

Background: Phosphatidylinositol 4,5-bisphosphate levels need to be replenished during calcium signaling, but how this is achieved is unclear.
Results: Ca2+ entry through Orai1 channels contributes to the resynthesis of phosphatidylinositol 4,5-bisphosphate and, thereby, helps sustain cytoplasmic Ca2+ oscillations.
Conclusion: Ca2+ influx sustains cytoplasmic Ca2+ oscillations by regulating phosphatidylinositol-4-phosphate 5-kinase.
Significance: Store-operated Ca2+ influx is important in maintaining phosphatidylinositol 4,5-bisphosphate levels.
Keywords: calcium channel, calcium intracellular release, calcium release-activated calcium channel protein 1 (ORAI1), phosphatidylinositol kinase (PI kinase), phosphatidylinositol signaling
Abstract
Oscillations in cytoplasmic Ca2+ concentration are a universal mode of signaling following physiological levels of stimulation with agonists that engage the phospholipase C pathway. Sustained cytoplasmic Ca2+ oscillations require replenishment of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2), the source of the Ca2+-releasing second messenger inositol trisphosphate. Here we show that cytoplasmic Ca2+ oscillations induced by cysteinyl leukotriene type I receptor activation run down when cells are pretreated with Li+, an inhibitor of inositol monophosphatases that prevents PIP2 resynthesis. In Li+-treated cells, cytoplasmic Ca2+ signals evoked by an agonist were rescued by addition of exogenous inositol or phosphatidylinositol 4-phosphate (PI4P). Knockdown of the phosphatidylinositol 4-phosphate 5 (PIP5) kinases α and γ resulted in rapid loss of the intracellular Ca2+ oscillations and also prevented rescue by PI4P. Knockdown of talin1, a protein that helps regulate PIP5 kinases, accelerated rundown of cytoplasmic Ca2+ oscillations, and these could not be rescued by inositol or PI4P. In Li+-treated cells, recovery of the cytoplasmic Ca2+ oscillations in the presence of inositol or PI4P was suppressed when Ca2+ influx through store-operated Ca2+ channels was inhibited. After rundown of the Ca2+ signals following leukotriene receptor activation, stimulation of P2Y receptors evoked prominent inositol trisphosphate-dependent Ca2+ release. Therefore, leukotriene and P2Y receptors utilize distinct membrane PIP2 pools. Our findings show that store-operated Ca2+ entry is needed to sustain cytoplasmic Ca2+ signaling following leukotriene receptor activation both by refilling the Ca2+ stores and by helping to replenish the PIP2 pool accessible to leukotriene receptors, ostensibly through control of PIP5 kinase activity.
Introduction
A rise in intracellular Ca2+ concentration ([Ca2+]i) is a universal signal used throughout the phylogenetic tree to activate a broad range of spatially and temporally distinct cellular responses ranging from exocytosis and fast-twitch muscle contraction to nuclear gene expression and cell proliferation (1, 2). Inherent to the use of a multifarious signal like Ca2+ is the problem of specificity. How can one Ca2+-dependent response within a cell be activated but not another? Evidence is now accumulating that the size, time course, and subcellular location of the rise in [Ca2+]i are all important factors that contribute to the selective recruitment of downstream targets (2, 3).
In many cells types, stimulation of surface receptors that couple to heterotrimeric Gq proteins and phospholipase Cβ and, thereby, hydrolyze the membrane phospholipid PIP23 generate the second messenger InsP3, which releases stored Ca2+ by opening ligand-gated Ca2+ channels on the endoplasmic reticulum (ER) (4). Low levels of stimulation of these G-protein-coupled receptors, considered to represent physiological levels of receptor activation, often result in the generation of repetitive oscillations in [Ca2+]i that present either as a series of baseline Ca2+ spikes or slower sinusoidal Ca2+ waves on an elevated plateau (5). Information is encoded in the amplitude, frequency, and spatial profile of the oscillation (3). Oscillations in [Ca2+]i, as opposed to a sustained rise in [Ca2+]i, enhance mitochondrial energy production (6), exocytosis (7), and Ca2+-dependent gene expression (8, 9) while avoiding the toxic effects that are associated with prolonged elevation of [Ca2+]i.
Oscillations in [Ca2+]i require either oscillations in the levels of InsP3 or biphasic gating of the InsP3 receptor by cytoplasmic Ca2+ in the presence of a steady increase in InsP3. Regardless of the mechanism, two conditions need to be satisfied for repetitive oscillations in [Ca2+]i to occur. First, the Ca2+ content of the stores must be maintained at a level sufficient for Ca2+ release. This is necessary because, following each Ca2+ release event, a fraction of the mobilized Ca2+ is extruded from the cell by plasma membrane Ca2+ATPase pumps (10). In the absence of refilling, store Ca2+ content would therefore drop below the level that supports InsP3-dependent Ca2+ release. Store refilling is accomplished through activation of store-operated CRAC channels in the plasma membrane that open following loss of Ca2+ from within the ER (11). CRAC channels are comprised of STIM and Orai proteins (12). STIM1 and STIM2 are ER Ca2+ sensors and migrate toward the plasma membrane upon store depletion (reviewed in Ref. 13). Orai1 is the pore-forming subunit of the CRAC channel and is gated by STIM binding to the C and N termini of the protein (reviewed in Ref. 14). The second criterion that needs to be satisfied for the generation of prolonged oscillations in [Ca2+]i is that PIP2 levels must be replenished following each Ca2+ spike to support production of the InsP3 that is needed for the next oscillation in [Ca2+]i. InsP3 has a lifetime of just a few seconds in the cytoplasm (15, 16) because of the presence of strong catabolic pathways. These breakdown pathways are well characterized and involve sequential dephosphorylation by phosphatases to myoinositol monophosphate, which, in turn, is dephosphorylated to myoinositol by inositol monophosphatases (IMPases), enzymes that are inhibited by Li+. Inositol combines with cytidine diphosphate diacylglycerol to form phosphatidylinositol (PI), which can then be phosphorylated by PI4 and PIP5 kinases to make PIP2.
Two IMPase genes have been identified in mammals: IMPase1 and IMPase2. IMPase2 is implicated in the pathogenesis of bipolar disorder, schizophrenia, and febrile seizures (17). Blocking of inositol monophosphatases, which would deplete PIP2 levels and, therefore, impair production of InsP3, is a possible mechanism to explain the mood-stabilizing effect of Li+ treatment on bipolar disorder (18). Although both IMPases are inhibited by Li+, the IMPase1 is more sensitive to blocking, with an IC50 of 0.7 mm (in 2 mm MgCl2) that is ∼30-fold left-shifted compared with IMPase2 inhibition (19).
PIP2 levels regulate CRAC channel activity in various ways. STIM1 trafficking to the cell periphery is expedited by the presence of a cytoplasmic Lys-rich domain that may bind to PIP2 and phosphatidylinositol 3,4,5-trisphosphate in the inner leaflet of the plasma membrane (20, 21). Furthermore, slow Ca2+-dependent inactivation of the channels requires Orai1 confinement to PIP2-rich domains of the plasma membrane (22). Here we explored the possibility that Ca2+ influx through CRAC channels, in addition to refilling the stores, regulates replenishment of the agonist-sensitive PIP2 pool during oscillations in [Ca2+]i. We find that the step converting PI4P to PIP2 is dependent on Ca2+ entry through the channels, providing an autoregulatory mechanism through which CRAC channels maintain their own activity by ensuring sufficient PIP2 levels in the plasma membrane for store depletion via InsP3 production.
Experimental Procedures
Reagents
Culture medium and salts were obtained from Sigma. LTC4 was purchased from Cambridge Bioscience Ltd. (Cambridge, UK). FCS, Fura-2/AM, Dulbecco's modified Eagle's medium, and penicillin-streptomycin were obtained from Invitrogen. Thapsigargin was purchased from Merck Chemicals Ltd. (Darmstadt, Germany). PI4P diC8 and unlabeled shuttle PIP carrier 3 were purchased from Echelon Biosciences Inc. Unlabeled shuttle PIP carrier 3 (catalog no. P-9C3) was used to deliver PI4P into cells. The carrier was reconstituted in distilled water and mixed with PI4P. The carrier-PI4P complex was vortexed and left at room temperature for 15–20 min. Following this, the complex was diluted in external solution and applied to intact cells for 7 min prior to stimulation with LTC4. The carrier final concentration was 20 μm.
Cell Culture
Rat basophilic leukemia RBL-2H3 cells were from the ATCC. Early experiments to see whether LTC4 evoked Ca2+ signals in this cell line were carried out on cells provided by Dr. Shamshad Cockcroft (University College London, London, UK). RBL-2H3 cells were used because of their strong secretory response, which is being investigated in a broader study within the group. Cells were routinely maintained in Dulbecco's modified Eagle's medium and supplemented with 10% FCS and 1% penicillin-streptomycin, as described previously (23). Cells were kept in an atmosphere containing 5% CO2 and maintained at 37 °C. For calcium imaging experiments, cells were plated onto 13-mm glass coverslips using 0.05% trypsin and used 48 h after passaging.
Cytoplasmic Ca2+ Measurements
A Nikon Eclipse TE2000-U inverted microscope equipped with an IMAGO charge-coupled device camera-based system from TILL Photonics (Gräfelfing, Germany) was used as described previously (23). Cells were alternately excited at 356/380 nm with exposure times of 20 ms and an acquisition rate of 0.5 Hz. Data were analyzed after import to IGOR Pro (Wave Metrics, Lake Oswego, OR). All experiments were performed at room temperature, and cells were kept in the dark when loaded. For cytoplasmic Ca2+ measurements, cells were loaded with Fura-2/AM (4 μm) for 40 min in an external solution comprised of the following: 145 mm NaCl, 2.8 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm d-glucose, and 10 mm HEPES (pH 7.4 with NaOH). Calcium-free solution was of a similar composition as the external solution, except that 0.1 mm ethylene glycol-bis-(β-aminoethylether)-N,N,N′,N′-tetraacetic acid was substituted for calcium chloride. The number of calcium oscillations was quantified using IGOR Pro. Ca2+ spikes were considered to be oscillations when the Rpeak − Rbase value was >0.1.
Li+ Treatment
LiCl dissolved in DMEM replaced normal DMEM on cultured cells growing on coverslips. Cells were then maintained in the incubator for 40 min. Thereafter, cells were loaded with Fura-2/AM in the continuous presence of LiCl. Cells on the first coverslip were exposed to LiCl for 85–90 min prior to stimulation with LTC4. The last coverslip in the set had been exposed to LiCl for ∼110 min before stimulation.
Knockdown and cDNA Transfection Experiments
For siRNA and cDNA transfection, the Amaxa electroporation system was used (23) with the Amaxa cell line nucleofactor kit T. Transfection efficiency, judged by GFP fluorescence following transfection with a GFP plasmid, was typically ∼50–60%.
STIM1 siRNA was purchased from Life Technologies (catalog no. 4390815). Orai1 siRNA was bought from Origene Technologies (catalog no. SR508429), and the sequences (5′ to 3′) were AGUUCUUACCGCUCAAGAGGCAGGC, CCAUAAGACGGACCGACAGUUCCAG, and AGGGCAGAGUGUGGAAGGAAGAGGC. Both STIM1 and Oria1 siRNAs were used at a final concentration of 50 nm.
PIP5K1α and PIP5K1γ siRNAs and scrambled siRNA were purchased from Dharmacon and used at a final concentration of 75 nm. The PIP5K1α sequences (5′ to 3′) were CGGCAAGAACAUACGAAUU, GCAUCCGGCCUGACGAUUA, GCCCAUGAACAGCGAAAACA, and GAAAAUAGGCCAUCGAAGU. The PIP5K1γ sequences (5′ to 3′) were UCUGGAGAGACUACGUAUA, GCUUCUAUGCCGAGCGCUU, GAGAGGAUGUGCAGUACGA, and GGAGGAGCUGCAUGCGGAA.
Talin1 and talin2 siRNAs were from Dharmacon and used at 50 nm. The Talin1 SMARTpool sequences were CGAGAACUAUGCAGGUAUU, CGAAUGACCAAGGGUAUUA, GUUCGUAGAUUAUCAGACA, and GAGAUGAAGAGUCUACUAU. The Talin2 SMARTpool sequences were UGUUAGUACUCAAGGCGAA, CCGCAAUAAGUGUCGAAUU, CCGCAAAGCUCUUGGCCGA, and GCUAGAAGCAGGUCGGACA.
Immunofluorescence
Cells were fixed using 4% paraformaldehyde and then permeabilized with 0.5% Triton X-100. Cells were then blocked using SuperBlock blocking buffer for 1 h at room temperature. Primary antibody (1:200) in PBS was added to the cells and left overnight at 4 °C. Cells were then washed at 10-min intervals in PBS with 0.1% Tween 20 (PBS-T) buffer. Alexa Fluor 488 anti-mouse or anti-rabbit or Alexa Fluor 568 anti-rabbit conjugated antibody was then added for 1 h at room temperature. Cells were mounted onto microscope slides using Vectashield antifade mounting medium with DAPI. Images were obtained using the inverted Olympus FV1000 confocal system equipped with a motorized stage, using a ×60 oil objective of 1.3 numerical aperture and excitation at 488 or 568 nm. All images were grouped according to an image size of 640 × 480 and a step size of 4 μm along the z axis. Fluorescence intensities were analyzed for each treatment and normalized to the maximum measured fluorescence using ImageJ.
Reverse Transcriptase Polymerase Chain Reaction
QIAshredder was used for homogenization of cell lysate, and total RNA was extracted using the Qiagen RNeasy mini kit. The process of reverse transcription of 1 μg of RNA was achieved using an iScript cDNA synthesis kit. The produced cDNA was amplified utilizing GoTaq Green master mix. The product of the polymerase chain reaction was separated by electrophoresis on 2% agarose gel. Primers were synthesized by Sigma: Talin-1, 5′-TCGGAAGTGGCTTGTGTAGT-3′ (sense) and 5′-GAGAACGCCCGAACTAAACG-3′ (antisense); Talin-2, 5′-GTGGCAGCTAGAGAAACAGC-3′ (sense) and 5′-GGCTTCTTGGATGAGCATGG-3′ (antisense).
Western Blot Analysis
Cells were lysed in radioimmunoprecipitation assay lysis buffer supplemented with protease inhibitor mixture, 0.1% Triton X-100, 10 mm sodium metavanadate, and 1 mm PMSF. 15 μg of protein was loaded into 10% SDS-PAGE gel. The protein was next transferred into a nitrocellulose membrane using a semidry protein transfer apparatus (Bio-Rad). 5% nonfat dry milk in phosphate-buffered saline was used to block the membrane. The blocked membrane was then incubated with either PIP5 kinase α or γ antibody (1:2000) for 2 h at room temperature. After washing, a secondary antibody (1:4000) was applied in 5% nonfat dry milk in phosphate-buffered saline solution for 2 h at room temperature. Visualization was accomplished by the use of enhanced chemiluminescence plus the Western blotting detection system. The relative band intensities were analyzed using ImageJ. All bands were normalized to the corresponding β-actin levels as a loading control.
Statistical Analysis
All experiments were performed on three independent occasions unless specified otherwise in the text. Independent sample groups were first assessed for normality and equality of variances. To compare single-group treatment, an unpaired Student's t test or Mann-Whitney U test was used (StatsDirect v2.6.2, Sale, UK). Differences were considered significant at p < 0.05. All data are presented in the text and figures as mean ± S.E. (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001).
Results
Oscillations in [Ca2+]i Are Sustained by Ca2+ Entry through CRAC Channels in RBL-2H3 Cells
Activation of cysteinyl leukotriene type I receptors with LTC4 in RBL-1 cells evokes a series of oscillations in [Ca2+]i that are supported by Ca2+ entry through CRAC channels (9). We repeated these experiments in the RBL-2H3 cell line, which has a stronger secretory phenotype. Stimulation with LTC4 resulted in numerous oscillations in [Ca2+]i that were sustained for the duration of the experiment (800 s, Fig. 1A), although the amplitude decreased gradually with time because of receptor desensitization (24). To see whether Ca2+ entry through CRAC channels was required to maintain the oscillatory response, we used both pharmacological and siRNA-based approaches. La3+ is an effective CRAC channel blocker (25) and inhibited the rate of thapsigargin-induced Ca2+ influx with an IC50 of ∼300 nm (Fig. 1, B and C). Pre-exposure to 10 μm La3+ accelerated the rundown of the oscillations in [Ca2+]i induced by LTC4 in 2 mm external Ca2+-containing solution (Fig. 1D). The rate of rundown in La3+ was similar to that seen in cells challenged with LTC4 in the absence of external Ca2+ (Fig. 1E; aggregate data from several experiments are summarized in Fig. 1F). Similar results were obtained when 20 μm BTP2 was used to block CRAC channels instead (Fig. 1F).
FIGURE 1.

CRAC channels maintain cytoplasmic Ca2+ oscillations in RBL-2H3 cells following leukotriene receptor stimulation. A, typical oscillatory response in [Ca2+]i to 160 nm LTC4 in the presence of external calcium. B, CRAC channel-mediated Ca2+ entry was blocked in a dose-dependent manner by pretreatment with La3+. Ca2+ influx was evoked by readmission of external Ca2+ to cells challenged with 2 μm thapsigargin in Ca2+-free solution. Only the Ca2+ influx component is shown. The x axis represents the time when Ca2+ release had returned to resting levels. La3+ was applied 3 min prior to thapsigargin exposure. C, graph summarizing the La3+ dose-inhibition curve. Each point is the mean of between 21 and 32 cells. D, oscillations in [Ca2+]i evoked by LTC4 in external Ca2+ run down in the presence of 10 μm La3+ (pretreatment for 3 min). E, typical response to LTC4 in the absence of external Ca2+. F, aggregate data (from >50 cells for each treatment) are shown from three independent experiments. Each bin represents a consecutive recording period of 200 s. For most averages, error bars (mean ± S.E.) are contained within the symbols. G, immunohistochemical staining of STIM1 in either control cells or in cells after knockdown of STIM1. An experiment with a higher than usual transfection efficiency is shown here and in I. KD, knockdown. H, semiquantitative measurements of mean florescence from two independent experiments for STIM1. ****, p < 0.0001. I, as in G, but Orai1 was measured instead. J, histogram showing aggregate data from three independent experiments (each column is the average of >50 cells). ****, p < 0.0001. K, cytoplasmic Ca2+ signals in cells stimulated with LTC4 after knockdown of STIM1 or Orai1. A scrambled siRNA control is included. L, data from three independent experiments are compared. Cells were stimulated with 160 nm LTC4. **, p < 0.01; ****, p < 0.0001.
To extend the pharmacological results, we knocked down either STIM1 or Orai1 using siRNA-based approaches. Although both proteins were robustly expressed in wild-type RBL-2H3 cells, knockdown significantly reduced expression (STIM1, Fig. 1, G and H; Orai1, Fig. 1, I and J). Oscillations in [Ca2+]i evoked by LTC4 in the presence of external Ca2+ ran down rapidly after knockdown of either STIM1 or Orai1 (Fig. 1K), and the responses resembled those obtained when agonist was applied in the absence of external Ca2+ (Fig. 1L).
Oscillations in [Ca2+]i in Response to Leukotriene Receptor Stimulation Run Down in the Presence of Li+
Li+ is an effective inhibitor of inositol monophosphatases and, therefore, should lead to gradual depletion of PIP2 levels (18). Preincubation with 10 mm LiCl accelerated the rundown of oscillations in [Ca2+]i evoked by LTC4 in the presence of external Ca2+ (Fig. 2A). The effect of LiCl was concentration-dependent (Fig. 2B), and the dose-inhibition curve yielded an IC50 of 15 mm (Fig. 2C), close to the value reported for IMPase2 (19).
FIGURE 2.

Li+ blocks agonist-mediated oscillations in [Ca2+]i. A, cytoplasmic Ca2+ oscillations evoked by 160 nm LTC4 are compared between a control cell and one pre-exposed to 10 mm LiCl for ∼90 min. B, graph comparing cytoplasmic Ca2+ oscillations evoked by LTC4 between control cells and cells exposed to different concentrations of LiCl prior to stimulation. C, graph plotting the number of cytoplasmic Ca2+ oscillations evoked by LTC4 over 800 s of stimulation in the presence of different LiCl concentrations. The number of oscillations is normalized to the number of oscillations observed in control cells. Values were fitted with a Hill-type equation, yielding an IC50 value of 15 mm. D, thapsigargin-evoked Ca2+ release and Ca2+ influx are compared between control cells and cells pre-exposed to 50 mm LiCl for 90 min. Error bars (mean ± S.E.) are within the symbols. E, comparison of ionomycin-induced Ca2+ release in control cells and cells pretreated with 15 mm of LiCl for 90 min. F, data from three independent experiments as in E. ns, not significant. G, cytoplasmic Ca2+ oscillations in Ca2+-free external solution following stimulation with 160 nm LTC4 are compared between a control cell and one pretreated with 15 mm LiCl for 90 min. H, aggregate data from three experiments measuring the total number of cytoplasmic Ca2+ oscillations over an 800-s stimulation period with LTC4 are compared. Cells were stimulated either in the presence or absence of external Ca2+. The LiCl groups refer to pretreatment with 15 mm LiCl for 90 min. The data represent >52 cells from four independent experiments for each condition. ****, p < 0.0001. I, aggregate data showing the number of cytoplasmic Ca2+ oscillations evoked by LTC4 in 50 mm LiCl in an external solution containing either 155 or 105 mm NaCl, both with 2 mm Ca2+. Each bin number represents a period of 200 s. Each data point is >25 cells.
The extent of Ca2+ release from the stores evoked by stimulation with either thapsigargin (Fig. 2D) or ionomycin (Fig. 2, E and F) in Ca2+-free external solution was unaffected by pretreatment with even high concentrations (50 mm) of Li+. Store-operated Ca2+ entry, induced by readmission of external Ca2+ to cells treated with thapsigargin, was also unaffected by Li+ (Fig. 2D). Pretreatment with Li+ also had no inhibitory effect on the transient oscillatory cytoplasmic Ca2+ signals evoked by LTC4 when agonist was applied in Ca2+-free solution (Fig. 2G, aggregate data are summarized in Fig. 2H). Collectively, these results rule out an action of Li+ on store Ca2+ content, store-operated Ca2+ entry, and agonist-induced InsP3 production.
The time course of rundown of cytoplasmic Ca2+ oscillations by LiCl in our experiments is broadly similar to the decline in InsP3 levels reported in Chinese hamster ovary cells following muscarinic receptor stimulation (26, 27). In these latter studies, pretreatment with 5–10 mm LiCl for 30 min had no effect on the initial rise in InsP3 levels but suppressed the sustained increase that was manifested several minutes after stimulation. We observed no change in the number of oscillations in [Ca2+]i evoked by LTC4 in Ca2+-free solution (Fig. 2, G and H), in agreement with these earlier reports showing little effect of LiCl on the initial increase in InsP3 levels.
To test for an osmotic effect of LiCl on oscillatory Ca2+ signals, we reduced external Na+ from 155 to 105 mm and then added 50 mm LiCl. The oscillations in [Ca2+]i evoked by LTC4 still ran down quickly (Fig. 2I). Therefore, changes in osmolarity are unlikely to explain the accelerated rundown of cytoplasmic Ca2+ oscillations in the presence of Li+.
Inositol Rescues Oscillations in [Ca2+]i in Li+-treated Cells but Only in the Presence of Ca2+ Entry through CRAC Channels
In the blowfly salivary gland, a seminal study by Fain and Berridge (28) has demonstrated that application of inositol rescued serotonin receptor-induced Ca2+ flux that had subsided in the continuous presence of agonist. We therefore examined whether the presence of exogenous inositol could rescue the cytoplasmic Ca2+ oscillations in Li+-treated cells. Although oscillations in [Ca2+]i evoked by LTC4 ran down in the presence of LiCl (Fig. 3, A and B), pre-exposure to 10 mm inositol for a few minutes prior to stimulation rescued the oscillations in [Ca2+]i (Fig. 3A; aggregate data are shown in Fig. 3B and compared with the control response). Interestingly, addition of inositol to control cells prior to stimulation with LTC4 in the presence of external Ca2+ resulted in an increase in the frequency of oscillations in [Ca2+]i after agonist was added (Fig. 3, B and C).
FIGURE 3.

Inositol rescues cytoplasmic Ca2+ oscillations in LiCl-treated cells. A, oscillations in cytoplasmic [Ca2+]i evoked by LTC4 are sustained in a LiCl-treated cell when 10 mm inositol is added shortly before stimulation. B, aggregate data comparing the number of oscillations over an 800-s recording period for the conditions shown. Each column is the average of >30 cells from four independent experiments. **, p < 0.01; ****, p < 0.0001; ns, not significant. C, exposure to 10 mm inositol increases the oscillation frequency in a control cell. D and E, inositol fails to rescue oscillations in [Ca2+]i evoked by LTC4 in a LiCl-treated cell when CRAC channels are blocked with either La3+ (D) or BTP2 (E). F, aggregate data are compared for >50 cells/point. Each bin number reflects 200 s of recording. G, cytoplasmic Ca2+ oscillations are maintained over 800 s when a cell is challenged with LTC4 in Ca2+-free solution containing 1 mm La3+. H, Ca2+ oscillations run down in 0 Ca2+solution containing 1 mm La3+ following pretreatment with 15 mm LiCl, and this cannot be rescued by inositol. I, aggregate data are compared. Cells were stimulated with LTC4 in 0Ca2+ solution containing 1 mm La3+. ****, p < 0.0001.
Although inositol rescued oscillations in [Ca2+]i in Li+-treated cells, Ca2+ entry through CRAC channels was still required to sustain the oscillatory response. Exposure to the CRAC channel blocker La3+ accelerated the rundown of cytoplasmic Ca2+ oscillations in Li+-treated cells despite the presence of inositol (Fig. 3D; aggregate data are shown in Fig. 3F). Similar results were obtained when BTP2 was used to block the CRAC channels instead (Fig. 3, E and F).
The rundown of oscillations in [Ca2+]i in Li+-treated cells when CRAC channels are blocked, despite the presence of inositol, could reflect a role for Ca2+ entry in the synthesis of PIP2 from inositol. Alternatively, loss of the oscillatory response might simply arise from compromised store refilling. To distinguish between these possibilities, we took advantage of a protocol that enables store refilling to occur but in the absence of Ca2+ entry through CRAC channels. Cytoplasmic Ca2+ oscillations can be sustained in the absence of external Ca2+ when Ca2+ extrusion across the plasma membrane is inhibited, for example with high (millimolar) concentrations of La3+ (9, 10). Under these conditions, Ca2+ released from the stores cannot be exported out of the cell and so is taken back into the ER, thereby maintaining store Ca2+ content in readiness for the next oscillatory cycle in [Ca2+]i. Stimulation with LTC4 in Ca2+-free solution supplemented with 1 mm La3+ resulted in the generation of numerous oscillations in [Ca2+]i (Fig. 3G). Preincubation with LiCl led to rundown of the cytoplasmic Ca2+ oscillations (Fig. 3H). However, inositol no longer rescued the oscillatory response in Li+-treated cells (Fig. 3H; aggregate data are shown in Fig. 3I). These results, therefore, reveal a major role for Ca2+ influx through CRAC channels in replenishment of the PIP2 pool from inositol.
Inhibition of Phosphatidylinositol 4 Kinase Accelerates the Rundown of Agonist-driven Oscillations in [Ca2+]i
The preceding results suggest that Ca2+ influx through CRAC channels helps replenish PIP2 levels during cytoplasmic Ca2+ oscillations. This would mean that significant amounts of PIP2 are hydrolyzed following stimulation with LTC4 and that, therefore, phosphatidylinositol 4- and 5-kinases should be active during stimulation. A simple prediction is therefore that inhibition of these kinases should accelerate the rundown of the oscillations in [Ca2+]i because of the loss of PIP2 production. At relatively high concentrations, wortmannin inhibits phosphatidylinositol 4- kinase (29, 30) and, therefore, impairs PIP2 resynthesis. Compared with control responses, pretreatment with 10 μm wortmannin resulted in faster rundown of cytoplasmic Ca2+ oscillations evoked by LTC4 (compare the control response in Fig. 4A with that evoked in the presence of wortmannin in Fig. 4B). The number of Ca2+ oscillations that were triggered by LTC4 in the presence of wortmannin (Fig. 4C) was similar to the number seen in Ca2+-free solution (Fig. 1L). A previous study reported that wortmannin inhibited store-operated Ca2+ entry through inhibition of phosphatidylinositol 4-kinase and, thereby, depletion of PIP2 (31). In that report, a prominent inhibition of Ca2+ influx was seen when wortmannin pretreatment was combined with exposure to the agonist methacholine to ensure PIP2 depletion. Pretreatment with wortmannin (20 μm) but in the absence of methacholine had no inhibitory effect on Ca2+ entry (31). These data are consistent with our previous report showing that wortmannin alone had no effect on CRAC channel activity in RBL cells (32). Furthermore, loss of cytoplasmic Ca2+ oscillations evoked by LTC4 in the presence of wortmannin could be rescued by exogenous PI4P (Fig. 4, B and C), consistent with an action by wortmannin on phosphatidylinositol 4-kinase. Wortmannin also blocks phosphatidyl inositol 3-kinase. However, pretreatment with the PI 3-kinase inhibitor LY294002 did not mimic the inhibitory effect of wortmannin on oscillations evoked by LTC4 (Fig. 4C).
FIGURE 4.

Involvement of PI4P in supporting oscillations in [Ca2+]i evoked by LTC4. A, a typical control recording showing oscillations in [Ca2+]i evoked by LTC4 in the presence of external Ca2+. B, wortmannin (10 μm, 10-min pretreatment) accelerates rundown of the cytoplasmic Ca2+ oscillations. Rundown is less pronounced when PI4P (70 μm, 7-min pretreatment) is applied prior to stimulation. C, aggregate data from three independent experiments are compared. Each column represents data from >30 cells. ***, p < 0.001; ns, not significant. D, pretreatment with PI4P increases the cytoplasmic Ca2+ oscillation frequency. E, pretreatment with PI4P rescues oscillations in [Ca2+]i in a cell pre-exposed to LiCl for 105 min. F, cytoplasmic Ca2+ oscillations evoked by LTC4 are not rescued in LiCl-treated cells when the carrier for PI4P is used instead. G, PI4P fails to rescue oscillations in [Ca2+]i in Li+-treated cells in the presence of BTP2. H, PI4P has no effect on the rundown of cytoplasmic Ca2+ oscillations in Ca2+-free solution containing 1 mm La3+ in a LiCl-treated cell. I, aggregate data comparing the number of Ca2+ oscillations over an 800-s recording period for the different conditions are shown. La3+ in 0 mm Ca2+ denotes 0 Ca2+ supplemented with 1 mm La3+. ****, p < 0.0001.
Application of Phosphatidylinositol 4-Phosphate Rescues Oscillations in [Ca2+]i Evoked by LTC4 in Li+-treated Cells
The results described above with wortmannin suggest that PI4P production is important for sustaining oscillations in [Ca2+]i evoked by LTC4. To test this more directly, we applied purified PI4P to Li+-treated cells to see whether this could prevent rundown of the cytoplasmic Ca2+ oscillations. In control cells not treated with LiCl, exposure to PI4P led to a small increase in the number of oscillations in [Ca2+]i induced by LTC4 (Fig. 4D). Pre-exposure to PI4P prevented the rundown of the oscillations in [Ca2+]i in Li+-treated cells (Fig. 4E). By contrast, the histone carrier for PI4P failed to rescue the oscillations in [Ca2+]i in Li+-treated cells (Fig. 4F). Blocking of CRAC channels with BTP2 prevented the rescue of the cytoplasmic Ca2+ oscillations by PI4P in Li+-treated cells (Fig. 4G), demonstrating that Ca2+ influx was still important for maintaining the response despite the presence of PI4P. Aggregate data for the various conditions are summarized in Fig. 4I. Importantly, as was the case with inositol (Fig. 3H), PI4P failed to rescue cytoplasmic Ca2+ oscillations evoked by LTC4 when agonist was applied to Li+-treated cells in Ca2+-free external solution containing 1 mm La3+ (Fig. 4, H and I). Rescue of oscillations in [Ca2+]i in Li+-treated cells by PI4P therefore requires Ca2+ influx and places the Ca2+-dependent site downstream of PI4P production.
Knockdown of PIP5 Kinase I Abolishes Oscillations in [Ca2+]i Evoked by LTC4
PI4P is converted to PIP2 by type I PIP kinases, which consist of three isoforms: PIP5 kinase 1α, 1β, and 1γ (33, 34). RT-PCR revealed the presence of PIP5 kinase1α and 1γ in RBL-2H3 cells but not the β isoform (Fig. 5, A and B). The presence of these proteins was confirmed using immunocytochemistry (Fig. 5, C and D) and Western blots (Fig. 5, E and F). SiRNA against either PIP5 kinase 1α or PIP5 kinase 1γ resulted in an ∼60% reduction in protein expression (Fig. 5, C–F). Knockdown of PIP5 kinase 1α had no effect on the expression of PIP5 kinase 1γ and vice versa (Fig. 5G). Following knockdown of PIP5 kinase 1α, the typical oscillatory response in [Ca2+]i evoked by LTC4 in the presence of external Ca2+ was abolished (Fig. 5, H and J). Similar results were observed when PIP5 kinase 1γ was knocked down instead (Fig. 5, I and J), suggesting redundancy between these isoforms. Knockdown of either PIP5 kinase isoform had only a modest effect on responses in Ca2+-free solution (Fig. 5, K–M). Neither the store Ca2+ content, as measured by the response to thapsigargin in Ca2+-free solution, nor the rate of store-operated Ca2+ influx were affected by knockdown of either PIP5 kinase isoform (Fig. 5, N and O).
FIGURE 5.

PIP5K1 isoforms are involved in maintaining LTC4-driven cytoplasmic Ca2+ oscillations. A, RT-PCR comparing the expression of PIP5K1 isoforms in RBL-2H3 cells. B, PIP5KIβ mRNA is absent from RBL-2H3 cells despite genomic DNA (gDNA) being detectable. C and D, confocal microscopy images comparing the expression of PIPK1α and γ protein between control cells and those in which either PIP5K1α (C) or PIP5K1γ (D) had been knocked down (KD). The corresponding histograms summarize data from >40 cells in each group. ****, p < 0.0001. E, Western blot comparing the expression of PIP5K1α in control (Ctrl) cells and after siRNA-directed knockdown. *, p < 0.05. F, Western blot comparing the expression of PIP5K1γ in control cells and after siRNA-directed knockdown. *, p < 0.05. G, Western blot comparing the expression of PIP5K1α after knockdown of PIP5Kγ (top panel) and vice versa (bottom panel). H and I, cytoplasmic Ca2+ oscillations evoked by LTC4 run down quickly after knockdown of PIP5K1α (H) or PIP5K1γ (I). Scrambled siRNA controls are included. J, histogram comparing the total number of cytoplasmic Ca2+ oscillations evoked by LTC4 in 2 mm external Ca2+ over 800 s of stimulation from three independent experiments. ****, p < 0.0001. K and L, the effect of knockdown of PIP5K1α (K) or PIP5K1γ (L) on responses evoked by LTC4 in Ca2+-free solution. M, histogram comparing the average number of Ca2+ oscillations in Ca2+-free solution for the conditions shown. ***, p < 0.001. N, Ca2+ release and Ca2+ influx evoked by thapsigargin are compared following knockdown of PIP5K1α and PIP5K1γ or after transfection with scrambled siRNA. O, the rates of Ca2+ entry following stimulation with thapsigargin as in K are compared for the conditions shown. ns, not significant.
Oscillations in [Ca2+]i in Li+-treated cells challenged with LTC4 in the presence of external Ca2+ run down within a few minutes (Fig. 2, A and B) but can be rescued by PI4P (Fig. 4, E and I). To see whether this rescue required conversion of PI4P to PIP2, we knocked down either PIP5 kinase 1α or 1γ and examined whether this prevented PI4P from rescuing the cytoplasmic Ca2+ oscillations evoked by LTC4. In Li+-treated cells in which PIP5 kinase 1α or 1γ had been knocked down, oscillations in [Ca2+]i ran down within 5 min (Fig. 6, A–C). However, under these conditions, application of PI4P no longer rescued the cytoplasmic Ca2+ oscillations (Fig. 6, D–F).
FIGURE 6.

PI4P fails to rescue oscillations in [Ca2+]i evoked by LTC4 following knockdown of PIP5K1α or PIP5K1γ in LiCl-treated cells. A and B, typical transient oscillations in [Ca2+]i evoked by LTC4 following knockdown (KD) of either PIP5K1α (A) or PIP5K1γ (B). C, aggregate data are summarized. ns, not significant. D and E, PI4P (70 μm) fails to rescue the cytoplasmic Ca2+ oscillations evoked by LTC4 in either PIP5K1α- (D) or PIP5K1γ-deficient (E) cells pre-treated with LiCl. F, aggregate data for the conditions shown are summarized. Each column represents results from >19 cells. ****, p < 0.0001.
Prolonged Agonist-evoked Oscillations in [Ca2+]i Require Talin1
To identify a potential mechanism linking Ca2+ influx to PIP2 replenishment, we sought Ca2+-dependent proteins that regulate PIP5 kinase activity. One candidate is talin, an adaptor protein that links integrin to the actin cytoskeleton (35). There are two homologous talin isoforms, talin1 and talin2. The NH2-terminal globular head has a band4.1/ezrin/radixin/moesin-like domain that binds actin, PIP2, and PIP5 kinase (35, 36). Talins can be cleaved by the Ca2+-dependent protease calpain (37), providing a possible link between cytoplasmic Ca2+ and PIP5 kinase activity. Talin1 mRNA was expressed in RBL-2H3 cells, whereas talin2 was undetectable (Fig. 7A). Immunocytochemical studies confirmed the presence of talin1, and this was reduced by ∼50% following knockdown (Fig. 7B). Oscillations in [Ca2+]i evoked by LTC4 ran down considerably more quickly following talin1 knockdown (Fig. 7, C and D). Neither store-operated Ca2+ influx, measured by stimulating cells with thapsigargin (Fig. 7E), nor Ca2+ release in the absence of Ca2+ influx (Fig. 7F) were impaired following talin1 knockdown. Pre-exposure to either inositol (Fig. 7G) or PI4P (Fig. 7H) did not rescue cytoplasmic Ca2+ oscillations to LTC4 following knockdown of talin1. Aggregate data from several such experiments are summarized in Fig. 7I.
FIGURE 7.

Knockdown of talin1 accelerates the rundown of oscillations in [Ca2+]i evoked by LTC4. A, RT-PCR reveals the presence of talin1 but not talin2 in RBL-2H3 cells. B, immunocytochemical detection of talin1 in control cells and after knockdown (KD). The histogram summarizes data from between 40 and 52 cells for each condition. ****, p < 0.0001. C, cytoplasmic Ca2+ oscillations evoked by LTC4 run down more quickly after talin1 knockdown. D, aggregate data from several experiments are summarized. Each column represents data from between 24 and 32 cells. E, thapsigargin-evoked cytoplasmic Ca2+ signals are unaffected by talin1 knockdown. F, the total number of cytoplasmic Ca2+ oscillations evoked by LTC4 in Ca2+-free solution are similar in control cells and those in which talin1 had been knocked down. ns, not significant. G, oscillations in [Ca2+]i evoked by LTC4 in talin1-deficient cells are not rescued by addition of inositol. Control refers to a wild-type cell stimulated with LTC4 in the presence of inositol. H, PI4P fails to rescue cytoplasmic Ca2+ oscillations in cells following knockdown of talin1. I, aggregate data from several independent experiments are compared. ****, p < 0.0001 compared with the corresponding controls. LTC4 was used to stimulate the cells. KD, knockdown of talin1.
Independence of P2Y and cysLT1 Receptors
CysLT1 receptor-dependent Ca2+ signaling is affected by the presence of caveolin-1 and is disrupted by removal of cholesterol from the plasma membrane by methyl-β-cyclodextrin (MβCD), suggesting the leukotriene receptors signal within lipid raft domains (23).
In RBL-1 cells, stimulation of P2Y receptors with ATP generates InsP3, which then triggers Ca2+ release from the stores. P2Y-dependent responses are unaffected by MβCD, indicating that they are not located close to cysLT1 receptors (23). To test whether P2Y and cysLT1 receptors coupled to the same pool of PIP2, we designed experiments to see whether ATP was able to evoke InsP3-dependent Ca2+ release after responses to LTC4 had ceased. We first confirmed that previous findings made in RBL-1 cells also occurred in the RBL-2H3 cell line. Immunocytochemical studies showed expression of cysLT1 receptors in the cell periphery, and this was not altered by MβCD (Fig. 8A). Oscillations in [Ca2+]i evoked by LTC4 were abolished by MβCD (Fig. 8, B and C). However, ATP still elicited a large Ca2+ transient in the presence of MβCD (Fig. 8D).
FIGURE 8.

Non-overlap of PIP2 pools in RBL-2H3 cells. A, confocal microscopic images compare the distribution of FLAG-tagged CysLT1 receptors following stimulation with 160 nm LTC4 in control cells and in cells pretreated with 10 mm MβCD for 30 min. The fluorescence profiles from the line scans are shown in the corresponding graph. B, the typical oscillatory Ca2+ signal induced by LTC4 in a control cell is lost following treatment with MβCD. C, aggregate data analyzing the number of oscillations from three independent experiments (34 cells for each condition). ****, p < 0.0001. D, the Ca2+ response induced by ATP is unaffected by the presence of MβCD (10 mm, 30 min pretreatment). E, after cytoplasmic Ca2+ oscillations evoked by LTC4 had run down in a cell pretreated with 15 mm LiCl for 90 min, application of ATP elicited prominent Ca2+ release. F, ATP application evoked Ca2+ release after the oscillatory response to LTC4 had run down in the presence of 10 μm wortmannin and LiCl. G, aggregate data from three independent experiments (between 26 and 30 cells/column) are compared. When LiCl (15 mm) was present, it was preincubated for 90 min prior to challenge with LTC4. ns, not significant.
After cytoplasmic Ca2+ oscillations evoked by LTC4 had run down in Li+-treated cells, we applied ATP to see whether Ca2+ release could still be evoked. A robust Ca2+ transient occurred (Fig. 8, E and G), revealing that P2Y receptors were able to couple to phospholipase C under conditions where cysLT1 receptors could not.
To ensure that there was no PIP2 resynthesis during the interval between LTC4 exposure and ATP stimulation, we pretreated cells with both LiCl and wortmannin. After oscillations in [Ca2+]i following challenge with LTC4 had run down (Fig. 8F), subsequent exposure to ATP elicited robust Ca2+ release (Fig. 8, F and G).
Discussion
Our findings reveal a role for Ca2+ entry through CRAC channels in regulating PIP2 replenishment and, therefore, in supporting oscillations in [Ca2+]i following physiological levels of cysLT1 receptor stimulation. A schematic summarizing our main findings and the sites of intervention is shown in Fig. 9.
FIGURE 9.
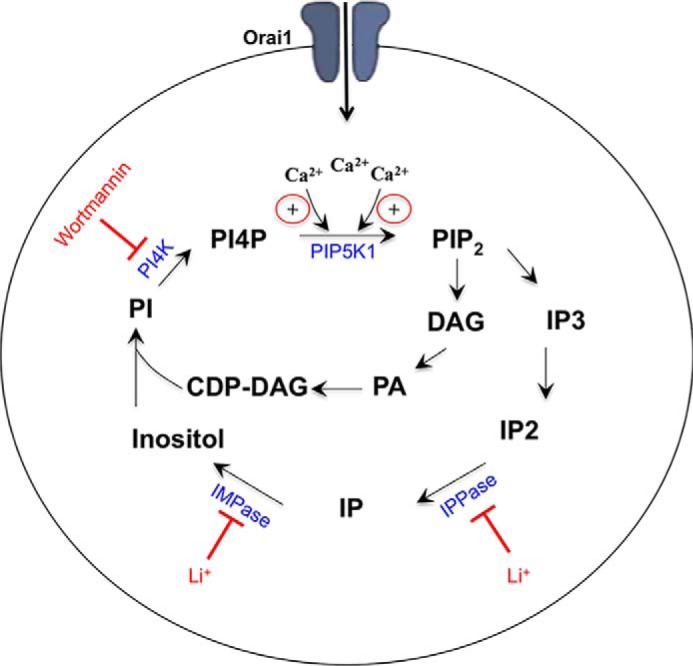
Schematic summarizing how calcium influx affects the PIP2 pathway. Both inositol polyphosphatase (IPPase) and IMPase are targets of Li+. CDP-DAG, cytidine diphosphate-diacylglycerol; DAG, diacylglycerol; PA, phosphatidic acid; IP, inositol phosphate.
Cytoplasmic Ca2+ oscillations evoked by LTC4 ran down in the presence of Li+, an inhibitor of inositol monophosphatases (18), demonstrating that PIP2 resynthesis was required for maintaining the Ca2+ response. Direct application of either inositol or phosphatidylinositol 4-phosphate was able to rescue the oscillatory Ca2+ response in the presence of Li+, consistent with an action of Li+ on inositol monophosphatase. However, recovery of the oscillations in [Ca2+]i evoked by LTC4 was prevented by block of CRAC channel activity. Our results suggest that Ca2+ influx is required to replenish the PIP2 pool that is targeted by cysLT1 receptors. Phosphatidylinositol 4-phosphate is phosphorylated to PIP2 by three isoforms of the type I PIP5 kinase (33) that are localized to various membrane compartments, including the plasma membrane, through a dilysine motif and another conserved lysine within the activation loop, a stretch of ∼20 amino acids in the C terminus (38). RBL-2H3 cells expressed both PIP5 kinase α and γ, and knockdown of either accelerated rundown of the cytoplasmic Ca2+ oscillations evoked by LTC4. The simplest explanation for our results is, therefore, that Ca2+ entry through CRAC channels stimulates PIP5 kinase α/γ to convert phosphatidylinositol 4-phosphate to PIP2 and, therefore, ensure adequate levels of the phospholipid for sustained cysLT1 receptor-dependent Ca2+ signaling. Type I PIP5 kinases are regulated by a variety of signals, including small G-proteins such as Rho, Rac, and Arf; phosphatidic acid; phosphorylation by tyrosine kinases; and the cytoskeletal protein talin 1 (33). Many of these pathways exhibit Ca2+ dependence, and it is therefore conceivable that Ca2+ entry modulates a regulatory pathway that serves to stimulate PIP5 kinase. Our data using an siRNA-based approach suggest a role for talin1, although how the protein is activated by Ca2+ influx and whether it is actively involved in supporting oscillations in [Ca2+]i or has more of a housekeeping role remains unclear.
Ca2+ influx might also stimulate PI transfer from the peripheral ER to the plasma membrane. Recent work has shown that extended synaptotagmins are activated by cytoplasmic Ca2+ and serve to bring the subplasmalemmal ER closer to the plasma membrane, where phosphatidylinositol transfer proteins, including Nir2, shuttle PI from the ER to the plasma membrane (39). Although such a mechanism might contribute to PIP2 replenishment under our conditions, our data suggest that the key step that is regulated by Ca2+ influx following cysLT1 receptor activation is the conversion of phosphatidylinositol 4-phosphate to PIP2 by PIP5 kinase, a reaction that takes place mainly in the plasma membrane.
The frequency of InsP3-dependent cytoplasmic Ca2+ oscillations can be altered by varying the rate or extent of Ca2+ entry (40). We now find that oscillation frequency evoked by LTC4 can also be increased by supply of exogenous inositol or PI4P. This suggests that PIP2 availability is rate-limiting. Providing more precursor accelerates its replenishment, and this increases the frequency of oscillations in [Ca2+]i.
Growing evidence points to the existence of local pools of PIP2 within the plasma membrane (41). Our functional studies are consistent with this view and suggest that these local pools exchange slowly. Oscillations in [Ca2+]i evoked by cysLT1 receptor activation ran down quickly in the presence of Li+ and wortmannin, which inhibit inositol monophosphatases and PI4P kinases, respectively. Nevertheless, subsequent stimulation of P2Y receptors, which also couple to phospholipase Cβ, elicited Ca2+ release that was similar in size and kinetics to responses obtained in the absence of the inhibitors. Therefore, P2Y receptors access a PIP2 pool that is distinct from that utilized by cysLT1 receptors.
In summary, our findings identify a role for Ca2+ entry through CRAC channels in maintaining a plasma membrane pool of PIP2 that is accessible to cysLT1 receptors. Ca2+ influx, therefore, has two important roles in controlling cytoplasmic Ca2+ oscillations: maintaining PIP2 levels and refilling the intracellular Ca2+ stores. The former will be important in initiating each oscillation in [Ca2+]i, whereas the latter will help set the interspike interval. Both will affect spike frequency but through distinct mechanisms. Stimulation of PIP2 production constitutes an interesting autoregulatory mechanism through which CRAC channels will be able to sustain their activity by ensuring that there are sufficient PIP2 levels in the plasma membrane to produce InsP3 and, therefore, store depletion, which is required for the opening of the channels. How Ca2+ influx activates PIP5 kinase and whether this is driven by local Ca2+ entry requires further investigation.
Author Contributions
A. A. and A. B. P. designed the study. A. A. performed and analyzed the experiments. A. B. P. wrote the manuscript. Both authors approved the final version of the manuscript.
This work was supported by MRC Program Grant L01047X (to A. B. P.). The authors declare that they have no conflict of interest with the contents of this article.
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- InsP3
- inositol trisphosphate
- ER
- endoplasmic reticulum
- IMPase
- inositol monophosphatase
- PI
- phosphatidylinositol
- PIP5
- phosphatidylinositol 4-phosphate 5
- PI4P
- phosphatidylinositol 4-phosphate
- LTC4
- leukotriene C4
- MβCD
- methyl-β-cyclodextrin
- CRAC
- Ca2+ release-activated Ca2+
- STIM
- stromal interaction molecule.
References
- 1.Berridge M. J., Bootman M. D., and Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 2.Clapham D. E. (2007) Calcium signaling. Cell 131, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 3.Parekh A. B. (2011) Decoding cytosolic Ca2+ oscillations. TiBS 36, 78–87 [DOI] [PubMed] [Google Scholar]
- 4.Berridge M. J. (1993) Inositol trisphosphate and calcium signalling. Nature 361, 315–325 [DOI] [PubMed] [Google Scholar]
- 5.Thomas A. P., Bird G. S., Hajnóczky G., Robb-Gaspers L. D., and Putney J. W. Jr. (1996) Spatial and temporal aspects of cellular calcium signalling. FASEB J. 10, 1505–1517 [PubMed] [Google Scholar]
- 6.Hajnóczky G., Robb-Gaspers L. D., Seitz M. B., and Thomas A. P. (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 [DOI] [PubMed] [Google Scholar]
- 7.Tse A., Tse F. W., Almers W., and Hille B. (1993) Rhythmic exocytosis stimulated by GnRH-induced calcium oscillations in rat gonadotropes. Science 260, 82–84 [DOI] [PubMed] [Google Scholar]
- 8.Dolmetsch R. E., Xu K., and Lewis R. S. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 [DOI] [PubMed] [Google Scholar]
- 9.Di Capite J., Ng S.-W., and Parekh A. B. (2009) Decoding of cytoplasmic Ca2+ oscillations through the spatial signature drives gene expression. Curr. Biol. 19, 853–858 [DOI] [PubMed] [Google Scholar]
- 10.Bird G. S., and Putney J. W. Jr. (2005) Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J. Physiol. 562, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoth M., and Penner R. (1992) Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356 [DOI] [PubMed] [Google Scholar]
- 12.Hogan P. G., Lewis R. S., and Rao A. (2010) Molecular basis of calcium signalling in lymphocytes:STIM and ORAI. Annu. Rev. Immunol. 28, 491–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soboloff J., Rothberg B. S., Madesh M., and Gill D. L. (2012) STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell. Biol. 13, 549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amcheslavsky A., Wood M. L., Yeromin A. V., Parker I., Freites J. A., Tobias D. J., and Cahalan M. D. (2015) Molecular biophysics of Orai store-operated Ca2+ channels. Biophys. J. 108, 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allbritton N. L., Meyer T., and Stryer L. (1992) Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science 258, 1812–1815 [DOI] [PubMed] [Google Scholar]
- 16.Wang S. S., Alousi A. A., and Thompson S. H. (1995) The lifetime of inositol 1,4,5-trisphosphate in single cells. J. Gen. Physiol. 105, 149–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshikawa T., Kikuchi M., Saito K., Watanabe A., Yamada K., Shibuya H., Nankai M., Kurumaji A., Hattori E., Ishiguro H., Shimizu H., Okubo Y., Toru M., and Detera-Wadleigh S. D. (2001) Evidence for association of the myo-inositol monophosphatase 2 (IMPA2) gene with schizophrenia in Japanese samples. Mol. Psychiatry 6, 202–210 [DOI] [PubMed] [Google Scholar]
- 18.Berridge M. J., Downes C. P., and Hanley M. R. (1982) Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem. J. 206, 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohnishi T., Ohba H., Seo K. C., Im J., Sato Y., Iwayama Y., Furuichi T., Chung S. K., and Yoshikawa T. (2007) Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J. Biol. Chem. 282, 637–646 [DOI] [PubMed] [Google Scholar]
- 20.Liou J., Fivaz M., Inoue T., and Meyer T. (2007) Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after calcium store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh C. M., Chvanov M., Haynes L. P., Petersen O. H., Tepikin A. V., and Burgoyne R. D. (2010) Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 425, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maléth J., Choi S., Muallem S., and Ahuja M. (2014) Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat. Commun. 5, 5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeh Y.-C., Tang M.-J., and Parekh A. B. (2014) Caveolin-1 alters the pattern of cytoplasmic Ca2+ oscillations and Ca2+-dependent gene expression by enhancing leukotriene receptor desensitization. J. Biol. Chem. 289, 17843–17853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng S. W., Bakowski D., Nelson C., Mehta R., Almeyda R., Bates G., and Parekh A. B. (2012) Cysteinyl leukotriene type I receptor desensitization sustains Ca2+-dependent gene expression. Nature 482, 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoth M., and Penner R. (1993) Calcium release-activated calcium current in rat mast cells. J. Physiol. 465, 359–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atack J. R., Prior A. M., Griffith D., and Ragan C. I. (1993) Characterization of the effects of lithium on phosphatidylinositol (PI) cycle activity in human muscarinic m1 receptor-transfected CHO cells. Br. J. Pharmacol. 110, 809–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkinson S., Nahorski S. R., and Challiss R. A. (1994) Disruption by Lithium of phosphatidylinositol-4–5-bisphosphate supply and inositol1,4,5-trisphosphate generation in Chinese hamster ovary cells expressing human recombinant m1 muscarinic receptors. Mol. Pharmacol. 46, 1138–1148 [PubMed] [Google Scholar]
- 28.Fain J. N., and Berridge M. J. (1979) Relationship between phosphatidylinositol synthesis and recovery of 5-hydroxytryptamine-responsive Ca2+ flux in blowfly salivary glands. Biochem. J. 180, 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willars G. B., Nahorski S. R., and Challiss R. A. (1998) Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J. Biol. Chem. 273, 5037–5046 [DOI] [PubMed] [Google Scholar]
- 30.Xie L. H., Horie M., and Takano M. (1999) Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U.S.A. 96, 15292–15297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broad L. M., Braun F.-J., Lievremont J.-P., Bird G. S., Kurosaki T., and Putney J. W. Jr. (2001) Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 276, 15945–15952 [DOI] [PubMed] [Google Scholar]
- 32.Straube S., and Parekh A. B. (2001) Effects of phosphatidylinositol kinase inhibitors on the activation of the store-operated calcium current ICRAC in RBL-1 cells. Pfluegers Archiv 442, 391–395 [DOI] [PubMed] [Google Scholar]
- 33.Doughman R. L., Firestone A. J., and Anderson R. A. (2003) phosphatidyl inositol phosphate kinases put PI4,5P2 in its place. J. Membr. Biol. 194, 77–89 [DOI] [PubMed] [Google Scholar]
- 34.Anderson R. A., Boronenkov I. V., Doughman S. D., Kunz J., and Loijens J. C. (1999) Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J. Biol. Chem. 274, 9907–9910 [DOI] [PubMed] [Google Scholar]
- 35.Di Paolo G., Pellegrini L., Letinic K., Cestra G., Zoncu R., Voronov S., Chang S., Guo J., Wenk M. R., and De Camilli P. (2002) Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 γ by the FERM domain of talin. Nature 420, 85–89 [DOI] [PubMed] [Google Scholar]
- 36.Ling K., Doughman R. L., Firestone A. J., Bunce M. W., and Anderson R. A. (2002) Type Iγ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 89–93 [DOI] [PubMed] [Google Scholar]
- 37.Bate N., Gingras A. R., Bachir A., Horwitz R., Ye F., Patel B., Goult B. T., and Critchley D. R. (2012) Talin contains a C-terminal Calpain 2 cleavage site important in focal adhesion dynamics. PLoS ONE 7, e34461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunz J., Wilson M. P., Kisseleva M., Hurley J. H., Majerus P. W., and Anderson R. A. (2000) The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol. Cell 5, 1–11 [DOI] [PubMed] [Google Scholar]
- 39.Chang C. L., Hsieh T. S., Yang T. T., Rothberg K. G., Azizoglu D. B., Volk E., Liao J. C., and Liou J. (2013) Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum-plasma membrane junctions. Cell Rep. 5, 813–825 [DOI] [PubMed] [Google Scholar]
- 40.Thorn P. (1995) Ca2+ influx during agonist and Ins(2,4,5)P3-evoked Ca2+ oscillations in HeLa epithelial cells. J. Physiol. 482, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balla T. (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]