMC4H-CC30 heterodimer
| Binding site | Parameters |
|||
|---|---|---|---|---|
| Kd | ΔG | ΔH | −TΔS | |
| nm | kcal mol−1 | kcal mol−1 | kcal mol−1 | |
| Primary | 22.3 | −10.4 | −1.26 | −9.18 |
| Secondary | 2500 | −7.64 | −4.78 | −2.86 |
HDX-MS of the hSmc2–4H with Middle Coiled Coils
We performed a HDX-MS analysis of hSmc2H-CC30/hSmc4H-CC30 to assess its conformation in a solution. In this analysis, both hSmc2H-CC30 and hSmc4H-CC30 were digested to over 500 peptides, and the peptides, which covered 97 and 98% of each amino acid length, respectively, were successfully identified. The exchange rates of each peptide at the different time points were mapped on the crystal structure of the heterodimer to visualize the structural features in a solution (Fig. 6, a–f). Basically, the HDX-MS results reflected the crystal structure. However, information on further structural features was also derived from the HDX-MS results. The interfaces between two subunits, such as interface 1 identified in the crystal structure (Met-633–Phe-641, Leu-652–Val-656, and His-660–Ala-667 in hSmc2H-CC30 and Ile-648–Gln-661 and Gly-664–Gly-670 in hSMC4h-scc) contained peptides with a slow deuterium exchange rate. On the other hand, the crystal structure of interface 2 (Thr-556–Gly-567 and Arg-571–Pro-577 in hSmc2H-CC30 and Ser-748–Ser-756 and Asn-720–Tyr-729 in hSmc4H-CC30) revealed a slow exchange rate for Arg-571–Pro-577 in hSmc2H-CC30 and Ser-748–Ser-756 and Asn-720–Tyr-729 in hSmc4H-CC30, whereas Thr-556–Gly-567 in hSmc2H-CC30 exhibited a more rapid exchange rate. Most of the peptides in the coiled coil arm of hSmc2H-CC30 (Ile-489–Leu-499 in Ile-489–Arg-502 and Gln-679–Ala-697 in Ile-674–Leu-702) showed a slow deuterium exchange rate, for which three-dimensional coordinates were unambiguously determined. On the other hand, regions of hSmc4H-CC30 (Phe-572–Ala-602 and Ser-765–Glu-795) in the coiled coil arm (Phe-572–Ser-609 and Ser-765–Val-804, respectively) observed based on the amino acid sequence alignment, showed rapid exchange rates. Because the electron density of one helix could not be confirmed for this arm, the observed rapid exchange rates suggest the flexible nature of the coiled coil arm of hSmc4H-CC30. Consistently, amino acid regions connecting hinge region and coiled coil arm were disordered in the crystal structure of yeast Smc2/Smc4 hinge with long coiled coil (1).
FIGURE 6.

The hydrogen uptake ratios of hSmc2H-CC30/hSmc4H-CC30 heterodimer at different time points. The time points correspond to the duration of hydrogen deuterium exchange reaction, which was produced by reducing the pH to 2.5 at different timing from the beginning of the reaction. a, hSmc2H-CC30. b, hSmc4H-CC30. Each horizontal color block indicates an analyzed digested peptide, and each block contains a number of time points. c–f, hSmc2H-CC30 (c), hSmc2H-CC30 under the presence of ssDNA (d), hSmc4H-CC30 (e), and hSmc4H-CC30 under the presence of ssDNA (f). The deuteration level of each peptide at each time point is color-coded. The figures in a–f were generated using all identified peptides that could be used for deuterium exchange analysis. g, commonly identified peptides in hSmc2H-CC30 for hSmc2H-CC30/hSmc4H-CC30 heterodimer in the absence and presence of ssDNA. h, those in hSmc4H-CC30 for hSmc2H-CC30/hSmc4H-CC30 heterodimer.
It should be noted that His-577–Leu-579 of hSmc4H-CC30, showing a reduced exchange rate, interacts with the transition region between the hinge domain and the coiled coil of hSmc2H-CC30 in the crystal structure (Fig. 4). In addition, the C-terminal small portion (Gln-796–Val-804) of hSmc4H-CC30 also shows a reduced exchange rate, suggesting the involvement of this region in the coiled coil formation, although the possible interacting partner (Thr-566–Asn-571) of hSmc4H-CC30 is unfortunately not detected. Although our HDX-MS shows some flexibility of the coiled coil of hSmc4H-CC30, these results suggested that several regions are partly stabilized to align to the hSmc2H-CC30 coiled coil in a parallel direction via the interaction between the transition region of hSmc2H-CC30 and the coiled coil of hSmc4H-CC30, in solution.
Interaction Site of the hSmc2–4H with Middle Coiled Coils with ssDNA Revealed by HDX-MS
The interaction of hSmc2H-CC30/hSmc4H-CC30 and ssDNA was analyzed by HDX-MS. Based on the ITC analysis, the concentrations of the heterodimer and ssDNA for HDX-MS analysis were adjusted to 2.8 and 5.6 μm, respectively, wherein 98% of the heterodimer existed as a 1:1 complex with ssDNA at deuterium exchange conditions.
In the presence of ssDNA, low quantities of longer peptides were detected. Sixty-six peptides of hSmc2H-CC30 and 56 peptides of hSmc4H-CC30, which covered 92.2 and 89.8% of the hSmc2H-CC30 and hSmc4H-CC30 sequences, respectively, were detected to be common, with enough intensity in the presence and absence of ssDNA (Fig. 6, g and h). To detect the subtle differences, we employed a plotting scheme that showed the differences in deuterium uptake in the presence and absence of ssDNA at each exposure time and the summation of the differences for each peptide (Fig. 7 and supplemental Figs. S2 and S3). In this plot, the y axis indicates the difference between two states of each peptide. Therefore, the upper lines indicate that the free heterodimer allows greater deuterium uptake compared with ssDNA-bound heterodimer; therefore, deuterium exchange was reduced by binding to ssDNA, from which candidates of the binding sites can be identified, although it should be noted the observed reduction of the exchange rate might be caused by any such conformational change. On the other hand, lower lines indicate the reverse pattern; solvent accessibility was higher in the presence of ssDNA, which is generally caused by a conformational change. To compare the heterodimer characteristics between different conditions, such as the absence and presence of ssDNA, we employed the statistical criteria proposed by Houde et al. (30) for the significant change in exchange rate (see “Experimental Procedures”).
FIGURE 7.
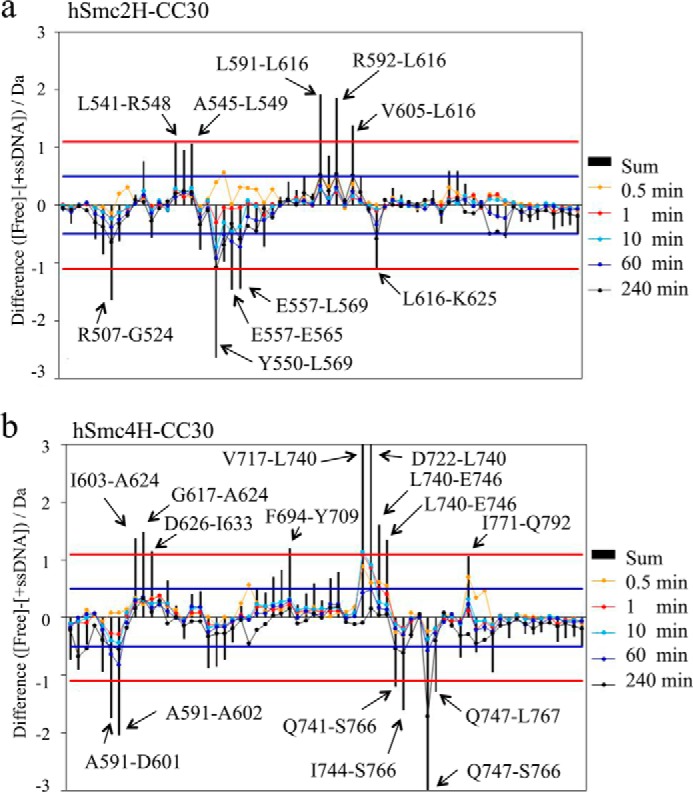
Differential map of the hSmc2–4 hinge with middle coiled coils. For each peptide, the deuterium uptake at each exposure time of hSmc2–4 hinge with middle coiled coils in the presence of ssDNA (+ssDNA) was subtracted from that in the absence of ssDNA (Free). The summation of changes in the uptake over all exposure times has also been indicated. Positive or negative values indicate that the uptake rates were reduced or enhanced, respectively, upon interaction with ssDNA. Red lines, when the sum of difference is over 1.1 Da between two states for a peptide, the result reflects differing amide proton environments of the region covered by the peptide in solution, with 98% confidence. Blue lines, a difference of over 0.5 Da (at a specific exposure time) in a peptide indicates the differing amide proton environments of the region. a, hSmc2H-CC30 in the heterodimer. b, hSmc4H-CC30 in the heterodimer.
Consequently, region 1 (Val-593–Leu-607 and Glu-615–Leu-616) of hSmc2H-CC30 and region 2 (Asp-722–Leu-740 and Gln-743–Glu-746) of hSmc4H-CC30 were confirmed to express a significant depression of deuterium incorporation in the presence of ssDNA (Figs. 7 and 8). Although some of these peptides were distantly located in the primary structure, they are classified into two spatial regions in the three-dimensional structure of the hSmc2H-CC30/hSmc4H-CC30 heterodimer (Fig. 8); the regions with enhanced exchange rates were also confirmed at regions Ala-509–Gly-524 and Val-552–Leu-569 in hSmc2H-CC30 and Asn-593–Ala-602 and Gln-747–Ser-766 in hSmc4H-CC30. Among these peptides, Val-552–Leu-569 of hSmc2H-CC30 and Asn-593–Ala-602 of hSmc4H-CC30 are involved in interface 2 of the crystal structure.
FIGURE 8.

Mapping of deuterium exchange rates onto the three-dimensional structure of the hSmc2–4 hinge with middle coiled coils. The cyan- and magenta-colored regions were derived from peptides that showed statistically significant differences in Fig. 7, except that two amino acids at the N terminus of each peptide were excluded. a and b, cyan-colored regions indicate the amino acid regions where the uptake rate was reduced upon interaction with ssDNA, whereas the magenta-colored regions indicate the regions where the uptake rate was enhanced upon interaction with ssDNA. The bright cyan-colored region (Leu-543–Leu-549) also showed a reduction of the exchange rate with the sum of difference marginally below 1.1 Da. c, surface charge distributions of the heterodimer.
The electron surface potential distribution, calculated for hSmc2H-CC30/hSmc4H-CC30, showed the characteristic neutral to positive charge distribution of the condensin Smc hinge complex. The examination of the surface clefts and grooves suggested the possible ssDNA binding mode of hSmc2H-CC30/hSmc4H-CC30, as shown by the dotted lines in Fig. 8c.
Discussion
Condensins are components of the metaphase chromosome scaffold (3–5, 36). Based on previous research conducted in biochemistry and cell biology, condensins are regarded as one of the protein complexes essential for the maintenance of chromosome shape by maintaining their length and width properly during mitosis. Involvement of condensins in DNA repair and transcription have also been revealed. Despite these studies, the functional mechanics of condensins, including their selective dimerization and interaction with DNA, remain to be elucidated.
In this study, the crystal structure of human condensin Smc2–4 hinge heterocomplex with middle coiled coils (hSmc2H-CC30/hSmc4H-CC30) was determined. This structure allowed for the complete description of two independent dimerization interfaces of the human condensin Smc hinges, with the specific interactions between outermost helices found specifically in the eukaryotic SMC hinges, in addition to the canonical β-sheet interactions commonly observed in prokaryotic and eukaryotic SMC hinge dimers. Therefore, the formation of the donut-shaped dimer of condensin SMC hinges has been well explained in this study. However, this model failed to discard the possibility of homodimeric interactions in the human condensin Smc hinges. Indeed, a homodimeric association of hSmc2H-CC30 was observed in solution, when expressed independently. In this context, it is interesting to note that the N-terminal long extended coil of hSmc4H-CC30 protrudes toward the bottom region of hSmc2H-CC30, resulting in easy contact between the two coiled coil arms of the Smc2–4 heterodimer in solution. This further stabilizes the dimerization interface and may contribute to the dimerization selectivity. Also, this characteristic arrangement of coiled coils could make the condensin SMC complex functionally unique among the SMC protein family, especially for binding to DNA molecule, as illustrated in Figs. 8 and 9.
FIGURE 9.

Schematic representation of the conformational changes in the hSmc2–4 hinge with middle coiled coils and its interaction with 30-mer ssDNA, based on this study. a, proposed model of the hSMC2h-scc/hSMC4h-scc heterodimer in solution, wherein the coiled coils of both subunits interact with each other. a′, crystal structure of hSmc2H-CC30/hSmc4H-CC30. A cartoon model of hSmc2H-CC30/hSmc4H-CC30 is presented in the right panel. a″, hypothetical V-shaped structure of hSmc2H-CC30/hSmc4H-CC30, in which a total of six basic residues, located in the basic patches, are designated by red circles. b, the initial encounter complex of hSmc2H-CC30/hSmc4H-CC30 and 30-mer ssDNA. c, the most stable complex of hSmc2H-CC30/hSmc4H-CC30 with ssDNA formed via a binding mechanism called “hooking.”
Configuration of the two coiled coil arms
So far, it has been debated whether the two coiled coil arms of SMC alter the angle depending on the combinations of the SMC subunits. Through the EM analyses of several groups, it was clearly revealed that cohesin complexes, such as the yeast Smc1/Smc3 complex, form V-shaped open ring configurations (9), whereas a majority of the human condensin 13S complex, composed of a Smc2/Smc4 heterodimer and three non-SMC subunits, forms a rod-like closed configuration, as detailed by Hirano and co-workers (10). The crystal structure of hSmc2H-CC30/hSmc4H-CC30 in this study supports the closed configuration of a human condensin complex, as the coiled coil arm of hSmc4H-CC30 snuggles up to coiled coil arm of hSmc2H-CC30; consequently, two arms express the same orientation, with an extensive hydrogen bond network (Fig. 4a). Similar parallel orientation of coiled coil arms of Smc2 and Smc4 was recently reported in the crystal structure of yeast Smc2–4 hinge with long coiled coils (1) and isolated chicken Smc2–4 complex (34), showing that this is a conserved feature of condensin complexes. The existence of coiled coil interactions in solution is also probed by the results of HDX-MS of hSmc2H-CC30/hSmc4H-CC30, which showed a relatively slow deuterium uptake in the corresponding regions. Because a major part of the hSmc4H-CC30 coiled coil arm showed a rapid exchange rate compared with the hinge domain, the arm of hSmc4H-CC30 could potentially have a flexible nature and/or be exposed to the solvent in solution; however, a portion of the arm is stabilized to form a closed configuration (Fig. 9a).
Interaction of the hSmc2–4H with Middle Coiled Coils with DNA
One of the aims of this study was to determine the DNA recognition mechanism of the SMC hinge domain. The previous DNA binding assays in vitro done by Griese et al. (14) have demonstrated that mSmc2H-sCC/mSmc4H-sCC preferentially binds to 30-mer ssDNA, whereas its interaction with 30-mer dsDNA is nonspecific. They also reported under the conditions of physiological ionic strength that the mouse Smc hinge fails to bind to dsDNA but recognizes ssDNA (14). Consistent with these data, the ITC experiments performed in our study clearly indicated the strong binding potency of hSmc2H-CC30/hSmc4H-CC30 to 30-mer ssDNA. The ITC results provided the binding model in which two Smc heterodimers bind to one 30-mer ssDNA with two different dissociation constants (22.3 nm and 2.5 μm). Because oligo(dT) DNA was used as a substrate, this could be reasonably interpreted, as the 30-mer ssDNA could be long enough to hold one heterodimer with a strong binding constant (Kd = 22.3 nm), with the residual portion of ssDNA interacting with another heterodimer molecule with lower affinity (Kd = 2.5 μm). The degree of cooperativity for the bindings is a future topic to be elusive.
The observed larger entropic contribution of the primary binding (TΔS, 9.18 kcal/mol) than the secondary binding (TΔS, 2.86 kcal/mol) could be originated from a release of water molecules existed on the surface of ssDNA. Instead, disruption of the hydrogen bond network mediated by water molecules resulted in the less enthalpy change of the primary binding than that of the secondary binding. Importantly, enthalpy changes of the primary and secondary bindings were both negative, although titration of ssDNA into the protein solution generated apparently exothermic heat and then endothermic heat. This result could be interpreted as follows: at initial titration of ssDNA, two proteins bind to one ssDNA; however, with ssDNA concentration increasing, one of the two protein molecules on a ssDNA was dissociated and then bound to the other free ssDNA, as depicted by the concentration dependence of three species on ssDNA concentration shown in Fig. 5c.
Subsequent HDX-MS measurements were performed taking this into consideration, at concentrations where a 1:1 DNA-protein complex is dominantly constructed, providing structural information on the primary interaction between the hSmc2H-CC30/hSmc4H-CC30 and ssDNA. The HDX-MS results display the unique structural alterations in the condensin hinge domain upon DNA binding, with slower or faster HDX kinetics depending on the region of binding, both of which are mapped on the crystal structure of hSmc2H-CC30/hSmc4H-CC30 (Fig. 8, a and b).
Previous mutational analyses (16) demonstrated that four basic residues: Lys-566 and Lys-613 of mSmc2H-sCC (which correspond to Arg-566 and Lys-613 of hSmc2H-CC30, respectively) and Lys-657 and Lys-698 of mSmc2H-sCC (which correspond to Lys-659 and Lys-700 of hSmc2H-CC30, respectively), located at the outer surface of the hinge domains, are responsible for ssDNA binding of mSmc2H-sCC/mSmc4H-sCC. Together with these data, the examination of the surface charge distribution and grooves and clefts of the hSmc2H-CC30/hSmc4H-CC30 surface structure allow the estimation of the potential DNA binding mode of the heterodimer (Figs. 8 and 9). When the ssDNA molecule is fitted onto several of the surface grooves, to wrap around the outer surface of the hinge domain where the surface potential is positively charged (Fig. 8), three of the four above-mentioned basic residues, i.e. Arg-566, Lys-659, and Lys-700, were found to be located in close proximity. These regions were correlated with regions showing slower HDX kinetics upon ssDNA binding (Fig. 8), except Arg-566. Although situated at the positively charged region, the peptide fragment containing Arg-566 somehow shows rapid HDX kinetics, which will be discussed later. In addition, one of the four basic residues (Lys-613) of hSmc2H-CC30 is located far from the others and is surrounded by a region displaying a negatively charged surface potential. Therefore, this residue may not be involved in the ssDNA binding. It should be noted that previous mutational analyses were performed utilizing a mSmc2H-sCCK613E/mSmc4H-sCCK698E double mutant but not a mSmc2H-sCCK613E/mSmc4H-sCC single mutant. Therefore, the observed reduction in the ssDNA binding ability of the double mutant could be attributed to the K698E mutation. Based on these results, it appears to be probable that the ssDNA interacted with these regions by wrapping around the protein, as previously suggested from mutational studies of the mouse Smc hinge heterodimer complex.
Possibility of Conformational Changes in the hSmc2–4H with Middle Coiled Coils
Despite these characteristic insights, it is also interesting to note that β7 of hSmc4H-CC30, located at the dimerization interface 2, unexpectedly increases its HDX rate upon ssDNA binding (Fig. 8a), although this region showed a positively charged surface and was involved in the dimerization. Of note, the Leu-543–Leu-549 region of hSmc2H-CC30, located at the inner cavity of the donut-shaped dimer (Figs. 6 and 8) showed a decrease in the exchange rate. This cavity had minimum and maximum diameters of ∼1 and 2 nm, respectively. Therefore, even the ssDNA was not directly accessible to the cavity. These results cannot be interpreted without allowing for conformational alternations in the heterodimer after the formation of the encounter complex and are explained when the dynamic conformational changes in the Smc hinges are considered. The conformational change induces the disruption of dimerization interface 2 to open the hinge dimer to provide an appropriate binding surface to ssDNA. After these conformational changes, ssDNA could be accessible to the inner region of a half-opened hinge dimer. Interestingly, in contrast to the β7 of hSmc4H-CC30 that showed rapid HDX kinetics, the β3 sheet, a binding counterpart of interface-2, showed no changes in its HDX profile upon DNA binding, indicating that the interaction of ssDNA at this region, which results in destabilization of the structure by hinge-hinge opening, is compensated by stable interactions with ssDNA. Intriguingly, this structural change allows ssDNA to interacts with initially “hidden” basic residues such as Arg-548 and Arg-572 that are located at the above stated regions, Leu-543–Leu-549 and the β3 sheet, respectively, with slow HDX kinetics further suggesting this notion.
The structural alteration resulting from hinge-hinge opening also affects the dynamics of some part of the long N-terminal coil of hSmc4H-CC30. The region (Asn-593–Ala-602) that interacted with the bottom region of interface 2 showed an increase in the HDX rate, indicating the destabilization of the structure in this region. Nevertheless, the negligible differences in the HDX profiles observed at the most part of the coiled coils suggest the existence of aligned coils even if ssDNA disrupts one of the dimerization interfaces of the heterodimer. Taken together, these results motivated us to propose the existence of a “hooking” mechanism, a unique binding mode for the interaction of a half-opened condensin Smc hinges with ssDNA (Fig. 9c).
The “hooking” mechanism was previously proposed by Hirano and Hirano (16) for bacterial BsSMC complexes. They demonstrated that three lysine residues (Lys-666, Lys-667, and Lys-668), located at the transition region so-called TR connecting the hinge domain and the long coiled coil, as well as one lysine residue (Lys-565) situated in close proximity to TR in BsSMC, are essential for the interaction with DNA. These lysine residues are located close to each other and form two clusters of positively charged surfaces, called basic patches, in the bacterial SMC homodimer. These basic patches are also conserved in the eukaryotic cohesin Smc1/Smc3 complex. Based on biochemical assays and mutation analyses, Chiu et al. (17) showed that the bovine Smc1/Smc3 hinge complex includes a TR and that the short coiled coils preferentially bind dsDNA over ssDNA at these regions, which include basic patches. The experiments using the artificially linked yeast Smc1/Smc3 complex with non-SMC subunits revealed that the entrapment of dsDNA within the closed ring formed by the five-subunit cohesin holo-complex could occur at the hinge domain. The results led to the postulation of a “hinge open model” for cohesin action proposed by Nasmyth and co-workers (19). This model requires that the ring-like structure open at the hinge domain during the entrapment of DNA, with the regions that include the basic patches acting as potential initial DNA binding sites. If the cohesin Smc1/Smc3 complex forms a V-shaped dimer as suggested in previous studies, the region that includes the basic patches is extensively exposed to the solvent region, allowing a stable interaction with ssDNA.
The sequence alignment of condensin shows that none of the four lysine residues is conserved in the human Smc2 subunit; however, two of the lysine residues are replaced by arginine residues (Arg-566 and Arg-668) that also have a positive charge. On the other hand, three among the four lysine residues in the human Smc4 subunit are conserved (Lys-659, Lys-757, and Lys-760). Jessberger and co-workers (17) reported that only one of the two TR would be sufficient to facilitate DNA binding. Therefore, it was initially expected that the hSmc2H-CC30/hSmc4H-CC30, which expressed at least one TR region rich in basic residues of hSmc4H-CC30, would show a strong binding preference to dsDNA and/or ssDNA when the heterodimer takes a V-shaped form (Fig. 9a″). However, the binding preference of condensin Smc hinge region on dsDNA has not been reported so far. Furthermore, the results of HDX-MS performed in this study revealed the enhanced exchange rates of peptides including the Arg-566 residue of hSmc2H-CC30 and Lys-757 and Lys-760 of hSmc4H-CC30 upon DNA binding. On the other hand, the peptides including the Arg-668 residue of hSmc2H-CC30 and Lys-659 residue of hSmc4H-CC30 showed no changes in the exchange rate, indicating the irrelevance of these residues to the hSmc2H-CC30/hSmc4H-CC30-DNA interactions. These results are well explained by the closed configuration of coiled coils of the condensin Smc complex. This would prevent providing the access for DNA molecules to interact with the basic patch, even if it expresses some basic residues at this site, indicating the functional differences between condensin and cohesin, in terms of DNA binding. Therefore, the previously proposed theory for cohesin action, wherein the DNA passes through its hinge-hinge interface, with the loading of DNA, if it occurs at all, onto the condensin complex utilizing the hinge-hinge interface as the entrance gate, appears to be unlikely. In this context, it is important to note that Barysz et al. (34) recently reported by the chemical cross-linking analysis of chicken condensin complex in situ that several cross-links were formed between Smc2 and Smc4 near the exact center of coiled coils, showing that the condensin on mitotic chromosomes does have closely aligned coiled coils during its action.
Functional Implications of Condensin Hinge-DNA Interaction
A previous study conducted by Hirano and Hirano revealed that the interaction between the bacterial SMC hinge and ssDNA triggers ssDNA-stimulated ATP hydrolysis, inducing head-head disengagement (16). Therefore, the initial loading of condensin Smc to DNA in vivo was speculated to occur via the SMC hinge-DNA interactions at the ssDNA region, which is created behind the replicative helicase and is believed to be necessary for the subsequent functionality of SMC (7). However, recent studies have suggested that the recruitments of condensins onto chromosomes are mediated by non-SMC subunits expressing HEAT repeat sequences (hCAP-D2 and -G for condensin I and hCAP-D3 and -G2 for condensin II (37)). The interaction between the non-SMC subunits and dsDNA consequently activates the Smc2-Smc4 ATPases, followed by the exertion of condensin functions. This study also reported that the condensin subcomplex, composed of non-SMC subunits, shows preference toward binding free DNA over nucleosomal DNA. Thus condensin hinge-DNA interactions might function after the initial loading of condensin to chromosomes. Akai et al. (7) suggested that the yeast condensin winds the DNA strands after DNA repair prior to mitosis. The higher sensitivity of nuclear chromatin from cut14 mutant cells against S1 nuclease supports the importance of condensin SMC binding to ssDNA (6).
In the series of studies for fission yeast condensin SMC, Yanagida and co-workers (6, 7) demonstrated that condensin antagonizes replication protein A activity by removing it from DNA in vitro and in vivo, which suggests that the DNA reannealing activity of condensin may facilitate the removal of proteins from chromosomes after DNA repair or prior to chromosome segregation. It was also speculated that promotion of the ssDNA annealing by Smc2/Smc4 could be required to remove “leftover” products of interphase processes, such as DNA-RNA hybrids, from mitotic chromosomes to facilitate their faithful segregation (38). This scenario might be related to the initiation of sister chromatid resolution during the S phase by condensin II (39).
Our present study together with previous studies on the interaction of SMC hinge with DNA has clearly shown that SMC hinge preferentially binds to ssDNA (8, 14, 18). On the other hand, our previous proteome analysis of human metaphase chromosomes indicated that topoisomerases I and II exist on the isolated metaphase chromosomes with their amount similar to those of condensin SMC2 and SMC4 subunits (4). These results present one scenario that, in the process of mitotic condensation and/or in condensed mitotic chromosome, a part of dsDNA dissociates into ssDNA, presumably by the help of topoisomerases that mediate catenation or decatenation of dsDNA, and condensin molecules hold multiple DNA strands via binding to ssDNA portion, contributing to the maintenance of condensed chromosome state.
Author Contributions
Y. H., Y. K., and H. O. expressed and purified recombinant proteins. K. K., S. F., S. N., and T. O. performed crystallization and structure determination. T. M. and Y. K. performed AUC and ITC analyses. Y. M., R. T., and M. N. performed HDX-MS experiments. S. U., K. K., and K. F. directed the research and wrote the paper with assistance from all other authors.
Supplementary Material
Acknowledgments
We thank Drs. Kamada and Ono (RIKEN) for critical discussions. The synchrotoron x-ray diffraction experiments were performed with the approval of the SPring-8 Proposal Review Committee (2010A1501, 2010B2007, 2013A1251, and 2013B1192) and the Photon Factory Advisory Committee (2010G071).
This work was supported by Grants-in-Aid 18681032, 90335381, 25121722, and 26102530 from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (to S. U.), the Japan Society for the Promotion of Science Japanese-German Graduate Externship (to S. U.), and the Senri-Life Science Foundation (to S. U.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4U4P) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Table S1 and Figs. S1–S3.
- SMC
- structure maintenance of chromosome
- HDX-MS
- hydrogen deuterium exchange mass spectrometry
- ssDNA
- single-stranded DNA
- TR
- transition region
- ITC
- isothermal titration calorimetry
- AUC
- analytical ultracentrifugation.
References
- 1.Soh Y. M., Bürmann F., Shin H. C., Oda T., Jin K. S., Toseland C. P., Kim C., Lee H., Kim S. J., Kong M. S., Durand-Diebold M. L., Kim Y. G., Kim H. M., Lee N. K., Sato M., Oh B. H., and Gruber S. (2015) Molecular basis for SMC rod formation and its dissolution upon DNA binding. Mol. Cell 57, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasmyth K., and Haering C. H. (2005) The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74, 595–648 [DOI] [PubMed] [Google Scholar]
- 3.Ono T., Losada A., Hirano M., Myers M. P., Neuwald A. F., and Hirano T. (2003) Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115, 109–121 [DOI] [PubMed] [Google Scholar]
- 4.Uchiyama S., Kobayashi S., Takata H., Ishihara T., Hori N., Higashi T., Hayashihara K., Sone T., Higo D., Nirasawa T., Takao T., Matsunaga S., and Fukui K. (2005) Proteome analysis of human metaphase chromosomes. J. Biol. Chem. 280, 16994–17004 [DOI] [PubMed] [Google Scholar]
- 5.Poonperm R., Takata H., Hamano T., Matsuda A., Uchiyama S., Hiraoka Y., and Fukui K. (2015) Chromosome scaffold is a double-stranded assembly of scaffold proteins. Sci. Rep. 5, 11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sutani T., and Yanagida M. (1997) DNA renaturation activity of the SMC complex implicated in chromosome condensation. Nature 388, 798–801 [DOI] [PubMed] [Google Scholar]
- 7.Akai Y., Kurokawa Y., Nakazawa N., Tonami-Murakami Y., Suzuki Y., Yoshimura S. H., Iwasaki H., Shiroiwa Y., Nakamura T., Shibata E., and Yanagida M. (2011) Opposing role of condensin hinge against replication protein A in mitosis and interphase through promoting DNA annealing. Open Biol. 1, 110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirano M., and Hirano T. (2002) Hinge-mediated dimerization of SMC protein is essential for its dynamic interaction with DNA. EMBO J. 21, 5733–5744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haering C. H., Löwe J., Hochwagen A., and Nasmyth K. (2002) Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell 9, 773–788 [DOI] [PubMed] [Google Scholar]
- 10.Anderson D. E., Losada A., Erickson H. P., and Hirano T. (2002) Condensin and cohesin display different arm conformations with characteristic hinge angles. J. Cell Biol. 156, 419–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y., Schoeffler A. J., Berger J. M., and Oakley M. G. (2010) The crystal structure of the hinge domain of the Escherichia coli structural maintenance of chromosomes protein MukB. J. Mol. Biol. 395, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ku B., Lim J. H., Shin H. C., Shin S. Y., and Oh B. H. (2010) Crystal structure of the MukB hinge domain with coiled-coil stretches and its functional implications. Proteins 78, 1483–1490 [DOI] [PubMed] [Google Scholar]
- 13.Griese J. J., and Hopfner K. P. (2011) Structure and DNA-binding activity of the Pyrococcus furiosus SMC protein hinge domain. Proteins 79, 558–568 [DOI] [PubMed] [Google Scholar]
- 14.Griese J. J., Witte G., and Hopfner K. P. (2010) Structure and DNA binding activity of the mouse condensin hinge domain highlight common and diverse features of SMC proteins. Nucleic Acids Res. 38, 3454–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurze A., Michie K. A., Dixon S. E., Mishra A., Itoh T., Khalid S., Strmecki L., Shirahige K., Haering C. H., Löwe J., and Nasmyth K. (2011) A positively charged channel within the Smc1/Smc3 hinge required for sister chromatid cohesion. EMBO J. 30, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirano M., and Hirano T. (2006) Opening closed arms: long-distance activation of SMC ATPase by hinge-DNA interactions. Mol. Cell 21, 175–186 [DOI] [PubMed] [Google Scholar]
- 17.Chiu A., Revenkova E., and Jessberger R. (2004) DNA interaction and dimerization of eukaryotic SMC hinge domains. J. Biol. Chem. 279, 26233–26242 [DOI] [PubMed] [Google Scholar]
- 18.Yoshimura S. H., Hizume K., Murakami A., Sutani T., Takeyasu K., and Yanagida M. (2002) Condensin architecture and interaction with DNA: regulatory non-SMC subunits bind to the head of SMC heterodimer. Curr. Biol. 12, 508–513 [DOI] [PubMed] [Google Scholar]
- 19.Gruber S., Arumugam P., Katou Y., Kuglitsch D., Helmhart W., Shirahige K., and Nasmyth K. (2006) Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 127, 523–537 [DOI] [PubMed] [Google Scholar]
- 20.Chan K. L., Roig M. B., Hu B., Beckouët F., Metson J., and Nasmyth K. (2012) Cohesin's DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 150, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huis in 't Veld P. J., Herzog F., Ladurner R., Davidson I. F., Piric S., Kreidl E., Bhaskara V., Aebersold R., and Peters J. M. (2014) Characterization of a DNA exit gate in the human cohesin ring. Science 346, 968–972 [DOI] [PubMed] [Google Scholar]
- 22.Sakai A., Hizume K., Sutani T., Takeyasu K., and Yanagida M. (2003) Condensin but not cohesin SMC heterodimer induces DNA reannealing through protein-protein assembly. EMBO J. 22, 2764–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawahara K., Nakamura S., Katsu Y., Motooka D., Hosokawa Y., Kojima Y., Matsukawa K., Takinowaki H., Uchiyama S., Kobayashi Y., Fukui K., and Ohkubo T. (2010) Cloning, expression, crystallization and preliminary X-ray crystallographic analysis of a human condensin SMC2 hinge domain with short coiled coil. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1067–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houtman J. C., Brown P. H., Bowden B., Yamaguchi H., Appella E., Samelson L. E., and Schuck P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 26.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lupas A., Van Dyke M., and Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 32.Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 33.Haering C. H., Schoffnegger D., Nishino T., Helmhart W., Nasmyth K., and Löwe J. (2004) Structure and stability of cohesin's Smc1-kleisin interaction. Mol. Cell 15, 951–964 [DOI] [PubMed] [Google Scholar]
- 34.Barysz H., Kim J. H., Chen Z. A., Hudson D. F., Rappsilber J., Gerloff D. L., and Earnshaw W. C. (2015) Three-dimensional topology of the SMC2/SMC4 subcomplex from chicken condensin I revealed by cross-linking and molecular modelling. Open Biol. 5, 150005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buczek P., and Horvath M. P. (2006) Thermodynamic characterization of binding Oxytricha nova single strand telomere DNA with the alpha protein N-terminal domain. J. Mol. Biol. 359, 1217–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirano T., Kobayashi R., and Hirano M. (1997) Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 89, 511–521 [DOI] [PubMed] [Google Scholar]
- 37.Piazza I., Rutkowska A., Ori A., Walczak M., Metz J., Pelechano V., Beck M., and Haering C. H. (2014) Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Nat. Struct. Mol. Biol. 21, 560–56838 [DOI] [PubMed] [Google Scholar]
- 38.Yanagida M. (2009) Clearing the way for mitosis: is cohesin a target? Nat. Rev. Mol. Cell. Biol. 10, 489–496 [DOI] [PubMed] [Google Scholar]
- 39.Ono T., Yamashita D., and Hirano T. (2013) Condensin II initiates sister chromatid resolution during S phase. J. Cell Biol. 200, 429–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.