

Background: NMDA receptor controls synaptic plasticity and memory function.
Results: Knockdown of Egr-1 blocks NMDAR-induced PSD-95 down-regulation and AMPA receptor endocytosis.
Conclusion: Egr-1 is a mediator of NMDA receptor signaling during the AMPA receptor trafficking.
Significance: Elucidation of the mechanism of AMPA receptor trafficking is critical to understand how our brain functions.
Keywords: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPA receptor, AMPAR); early growth response protein 1 (EGR1); endocytosis; neurobiology; transcription regulation; PSD95
Abstract
The N-methyl-d-aspartate receptor (NMDAR) controls synaptic plasticity and memory function and is one of the major inducers of transcription factor Egr-1 in the hippocampus. However, how Egr-1 mediates the NMDAR signal in neurons has remained unclear. Here, we show that the hippocampus of mice lacking Egr-1 displays electrophysiology properties and ultrastructure that are similar to mice overexpressing PSD-95, a major scaffolding protein of postsynaptic density involved in synapse formation, synaptic plasticity, and synaptic targeting of AMPA receptors (AMPARs), which mediate the vast majority of excitatory transmission in the CNS. We demonstrate that Egr-1 is a transcription repressor of the PSD-95 gene and is recruited to the PSD-95 promoter in response to NMDAR activation. Knockdown of Egr-1 in rat hippocampal primary neurons blocks NMDAR-induced PSD-95 down-regulation and AMPAR endocytosis. Likewise, overexpression of Egr-1 in rat hippocampal primary neurons causes reduction in PSD-95 protein level and promotes AMPAR endocytosis. Our data indicate that Egr-1 is involved in NMDAR-mediated PSD-95 down-regulation and AMPAR endocytosis, a process important in the expression of long term depression.
Introduction
Excitatory synapses are formed between presynaptic axon terminals and the postsynaptic surface of dendritic spines. In the presynaptic terminal, synaptic vesicles are concentrated at the active zone and, upon release, bind to specific target receptors on the postsynaptic membrane. This occurs in an electron dense region known as the postsynaptic density (PSD),2 located at the head of the spine (1). The PSD is composed of cytoskeletal proteins, cellular adhesion proteins, and signaling molecules, as well as membrane receptors (2). Synaptic plasticity is the biological process of learning and memory and occurs at the PSD of excitatory synapses (3). PSD is the primary postsynaptic site for signal transduction and processing and undergoes marked changes in shape in response to synaptic signals. Synaptic activity-dependent changes in PSD have been suggested to be a mechanism regulating synaptic plasticity (4, 5). Biochemical and structural changes in the PSD occur via activity-dependent gene expression, protein synthesis, protein degradation, and/or protein trafficking (3, 6).
Early growth response 1 (Egr-1), a zinc finger transcription factor (also known as krox-24 and Ziff-68) regulates genes that have diverse cellular functions, including cell proliferation, female reproduction, immune response, apoptosis, vascular function, cell growth, learning, and memory (7, 8). The basal level of Egr-1 in the brain is relatively low but is rapidly induced by growth factors, hormones, cytokines, neurotransmitters, brain injury, ischemia, and other stressors. Once induced, Egr-1 regulates the expression of target genes. Egr-1 is a Ca2+-dependent transcription factor. Elevation of intracellular Ca2+ is a key stimulator of Egr-1 expression (9). N-Methyl-d-aspartate (NMDA) receptors (NMDARs) are one of the main activators of Egr-1 in the CNS (10). NMDARs are ligand-gated Ca2+ channels and bind to glutamate, the major neurotransmitter of excitatory synapses. Activity of NMDAR and subsequent Ca2+ influx into the postsynaptic cells triggers a number of synaptic events leading to long lasting synaptic strength (3). Although NMDAR activation is well known to induce Egr-1 expression (10), the specific molecular mechanisms downstream of Egr-1 are poorly defined.
Recently, during our ongoing studies on Egr-1, we found that hippocampal slice cultures from Egr-1 knock-out (KO) mice display a higher frequency of miniature excitatory postsynaptic current (mEPSC) and larger PSD than the WT. Previous studies have shown that overexpression of PSD-95 enhances mEPSC frequency and enlargement of the PSD area in rat hippocampal and cortical slice cultures, respectively (11, 12). PSD-95 is one of the major scaffolding proteins of PSD (2) and an important regulator of synaptic strength and plasticity and is involved in synapse formation and stabilization (13, 14). Our observations suggested that the hippocampus of Egr-1 KO mice displays similar electrophysiology and PSD ultrastructure to those overexpressing PSD-95. This led us to examine whether Egr-1 plays any role in the regulation of PSD-95 levels at synapses. Furthermore, PSD-95 is involved in synaptic targeting of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) (15, 16), which are the mediators of a vast majority of excitatory synaptic transmissions in the CNS (17, 18). AMPARs undergo activity-dependent endocytosis followed by degradation or recycling (19–21). This dynamic trafficking of AMPARs plays a key role in the synaptic plasticity that is thought to underlie the aspects of learning, memory, and cognition (17, 18). We have therefore also examined the potential role of Egr-1 in NMDAR-dependent AMPAR endocytosis. Here, we show that in cultured hippocampal primary neurons, NMDAR activation causes induction and recruitment of Egr-1 to the PSD-95 promoter. We demonstrate that Egr-1 suppresses PSD-95 transcription and promotes AMPAR endocytosis in response to NMDAR activation. Our data indicate that Egr-1 is a transcription suppressor of PSD-95 and regulates NMDAR-dependent AMPAR trafficking.
Materials and Methods
Animals
All experiments were performed following the Guidelines of the Canadian Council of Animal Care and The Lady Davis Institute for Medical Research. Adult WT (Egr-1+/+) and KO (Egr-1−/−) mice in a C57BL/6 background were described previously (8, 22) and were genotyped by PCR. Mice were euthanized by cervical dislocation, and their hippocampi were immediately homogenized in the extraction buffer (0.1% SDS, 0.5% deoxycholic acid, 1% Triton X-100, 50 mm NaPO4, 150 mm NaCl, 2 mm EDTA, and protease inhibitor mixture). Each sample was centrifuged, and the supernatant was analyzed.
Isolation of Total RNA and Quantitative Real Time PCR (qPCR)
Total RNA was purified from brain extract or neurons by using RNeasy mini kit (Qiagen). Reverse transcriptase II-catalyzed first stand synthesis was performed using total RNA and oligo(dT) or random primer. SYBR Green-based (Qiagen) real time PCR was carried out with gene-specific primer for PSD-95, Egr-1, GAPDH, and actin (see list of primers in Table 1). Real time PCR software 7500 version 2.0.4 (Applied Biosystems) was used for data analysis. The relative RNA expression of the gene of interest was determined using comparative ΔΔCt method with GAPDH or actin as the internal control as described previously (23).
TABLE 1.
List of primers used for qPCR and ChIP assays
| qPCR in general | |
| Mouse Egr-1 forward | 5′CGAACAACCCTATGAGCACCTG3′ |
| Mouse Egr-1 reverse | 5′CAGAGGAAGACGATGAAGCAGC3′ |
| Mouse PSD-95 forward | 5′GACGCCAGCGACGAAGAG3′ |
| Mouse PSD-95 reverse | 5′CTCGACCCGCCGTTTG3′ |
| Mouse β-actin forward | 5′AGGTATCCTGACCCTGAAG3′ |
| Mouse β-actin reverse | 5′GCTCATTGTAGAAGGTGTGG3′ |
| Rat Egr-1 | Qiagen QT02423414 |
| at PSD-95 | Qiagen QT00183414 |
| Rat GAPDH | Qiagen QT00199633 |
| PSD-95 promoter constructs | |
| PSD-95 promoter −346 forward | 5′ATATACGCGTCGTCCCTCTTGTCACACACA3′ |
| PSD-95 promoter −286 forward | 5′ATATACGCGTCTGCATCTCCGAATCTCTCC3′ |
| PSD-95 promoter reverse | 5′CGCGAGATCTCCTATCCCATCCCCGTCGA3′ (for both − 346 and − 286) |
| PSD-95 promoter −mut forward | 5′TGTCACACACAGACCTTCCGAAAAAGAGCCTCAGCAGCTGCCGCCCCAAG3′ |
| PSD-95 promoter −mut reverse | 5′TTTTTCGGAAGGTCTGTGTGTCAAGGGACGGTGGGCAGCGG3′ |
| ChIP qPCR for COS-7 cells | |
| PSD-95 forward | 5′CGTCCCTCTTGTCACACACA3′ |
| PSD-95 reverse | 5′GCAGCTTGGAGATCCCTCTA3′ |
| ChIP qPCR for rat primary neurons | |
| PSD-95 forward | 5′CAAGATGGGTATGGGAACAC3′ |
| PSD-95 reverse | 5′AAGCCAGAGACCCTCTCCTC3′ |
Lentiviral Production, Neuronal Culture, and Viral Infection
Human Egr-1 with a Myc tag at C terminus was subcloned into pcDNA3 vector at EcoRI/XbaI sites using forward (5′-ATA TGA ATT CAT GGC CGC GGC CAA GGC CGA GAT GC-3′) and reverse (5′-CGC GTC TAG ACT CGA GTC AGC AAA TTT CAA TTG TCC TGG GAG-3′) primers. The pcDNA-Egr-1 and pcDNA3 vectors were subsequently cloned into lentiviral pLVX-IRWS-ZsGreen1 vector (Clontech) at the EcoRI and XbaI site by PCR and confirmed by DNA sequencing as described previously (22). Lentivirus was produced as per manufacturer's protocol. Briefly, the DNA was transfected along with a Lenti-X HTX packaging mix into the Lenti-X 293T cells. The virus was purified from the supernatant 48 and 72 h post-transfection following manufacturer's protocol. The titer of purified virus was determined by flow cytometry, and the virus was stored frozen at −80 °C. Hippocampal neurons were prepared from rat or mouse pups P0 (Charles River) as described previously (22, 23). Cells maintained in Neurobasal medium supplemented with B27 (Invitrogen), streptomycin, penicillin, glutamate, and glutamine were treated with cytosine arabinoside (5 μg/ml) to inhibit glia proliferation after 3 days of plating. After 2 weeks, cultures were infected with Ln-Egr-1 or Ln vector (multiplicity of infection of 20 each) (23). After 72 h, neurons were fixed for immunocytochemistry or homogenized in extraction buffer for Western blot analysis.
Electron Microscopy (EM)
Age-matched (6-month-old) KO and WT mice (n = 3 for each genotype), anesthetized with halothane followed by a lethal injection of euthasol, were perfused transcardially with 0.9% saline followed by 25 ml of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m sodium cacodylate buffer at 37 °C. The brains were removed and postfixed overnight at 4 °C. Blocks were treated with 1% osmium in potassium ferrocyanate and uranyl acetate, and thin sections (80 nm) were cut and placed on Formvar grids. Grids were treated with uranyl acetate and lead acetate. EM images were taken at ×13,000 and ×23,000 magnifications in stratum radiatum at a distance of 50–100 μm from stratum pyramidale. To clearly separate postsynaptic elements, photographs were enlarged to ×50,000. For quantification, five EM photographs from each KO and WT (n = 3 each) were taken, and 20 synapses from each animal were randomly selected and analyzed for length, width, and area of the PSD zone using Volocity analysis system (Improvision). Assessment of the size of synaptic elements was measured on digital micrographs across six serial sections taking measurement of every other section. The numbers were then averaged to ensure that measurements were not influenced by the two-dimensional position within the synapse. Synaptic vesicles were manually counted in high magnification images and were classified according to their location within the presynaptic bouton.
Immunocytochemistry, Image Acquisition, and Quantification
Immunocytochemistry was carried out as described previously (22, 23). Briefly, Ln-Egr-1 or Ln vector-infected neurons were fixed with 4% paraformaldehyde in PBS at room temperature for 30 min. Cultures were washed and permeabilized via incubation with PBS containing 0.1% Triton X-100 and 1% BSA. After 30 min of incubation at room temperature, neurons were blocked in PBS containing 10% normal goat serum for 1 h and then incubated overnight at 4 °C with one of the following antibodies in blocking buffer: PSD-95 (rabbit 1:200, Cell Signaling), GluA2 (mouse,1:200 Millipore), and GFP (mouse1:1000 Cell Signaling). Incubated cultures were washed and labeled via incubation with Alexa Fluor 532 or 488 goat anti-rabbit or goat anti-mouse secondary antibody for 1 h at room temperature. Labeled neurons were mounted in ProLong Gold anti-fade reagent (Invitrogen). Confocal images were obtained using LSM Pascal (Zeiss, Germany) confocal microscopy system. Light and camera settings were controlled using the AxioVision Version 4.6 (Zeiss, Germany) software, resulting in average background values of 63 ± 13 ms (mean ± standard deviation) for the red, green, and blue channels. Images were captured at 10, 20, 40, 60, or 100× oil objective lenses. At least 15 labeled neurons were randomly chosen from each group for quantification from three to four coverslips. Number of puncta and puncta intensity were analyzed along 30–40 μm of dendritic length of each neuron by Volocity image analysis software (Improvision).
Histology
Procedure for immunohistochemistry was described previously (22). Brains of KO and WT mice were removed and immediately immersed in Na2HPO4 and NaH2PO4 (81:19) for 24 h and then fixed in 20% followed by 10% sucrose for 24 h each. Fixed brains were sectioned at 40 mm with a vibratome and immunolabeled with rabbit anti-PSD-95 (1:50 Cell Signaling) followed by FITC-conjugated secondary antibody. Sections were developed using a diaminobenzidine histochemistry kit (Molecular Probes), counterstained with hematoxylin-eosin, and viewed under a microscope. Slides were scanned at ×400 magnification (resolution of 0.25 μm/pixel (100,000 pixels/inch)) using an Aperio ScanScope AT Turbo (Leica Biosystems). The background illumination levels were calibrated using a prescan procedure. Acquired digital images representing whole tissue sections were analyzed applying the Spectrum Analysis algorithm package and ImageScope analysis software (version 11.2, Aperio Technologies). The area of immunoreactivity in each section was averaged to generate a mean immunoreactive value for each animal.
Immunocytochemical Measurement for Surface and Endocytosed AMPARs
Endocytosis of AMPARs was monitored using the antibody feeding protocol as described previously (19, 24). Rat primary hippocampal cultures at 14 days in vitro were infected with Ln-Egr-1 or Ln vector. After 3 days of infection, surface GluA2 receptors were labeled with anti-mouse GluA2 antibody on living neurons (Millipore MAB397; 1:200) at 37 °C for 30 min and fixed in 4% paraformaldehyde for 20 min. Surface GluA2s were then labeled with Alexa Fluor 532-conjugated (red) secondary antibodies for 2 h (1:200). Cultures were next permeabilized for 1 min with 100% methanol, rinsed with PBS, and stained with Alexa Fluor 647-conjugated (blue) secondary antibody (1:200) for 1 h to label internalized GluA2s. Cultures were labeled finally with anti-rabbit GFP antibodies (1:1000) for 1 h and Alexa Fluor 488-conjugated (green) secondary antibodies (1:500) for 1 h to identify infected neurons.
To monitor endocytosis of AMPARs in NMDA-treated neurons, hippocampal neurons were prepared from mice P0 pups. Neurons were infected with Ln-shRNA-Egr-1 or Ln-shRNA control (Ln-shRNA-ctl) for 48 h. Surface GluA2 receptor was labeled with anti-mouse GluA2 antibody on living neurons (Millipore MAB397; 1:200) at 37 °C for 30 min. Neurons were treated with 20 μm of NMDA for 3 min to induce AMPAR endocytosis (24). After treatment, cultures were immediately rinsed with PBS, returned to the incubator, and kept for 27 min to allow endocytosis to occur. Cultures were then fixed in 4% paraformaldehyde for 20 min and then 0.1% Triton X-100 for 20 min. Surface and internalized GluA2s were labeled with Alexa Fluor 532-conjugated (red) and Alexa Fluor 647-conjugated (blue) secondary antibodies, as described above. Lentivirus-infected neurons were identified by immunolabeling with anti-rabbit GFP antibody (1:1000). Images were captured by using a laser confocal microscope. Data were acquired using 15 labeled neurons from each group from three separate coverslips. The number of puncta were counted along 30–40 μm of dendrite of each neuron using Volocity software.
Surface Receptor Labeling and Analysis
Surface receptor biotinylation experiments were performed using a surface receptor labeling kit (Pierce) following the manufacturer's instructions as described previously (19, 24). High density dissociated rat hippocampal neurons infected with lentivirus or treated with NMDA (20 μm for 3 min) were placed on ice to halt receptor trafficking. Medium was removed, and cells were washed twice with ice-cold PBS. Cultures were then incubated with Sulfo-NHS-LC-Biotin (250 μg/ml in PBS) for 30 min on ice. Reaction was stopped by adding quenching solution. Cultures were rinsed with TBS twice, lysed in 500 μl of Lysis buffer, and centrifuged at 10,000 × g for 2 min at 4 °C. The supernatant (500 μl) was mixed with 500 μl of NeutrAvidin-agarose beads previously equilibrated in TBS and incubated for 2 h at room temperature. Beads were recovered by centrifugation and washed three times, and bead-bound proteins were eluted in SDS-PAGE Sample Buffer and analyzed by Western blot analysis using anti-GluA2 and anti-NMDAR NR2A subunit antibodies. Surface expression of each receptor was quantified by normalizing the total amount of that receptor as described (19, 25).
Constructions of Plasmids for Luciferase Activity
A 346-bp PSD-95 promoter construct containing most of 5′UTR and the putative Egr-1-binding sequence (fragment 1), one 286-bp deletion construct with the putative Egr-1-binding site deleted (fragment 2), and one mutant construct, putative Egr-1-binding site mutated (fragment 3) were cloned by PCR using human genomic DNA as the template. The primers used for the plasmid constructs are listed in Table 1. Mutations were introduced into the plasmid by overlap extension PCR. Genetic constructs were assembled through site-specific promoter swapping approaches involving successive PCRs, as outlined previously (26). First, two DNA fragments with an overlap containing the mutant site were amplified by PCR. The promoter fragment containing the mutated Egr-1-binding site was obtained by mixing the two DNA fragments produced from the first PCR cycle and then using them as the template in the second PCR with the outermost primers containing MluI/BglII restriction sites. PCR was performed with an increased extension time of 5 min per cycle. All subsequent PCR products were subcloned into pGL3-Promoter Vector (Promega). Amplification was accomplished using the high fidelity Pwo DNA polymerase (Roche Diagnostics). All constructs were confirmed by DNA sequencing (McGill University and Genome Quebec Innovation Centre). For luciferase activity, COS-7 cells were co-transfected with 1 μg of pcDNA3-Egr-1 or pcDNA3 vector control, 20 ng of pGL3 plasmid expressing one of the constructs, and 10 ng of the pRL-CMV Renilla luciferase reporter plasmid (Promega) using Lipofectamine. Cells were harvested 24 h post-transfection, lysed, and assayed for luciferase activity (Promega kit). Firefly luciferase activity was normalized to Renilla luciferase activity.
Chromatin Immunoprecipitation (ChIP) Assay
Rat hippocampal primary neurons were treated with NMDA (20 μm) for 3 min, or COS-7 cells were treated with phorbol 12-myristate 13-acetate (PMA) (30 ng/ml) for 1 h. ChIP assay was performed on these cells using Magna ChIP T/MA/G kit (Millipore) following the manufacturer's instructions. Cells were cross-linked, and the DNA was fragmented to 200–1000 bp in length. Chromatin solution (100 μl each) was immunoprecipitated using 5 μg of either monoclonal anti-Egr-1 (Millipore) or IgG control. Input and immunoprecipitated samples were digested with proteinase K to reverse the cross-linking. Finally, the DNA was purified and analyzed by qPCR using primers against the PSD-95 promoter (Table 1).
Mouse Ln-shRNA-Egr-1 Construction
Four unique siRNA cassettes were obtained through GeneCopoeia (MSH032227). Each of these constructs, in a lentivirus GFP vector, expressed a 19-mer, either a non-effective scrambled control sequence (5′-CAA AGC CAA GCA AAC CAA T-3′) or one of the three Egr-1-targeting sequences, Sh-1 (5′-CGC TCT GAA TAA TGA GAA G-3′), Sh-2 (5′-ACT GGT CTT TCA GAC ATG A-3′), or Sh-3 (5′-AGC GCT AGA CCA TCA AGT T-3′) driven by histone H1 promoter. These shRNAs were validated by transfection into the mouse cell line, 3T3L1 followed by Western blot analysis for Egr-1 expression. Our Western blots showed that Sh-3 down-regulated Egr-1 expression by about ∼80% (see Fig. 8A). The lentiviruses expressing scramble (shRNA-control) and Sh-3 (shRNA-Egr-1) were produced as described above and were used to infect mouse hippocampal primary neurons to knock down Egr-1.
FIGURE 8.

Knockdown of Egr-1 inhibits NMDA-induced down-regulation of PSD-95 and surface expression of AMPARs in mouse hippocampal cultured neurons. Hippocampal primary neurons were infected with Ln-shRNA-Egr-1 or Ln-shRNA-ctl. Infected cultures were treated with NMDA or vehicle and analyzed by Western blotting for total proteins and biotinylation assay for surface receptors. Based on blot band intensities, various proteins were quantified. A, construct design and in vitro testing of lenti-shRNA constructs. Upper panel is a diagrammatic representation of mouse siEgr-1 targeting sequence locations. Lower panel shows confirmation by Western blot analysis of silencing of endogenous Egr-1 in mouse 3T3L1 cell line. Compared with control shRNA, all three shRNAs against Egr-1 down-regulate endogenous Egr-1. Sh-3 was most effective and hence was used to silence Egr-1 in hippocampal primary neurons. *, p < 0.05 (n = 3), and **, p < 0.01 (n = 3), with respect to Ln-shRNA-ctl-infected cells. B, knockdown of Egr-1 inhibits NMDA-induced down-regulation of PSD-95. Upper panel is a representative Western blot. Lower panels show quantification data. Values are from three different experiments and are expressed as the % of Ln-shRNA-ctl- and vehicle-treated neurons. *, p < 0.05 (n = 3) with respect to Ln-shRNA-ctl-infected and vehicle-treated neurons. **, p < 0.01 (n = 3) with respect to Ln-shRNA-ctl-infected and NMDA-treated neurons. C, knockdown of Egr-1 inhibits NMDA-induced surface expression of AMPAR GluA2. Neurons were analyzed for expression of total and surface GluA2 and NMDAR N2A by Western blotting and biotinylation assay, respectively. The upper panel shows representative Western blots. Lower panel represents quantifications from three different cultures. Quantification of each receptor was performed as described under “Materials and Methods.” *, p < 0.05 (n = 3) with respect to Ln-shRNA-ctl- and vehicle-treated neurons. **, p < 0.05 (n = 3) with respect to Ln-shRNA-ctl- and NMDA-treated neurons. D, overexpression of Egr-1 rescues shRNA-Egr-1-induced surface accumulation of GluA2. Neurons co-infected with Ln-shRNA-Egr-1 and Ln-Egr-1 were analyzed by biotinylation assay. *, p < 0.05 (n = 3) with respect to Ln vector control.
Hippocampal Slice Preparation and Electrophysiological Recording
Procedure for hippocampal slice preparation was described previously (27). WT or Egr-1 KO mice were anesthetized with isoflurane before being decapitated by a guillotine. The brain was quickly removed from the skull and immersed in ice-cold carbogenated (95% O2 and 5% CO2) sucrose-substituted hyperosmotic artificial cerebrospinal fluid (S-ACSF) containing the following (in mm): 252 sucrose, 2.5 KCl, 0.1 CaCl2, 4 MgCl2, 10 glucose, 26 NaHCO3, 1.25 NaH2PO4 (pH 7.35, 360–370 mosm). The brain was cut into 350-μm-thick coronal slices using a Vibratome (Leica, Concord, Ontario, Canada). Slices were immediately placed in carbogenated normal ACSF (125 mm NaCl instead of sucrose; 310–320 mosm) at 32 °C for 1 h and then maintained at room temperature for at least 30 min before electrophysiological recording.
The general procedures for electrophysiological recording were described previously (27). Briefly, patch pipettes were pulled from borosilicate glass capillaries and filled with intracellular solution (pH 7.25, 280–290 mosm) composed of the following (in mm): 110 cesium gluconate, 17.5 CsCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 4 ATP and 5 QX-314 (pH 7.2). Slices were transferred to a recording chamber perfused continuously with carbogenated ACSF. All recordings were done at room temperature. Action potential independent miniature synaptic activities were isolated by tetrodotoxin (0.5 μm), which blocks voltage-gated sodium channels. A GABAA receptor antagonist bicuculline (5 μm) was used to block inhibitory synaptic activities. The access resistance of the patch pipette was monitored throughout each experiment, and only recordings with stable and low access (<20 megohms) were analyzed. No electronic compensation for series resistance was used. Hippocampal CA1 neurons were voltage-clamped at −60 mV to isolate spontaneous miniature excitatory synaptic currents (mEPSCs). A Multi-Clamp 700B amplifier (Molecular Devices, Palo Alto, CA) was used for all recordings. Recordings were low pass filtered at 2 kHz, sampled at 10 kHz, digitized, and stored with a PC using Clampex 10.1 (Molecular Devices). Electrophysiological data were analyzed off-line using the Mini Analysis Program 6.0.3 (Synaptosoft, Decatur, GA) for synaptic events examination.
Statistics
All data were analyzed by one- or two-way analysis of variance followed by Bonferroni's post hoc test for multigroup comparisons and Student's t test for comparisons between two groups. All data are presented as means ± S.E.
Results
Frequency of mEPSC Is Higher in Egr-1 KO Mice Hippocampus
We recorded mEPSC in the hippocampal slices of Egr-1 WT and KO mice (Fig. 1). We found that the frequency of mEPSCs in Egr-1 KO mice is significantly higher than that of WT mice (0.76 ± 0.17 Hz (n = 6) versus 0.41 ± 0.08 Hz (n = 6)). No difference in mEPSC amplitude was found between these two groups (8.69 ± 1.15 pA in WT versus 9.54 ± 0.75 pA in KO). These findings suggest that the number of functional excitatory synaptic inputs or the probability of presynaptic glutamate release onto CA1 hippocampal neurons in KO mice is higher than that in WT mice.
FIGURE 1.
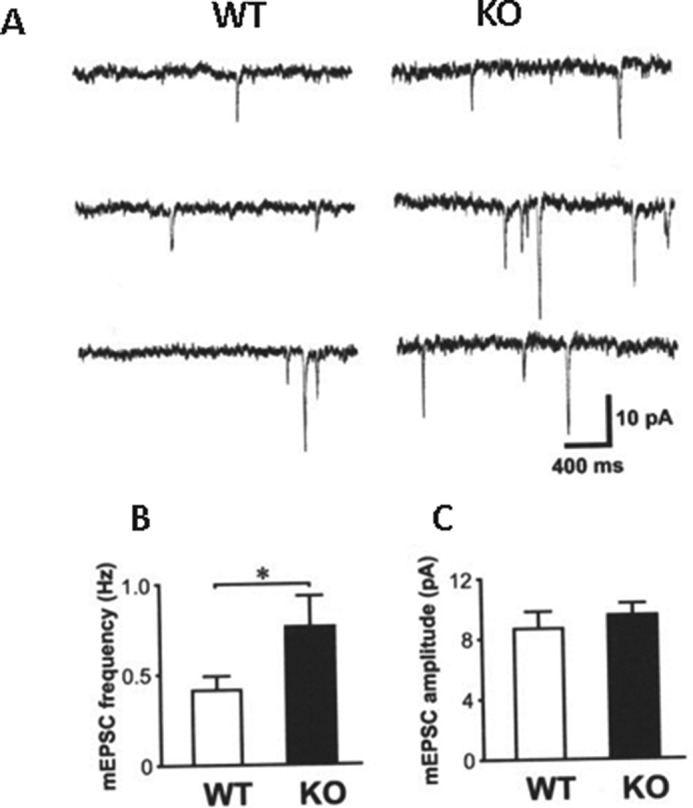
Enhanced mEPSC frequency in Egr-1 KO mice. A, sample traces recorded from WT (left panel) and Egr-1 KO mice (right panel). B, histogram shows higher mEPSC frequency in KO mice. Data are average from WT (n = 6) and KO (n = 6). *, p < 0.05. C, histogram shows no difference in mEPSC amplitude between WT and KO groups.
Distribution of Presynaptic Vesicles Is Altered in Egr-1 KO Mouse Hippocampus
The amplitude of the mEPSC corresponds to the glutamate sensitivity of the postsynaptic glutamate receptors. The frequency of mEPSC, however, reflects the probability of vesicle release from the presynaptic terminal and the number of release sites, which may reflect the number of synapses (28). Therefore, to determine the cause of higher mEPSC frequency in Egr-1 KO hippocampal slices, we examined single plane sections of stratum radiatum in CA1 of adult Egr-1 KO and WT mice by EM. Interestingly, there were 49.7% more synaptic vesicles per synaptic bouton of KO mice than in the WT (Fig. 2, A–C). Moreover, in the WT, the average number of readily releasable vesicles, docked to the active zone, was 4.2 ± 0.6 (Fig. 2, A, white arrowhead, and C). In KO mice, this number doubled to 8.5 ± 0.8 (Fig. 2, B, white arrowhead, and C).
FIGURE 2.
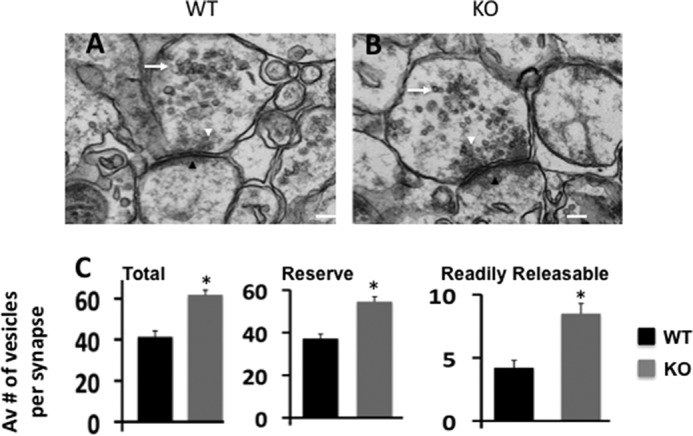
Organization of hippocampal presynaptic vesicle pool is altered in Egr-1 KO mice. A–C, representative EM images of stratum radiatum of CA1 hippocampus of WT (A) and Egr-1 KO (B) mice showing the organization of hippocampal presynaptic vesicle pools. The white arrowheads show presynaptic active zones where dense synaptic vesicles are shown. Black arrowheads show postsynaptic density zone. In KO mice, the number of presynaptic vesicles are higher and located closer to the active zone when compared with the WT. In addition, KO mice have more readily released vesicles docked to the active zone. C, quantification data are from WT and KO (three each). Twenty synapses from each animal were randomly selected and analyzed by Volocity software. Magnifications are ×23,000 (A and B) with corresponding scale bars of 100 nm each. *, p < 0.05 (n = 3) with respect to the WT.
Postsynaptic Organization Is Altered in Egr-1 KO Mice Hippocampi
When postsynaptic structure was analyzed, the PSDs of KO mouse hippocampus appeared more electron dense and bigger in size than the WT (Fig. 3, A and B, white arrows). Although the average lengths of PSD of the WT and KO were similar, the average width of the WT was 10.8 ± 0.8 nm compared with 15.0 ± 1.7 nm of the KO mice. As a consequence, the average PSD area of the KO mice was 1.5 times higher than that of the WT (Fig. 3C). Thus, Egr-1 KO mice display both structural and functional differences in the hippocampus when compared with the WT.
FIGURE 3.

Postsynaptic structure is altered in Egr-1 KO hippocampus. EM of WT (A) and KO (B) stratum radiatum of CA1 hippocampus shows the postsynaptic structures. White arrows show PSD, which is generally larger in the KO than in the WT. Scale bars, 500 nm each. C, quantification of synapses. Data represented are measurement from 20 randomly chosen synapses from each genotype. *, p < 0.01 (n = 3) with respect to the WT.
PSD-95 Protein and mRNA Levels Are Up-regulated in Egr-1 KO Mouse Hippocampus
Previously, PSD-95 overexpression in cortical and hippocampal slices was shown to increase the mEPSC frequency and PSD size, respectively (11, 12). Therefore, to determine the molecular basis of the altered synapse structure of the Egr-1 KO mouse, we analyzed levels of PSD-95 in the hippocampal extracts of KO and WT mice. As controls, we examined protein levels of presynaptic proteins, synaptophysin, and the intersynaptic adhesion protein, N-cadherin. The levels of synaptophysin and N-cadherin were similar (Fig. 4A), but the protein level of PSD-95 was significantly higher in mutant mice when compared with WT (Fig. 4B). When hippocampal sections were immunostained, both WT and KO mice showed homogeneous PSD-95 expression in the pyramidal neurons in the CA1-CA3 regions of hippocampal formation. However, compared with WT, KO mice displayed significantly higher PSD-95 immunostaining in all parts of hippocampal formation, including in the dentate gyrus and CA1 subfields (Fig. 4C, lower panel). Moreover, PSD-95 mRNA levels in the hippocampus were 1.55-fold higher in the KO compared with the WT (Fig. 4B, lower panel). These data indicate that Egr-1 regulates levels of PSD-95 mRNA and protein in the hippocampus.
FIGURE 4.

PSD-95 protein and mRNA levels are elevated in Egr-1 KO mouse hippocampi. Hippocampi of WT and KO mice (each 6 months old) were analyzed by Western blotting, qPCR, and immunohistochemistry. A, levels of synaptophysin and N-cadherin are not altered in Egr-1 KO mouse hippocampus. Hippocampal extracts of adult WT and Egr-1 KO mice (n = 3 in each group) were analyzed by quantitative Western blot analysis. Based on band intensities, relative amount of each protein was determined and compared. Upper panel is a representative Western blot. Lower panel indicates quantification data from three animals in each group. B, quantitative Western blot and qPCR analyses for PSD-95. Upper panel shows a representative Western blot. Middle panel is the result of Western blot quantification. Data presented are from three animals from each group. *, p < 0.05 (n = 3) with respect to WT. Lower panel shows quantitative RT-PCR analysis of PSD-95 mRNA (normalized to β-actin) in hippocampus of KO and WT genotypes (see Table 1 for primers used). The data are from three mice in each group. *, p < 0.04 with respect to WT. C, immunohistochemistry. Upper panel is a representative micrograph. Middle panels are corresponding insets at higher magnification. Immunohistochemical staining revealed widespread expression of PSD-95 in the hippocampal formation in both genotypes. Compared with WT, KO mice show higher PSD-95 immunostaining in CA1, CA2, and DG subfields. Scale bars, 100 μm in upper panel and 20 μm in middle panels. Lower panel shows analysis of PSD-95 immnoreactivity (optical density) in the CA1 and CA2 of hippocampus. Quantification of immunohistochemical staining was performed using the Spectrum Analysis algorithm package and ImageScope analysis software (version 11.2, Aperio Technologies). A rectangular region of interest, 25 μm2 in size, was defined. Immunoreactivity value for each region of interest was obtained. The immunoreactivity within each regions of interest in each section was averaged to generate a mean immunoreactive value for each animal. Data were averaged from three mice each with four regions (n = 12) for each genotype and are expressed as the fold of WT. ***, p < 0.001 (n = 3) with respect to WT.
Overexpression of Egr-1 Down-regulates PSD-95 in Rat Hippocampal Primary Neurons in Culture
To substantiate the brain data, we infected hippocampal primary neurons with Lenti-Myc-Egr-1 (Ln-Egr-1) and control Ln vector and analyzed them. In Ln vector-infected neurons, dense PSD-95 puncta in soma and dendrites were observed (Fig. 5A, red). In Ln-Egr-1-infected neurons, puncta intensity in both soma and dendrites was significantly decreased when compared with Ln vector-infected neurons (Fig. 5A, bar graph). In addition, the number of PSD-95 puncta was significantly less in the dendrites of Ln-Egr-1-infected than Ln vector neurons (Fig. 5A, bar graph). Western blotting confirmed the immunocytochemistry data and determined that the PSD-95 protein level was 45% less in Ln-Egr-1-infected neurons compared with Ln vector-infected controls (Fig. 5B). Likewise, PSD-95 mRNA levels were 52% less in Ln-Egr-1-infected neurons when compared with those infected with Ln vector (Fig. 5C). Thus, overexpression of Egr-1 in rat hippocampal primary neurons in culture down-regulated PSD-95 protein and mRNA levels.
FIGURE 5.

Overexpression of Egr-1 suppresses PSD-95 levels in rat hippocampal primary neurons. Rat hippocampal neurons in culture for 14 days were infected with Ln-Egr-1 of Ln vector. After 72 h, infected neurons were analyzed by immunocytochemistry, Western blotting, and qPCR. A, immunocytochemistry. Representative Confocal images showing GFP (green) and PSD-95 (red) immunostaining. Right-hand panel shows corresponding inset in higher magnification. Quantification of PSD-95 puncta (top right) was done from 15 randomly chosen infected neurons from three different cultures in each group along at least 30 μm of each neurite. *, p < 0.02 (n = 3) with respect to Ln vector-infected control. B, Western blot analysis of rat hippocampal extract using antibody against PSD-95, Myc-Egr-1 (anti-Myc), and tubulin (loading control). Based on the band intensities, the relative amounts of different proteins were calculated. Values are from three different cultures. *, p < 0.05 (n = 3) with respect to Ln vector-infected control. C, qPCR was performed to determine relative mRNA of PSD-95 and Egr-1 in infected neurons. Data are from three different cultures. *, p < 0.03 (n = 3), and ***, p < 0.0001 (n = 3), with respect to Ln vector-infected neurons.
Egr-1 Regulates PSD-95 Promoter Activity
We noted a putative Egr-1 binding region 286 bp upstream from the transcription initiation site of the PSD-95 promoter (Fig. 6A) (29, 30). To determine whether Egr-1 binds to this site, we cloned three corresponding PSD-95 promoter fragments upstream of the luciferase reporter vector (pGL3) (Fig. 6A) as follows: fragment 1, 346 bp long containing the putative Egr-1-binding site and most of the 5′ UTR sequence; fragment 2, with the putative Egr-1-binding site deleted; and fragment 3, with the putative Egr-1 site mutated. These constructs were individually co-transfected in COS-7 cells with Egr-1 or vector control, and luciferase activity in each cell extract was assayed.
FIGURE 6.

Egr-1 binds to PSD-95 promoter. Luciferase and ChiP assays were performed to evaluate the binding of Egr-1 onto the PSD-95 promoter. A, luciferase assay. Schematic diagram of the three constructs used to identify the Egr-1-binding site on PSD-95 promoter by luciferase activity assay. Putative Egr-1-binding site and mutations used to disrupt the binding sequence are shown. These constructs were co-transfected with luciferase reporter plasmid and Egr-1 or vector control in COS-7 cells, and luciferase activity was assayed and quantified. Bar graph in the lower panel represents results from three independent experiments. *, p < 0.05 (n = 3) with respect to vector and fragment 1-transfected cells. B, ChiP assay. ChiP assay was performed to confirm the Egr-1-binding site identified above. COS-7 cells were treated with PMA to induce Egr-1 expression for 1 h and then subjected to ChiP assay using anti-Egr-1 antibody or IgG control. Upper panel shows kinetics of Egr-1 induction in these cells. Lower panel shows the result of ChiP assay using primer specific for the above PSD-95 site. The values are the average of three independent determinations. ***, p < 0.001 (n = 3) with respect to IgG control.
In the cells co-transfected with Egr-1 and fragment 1, the luciferase activity was 54.3% of cells co-transfected with vector and fragment 1 (Fig. 6A, lower panel). However, when cells co-transfected with fragment 2 or 3 were assayed, the luciferase activity was similar in Egr-1 or vector control expressing cells. Thus, either deletion or mutation of the putative Egr-1-binding site abolished Egr-1-mediated inhibition of luciferase activity. These data determine that Egr-1 binds the putative site and inhibits PSD-95 promoter activity.
To substantiate the luciferase data, we exposed COS-7 cells to PMA to activate endogenous Egr-1 expression (31). PMA exposure increased Egr-1 level 2.5-fold in 1 h, peaked in 2 h, and then slowly declined over the next 24 h (Fig. 6B, upper panel). Using cells exposed for 1 h, we performed a ChIP assay using anti-Egr-1 antibody and primers flanking the Egr-1-binding site on the PSD-95 promoter (Table 1) that we identified by luciferase assay. We observed 3.4-fold higher amplification of promoter in anti-Egr-1 than in IgG control (Fig. 6B, lower panel). This result confirmed that Egr-1 binds to the site identified by luciferase assay on the PSD-95 promoter.
NMDAR Activation Recruits Egr-1 to PSD-95 Promoter in Hippocampal Primary Neurons
Egr-1 binds to the promoter of the target gene and couples extracellular signal to changes in cellular gene expression (9). NMDAR activation induces Egr-1 in neurons (10, 32). Therefore, we examined whether Egr-1 binds to the PSD-95 promoter in response to NMDAR activation by ChIP assay of NMDA-treated hippocampal primary neurons. In vehicle-treated neurons, we observed a 1.5-fold higher amplification of the PSD-95 promoter from anti-Egr-1 than from IgG (Fig. 7A). In NMDA-exposed neurons, this amplification rose to 2.7-fold (Fig. 7A). More importantly, compared with vehicle-treated neurons, NMDA-treated neurons displayed 5-fold higher amplification of the PSD-95 promoter (Fig. 7B). No significant difference was observed in the GAPDH promoter in the two groups. These data showed that when compared with vehicle-treated neurons, 5-fold more Egr-1 was complexed with the PSD-95 promoter in NMDA-treated counterpart. This, in turn, indicates that NMDA exposure recruits Egr-1 onto PSD-95 promoter.
FIGURE 7.
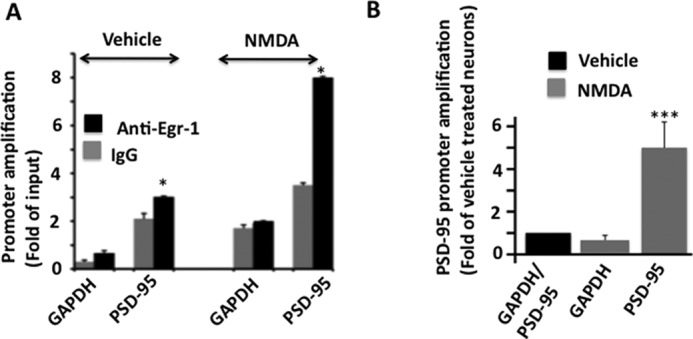
NMDA exposure recruits Egr-1 onto PSD-95 promoter in rat hippocampal cultured neurons. A, ChiP assay using antibody against Egr-1 was performed on neurons treated with NMDA or vehicle to monitor the recruitment of Egr-1 onto the PSD-95 promoter. Treated cultures were immunoprecipitated with anti-Egr-1 or IgG control. PSD-95 promoter was amplified by qPCR using primers against the Egr-1-binding site identified in Fig. 6. GAPDH DNA level was monitored as the internal control. Bar graphs were generated using qPCR data from three independent experiments. *, p < 0.05 (n = 3) with respect to corresponding IgG control. B, comparison of PSD-95 promoter amplification in vehicle- and NMDA-treated neurons. To compare between the two groups, qPCR values obtained from IgG from A were subtracted from the corresponding values from anti-Egr-1. Resulting values were used to generate the graph. Values are from three determinations and are presented as the fold of vehicle-treated controls. ***, p < 0.0001 (n = 3) with respect to vehicle-treated neurons.
Egr-1 Regulates NMDAR-dependent PSD-95 Levels
NMDAR activation down-regulates synaptic PSD-95 levels (24, 33, 34). Because NMDARs activate Egr-1 (10, 32), which subsequently binds to the PSD-95 promoter (Fig. 7), we investigated whether Egr-1 has any role in NMDAR-mediated PSD-95 down-regulation.
We used three siRNA targeting sequences from the open reading frame of the mouse Egr-1 expression cassette driven by the histone H1 promoter to drive shRNA expression, and we validated the efficiency of these three shRNA constructs in mouse T3L1 cells (Fig. 8A). We infected neurons with the most effective Ln-shRNA-Egr-1 (Sh-3) or Ln-shRNA-ctl. Infected neurons were treated with NMDA and analyzed (Fig. 8B). In Ln-shRNA-ctl-infected neurons, NMDA exposure enhanced Egr-1 levels by 1.8-fold and reduced PSD-95 levels by 44% (Fig. 8B, compare lanes 1 and 2) as reported (10, 24). In Ln-shRNA-Egr-1-infected neurons, Egr-1 levels were significantly reduced in both groups compared with Ln-shRNA-ctl-infected neurons demonstrating the effectiveness of the siRNA-Egr-1 (Fig. 8B, compare lane 1 or 2 with lane 3 or 4). Importantly, in Ln-shRNA-Egr-1-infected and NMDA-treated neurons, PSD-95 levels increased by 2.3-fold when compared with Ln-shRNA-ctl-infected and NMDA-treated neurons (Fig. 8B, compare lanes 2 and 4) and became similar to Ln-shRNA-ctl- or Ln-shRNA-Egr-1-infected neurons and were treated with vehicle (Fig. 8B, compare lane 4 with lane 1 or lane 3). Thus, knocking down endogenous Egr-1 significantly blocks NMDA-mediated PSD-95 down-regulation in neurons.
Knockdown of Egr-1 Blocks NMDAR-mediated Loss of AMPAR Surface Expression
NMDAR activation down-regulates PSD-95 and lowers the number of surface AMPARs in cultured rat hippocampal neurons (19, 24, 33). NMDAR activation also induces Egr-1 expression (10) and recruits Egr-1 to the PSD-95 promoter (Fig. 7A). We therefore examined whether Egr-1 has any role in NMDAR-mediated AMPAR surface expression.
We analyzed AMPAR subunit GluA2 surface expression in Ln-shRNA-Egr-1-infected and NMDA-treated neurons by biotinylation assay (Fig. 8C) (19, 24). In neurons infected with Ln-shRNA-ctl, NMDA treatment reduced surface expression of GluA2 by 45% without any significant effect on NMDAR subunit NR2A surface expression (Fig. 8C, lane 2) as expected (19, 24). In Ln-shRNA-Egr-1-infected neurons, we found no difference in the surface expression of GluA2 between NMDA- and vehicle-treated neurons (Fig. 8C, compare lanes 3 and 4). Thus, knocking down Egr-1 significantly blocks NMDA-mediated down-regulation of GluA2 surface expression.
To confirm the specificity of shRNA-Egr-1, we co-infected hippocampal primary neurons with Ln-shRNA-Egr-1 and Ln-Egr-1 and subjected them to biotinylation assay for surface GluA2 (Fig. 8D). Neurons infected with Ln-shRNA-Egr-1 displayed 1.5-fold more surface GluA2 than those infected with Ln-shRNA-ctl. In contrast, neurons co-infected with Ln-shRNA-Egr-1 and Ln-Egr-1 had surface GluA2 content similar to those observed in neurons infected with Ln-shRNA-ctl. Thus, shRNA-Egr-1 caused accumulation of GluA2 at the surface, and this effect was rescued by overexpression of Egr-1. These data indicated that shRNA-Egr-1 is specific for Egr-1.
Knockdown of Egr-1 Blocks NMDA-induced AMPAR Endocytosis
The density of AMPARs at postsynapse is regulated by lateral diffusion, endocytosis, and exocytosis (17, 18). We found that in mouse hippocampal neurons, NMDA-induced loss of surface GluA2 is significantly blocked by knockdown of Egr-1 (Fig. 8C). To determine the cause of this phenomenon, we monitored GluA2 endocytosis using the antibody feeding protocol that allows visual observation of AMPAR trafficking (19, 24).
In Ln-shRNA-ctl-infected neurons, GluA2 was found in both surface and internal pools (Fig. 9, A and B). When these neurons were treated with NMDA, the density of surface GluA2 was significantly reduced with concomitant increase in the internal pool (Fig. 9, A–C). In shRNA-Egr-1-infected and vehicle-treated neurons, relatively more GluA2s were on the surface than in the internal pool and remained on the surface when neurons were treated with NMDA (Fig. 9, A and B). Quantification data indicated that in the dendrites of the Ln-shRNA-ctl-infected and vehicle-treated neurons, distribution of GluA2 between surface and internal was 59.1 and 40.9%, respectively, of the total (Fig. 9C, upper left panel). When neurons were treated with NMDA, the surface and internal GluA2 ratio became 19.5 and 80.5%, respectively, of the total (Fig. 9C, lower left panel). Thus, compared with vehicle-treated neurons, NMDA-treated neurons had 39.9% less of the total surface receptor (39.9% more of the total internal surface receptor). As shown in Fig. 9C, upper left panel, there was 59.1% of the total GluA2 at the surface in vehicle-treated neurons. These data therefore indicated that out of 59.1%, 39.9% of the total (67.5%) surface GluA2 has moved from surface to internal pool upon NMDA exposure in Ln-shRNA control-infected neurons.
FIGURE 9.

Knockdown of Egr-1 blocks NMDA-induced AMPAR endocytosis in mouse hippocampal neurons in culture. Neurons infected with Ln-shRNA-ctl or Ln-shRNA-Egr-1 were treated with NMDA or vehicle and subjected to antibody feeding protocol to analyze GluA2 endocytosis by confocal microscopy. Images were captured, and GluA2 puncta were quantified. Based on puncta numbers, relative distributions and relative endocytosis values were determined. A, representative micrographs of neurons infected and treated as indicated showing GFP, surface GluA2, and internal GluA2. In Ln-shRNA-ctl-infected neurons, GluA2 is found on both surface and internal pool. When these neurons are treated with NMDA, the number of surface GluA2 is significantly reduced with a concomitant increase in the internal pool. In shRNA-Egr-1-infected and vehicle-treated neurons, more GluA2s are on the surface than in the internal pool; and they remain on the surface when neurons are treated with NMDA. Thus, knockdown of Egr-1 blocks NMDA-induced GluA2 endocytosis. B, representative micrographs of dendrites of infected neurons in higher magnifications. C, relative distribution. Fifteen GFP-positive (hence infected) neurons were randomly chosen from three different cultures from each group. Number of puncta along 30 μm of dendrite in each neuron was scored using Volocity software. The average number of puncta within 10 μm of the dendrite from three independent experiments were 3.9 ± 0.3 (surface) and 2.7 ± 0.5 (internal) in Ln-shRNA-ctl-infected and vehicle-treated neurons, 1.4 ± 0.25 (surface) and 5.8 ± 1.5 (internal) in Ln-shRNA-ctl- and NMDA-treated neurons, 4.5 ± 0.4 (surface) and 1.3 ± 0.2 (internal) in Ln-shRNA-Egr-1 infected and vehicle-treated neurons, and 3.8 ± 0.4 (surface) and 1.6 ± 0.3 (internal) in NMDA-treated neurons. Based on these numbers, relative distribution values were determined. The relative distribution value is the number of puncta in a fraction (surface or internal) of a group divided by the total (sum of the puncta in the two fractions (surface and internal)) of that group. Values are expresses as the % of the total. D, relative GluA2 endocytosis. To determine the relative amount of NMDA-induced GluA2 endocytosis, the % value of surface GluA2 from NMDA-treated neurons from panel C of a group was subtracted from the corresponding % value of surface GluA2 from vehicle-treated neurons of that group. Resulting value is then expressed as the % of the total GluA2 (% value of surface GluA2 in corresponding vehicle-treated neurons of that group). **, p < 0.005 (n = 3) with respect to Ln-shRNA-ctl-infected cells.
In Ln-shRNA-Egr-1-infected and vehicle-treated neurons, the ratio of surface and internal GluA2 was 77.6 and 22.4%, respectively, of the total (Fig. 9C, upper right panel). Upon treatment with NMDA, the ratio became 70.4 and 29.6%, respectively, of the total (Fig. 9C, lower right panel). There was 77.6% of the total GluA2 at the surface in vehicle-treated neurons in this group (Fig. 9C, upper right panel). These data therefore indicated that out of 77.6%, 7.2% of the total (9.2%) surface GluA2 was endocytosed upon NMDA exposure in Ln-shRNA-Egr-1-infected neurons.
As discussed above, NMDA exposure caused 67.5 and 9.2% of the total surface GluA2 endocytosis in Ln-shRNA-ctl- and Ln-shRNA-Egr-1-infected neurons, respectively. This means that NMDA exposure caused only 13.6% of total surface GluA2 endocytosis in Ln-shRNA-Egr-1-infected neurons when compared with Ln-shRNA-ctl-infected neurons (Fig. 9D). Thus, knockdown of Egr-1 blocked 86.4% of NMDA-induced GluA2 endocytosis in mouse hippocampal neurons.
Overexpression of Egr-1 Reduces Surface AMPARs in Rat Hippocampal Primary Neurons in Culture
NMDARs activate a number of signaling molecules in neurons (35). To ascertain the role of Egr-1 in NMDAR-induced down-regulation of PSD-95 and AMPAR endocytosis, we asked whether the effect of NMDAR activation could be mimicked by Egr-1 overexpression. For this, we infected rat hippocampal primary neurons with Ln-Egr-1 or Ln vector and analyzed them for PSD-95 levels and surface GluA2. Neurons overexpressing Egr-1 had a 55% reduction in PSD-95 levels but similar levels GluA2 when compared with those expressing vector (Fig. 10A). These data show that Egr-1 overexpression down-regulates PSD-95 protein levels but does not affect AMPAR content. When surface expression of GluR2 was analyzed, neurons overexpressing Egr-1 had 48.8% less GluA2 than those expressing vector (Fig. 10B). However, the level of surface NR2A remained unchanged in both groups. Thus, Egr-1 overexpression down-regulated PSD-95 levels and reduced expression of surface GluA2 in hippocampal primary neurons in culture.
FIGURE 10.

Overexpression of Egr-1 in rat hippocampal cultured neurons promotes AMPAR trafficking. A and B, overexpression of Egr-1 reduces levels of PSD-95 and surface GluA2. Rat hippocampal primary neurons in culture were infected with Ln-Egr-1 or Ln vector and analyzed by Western blot for proteins and biotinylation assay for surface receptors. A, Western blot. Proteins were separated and analyzed using indicated antibodies. Based on blot bands, the relative amounts of protein in each lane were quantified. Values are the average from three independent experiments. *, p < 0.05 (n = 3) with respect to Ln vector-infected neurons. B, biotinylation assay. Biotinylated samples were Western-blotted, and bands were quantified as in Fig. 7. Values are from three independent determinations from three different cultures. **, p < 0.005 (n = 3) with respect to Ln vector-infected cultures. C–F, overexpression of Egr-1 in rat hippocampal primary neurons promotes AMPAR endocytosis and reduced surface AMPAR levels. Neurons infected with Ln-Egr-1 or Ln vector were subjected to antibody feeding protocol for surface and internal GluA2 receptors. Images were captured by confocal microscope and quantified for surface and internal GluA2 as in Fig. 9C. Based on the numbers, relative distribution and relative endocytosis values were determined. C, representative micrographs showing infected neurons (GFP), surface GluA2, and internal GluA2. D, representative micrographs of dendrites of infected neurons under higher magnification. Fifteen GFP-positive neurons were randomly chosen from three different cultures, and the number of puncta along 30–40 μm of each dendrite was determined by Volocity software. The average number of surface and internal puncta within the 20 μm of dendrites from three independent experiments were 10.8 ± 3.3 and 4.5 ± 1.3, respectively, in Ln vector-infected neurons and 6.4 ± 2.2 and 10.3 ± 1.6, respectively, in Ln-Egr-1-infected neurons. Based on these numbers, relative distribution and relative endocytosis values were calculated. E, relative distribution. To calculate the relative distribution of GluA2 between the surface and internal portion of a group, the number of puncta in a fraction (surface or internal) was divided by the total (sum of the number of puncta in the two fractions of that group). The resulting value is expressed as the % of the total. F, relative endocytosis. The relative GluA2 endocytosis is the % value of internal GluA2 in each group from E and is expressed as the fold of Ln vector control. Values are from three determinations. **, p < 0.05 (n = 3) with respect to Ln vector-infected neurons.
Egr-1 Overexpression Promotes Endocytosis of AMPARs in Rat Primary Neurons in Culture
Overexpression of Egr-1 did not affect GluA2 protein levels but reduced its surface expression (Fig. 10, A and B). To substantiate the data and to determine the mechanism, we examined the effects of Egr-1 overexpression on the surface trafficking of GluA2 (Fig. 10, C–F). In both the Ln vector- and Ln-Egr-1-infected neurons (GFP-positive), surface GluA2 puncta and internal GluA2 were detected (Fig. 10, C and D). In Ln vector-infected neurons, 70.6% of total GluA2 was on the surface and 29.4% of the total internal (Fig. 10E). In the Ln-Egr-1-infected neurons, 38.3% of the total was detected on the surface and 61.7% was internal. Thus, compared with Ln vector-infected neurons, Ln-Egr-1-infected neurons display 2.6-fold more GluA2 in the internal pool (Fig. 10F). These data indicate that Egr-1 overexpression promotes AMPAR endocytosis in rat primary hippocampal neurons in culture.
Discussion
Previous studies have shown that NMDAR activation down-regulates synaptic PSD-95 levels in hippocampal primary neurons (24, 34). In this study, we found that protein and mRNA levels of PSD-95 are elevated in the Egr-1 KO mouse hippocampus (Fig. 4, B and C). Egr-1 overexpression in primary neurons in culture down-regulates PSD-95 protein and mRNA levels (Fig. 5, A–C). By luciferase assay, we identified the Egr-1-binding site on the PSD-95 promoter and demonstrated both in COS-7 cells and primary neurons in culture that the Egr-1 binds to the PSD-95 promoter (Figs. 6 and 7). Finally, our data showed that Egr-1 binds to the PSD-95 promoter in response to NMDAR activation and suppresses PSD-95 expression (Fig. 7). Our study for the first time indicates that Egr-1 is an in vivo transcriptional repressor of PSD-95 and is involved in NMDAR-induced regulation of PSD-95 levels in neurons.
NMDAR controls synaptic plasticity and memory function in the brain. In the CA1 region of the hippocampus, long term potentiation and long term depression, two forms of synaptic plasticity, are induced by activation of NMDARs and are expressed by changes in the number of postsynaptic AMPARs, which mediate the vast majority of fast excitatory synaptic transmission (17, 18). Long term potentiation involves recruitment of AMPARs to the synaptic membrane, whereas long term depression is associated with removal of synaptic AMPARs (17, 18). In hippocampal neurons, NMDAR activation reduces surface AMPARs by causing AMPAR endocytosis, and evidence suggests that this process plays a key role in the expression of long term depression (19, 21, 24). In this study, we demonstrated that knockdown of Egr-1 blocks NMDAR-induced AMPAR endocytosis (Fig. 9). Likewise, overexpression of Egr-1 promotes AMPAR endocytosis and reduces surface levels of AMPARs (Fig. 10). Our data indicate that Egr-1 is involved in NMDAR-mediated AMPAR endocytosis in neurons.
We do not know the mechanism by which Egr-1 regulated AMPAR endocytosis. However, previous studies have shown that AMPARs at the synapses are bound to PSD-95 via the transmembrane AMPAR regulator protein Stargazing (15, 16). Overexpression of PSD-95 in dissociated hippocampal neurons causes selective enhancement of AMPARs at the PSD (13, 36). Likewise, in hippocampal slice cultures, overexpression of PSD-95 causes an increase in AMPAR-mediated synaptic response without affecting NMDAR response (16, 37). Conversely, acute inactivation of PSD-95 destabilizes AMPARs at synapses (36, 38). Increasing the level of synaptic PSD-95 recruits new AMPARs to synapses (16). This study and a number of other studies have shown that the levels of PSD-95 correlate with synaptic AMPAR content (17, 39, 40). NMDAR activates Egr-1 (10), which subsequently represses transcription of PSD-95 gene and reduces the cellular PSD-95 level (this study). These observations together suggest that Egr-1 promotes AMPAR endocytosis by suppressing PSD-95 levels in neurons.
The amount of PSD-95 is similar but that of surface GluA2 is 2.1-fold more in neurons infected with Ln-shRNA-Egr-1 and treated with vehicle than the basal level observe in neurons infected with Ln-shRNA-ctl and treated with vehicle (Fig. 8C). The data suggest that Egr-1 also regulates surface trafficking of AMPARs independent of NMDARs. Phosphorylation of PSD-95 by Cdk5 reduces PSD-95 surface clustering in neurons (41). Egr-1 activates Cdk5 via inducing expression of p35 subunit (42). Egr-1 by promoting Cdk5-mediated PSD-95 phosphorylation may reduce surface clustering of PSD-95 and thus the number of surface AMPARs.
Degradation of PSD-95 occurs via the ubiquitin-dependent proteosomal pathway (24, 33, 43). Initially, ubiquitin-dependent proteosomal degradation was suggested as the means of PSD-95 reduction in neurons, in response to NMDAR activation (24). However, a subsequent study failed to detect PSD-95 ubiquitination in NMDA-treated hippocampal neurons (33). It was also reported that NMDAR activation promoted PSD-95 ubiquitination but not degradation (44). More studies will be required to clarify the role of ubiquitin-dependent proteosomal degradation of PSD-95 in NMDAR-induced AMPARs endocytosis.
Egr-1 KO mice hippocampi display higher frequency but not amplitude of mEPSC when compared with WT (Fig. 1). In addition, the number of readily releasable docked vesicles at the synaptic cleft is significantly higher in the KO than in the WT mice (Fig. 2). The frequency with which mEPSC occurs was suggested to reflect the probability of vesicle release from the presynaptic terminals and/or number of synapses (28). Because we observed a higher number of vesicles in synapses, our results suggest that the higher frequency of mEPSC may be due to increased probability of vesicle release in the hippocampus of KO mice.
During synaptic transmission, synaptic vesicles at presynaptic terminals release neurotransmitter into the synaptic cleft in response to Ca2+ signaling (45). Cdk5 and protein phosphatase calcineurin together were shown to control the distribution and size of the presynaptic vesicle pools in the CNS (46, 47). In addition, these two enzymes also regulate synaptic vesicle endocytosis (48, 49). Although inhibition of calcineurin reduces the size of the total pool, inhibition of Cdk5 activity causes the opposite effect (46, 47). In this study, we showed that the total number of vesicles are significantly more in the KO mouse than the WT (Fig. 2). Thus, Egr-1 KO mice appear to display organization of presynaptic vesicles similar to neurons treated with the Cdk5 inhibitor (46). Cdk5 is composed of a catalytic Cdk5 and activating p35 subunits (50). Egr-1 activates Cdk5 by inducing p35 transcription in the brain (42, 51). These observations together suggest that Egr-1 may also regulate synaptic transmission by controlling Cdk5 activity.
Excitatory synapses are dynamic structures and undergo activity-dependent rearrangement, stabilization, and elimination (52). PSD-95 is involved in the organization of PSD and synapse stabilization (13, 36, 43). PSD-95 levels in neurons are regulated by transcription, translation, and degradation (6, 24, 33, 43, 53). In addition, PSD-95 is trafficked in and out of the synapses, and this process is regulated by PSD-95 post-translational modifications, including phosphorylation (41, 52, 54–56). NMDAR activation plays a key role in activity-dependent formation of synaptic connectivity (3). Egr-1 is induced by NMDAR activation (10) and is a transcription suppresser of the PSD-95 gene (this study) and activator of Cdk5 (42, 51), which phosphorylates PSD-95 (41). These observations together suggest that Egr-1 may regulate activity-dependent remodeling and stabilization and perhaps elimination of synapses in the CNS.
Egr-1 KO mice hippocampi have elevated PSD-95 levels and larger PSD than that of the WT (Figs. 3 and 4). Overexpression of PSD-95 causes an increase in the size of PSD in neurons (12). It is therefore possible that enlarged PSD observed in Egr-1 KO mouse brain may be due to elevated levels of PSD-95. It will be interesting to test this hypothesis by knockdown of PSD-95 in Egr-1 KO mice. However, in addition to PSD-95, a number of other synaptic proteins are targeted to synapses and are assembled at PSD during synaptic activities (2, 52). Further studies will be required to determine how Egr-1 regulates PSD organization by regulating PSD-95 levels and phosphorylation in neurons.
A previous study reported that both Egr-1 and PSD-95 are among a number of genes that are induced during an acute electroconvulsive stimulation of rat brain (57). Although the induction of Egr-1 is rapid and transient, the induction of PSD-95 is gradual and occurs only after 4 h and peaks at 16 h post-electroconvulsive stimulation. This lack of correlation between the inductions of Egr-1 and PSD-95 suggests that Egr-1 may not be involved in PSD-95 expression during electroconvulsive stimulation.
Author Contributions
H. K. P. conceived the project, designed the experiments, supervised the study, analyzed the data, and wrote the manuscript. X. Q. designed and performed most of the experiments and assisted in data analysis. Y. J. performed the luciferase assay and ChiP assay involving COS-7 cells. Y. C. T. and T. P. W. performed the electrophysiology experiment. Y. W. designed the shRNA-Egr-1 construct and prepared the lentivirus.
This work was supported by grants from Canadian Institute for Health Research and Alzheimer's Society of Canada. The authors declare that they have no conflicts of interest with the contents of this article.
- PSD
- post-synaptic density
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor
- Egr-1
- early growth response 1
- NMDA
- N-methyl-d-aspartate
- NMDAR
- N-methyl-d-aspartate receptor
- ACSF
- artificial cerebrospinal fluid
- PMA
- phorbol 12-myristate 13-acetate
- mEPSC
- miniature excitatory postsynaptic current
- qPCR
- quantitative PCR.
References
- 1.Alvarez V. A., and Sabatini B. L. (2007) Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97 [DOI] [PubMed] [Google Scholar]
- 2.Chen X., Vinade L., Leapman R. D., Petersen J. D., Nakagawa T., Phillips T. M., Sheng M., and Reese T. S. (2005) Mass of the postsynaptic density and enumeration of three key molecules. Proc. Natl. Acad. Sci. U.S.A. 102, 11551–11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lüscher C., Nicoll R. A., Malenka R. C., and Muller D. (2000) Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat. Neurosci. 3, 545–550 [DOI] [PubMed] [Google Scholar]
- 4.Kennedy M. B. (2000) Signal-processing machines at the postsynaptic density. Science 290, 750–754 [DOI] [PubMed] [Google Scholar]
- 5.Sheng M., and Kim M. J. (2002) Postsynaptic signaling and plasticity mechanisms. Science 298, 776–780 [DOI] [PubMed] [Google Scholar]
- 6.Bao J., Lin H., Ouyang Y., Lei D., Osman A., Kim T. W., Mei L., Dai P., Ohlemiller K. K., and Ambron R. T. (2004) Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat. Neurosci. 7, 1250–1258 [DOI] [PubMed] [Google Scholar]
- 7.Knapska E., and Kaczmarek L. (2004) A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog. Neurobiol. 74, 183–211 [DOI] [PubMed] [Google Scholar]
- 8.Lee S. L., Sadovsky Y., Swirnoff A. H., Polish J. A., Goda P., Gavrilina G., and Milbrandt J. (1996) Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1). Science 273, 1219–1221 [DOI] [PubMed] [Google Scholar]
- 9.Thiel G., Mayer S. I., Müller I., Stefano L., and Rössler O. G. (2010) Egr-1-A Ca2+-regulated transcription factor. Cell Calcium 47, 397–403 [DOI] [PubMed] [Google Scholar]
- 10.Cole A. J., Saffen D. W., Baraban J. M., and Worley P. F. (1989) Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature 340, 474–476 [DOI] [PubMed] [Google Scholar]
- 11.Béïque J. C., and Andrade R. (2003) PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J. Physiol. 546, 859–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikonenko I., Boda B., Steen S., Knott G., Welker E., and Muller D. (2008) PSD-95 promotes synaptogenesis and multiinnervated spine formation through nitric oxide signaling. J. Cell Biol. 183, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Husseini A. E., Schnell E., Chetkovich D. M., Nicoll R. A., and Bredt D. S. (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368 [PubMed] [Google Scholar]
- 14.Han K., and Kim E. (2008) Synaptic adhesion molecules and PSD-95. Prog. Neurobiol. 84, 263–283 [DOI] [PubMed] [Google Scholar]
- 15.Chen L., Chetkovich D. M., Petralia R. S., Sweeney N. T., Kawasaki Y., Wenthold R. J., Bredt D. S., and Nicoll R. A. (2000) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408, 936–943 [DOI] [PubMed] [Google Scholar]
- 16.Schnell E., Sizemore M., Karimzadegan S., Chen L., Bredt D. S., and Nicoll R. A. (2002) Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci. U.S.A. 99, 13902–13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bredt D. S., and Nicoll R. A. (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40, 361–379 [DOI] [PubMed] [Google Scholar]
- 18.Henley J. M., and Wilkinson K. A. (2013) AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 15, 11–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehlers M. D. (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525 [DOI] [PubMed] [Google Scholar]
- 20.Lin J. W., Ju W., Foster K., Lee S. H., Ahmadian G., Wyszynski M., Wang Y. T., and Sheng M. (2000) Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci. 3, 1282–1290 [DOI] [PubMed] [Google Scholar]
- 21.Beattie E. C., Carroll R. C., Yu X., Morishita W., Yasuda H., von Zastrow M., and Malenka R. C. (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 22.Lu Y., Li T., Qureshi H. Y., Han D., and Paudel H. K. (2011) Early growth response 1 (Egr-1) regulates phosphorylation of microtubule-associated protein tau in mammalian brain. J. Biol. Chem. 286, 20569–20581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qureshi H. Y., Han D., MacDonald R., and Paudel H. K. (2013) Overexpression of 14-3-3z promotes tau phosphorylation at Ser262 and accelerates proteosomal degradation of synaptophysin in rat primary hippocampal neurons. PLoS ONE 8, e84615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colledge M., Snyder E. M., Crozier R. A., Soderling J. A., Jin Y., Langeberg L. K., Lu H., Bear M. F., and Scott J. D. (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40, 595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mammen A. L., Huganir R. L., and O'Brien R. J. (1997) Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J. Neurosci. 17, 7351–7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 27.Tse Y. C., Bagot R. C., Hutter J. A., Wong A. S., and Wong T. P. (2011) Modulation of synaptic plasticity by stress hormone associates with plastic alteration of synaptic NMDA receptor in the adult hippocampus. PLoS ONE 6, e27215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerchner G. A., and Nicoll R. A. (2008) Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 9, 813–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubosaki A., Tomaru Y., Tagami M., Arner E., Miura H., Suzuki T., Suzuki M., Suzuki H., and Hayashizaki Y. (2009) Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation. Genome Biol. 10, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stathakis D. G., Udar N., Sandgren O., Andreasson S., Bryant P. J., Small K., and Forsman-Semb K. (1999) Genomic organization of human DLG4, the gene encoding postsynaptic density 95. J. Neurochem. 73, 2250–2265 [DOI] [PubMed] [Google Scholar]
- 31.Beck H., Semisch M., Culmsee C., Plesnila N., and Hatzopoulos A. K. (2008) Egr-1 regulates expression of the glial scar component phosphacan in astrocytes after experimental stroke. Am. J. Pathol. 173, 77–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bading H., Segal M. M., Sucher N. J., Dudek H., Lipton S. A., and Greenberg M. E. (1995) N-Methyl-d-aspartate receptors are critical for mediating the effects of glutamate on intracellular calcium concentration and immediate early gene expression in cultured hippocampal neurons. Neuroscience 64, 653–664 [DOI] [PubMed] [Google Scholar]
- 33.Bingol B., and Schuman E. M. (2004) A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47, 755–763 [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharyya S., Biou V., Xu W., Schlüter O., and Malenka R. C. (2009) A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat. Neurosci. 12, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinheiro P. S., and Mulle C. (2008) Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat. Rev. Neurosci. 9, 423–436 [DOI] [PubMed] [Google Scholar]
- 36.Ehrlich I., Klein M., Rumpel S., and Malinow R. (2007) PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. U.S.A. 104, 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stein V., House D. R., Bredt D. S., and Nicoll R. A. (2003) Postsynaptic density-95 mimics and occludes hippocampal long term potentiation and enhances long term depression. J. Neurosci. 23, 5503–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yudowski G. A., Olsen O., Adesnik H., Marek K. W., and Bredt D. S. (2013) Acute inactivation of PSD-95 destabilizes AMPA receptors at hippocampal synapses. PLoS ONE 8, e53965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehrlich I., and Malinow R. (2004) Postsynaptic density 95 controls AMPA receptor incorporation during long term potentiation and experience-driven synaptic plasticity. J. Neurosci. 24, 916–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elias G. M., Funke L., Stein V., Grant S. G., Bredt D. S., and Nicoll R. A. (2006) Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron 52, 307–320 [DOI] [PubMed] [Google Scholar]
- 41.Morabito M. A., Sheng M., and Tsai L. H. (2004) Cyclin-dependent kinase 5 phosphorylates the N terminal domain of the postsynaptic density protein PSD-95 in neurons. J. Neurosci. 24, 865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harada T., Morooka T., Ogawa S., and Nishida E. (2001) ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 3, 453–459 [DOI] [PubMed] [Google Scholar]
- 43.Tsai N. P., Wilkerson J. R., Guo W., Maksimova M. A., DeMartino G. N., Cowan C. W., and Huber K. M. (2012) Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bianchetta M. J., Lam T. T., Jones S. N., and Morabito M. A. (2011) Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J. Neurosci. 31, 12029–12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizzoli S. O., and Betz W. J. (2005) Synaptic vesicle pools. Nat. Rev. Neurosci. 6, 57–69 [DOI] [PubMed] [Google Scholar]
- 46.Kim S. H., and Ryan T. A. (2010) CDK5 serves as a major control point in neurotransmitter release., Neuron 67, 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marra V., Burden J. J., Thorpe J. R., Smith I. T., Smith S. L., Häusser M., Branco T., and Staras K. (2012) A preferentially segregated recycling vesicle pool of limited size supports neurotransmission in native central synapses. Neuron 76, 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai M. M., Hong J. J., Ruggiero A. M., Burnett P. E., Slepnev V. I., De Camilli P., and Snyder S. H. (1999) The calcineurin-dynamin 1 complex as a calcium sensor for synaptic vesicle endocytosis. J. Biol. Chem. 274, 25963–25966 [DOI] [PubMed] [Google Scholar]
- 49.Tan T. C., Valova V. A., Malladi C. S., Graham M. E., Berven L. A., Jupp O. J., Hansra G., McClure S. J., Sarcevic B., Boadle R. A., Larsen M. R., Cousin M. A., and Robinson P. J. (2003) Cdk5 is essential for synaptic vesicle endocytosis. Nat. Cell Biol. 5, 701–710 [DOI] [PubMed] [Google Scholar]
- 50.Dhavan R., and Tsai L. H. (2001) A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2, 749–759 [DOI] [PubMed] [Google Scholar]
- 51.Li T., Chalifour L. E., and Paudel H. K. (2007) Phosphorylation of protein phosphatase 1 by cyclin-dependent protein kinase 5 during nerve growth factor-induced PC12 cell differentiation. J. Biol. Chem. 282, 6619–6628 [DOI] [PubMed] [Google Scholar]
- 52.Okabe S., Kim H. D., Miwa A., Kuriu T., and Okado H. (1999) Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat. Neurosci. 2, 804–811 [DOI] [PubMed] [Google Scholar]
- 53.Akama K. T., and McEwen B. S. (2003) Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J. Neurosci. 23, 2333–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steiner P., Higley M. J., Xu W., Czervionke B. L., Malenka R. C., and Sabatini B. L. (2008) Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 60, 788–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perez de Arce K., de Arce K. P., Varela-Nallar L., Farias O., Cifuentes A., Bull P., Couch B. A., Koleske A. J., Inestrosa N. C., and Alvarez A. R. (2010) Synaptic clustering of PSD-95 is regulated by c-Abl through tyrosine phosphorylation. J. Neurosci. 30, 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El-Husseini Ael-D., Schnell E., Dakoji S., Sweeney N., Zhou Q., Prange O., Gauthier-Campbell C., Aguilera-Moreno A., Nicoll R. A., and Bredt D. S. (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863 [DOI] [PubMed] [Google Scholar]
- 57.Dyrvig M., Christiansen S. H., Woldbye D. P., and Lichota J. (2014) Temporal gene expression profile after acute electroconvulsive stimulation in the rat. Gene 539, 8–14 [DOI] [PubMed] [Google Scholar]