

Background: MageB2 is a tumor-specific antigen with unknown function.
Results: Through a mechanism involving histone deacetylases, MageB2 enhances E2F activity and resistance to Actinomycin D.
Conclusion: MageB2 is a protein conferring proliferation properties and resistance to ribotoxic stress to tumor cells.
Significance: Expression of MageB2 in human tumors could be a marker of chemotherapy refraction.
Keywords: cancer biology, cell proliferation, E2F transcription factor, histone deacetylase 1 (HDAC1), molecular cell biology
Abstract
MageB2 belongs to the melanoma antigen gene (MAGE-I) family of tumor-specific antigens. Expression of this gene has been detected in human tumors of different origins. However, little is known about the protein function and how its expression affects tumor cell phenotypes. In this work, we found that human MageB2 protein promotes tumor cell proliferation in a p53-independent fashion, as observed both in cultured cells and growing tumors in mice. Gene expression analysis showed that MageB2 enhances the activity of E2F transcription factors. Mechanistically, the activation of E2Fs is related to the ability of MageB2 to interact with the E2F inhibitor HDAC1. Cellular distribution of MageB2 protein includes the nucleoli. Nevertheless, ribotoxic drugs rapidly promote its nucleolar exit. We show that MageB2 counteracts E2F inhibition by ribosomal proteins independently of Mdm2 expression. Importantly, MageB2 plays a critical role in impairing cell cycle arrest in response to Actinomycin D. The data presented here support a relevant function for human MageB2 in cancer cells both under cycling and stressed conditions, presenting a distinct functional feature with respect to other characterized MAGE-I proteins.
Introduction
Melanoma antigen gene B2 (MageB2) is a member of the cancer testis antigens, whose expression is restricted to normal testis but aberrantly expressed in a broad number of human tumors and silent in somatic cells (1). Type I MAGE (MAGE-I) proteins contain an ∼200-amino acid-long conserved domain (MAGE homology domain, MHD)6 that identifies the family. MAGE-I genes are grouped in three clusters of human chromosome X, named MAGE-A, MAGE-B, and MAGE-C. In normal cells, repression of MAGE-I gene transcription is mainly maintained by promoter methylation (2).
Since their discovery as tumor antigens (3), MAGE-A genes have been mainly studied as potential targets for cancer vaccines (4). In the last years, our and other groups have started studying the cellular function of different MAGE-A proteins, showing their associated pro-oncogenic functions mostly through regulation of specific transcription factors. For example, MageA1 regulates Skip activity (5), MageA11 expression enhances androgen receptor transactivation function (6–9), and MageA2 associates with and regulates p53 tumor suppressor activity (10, 11) and with Pml, causing an impairment in oncogene-induced senescence (12). Overall, emerging data strongly suggest that MAGE-A proteins counteract pro-apoptotic or cell cycle-arresting stimuli (13). Accordingly, MAGE-A expression has been frequently correlated to a poor prognosis in cancer patients (14–17).
With respect to MAGE-B, a vaccine raised against murine Mageb2 has been shown to prevent metastasis formation in a mouse model of breast cancer (18). The mouse mageb1, mageb2, and mageb3 genes encode for proteins that are 97–100% identical and, therefore, almost indistinguishable. siRNA-mediated silencing of those murine mage-b genes reduces cell viability in the P815 mast cell line (19) and in the S91 melanoma cell line (20). Moreover, murine mage-b silencing has been associated with p53-dependent apoptosis (20). More recently, it has been shown that murine Mageb18 silencing induces apoptosis and increases p53 protein levels (21).
Little is known about human MAGE-B proteins. Human MageB2 presents 50% amino acid identity compared with mouse Mage-b proteins. MageB2 gene expression starts as early as that of MAGE-A genes during carcinogenesis (22). Recently, a correlation between human MageB2 promoter demethylation and MageB2 gene expression in primary head and neck squamous cell carcinoma has been reported (23). In addition, MageB2 overexpression showed growth-promoting effect in a transformed oral keratinocyte cell line (23). Even though human MageB2 expression is found in a variety of human cancers, such as lung carcinoma (22), head and neck squamous cell carcinoma (23), multiple myeloma (24), and renal cell carcinoma (25), its function in tumor cells is mostly unknown.
We report here that human MageB2 expression regulates tumor cell proliferation independently of p53 status. We found that MageB2 is able to enhance E2F transcription factor activity through HDAC interaction. MageB2 protein is able to enter the nucleolus through an N-terminal nucleolar localization signal (NoLS), whereas the ribotoxic drug Actinomycin D stimulates its nucleolar exit. We show that MageB2 endogenous expression correlates with a reduced cytostatic effect under ribotoxic stress conditions.
Experimental Procedures
Cell Culture and Reagents
The U2OS, HCT116, H1299, HEK293T, and B16F0 cell lines were obtained from the ATCC and cultured as recommended. HCT116 p53−/− cells were provided by Bert Vogelstein. Mouse embryonic fibroblast double knockout p53−/− mdm2−/− cells were cultured as recommended. U24 cells are HA-MageB2-inducible U2OS cells using the ecdysone-inducible expression system (Invitrogen) and were maintained in medium with an additional 1 mg/ml G418 and 250 μg/ml Zeocin (Invitrogen). U24 cells were induced by adding 5 μm Ponasterone A (Invitrogen) for 24 h. TSA and Actinomycin D were purchased from Sigma-Aldrich.
Growth Curves
Cells were fixed in 10% formalin at the indicated times. Crystal violet (0.1%, Sigma-Aldrich) was used to stain cells. Cell-associated dye was extracted in 10% acetic acid, and the optical density was measured at 590 nm with each value normalized to day 0. Each point was done in duplicate.
Tumor Growth
1 × 105 B16F0-GFP or B16F0-GFP-MageB2 cells were resuspended in 0.2 ml of PBS and injected subcutaneously into the left flank of 8-week-old C57BL/6 mice (5 mice/group). Mice were visited daily. When the tumors reached measurable size, their length and width were measured every 2 days with a caliper, and the mean tumor volume was estimated using the formula (length + width)½. Mice whose tumors reached a volume of 2 cm3 were killed by cervical dislocation. The protocol was approved by the Institutional Animal Care and Utilization Committee of School of Exact and Natural Science, University of Buenos Aires.
Plasmids, siRNA, and Transfections
U2OS cells were transfected using TransIT-LT1 reagent (Mirus) according to the instructions of the manufacturer. HEK293T, mouse embryonic fibroblast double knockout p53−/− mdm2−/−, and B16F0 cells were transfected with PEI (Polysciences). Unless stated otherwise, cells were analyzed 24 h after transfection. HA-MageB2 was cloned in pCDNA3 (Invitrogen). GFP-MageB2 was obtained by subcloning in pLPC. GFP-MageB2 (1–109), GFP-MageB2ΔNoLS, GFP-mageb2, and GFP-mageb18 were cloned in pEGFP (Clontech). Myc-MageB2 was cloned in pCMV (Clontech). pCDNA3-HA-MageA2, FLAG-HDAC1, FLAG-HDAC3, and HA-HDAC4 have been described previously (10). MageB2 siRNA was a pool of two different siRNAs (#1 sequence, GCGAAGAUCCUCUAACCAGTT; #2 sequence, AACUCAGCUACUGAGGAAGAG) and were purchased from Eurofins Genomics. Control siRNA (siC) was the AllStars Negative control siRNA from Qiagen. Cells were transfected with siRNAs using Lipofectamine RNAiMAX reagent (Invitrogen) as recommended by the manufacturer and analyzed after 72 h.
Gene Reporter Assays
The 6×E2F-LUC reporter plasmid (pGL3 containing a TATA box and six E2F binding sites) (26) was used to test E2F activity. For luciferase normalization, the pRL-CMV reporter (Promega) constitutively expressing Renilla luciferase was cotransfected. After 24 h, cells were lysed, and luciferine and coelenterazine-h were used to assay firefly and Renilla luciferase activity, respectively.
Antibodies
Western blot analysis was performed according to standard procedures using the following primary antibodies: for MageB2 detection, affinity-purified anti-MageB2 raised against a GST-tagged N-terminal fragment corresponding to amino acids 30–116 (GST-MageB2(30–116)). Other primary antibodies were as follows: anti-p53 polyclonal (catalog no. FL-393) antibody, anti-E2F1 monoclonal (catalog no. KH95) antibody, anti-AR monoclonal (catalog no. 441) antibody, and anti-c-myc monoclonal (catalog no. C-33) antibody from Santa Cruz Biotechnology; anti-NCL monoclonal antibody from Zymed Laboratories Inc.; and anti-vinculin monoclonal antibody and anti-tubulin monoclonal antibody from Sigma-Aldrich. For tags we used the following: anti-HA 12CA5 monoclonal antibody (Roche), anti-HA (catalog no. Y-11) polyclonal antibody (Santa Cruz Biotechnology), anti-FLAG M2 monoclonal antibody (Sigma-Aldrich), and anti-myc tag (catalog no. 9B11) monoclonal antibody (Cell Signaling Technology). Anti-GFP was an affinity-purified polyclonal antibody raised against GST-GFP.
Immunofluorescence
Immunostaining was performed as described previously (12). Glass slides were analyzed using a laser scan confocal microscope (Zeiss) or an epifluorescence microscope (Leica). Images were obtained at ×63 magnification.
Immunoprecipitation
The immunoprecipitation assay was performed as described previously (12).
Quantitative RT-PCR
Total RNA was extracted with Qiazol reagent (Qiagen), and cDNA was transcribed with a QuantiTect reverse transcription kit (Qiagen) according to the instructions of the manufacturer. Real-time PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems) and a StepOnePlus real time PCR machine (Applied Biosystems). Primer sequences were as follows: MageB2, 5′-CCTGACTTCCGCTTTGGAGGCG-3′ (forward) and 5′-ATCTCGGGCCTTGCGGCGTT-3′ (reverse); MCM6, 5′-ATCCCTCTTGCCAAGGATTT-3′ (forward) and 5′-GAAAAGTTCCGCTCACAAGC-3′ (reverse); CycD1, 5′-ACGGCCGAGAAGCTGTGCATC-3′ (forward) and 5′-CCTCCGCCTCTGGCATTTTGGAG-3′ (reverse); CDK1, 5′-CATGGCTACCACTTGACCTGT-3′ (forward) and 5′-AAGCCGGGATCTACCATACC-3′ (reverse); CycE1, 5′-TGAGCCGAGCGGTAGCTGGT-3′ (forward) and 5′-GGGCTGGGGCTGCTGCTTAG-3′ (reverse); MCM7, 5′-CACGGAGTCTCTCAGCACAG-3′ (forward) and 5′-AACATCTGTCTGATGGGGGA-3′ (reverse); and B-actin, 5′-CCAACCGCGAGAAGATGA-3′ (forward) and 5′-CCAGAGGCGTACAGGGATAG-3′ (reverse).
Cellular Fractionation
HCT116 cells were fractionated using a hypotonic buffer and a series of centrifugations over sucrose cushions.
BrdU Incorporation Assay
Cells were plated in a 96-well plate at a density of 1.2 × 105 cells/well in triplicate. 24 h later, cells were treated with 10 nm Act-D for 16 h or left untreated. Cells were subsequently pulsed with 30 μm BrdU (Sigma-Aldrich) for 1 h, fixed with 3% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS, and RNA-denatured with 50 mm NaOH for 20 s. BrdU incorporation was measured by immunofluorescence using an anti-BrdU monoclonal antibody (GE Healthcare Biosciences). and the nuclei were stained with Hoechst. Image acquisition and analysis were performed using an ImageXpress Micro automated high-content screening fluorescence microscope (Molecular Devices). An average of 4.5 × 103 cells for each point were scored for BrdU incorporation in at least three independent experiments.
Results
p53-independent Effect of MageB2 on Cell Proliferation
We started studying the requirement of p53 on cell proliferation induced by MageB2 expression because p53 function has been associated previously with human MageA2 (10) and murine Mageb (20) in apoptosis resistance. To this aim, we knocked down MageB2 expression in the human colorectal cancer cell line HCT116, both in the WT p53 and p53 knockout versions. By following cell proliferation for 7 days, we observed that MageB2 KD similarly affects the proliferation rate in both cell lines (Fig. 1, A and B). Also, MageB2 KD reduced the number of colonies in both HCT116 cell lines independent of p53 status (Fig. 1C). Consistently, MageB2 KD also correlated with reduced tumor cell proliferation in U2OS cells (WT p53) (Fig. 1, D and E). The efficiency of siRNA-mediated KD of MageB2 expression is shown in Fig. 1F. In addition, we observed that, opposite to MageA2 (10), MageB2 failed to form a protein complex with p53 even when it was overexpressed (Fig. 1G). All of these data confirm that MageB2 of human origin regulates tumor cell proliferation and demonstrate that its effect is independent of p53 status.
FIGURE 1.
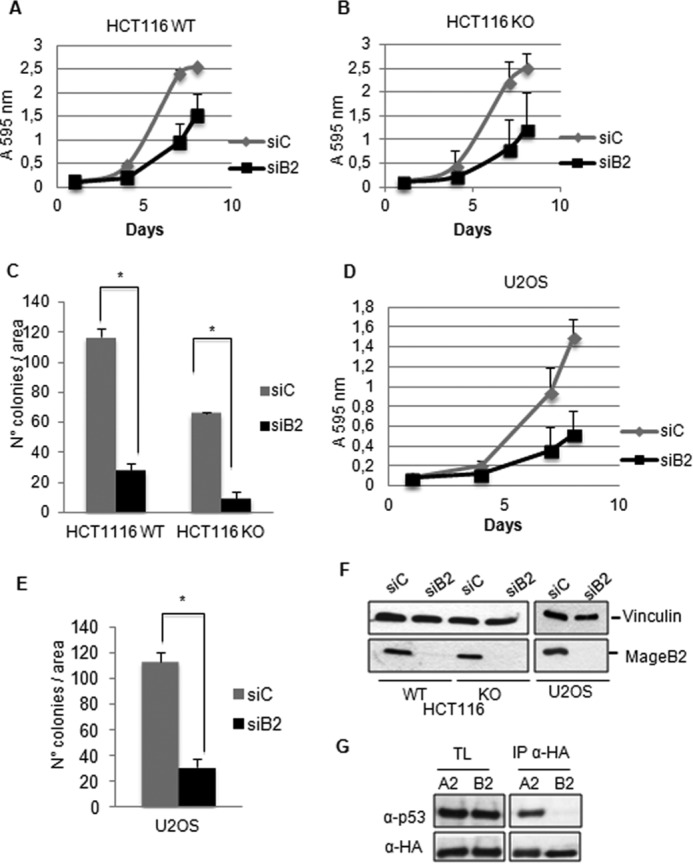
The p53-independent effect of MageB2 expression on tumor cell proliferation. A, determination of cell proliferation in HCT116 cells (HCT116 WT) during 7 days after siRNA transfection for MageB2 KD (siB2) or a control siRNA (siC). B, the same protocol as in A but using HCT116 p53−/− (HCT116 KO) cells. C, quantification of the colony formation assay in both HCT116 WT and KO cells after siRNA transfection for MageB2 KD or a control siRNA. *, p < 0.001. D, determination of cell proliferation in U2OS cells during 7 days after siRNA transfection for MageB2 KD or a control siRNA. E, quantification of the colony formation assay in U2OS cells after siRNA transfection for MageB2 KD or a control siRNA. *, p < 0.001. F, Western blot analysis of siRNA-mediated KD efficiency in HCT116 WT, KO, and U2OS cells 48 h after siRNA transfection for MageB2 or a control siRNA. G, immunoprecipitation of HA-tagged MageA2 and MageB2 coexpressed with p53 in H1299, p53-null cells. After transfection, immunoprecipitated material was analyzed by Western blot using anti-HA and anti-p53 antibody. Error bars indicate mean ± S.D. Student's t test was used for statistical analysis. TL, total lysates.
To corroborate the potential role of human MageB2 in stimulating tumor growth, we generated B16(F0) mouse melanoma cells lines stably expressing GFP or GFP-MageB2 (B16-GFP and B16-GFP-B2). Tumors were generated from B16-GFP and B16-GFP-B2 cells through subcutaneous injection in C57BL/6 mice. Tumor growth was examined regularly until the mass was measurable. From then on, we started to follow each mouse independently. As expected, B16-GFP-B2 tumors showed an increased growth rate compared with B16-GFP tumors (Fig. 2, A and B), indicating that the growth-promoting activity of human MageB2 could also be observed in vivo. After animal sacrifice, GFP and GFP-MageB2 expression was confirmed in random tumor samples (Fig. 2C).
FIGURE 2.
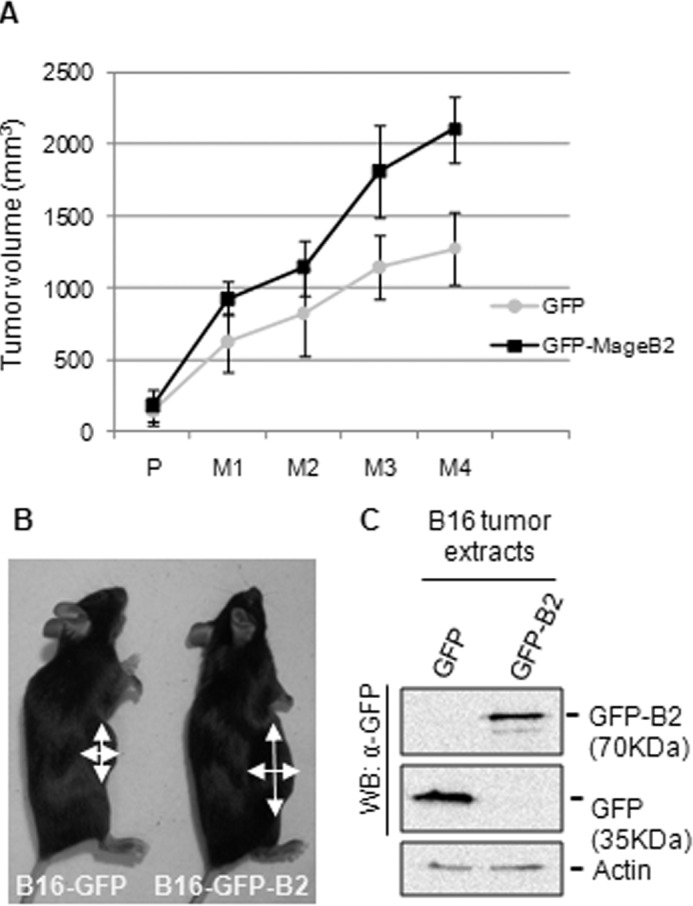
Overexpression of human MageB2 enhances tumor growth. A, determination of in vivo tumor growth after subcutaneous inoculation of melanoma B16(F0) cells stably expressing GFP (GFP) or GFP-tagged human MageB2 (GFP-MageB2). When the tumors were palpable (P), they were measured regularly (M) every 2 days for 8 days. Each group consisted of five C57/B mice. The experiment is representative of three experiments. B, representative mice belonging to the melanoma B16(F0) cells stably expressing GFP (B16-GFP) and GFP-tagged human MageB2 (B16-GFP-B2) groups. Arrows indicate the two measurements taken from mice to determine the tumor volume in A. C, Western blot (WB) analysis of B16-F0 tumor extracts expressing GFP or GFP-tagged human MageB2. To analyze protein expression after tumor growth, a polyclonal anti-GFP antibody was used. Error bars indicate mean ± S.D.
Human MageB2 Expression Stimulates E2F activity
To elucidate the mechanisms by which human MageB2 regulates cell proliferation independently of p53 status, we investigated the E2F pathway because it has been reported that MAGE-II proteins can target and inhibit E2F function (27, 28). To this aim, we silenced MageB2 gene expression in U2OS cells by siRNA. We quantified known cell cycle-associated E2F target genes by quantitative RT-PCR and detected a consistently significant down-regulation associated with MageB2 knockdown (Fig. 3A). A ChIP-quantitative PCR assay under these conditions indicated that endogenous MageB2 does not affect E2F1 association with chromatin, as assessed on the E2F1 promoter, a well established E2F1 target (data not shown).
FIGURE 3.

Human MageB2 induces E2F activity. A, determination of E2F target gene expression through quantitative RT-PCR in U2OS cells with normal (siC) or reduced (siB2) MageB2 expression. mRNA levels under the siB2 condition were normalized to the siC condition. Each individual mRNA value from the E2F targets was normalized to the actin mRNA value. B, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct and the indicated HA-tagged E2F protein. *, p < 0.05; **, p < 0.001. The assay was performed in U2OS cells. A Western blot of the expressed proteins is shown below the histogram. #, unspecific band. C, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct and GFP-tagged MageB2 of human (MageB2) or mouse (Mageb2) origin and mouse Mageb18. EV, GFP empty vector; Hn, human; Mo, mouse. *, p < 0,05. The assay was performed in U2OS cells. A Western blot (WB) of the expressed proteins is shown below the histogram. #, nonspecific band. Error bars indicate mean ± S.D. Student's t test was used for statistical analysis.
To verify the specific effect on E2F transcriptional activity, we analyzed whether MageB2 could regulate E2F activity by reporter gene assay using the 6×E2F-Luc construct. We tested the effect of MageB2 on E2F1, E2F2, and E2F3. As shown in Fig. 3B, MageB2 induced all E2Fs tested, suggesting that the effect observed in the quantitative RT-PCR approach could be the result of the regulation of different E2F members.
As mentioned above, the highly conserved murine Mageb1, Mageb2, and Mageb3 proteins as well as murine Mageb18 have been associated with cell proliferation. To elucidate whether these related mouse Mage-B proteins could similarly regulate E2F activity, we compared their effect on E2F1 transcriptional activity. Interestingly, only human MageB2 expression up-regulated E2F1 transcriptional activity, whereas murine proteins did not cause a significantly detectable effect (Fig. 3C). Different from what it has been reported in mouse cells (19), human MageB2 KD did not result in massive cell death but in cell growth with limited rate respect to control and independently of p53 status (Fig. 1, A, B, and D). These results suggest that, even when murine and human MAGE-B gene expression is associated with tumor cell proliferation, they probably make use of different molecular strategies to exert their functions.
MAGE-A proteins have been shown to interact with HDACs (5, 10), enzymes known to regulate gene transcription. Regulation of E2F transcriptional activity through HDACs has been studied extensively. Different E2F regulators, such as pRb (29) and its family of pocket proteins, Sin3B (30), Kap1 (31), Ebp1 (32) and Ell, (33) inhibit E2F activity by recruiting HDACs.
Our next step was to elucidate whether MageB2 could exert its function by targeting HDACs or E2F1 itself. To this aim, we produced the GST-MageB2 fusion protein in bacteria and immunopurified HA-tagged HDAC1 and E2F1 from HEK293T cell extracts. Tagged proteins were immunoadsorbed using the anti-HA antibody and protein A-Sepharose beads. Potential coadsorbed endogenous proteins were dissociated by supplementing the buffer with 500 mm NaCl (34). HA-tagged immunopurified proteins were then eluted from the HA-antibody/protein-A-Sepharose beads using commercial HA peptide. Next, an in vitro binding assay was performed by incubating immunopurified proteins with GST or GST-MageB2. The results from the in vitro binding assay indicate that, under these conditions, MageB2 strongly interacts with HDAC1, but no interaction was detected between MageB2 and E2F1 (Fig. 4A).
FIGURE 4.

MageB2 interacts with HDAC1 and interferes with E2F1-HDAC1 complex formation. A, in vitro binding assay between HA-tagged proteins produced and purified from HEK293T cells and GST or GST-MageB2 produced and purified from bacteria. B, immunoprecipitation of HA-tagged HDAC1, pRb, and p107 in HEK293T cells co-transfected with myc-MageB2. Immunoprecipitated protein complexes obtained with the anti-HA antibody (IP αHA) was assessed by Western blot (WB) and probed with the indicated antibodies. TL, total lysates. C, immunoprecipitation of E2F1 in HEK293T cells co-transfected with FLAG-HDAC1 in the presence or absence of Myc-MageB2. The immunoprecipitated material obtained when using the anti-E2F1 antibody (IP αE2F1) was assessed by Western blot and probed with the indicated antibodies. Left, total lysates. D, immunoprecipitation of FLAG-HDAC1 in HEK293T cells co-transfected with HA-E2F1 and Myc-MageB2. Immunoprecipitated material (IP αFLAG) was assessed by Western blot and probed with the indicated antibodies. Left, total lysates. E, immunoprecipitation of FLAG-HDAC1 in U2OS cells co-transfected with the AR vector (AR) and Myc-MageB2. Immunoprecipitated material (IP αFLAG) was assessed by Western blot and probed with the indicated antibodies. Left, total lysates. #, IgG heavy chains.
We then performed an assay to verify the MageB2/HDAC1 association in cells. In addition, we compared this association with that of other potential targets of the E2F1-regulating complex, such as the pocket proteins pRb and p107. An immunoprecipitation (IP) assay performed at the same time and under the same conditions confirmed HDAC1 as a strong MageB2 interactor, whereas pRb or p107 showed low or undetectable interaction with MageB2 (Fig. 4B). An additional IP assay showed that MageB2 can also interact with HDAC3 but not with HDAC4 and HDAC2 (data not shown).
Given that HDAC1 forms part of the E2F1 protein complex and represses E2F1 activity, we assessed the effect of MageB2 in such complex formation. The results shown in Fig. 4C indicate that, in the absence of MageB2 expression, E2F1 and HDAC1 form part of a protein complex. However, MageB2 expression strongly reduced the amount of HDAC1 associated with the E2F1 complex.
The results were confirmed by reverse IP showing the amount of E2F1 associated with HDAC1 when MageB2 was expressed. Accordingly, immunoprecipitated HDAC1 recruited lower amounts of E2F1 in the presence of MageB2 (Fig. 4D). Of note, MageB2 coimmunoprecipitates with HDAC1 (Fig. 4D, third lane) but is not detectable when E2F1 is immunoprecipitated (Fig. 4C, fourth lane), reinforcing the hypothesis that MageB2 interacts with HDAC1. When the effect of MageB2 expression was tested on the unrelated AR-HDAC1 complex, no evidence of reduced AR recruitment to HDAC1 (Fig. 4E) was observed, suggesting that MageB2 could preferentially target the E2F/HDAC association. These data suggest that MageB2 can associate to HDACs, releasing E2F from such interaction and enhancing its free and transcriptionally active fraction.
We reasoned that if MageB2 enhances E2F1 activity by reducing the amount of HDAC1 from the E2F1 protein complex, then enhanced expression of HDAC1 should reverse the MageB2 effect on E2F1 activity. We assessed the effect of MageB2 on E2F1 activity when high amounts of HDACs were available through ectopic expression. Increased HDAC1 availability clearly reversed the effect of MageB2 on E2F1 activity but not in the absence of MageB2 (Fig. 5A). Because HDAC overexpression could repress general transcription, we further controlled the specificity of this experiment by transfecting HDAC4, a class II histone deacetylase that does not interact with MageB2. The results shown in Fig. 5B indicate that HDAC4 does not affect the regulation of E2F1 by MageB2 expression (Fig. 5B).
FIGURE 5.

The HDAC-dependent effect of MageB2 on E2F1 activity. A, reporter gene assay for E2F1 activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed with increasing amounts of FLAG-HDAC1. *, p < 0.05. The assay was performed in U2OS cells. A Western blot of the expressed proteins is shown below the histogram. EV, empty vector. B, reporter gene assay for E2F1 activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed with increasing amounts of the vector expressing HA-HDAC4. *, p < 0.05. The assay was performed in U2OS cells. A Western blot of the expressed proteins is shown below the histogram. C, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed in the presence (TSA) and absence (not treated, NT) of 300 nm TSA during 24 h. Two different amounts of E2F1 were used, as indicated by gray and black columns. *, p < 0.05. The assay was performed in U2OS cells. A Western blot of the expressed proteins is shown below the histogram. Error bars indicate mean ± S.D. Student's t test was used for statistical analysis. #, nonspecific band.
Finally, to further highlight the relevance of HDAC in our mechanism, we assessed the effect of MageB2 on E2F1 activity when HDACs were inhibited by Trichostatin A (TSA). As expected, inhibition of HDAC activity by TSA enhanced E2F1 activity. However, under this condition, E2F1 activity was no longer induced significantly by MageB2 expression (Fig. 5C).
All of these data suggest that the mechanism by which MageB2 regulates E2F1 activity could involve the direct interaction between MageB2 and HDAC1. Then MageB2/HDAC1 association prevents efficient recruiting of HDAC1 to the E2F1 complex, resulting in enhanced E2F1 activity.
MageB2 Cellular Localization
MageB2 is a 319-amino acid-long protein with an MHD sharing ∼48% homology with that of MAGE-A proteins (MHD, amino acids 110–290). It has been reported previously that MAGE-A proteins can be found in different cell compartments, including the nucleus (10), cytoplasm (35), and PML nuclear bodies (12). We observed here that endogenous MageB2, in addition to nuclear and cytoplasm fractions, is also present in the nucleolar fraction (Fig. 6A). Accordingly, GFP-tagged MageB2 is observed in the cytoplasm, nucleus, and nucleoli, where it colocalizes with nucleolin staining (Fig. 6B, left panels). The MHD domain of MAGE-I is conserved throughout all family members, whereas the N-terminal domain is less conserved and usually unstructured. Studies performed with MageB2 deletion mutants indicate that the region comprised of amino acids 1–109 is sufficient to relocalize the GFP protein to the nucleolus (Fig. 6B, center panels). Moreover, by using bioinformatic tools, a 29-amino acid-long sequence rich in arginine and lysine was detected as a potential NoLS at the MageB2 N-terminal region. To assess its function, we generated the ΔNoLS-MageB2 construct carrying the MageB2 gene with the deleted NoLS sequence. We observed that ΔNoLS-MageB2 protein clearly delocalizes from the nucleoli (Fig. 6B, right panels). These results indicate that MageB2 can be driven to the nucleoli depending on a canonical NoLS sequence located at the N terminus, supporting the concept that even if MAGE-I proteins share the conserved MHD, then their localization/function is also dependent on short motifs present outside of this domain.
FIGURE 6.

Cellular localization of MageB2. A, Western blot assay of cytoplasmic (Cytop), nuclear, and nucleolar fractions obtained from HCT116 cells. Nucleolin (NCL), Tubulin (Tub), and c-myc antibodies were used as markers of nucleolar, cytoplasmic, and nuclear fractions, respectively. B, confocal microscopy images of U2OS cells transfected with GFP-tagged MageB2 constructs (top panels) containing wild-type MageB2 (GFP-MageB2 WT), amino acids 1–109 of MageB2 (GFP-MageB2 1–109), and MageB2 with a deleted nucleolar localization signal sequence (GFP-MageB2 ΔNoLS). Bottom panels, endogenous nucleolin staining as a nucleolar marker.
MageB2 Expression and Ribotoxic Stress
Ribotoxic stress triggers cell cycle arrest through the regulation of master signaling pathways such as those involving E2F, p53, and c-myc proteins. It has been shown extensively that drugs triggering ribotoxic stress relocalize specific ribosomal proteins (RPs) from the nucleoli to the nucleoplasm as a feedback loop to limit high translational demand and, consequently, cell proliferation. In the nucleus, ribosomal proteins have been found to reduce c-myc activity (36) and target Mdm2, enhancing p53 activity (37) and reducing E2F activity (38).
We observed that ribosomal stress, as induced by low Act-D concentrations (5–10 nm), does not significantly affect MageB2 protein levels compared with the established effect on c-myc (Fig. 7A). However, Act-D clearly induces a striking and early exit of MageB2 from nucleoli (Fig. 7B). This behavior is not associated with a general stress response because MageB2 does not change its nucleolar localization after treatments with other types of stressors, such as ultraviolet radiation (DNA damage), high temperature (heat shock), or thapsigargin (endoplasmic reticulum stress) (data not shown).
FIGURE 7.

MageB2 expression counteracts ribosomal protein-driven inhibition of E2F1. A, Western blot of a time course assay after addition of 5 nm Actinomycin D to U2OS cells. The membrane was probed with c-myc, MageB2, and Vinculin (loading control) antibodies. B, confocal microscopy images of HA-MageB2 and Myc-L11 constructs before (left panel, top row, untreated) and after (left panel, bottom row, Act-D, 5 nm) 3-h Actimomycin D treatment of U2OS cells. Right panel, the percentage of cells displaying MageB2 with nuclear or nucleolar localization before and after treatment with 5 nm Act-D. 200 cells in total were counted under each experimental condition. C, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed when co-transfected with Myc-L11 and FLAG-L23 ribosomal proteins constructs. ***, p < 0.05; **, p < 0.01; *, p < 0.005. The assay was performed in U2OS cells. A Western blot of the expressed proteins is shown below the histogram. EV, empty vector; #, nonspecific band. D, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed when cotransfected with Myc-L11 and FLAG-L23 ribosomal proteins constructs. **, p < 0.05; *, p < 0.005. The assay was performed in double knockout (p53−/−; Mdm2−/−) mouse embryonic fibroblasts (DKO cells). A Western blot of the expressed proteins is shown below the histogram. Error bars indicate mean ± S.D. Student's t test was used for statistical analysis.
We then evaluated whether MageB2 can enhance E2F activity in the presence of RPs known to inhibit E2F activity. We observed that MageB2 can still enhance E2F1 activity in the presence of RPL11 and RPL23 (Fig. 7C). However, the effect of MageB2 seems to be independent of Mdm2 expression because it is still observed in double knockout cells (mouse embryonic fibroblast p53−/−; mdm2−/− cells), where RPL11 and RPL23 failed to regulate E2F activity, as expected (Fig. 7D). These data suggest that, upon nucleolar stress, MageB2 expression could counteract the negative regulation of RPs on E2F, therefore avoiding part of their cell cycl-repressive function.
Act-D treatment promotes a prevalent nucleoplasmic localization of MageB2 but does not significantly affect MageB2 function on E2F1 activity (Fig. 8A) or MageB2 interaction with endogenous HDAC1 (Fig. 8B), suggesting that nucleoplasmic MageB2 should still be able to enhance E2F activity. To address the relevance of MageB2 localization on E2F activity, we expressed ΔNoLS-MageB2 missing the nucleolar localization. We observed that ΔNoLS-MageB2 is able to interact with endogenous HDAC1 as the wild-type protein (Fig. 8C) and displays E2F1 activating function similar to wild-type MageB2 (Fig. 8D). These data suggest that nuclear MageB2 is also able to regulate E2F activity.
FIGURE 8.

MageB2 induces E2F1 activity under ribotoxic conditions. A, reporter gene assay for E2F activity using the 6×E2F-LUC reporter construct. The effect of MageB2 was assessed in U2OS cells treated or not treated with 10 nm Act-D for 24 h. *, p < 0.005. B, immunoprecipitation of HA-MageB2 in U2OS cells stably expressing inducible HA-MageB2. Complex formation with endogenous HDAC1 was assessed in the presence (+) or absence (−) of 10 nm Act-D with induced (I) and not induced (NI) MageB2 expression. Immunoprecipitated material (IP αHA) was assessed by Western blot and probed with the indicated antibodies. Left, total lysates (TL). C, immunoprecipitation of FLAG-HDAC1 in HEK293T cells cotransfected with GFP-tagged MageB2 with a deleted nucleolar localization signal sequence (GFP-ΔNoLS-MageB2). Immunoprecipitated material (IP αFLAG) was assessed by Western blot and probed with the indicated antibodies. Left, total lysates. D, reporter gene assay for E2F1 activity using the 6×E2F-LUC reporter construct. The effect of GFP-tagged wild-type MageB2 (MageB2) and GFP-tagged MageB2 with deleted nucleolar localization signal sequence (ΔNoLS-MageB2) on E2F1 transcriptional activity was investigated in U2OS cells. ***, p < 0.01; **, p < 0.05; *, p < 0.005. A Western blot of the expressed proteins is shown below the histogram. EV, GFP empty vector. E, BrdU incorporation in U2OS cells transfected previously with siRNA for MageB2 (siB2) or a control siRNA (siC) and treated or not treated (NT) with 10 nm Act-D for 16 h. Shown are representative merged fluorescence microscopy images of BrdU (green) and Hoechst (blue) staining for each experimental condition. Right panel, determination of cell proliferation as the percentage of BrdU with respect to Hoechst-stained cells by an automated high-content screening (∼4.5 × 103 events/condition). ***, p < 0.05; **, p < 0.005; *, p < 0.001. Error bars indicate mean ± S.D. Student's t test was used for statistical analysis.
Our data indicating that, under ribotoxic stress conditions, MageB2 is mainly localized to the nucleoplasm maintaining E2F activity prompted us to study whether endogenous MageB2 could enhance cell proliferation under the reported stress condition. To test this relevant biological effect, we assessed BrdU incorporation in U2OS cells with KD endogenous MageB2 in the presence or absence of 10 nm Act-D. The analysis was performed by using an automated high-content screening fluorescence microscope. Importantly, MageB2 KD enhanced ribotoxic stress-induced cell cycle arrest, as shown in Fig. 8E, left panel. An average of 4.5 × 103 cells was analyzed automatically to quantify the percentage of BrdU-positive cells under each condition (Fig. 8E, right panel). The data obtained from this experiment indicate that endogenous MageB2 expression plays a role in enhancing cell cycle progression under ribotoxic stress.
Discussion
MAGE-I proteins are expressed in a variety of human tumors, and even though they are considered promising antigens to produce anti-tumor vaccines, their expression commonly correlates with a poor prognosis in untreated patients. Because of their high sequence homology, it has been considered a functionally redundant protein family. However, data obtained recently suggest that specific MAGE-I members (or at least a discrete subset of them) could interfere with normal cell function by affecting distinct signaling pathways (13).
Mouse mage-b1, -b2, and -b3 gene expression has been associated with anti-apoptotic/pro-survival balance (19, 20). In addition, mouse Mageb18 protein expression has been reported to have a similar phenotype as that of mouse Mage-b (21). The correlation between human MageB2 expression and cell proliferation has been suggested in normal oral keratinocytes (23). Here we extended the study to human cancer cell lines such as osteosarcoma (U2OS) and colon carcinoma (HCT116) bearing WT or deleted p53 loci. In addition, we assessed the proliferative potential of human MageB2 expression in vivo in B16 (F0) melanoma cells. All reported results are consistent with a significant role of human MageB2 in enhancing cell proliferation independently of p53 status, cell type, or species, suggesting its involvement in targeting a general mechanism of cell proliferation.
The first MAGE protein reported to regulate cell proliferation through E2F transcription factors was Necdin, a type II MAGE protein (27) able to associate and repress E2F1 transcriptional activity. Later, it was reported that another type II MAGE protein, MageG1 (necdin-like 2), also inhibited the E2F1 transcription factor through direct association (28). More recently, it has been reported that a type I MAGE (MageA11) interacts with the pocket protein p107 and enhances E2F1 activity (39) as part of its pro-oncogenic activity in prostate cancer.
As mentioned above, E2F activity is highly sensitive to HDAC activity because a number of proteins inhibit its activity through HDAC recruitment, including pRb (29), Sin3B (30), Kap1 (31), Ebp1 (32) and ELL (33). We and other groups have demonstrated that MageA1 and MageA2 associate with HDACs to exert their function (5, 10). On the basis of the abovementioned data, we investigated whether MageB2 could regulate E2F activity as part of its mechanism responsible for enhanced cell proliferation. We report here that MageB2 interacts with HDAC1, weakening its recruitment to the E2F1 protein complex and, therefore, increasing the fraction of free and active E2F. Similar mechanisms have been reported previously for the LEF1 (40) and HMGA2 oncoproteins, both increasing E2F activity (41). Moreover, potent oncogenes such as Ha-ras, c-myc, and E2Fs enhance E2F mRNA and protein levels (42, 43), confirming their key role in cell proliferation.
However, under specific conditions, E2F1 has been involved in triggering cell death by inducing the transcription of pro-apoptotic genes (44, 45). JAB1 could be a critical protein in this dichotomous role of E2F. It has been reported that JAB1 binds E2F, is found to be associated to E2F in promoters of apoptotic targets (and not in DNA-replication E2F targets), and is required for E2F-induced apoptosis (46, 47). Moreover, hyperactivation of the PI3K/AKT pathway specifically abolishes E2F-induced apoptosis (48), in part by interfering with E2F-JAB1 complex formation (47). Even if E2F expression can promote apoptosis in DNA-damaged cells (49), a number of clinical assays suggest that enhanced E2F activity is associated with invasion (50), metastasis formation (51), and, clinically, with poor survival (52). Along these lines, induction of E2F activity by MageB2 in a tumor-specific context could be relevant for the final clinical outcome. Other proteins, such as EAPP, known to enhance E2F activity, are also overexpressed in tumor cells (53, 54).
We observed that MageB2 is localized in the nucleus, cytoplasm, and nucleoli. This unusual localization for a MAGE protein is quickly lost upon ribotoxic stress as induced by low-concentration Act-D treatment. Contrary to RPs that relocalize from the nucleoli to the nucleus under ribotoxic stress to collaborate with cell cycle arrest, endogenous MageB2 exits the nucleolus but promotes cell cycle progression. Of note, MageB2 is still associated to HDAC1 and enhances E2F activity in Act-D-treated cells. Importantly, MageB2 can counteract the repressive effect of RPs on E2F activity. It has been reported that Mdm2 plays a central role in this nucleolar checkpoint because it is targeted by RPs, therefore down-regulating E2F activity and activating p53. However, MageB2 can regulate E2F activity in the absence of Mdm2, whereas RPs require its expression, demonstrating that MageB2 and RPs oppositely regulate E2F activity through different molecular mechanisms.
Taking into account all of the data presented here, we suggest that human MageB2 behaves as a cell proliferation-promoting protein because it expression enhances the proliferative rate in a variety of cell lines and in a melanoma mouse model. More specifically, we demonstrate that, different from MageA2, MageB2 function does not depend on p53 expression. However, MageA2 and MageB2 share the ability to associate to HDACs, therefore exerting specific regulatory activity on different transcription factors. In this study, we observed that MageB2-HDAC1 complex formation enhances E2F activity. In addition, we report, for the first time, a MAGE protein involved in nucleolar events, a function that has not been reported for any MAGE-I member. Interestingly, mouse proteins such as Mageb2 or Mageb18, which have been reported to display tumor cell survival properties, are not involved in E2F regulation. The functional homology between human and mouse MAGE proteins should be reconsidered carefully in light of the overall differences in basal metabolic rate of human and mouse (55), possibly linked to the rewiring of both p53 and E2F regulatory networking.
The striking difference between MAGE-II (ubiquitous expression) and MAGE-I (tumor-specific expression) could be the base for proposing a novel hypothesis suggesting that enhanced expression of MAGE-I members during tumor cell transformation could play a role in counteracting MAGE-II function. As shown here, MageB2 plays an opposite role as that reported for Necdin and MageG1 on E2F activity. Accordingly, and with respect to p53, MageA2 represses p53 activity (10, 11), whereas MageD1/Nrage activates p53 to stop cell proliferation (56). Therefore, this hypothesis could promote future experiments to shed light on the basic networking of MAGE gene expression in cancer cells.
A growing body of evidence suggests that different MAGE proteins play distinct roles in cancer cells. Therefore, it should be critical to precisely determine the identity of the different MAGE genes expressed in different tumor biopsies.
Author Contributions
M. Monte and C. S. conceived and coordinated the study, and M. Monte wrote the paper. L. Y. P. and M. F. L. designed, performed, and analyzed the experiments shown in Figs. 1–6. M. F. T. and J. E. L. designed, performed, and analyzed the experiments shown in Fig. 7. L. Y. P. and M. Mano designed, performed, and analyzed the experiments shown in Fig. 8. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Ramiro Mendoza Maldonado (ICGEB, Trieste, Italy) for the HA-E2F1 and pGL3-TATA-6xE2F-Luc plasmids. HA-E2F2 and HA-E2F3 were provided by Eduardo Cánepa (Dpto Qca Biológica, FCEN, UBA, Argentina). The AR plasmid was provided by Mario Galigniana (IByME-CONICET, Argentina).
This work was supported by Ministry of Science and Technology-ANPCyT Grant PICT12/866, University of Buenos Aires Grant UBACyT13/0013BA, and CONICET Grant PIP13/0411 (to M. Monte) and FIRB “Oncodiet” Grant RBAP11LP2W_002 and Associazione Italiana per la Ricerca sul Cancro Grant AIRC/IG10433 (to C. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- MHD
- MAGE homology domain
- HDAC
- histone deacetylase
- NoLS
- nucleolar localization signal
- TSA
- Trichostatin A
- AR
- androgen receptor
- Act-D
- Actinomycin D
- KD
- knockdown
- IP
- immunoprecipitation
- RP
- ribosomal protein.
References
- 1.Scanlan M. J., Simpson A. J., and Old L. J. (2004) The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 4, 1. [PubMed] [Google Scholar]
- 2.De Smet C., Lurquin C., Lethé B., Martelange V., and Boon T. (1999) DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell. Biol. 19, 7327–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B., Knuth A., and Boon T. (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 [DOI] [PubMed] [Google Scholar]
- 4.Sang M., Lian Y., Zhou X., and Shan B. (2011) MAGE-A family: attractive targets for cancer immunotherapy. Vaccine 29, 8496–8500 [DOI] [PubMed] [Google Scholar]
- 5.Laduron S., Deplus R., Zhou S., Kholmanskikh O., Godelaine D., De Smet C., Hayward S. D., Fuks F., Boon T., and De Plaen E. (2004) MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 32, 4340–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Askew E. B., Bai S., Hnat A. T., Minges J. T., and Wilson E. M. (2009) Melanoma antigen gene protein-A11 (MAGE-11) F-box links the androgen receptor NH2-terminal transactivation domain to p160 coactivators. J. Biol. Chem. 284, 34793–34808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai S., Grossman G., Yuan L., Lessey B. A., French F. S., Young S. L., and Wilson E. M. (2008) Hormone control and expression of androgen receptor coregulator MAGE-11 in human endometrium during the window of receptivity to embryo implantation. Mol. Hum. Reprod. 14, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai S., He B., and Wilson E. M. (2005) Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell. Biol. 25, 1238–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai S., and Wilson E. M. (2008) Epidermal-growth-factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol. Cell. Biol. 28, 1947–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monte M., Simonatto M., Peche L. Y., Bublik D. R., Gobessi S., Pierotti M. A., Rodolfo M., and Schneider C. (2006) MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. U.S.A. 103, 11160–11165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcar L., Maclaine N. J., Hupp T. R., and Meek D. W. (2010) Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 70, 10362–10370 [DOI] [PubMed] [Google Scholar]
- 12.Peche L. Y., Scolz M., Ladelfa M. F., Monte M., and Schneider C. (2012) MageA2 restrains cellular senescence by targeting the function of PMLIV/p53 axis at the PML-NBs. Cell Death Differ. 19, 926–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladelfa M. F., Peche L. Y., Toledo M. F., Laiseca J. E., Schneider C., and Monte M. (2012) Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 325, 11–17 [DOI] [PubMed] [Google Scholar]
- 14.Gure A. O., Chua R., Williamson B., Gonen M., Ferrera C. A., Gnjatic S., Ritter G., Simpson A. J., Chen Y. T., Old L. J., and Altorki N. K. (2005) Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin. Cancer Res. 11, 8055–8062 [DOI] [PubMed] [Google Scholar]
- 15.Olarte I., Martinez A., Ramos-Peñafiel C., Castellanos-Sinco H., Zamora J., Collazo-Jaloma J., Gutiérrez M., Gutiérrez-Kobeh L., Chavez-Olmos P., Manzanilla H., Garrido-Guerrero E., Ordoñez-Razo R. M., and Miranda E. I. (2011) MAGE-A3 expression is an adverse prognostic factor in diffuse large B-cell lymphoma. Hematology 16, 368–372 [DOI] [PubMed] [Google Scholar]
- 16.Ogata K., Aihara R., Mochiki E., Ogawa A., Yanai M., Toyomasu Y., Ando H., Ohno T., Asao T., and Kuwano H. (2011) Clinical significance of melanoma antigen-encoding gene-1 (MAGE-1) expression and its correlation with poor prognosis in differentiated advanced gastric cancer. Ann. Surg. Oncol. 18, 1195–1203 [DOI] [PubMed] [Google Scholar]
- 17.Lian Y., Sang M., Ding C., Zhou X., Fan X., Xu Y., Lü W., and Shan B. (2012) Expressions of MAGE-A10 and MAGE-A11 in breast cancers and their prognostic significance: a retrospective clinical study. J. Cancer Res. Clin. Oncol. 138, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sypniewska R. K., Hoflack L., Tarango M., Gauntt S., Leal B. Z., Reddick R. L., and Gravekamp C. (2005) Prevention of metastases with a Mage-b DNA vaccine in a mouse breast tumor model: potential for breast cancer therapy. Breast Cancer Res. Treat. 91, 19–28 [DOI] [PubMed] [Google Scholar]
- 19.Yang B., O'Herrin S., Wu J., Reagan-Shaw S., Ma Y., Nihal M., and Longley B. J. (2007) Select cancer testes antigens of the MAGE-A, -B, and -C families are expressed in mast cell lines and promote cell viability in vitro and in vivo. J. Invest. Dermatol. 127, 267–275 [DOI] [PubMed] [Google Scholar]
- 20.Yang B., O'Herrin S. M., Wu J., Reagan-Shaw S., Ma Y., Bhat K. M., Gravekamp C., Setaluri V., Peters N., Hoffmann F. M., Peng H., Ivanov A. V., Simpson A. J., and Longley B. J. (2007) MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 67, 9954–9962 [DOI] [PubMed] [Google Scholar]
- 21.Lin Y., Wen T., Meng X., Wu Z., Zhao L., Wang P., Hong Z., and Yin Z. (2012) The mouse Mageb18 gene encodes a ubiquitously expressed type I MAGE protein and regulates cell proliferation and apoptosis in melanoma B16-F0 cells. Biochem. J. 443, 779–788 [DOI] [PubMed] [Google Scholar]
- 22.Jang S. J., Soria J. C., Wang L., Hassan K. A., Morice R. C., Walsh G. L., Hong W. K., and Mao L. (2001) Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis. Cancer Res. 61, 7959–7963 [PubMed] [Google Scholar]
- 23.Pattani K. M., Soudry E., Glazer C. A., Ochs M. F., Wang H., Schussel J., Sun W., Hennessey P., Mydlarz W., Loyo M., Demokan S., Smith I. M., and Califano J. A. (2012) MAGEB2 is activated by promoter demethylation in head and neck squamous cell carcinoma. PLoS ONE 7, e45534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Duin M., Broyl A., de Knegt Y., Goldschmidt H., Richardson P. G., Hop W. C., van der Holt B., Joseph-Pietras D., Mulligan G., Neuwirth R., Sahota S. S., and Sonneveld P. (2011) Cancer testis antigens in newly diagnosed and relapse multiple myeloma: prognostic markers and potential targets for immunotherapy. Haematologica 96, 1662–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krämer B. F., Schoor O., Krüger T., Reichle C., Müller M., Weinschenk T., Hennenlotter J., Stenzl A., Rammensee H. G., and Stevanovic S. (2005) MAGED4-expression in renal cell carcinoma and identification of an HLA-A*25-restricted MHC class I ligand from solid tumor tissue. Cancer Biol. Ther. 4, 943–948 [DOI] [PubMed] [Google Scholar]
- 26.Müller H., Moroni M. C., Vigo E., Petersen B. O., Bartek J., and Helin K. (1997) Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol. 17, 5508–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taniura H., Taniguchi N., Hara M., and Yoshikawa K. (1998) Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J. Biol. Chem. 273, 720–728 [DOI] [PubMed] [Google Scholar]
- 28.Kuwako K., Taniura H., and Yoshikawa K. (2004) Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem. 279, 1703–1712 [DOI] [PubMed] [Google Scholar]
- 29.Magnaghi-Jaulin L., Groisman R., Naguibneva I., Robin P., Lorain S., Le Villain J. P., Troalen F., Trouche D., and Harel-Bellan A. (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391, 601–605 [DOI] [PubMed] [Google Scholar]
- 30.Grandinetti K. B., and David G. (2008) Sin3B: an essential regulator of chromatin modifications at E2F target promoters during cell cycle withdrawal. Cell Cycle 7, 1550–1554 [DOI] [PubMed] [Google Scholar]
- 31.Wang C., Rauscher F. J. 3rd, Cress W. D., and Chen J. (2007) Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 282, 29902–29909 [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., Woodford N., Xia X., and Hamburger A. W. (2003) Repression of E2F1-mediated transcription by the ErbB3 binding protein Ebp1 involves histone deacetylases. Nucleic Acids Res. 31, 2168–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W., Ji W., Liu X., Ouyang G., and Xiao W. (2014) ELL inhibits E2F1 transcriptional activity by enhancing E2F1 deacetylation via recruitment of histone deacetylase 1. Mol. Cell. Biol. 34, 765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galigniana M. D., Harrell J. M., O'Hagen H. M., Ljungman M., and Pratt W. B. (2004) Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J. Biol. Chem. 279, 22483–22489 [DOI] [PubMed] [Google Scholar]
- 35.Cheng S., Liu W., Mercado M., Ezzat S., and Asa S. L. (2009) Expression of the melanoma-associated antigen is associated with progression of human thyroid cancer. Endocr. Relat. Cancer 16, 455–466 [DOI] [PubMed] [Google Scholar]
- 36.Zhou X., Hao Q., Liao J. M., Liao P., and Lu H. (2013) Ribosomal protein S14 negatively regulates c-Myc activity. J. Biol. Chem. 288, 21793–21801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohrum M. A., Ludwig R. L., Kubbutat M. H., Hanlon M., and Vousden K. H. (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3, 577–587 [DOI] [PubMed] [Google Scholar]
- 38.Donati G., Brighenti E., Vici M., Mazzini G., Treré D., Montanaro L., and Derenzini M. (2011) Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 124, 3017–3028 [DOI] [PubMed] [Google Scholar]
- 39.Su S., Minges J. T., Grossman G., Blackwelder A. J., Mohler J. L., and Wilson E. M. (2013) Proto-oncogene activity of melanoma antigen-A11 (MAGE-A11) regulates retinoblastoma-related p107 and E2F1 proteins. J. Biol. Chem. 288, 24809–24824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou F., Zhang L., Gong K., Lu G., Sheng B., Wang A., Zhao N., Zhang X., and Gong Y. (2008) LEF-1 activates the transcription of E2F1. Biochem. Biophys. Res. Commun. 365, 149–153 [DOI] [PubMed] [Google Scholar]
- 41.Fedele M., Pierantoni G. M., Visone R., and Fusco A. (2006) E2F1 activation is responsible for pituitary adenomas induced by HMGA2 gene overexpression. Cell Div. 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berkovich E., and Ginsberg D. (2001) Ras induces elevation of E2F-1 mRNA levels. J. Biol. Chem. 276, 42851–42856 [DOI] [PubMed] [Google Scholar]
- 43.Matsumura I., Tanaka H., and Kanakura Y. (2003) E2F1 and c-Myc in cell growth and death. Cell Cycle 2, 333–338 [PubMed] [Google Scholar]
- 44.Stanelle J., and Pützer B. M. (2006) E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol. Med. 12, 177–185 [DOI] [PubMed] [Google Scholar]
- 45.Iaquinta P. J., and Lees J. A. (2007) Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 19, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hallstrom T. C., and Nevins J. R. (2006) Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 20, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu H., Liang X., Issaenko O. A., and Hallstrom T. C. (2011) Jab1/CSN5 mediates E2F dependent expression of mitotic and apoptotic but not DNA replication targets. Cell Cycle 10, 3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hallstrom T. C., Mori S., and Nevins J. R. (2008) An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 13, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engelmann D., and Pützer B. M. (2010) Translating DNA damage into cancer cell death-A roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist. Updat. 13, 119–131 [DOI] [PubMed] [Google Scholar]
- 50.Lee T. J., Yao G., Bennett D. C., Nevins J. R., and You L. (2010) Stochastic E2F activation and reconciliation of phenomenological cell-cycle models. PLoS Biol. 8, e1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imai T., Horiuchi A., Shiozawa T., Osada R., Kikuchi N., Ohira S., Oka K., and Konishi I. (2004) Elevated expression of E-cadherin and α-, β-, and γ-catenins in metastatic lesions compared with primary epithelial ovarian carcinomas. Hum. Pathol. 35, 1469–1476 [DOI] [PubMed] [Google Scholar]
- 52.Hans C. P., Weisenburger D. D., Vose J. M., Hock L. M., Lynch J. C., Aoun P., Greiner T. C., Chan W. C., Bociek R. G., Bierman P. J., and Armitage J. O. (2003) A significant diffuse component predicts for inferior survival in grade 3 follicular lymphoma, but cytologic subtypes do not predict survival. Blood 101, 2363–2367 [DOI] [PubMed] [Google Scholar]
- 53.Novy M., Pohn R., Andorfer P., Novy-Weiland T., Galos B., Schwarzmayr L., and Rotheneder H. (2005) EAPP, a novel E2F binding protein that modulates E2F-dependent transcription. Mol. Biol. Cell 16, 2181–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andorfer P., and Rotheneder H. (2011) EAPP: gatekeeper at the crossroad of apoptosis and p21-mediated cell-cycle arrest. Oncogene 30, 2679–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demetrius L. (2005) Of mice and men: when it comes to studying ageing and the means to slow it down, mice are not just small humans. EMBO Rep. 6, S39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wen C. J., Xue B., Qin W. X., Yu M., Zhang M. Y., Zhao D. H., Gao X., Gu J. R., and Li C. J. (2004) hNRAGE, a human neurotrophin receptor interacting MAGE homologue, regulates p53 transcriptional activity and inhibits cell proliferation. FEBS Lett. 564, 171–176 [DOI] [PubMed] [Google Scholar]