

Background: It is unclear whether Smoothened protein is the only G protein-coupled receptor (GPCR) that stimulates vertebrate Hedgehog (Hh) signaling.
Results: Knockdown of Gpr175, a Hh-dependent ciliary GPCR, reduces Hh signaling in multiple cell lines.
Conclusion: Gpr175 positively modulates Hh signaling by decreasing cAMP and Gli3 repressor levels via Gαi1.
Significance: Regulation of Hh signaling by GPCRs is more complicated than thought.
Keywords: cyclic AMP (cAMP), G protein-coupled receptor (GPCR), Hedgehog signaling pathway, primary cilium, protein kinase A (PKA), Gli3, Gpr175, Smoothened, Tpra40
Abstract
The Hedgehog (Hh) signaling pathway plays an essential role in vertebrate embryonic tissue patterning of many developing organs. Signaling occurs predominantly in primary cilia and is initiated by the entry of the G protein-coupled receptor (GPCR)-like protein Smoothened into cilia and culminates in gene transcription via the Gli family of transcription factors upon their nuclear entry. Here we identify an orphan GPCR, Gpr175 (also known as Tpra1 or Tpra40: transmembrane protein, adipocyte associated 1 or of 40 kDa), which also localizes to primary cilia upon Hh stimulation and positively regulates Hh signaling. Interaction experiments place Gpr175 at the level of PKA and upstream of the Gαi component of heterotrimeric G proteins, which itself localizes to cilia and can modulate Hh signaling. Gpr175 or Gαi1 depletion leads to increases in cellular cAMP levels and in Gli3 processing into its repressor form. Thus we propose that Gpr175 coupled to Gαi1 normally functions to inhibit the production of cAMP by adenylyl cyclase upon Hh stimulation, thus maximizing signaling by turning off PKA activity and hence Gli3 repressor formation. Taken together our data suggest that Gpr175 is a novel positive regulator of the Hh signaling pathway.
Introduction
The Hedgehog (Hh)2 signaling pathway is conserved from fly to man and plays a pivotal role in several aspects of embryonic development. In vertebrates Hh signaling plays a crucial role in the development of many organs and tissues, including the neural tube, cerebellum, gut, and lungs (1, 2). However, in adults the pathway plays a limited role in maintenance of normal homeostasis, whereas aberrant pathway activation can cause cancers such as medulloblastoma and basal cell carcinoma (3, 4).
In vertebrates, Hh signaling requires the presence of primary cilia (5, 6). Primary cilia are solitary microtubule-based protrusions that project from the surface of most quiescent cells and act to sense the surrounding environment and transmit signals to the cell body. In the absence of Hh ligand, the 12-transmembrane Hh receptor Patched (Ptch1) is enriched in the primary cilium and biochemically inhibits the activity of Smoothened (Smo), a member of the Frizzled (Fz) family of seven-pass transmembrane proteins that is thought to act like a G-protein coupled receptor (GPCR) to transmit the Hh signal. In this “OFF” state, Smo is excluded from the cilium and Suppressor of Fused (SuFu, probably along with Kif7) forms a complex with the Gli family of transcription factors (Gli1, Gli2, and Gli3). SuFu retains the Glis in the cytoplasm, where they are phosphorylated by the concerted effort of three kinases (7). In the absence of Hh, cAMP-dependent protein kinase A (PKA) phosphorylates key residues on Gli2/Gli3 to prime phosphorylation by glycogen synthase kinase 3β and casein kinase 1. These phosphorylation events recruit βTrCP and SCF1, leading to proteasomal cleavage of full-length Gli2 and Gli3 (Gli-FL) transcription factors into repressors (Gli-R), which inhibit transcription of Hh target genes (8–10).
In the presence of Hh, Ptch1 exits the cilium (11), thereby relieving its inhibition of Smo, which now moves into the cilium to initiate signaling (12). Downstream of Smo, the SuFu·Gli complex (likely with Kif7) also accumulates in the cilium (13–15), and Gli phosphorylation by PKA is somehow inhibited, preventing cleavage of Gli-FL into Gli-R. There, the SuFu·Gli complex dissociates via unknown mechanism(s) (16), thereby leading to the activation of Gli-FL into Gli-A, which can activate Hh target gene transcription in the nucleus. The Gli1 family member is not processed and functions solely as a transcriptional activator to enhance signaling, itself being an early Hh target gene that is up-regulated within 3 h upon Hh stimulation (15, 17). Gli2 is the initial activator when present in the Gli2-A form, whereas Gli3-A has less pronounced activity and functions more strongly as a repressor (Gli3-R) (9, 18, 19).
GPCRs are one of the largest gene families of signaling receptors that respond to diverse extracellular stimuli such as morphogens, neurotransmitters, neuropeptides, hormones, lipids, nucleotides, and other biological molecules. They are coupled to heterotrimeric G-proteins (Gαβγ), which are classified into four subfamilies based on their Gα subunit: Gαs, Gαi, Gαq, and Gα12. GPCR activation leads to guanine nucleotide exchange factor (GEF) activity that replaces GDP with GTP, resulting in dissociation of the heterotrimeric complexes into their Gβγ and Gα subunits. The liberated Gα protein initiates a cascade of downstream second messenger pathways that eventually induces gene transcription. One of the second messengers regulated by GPCR signaling is adenylyl cyclase, which is stimulated by Gαs and inhibited by Gαi. Activated adenylyl cyclase converts cellular ATP to cAMP, a ubiquitous second messenger, which, among other roles, increases the activity of PKA, an enzyme that plays a crucial negative role in the Hh pathway. Inactivation of PKA in mice leads to midgestation lethality and a completely ventralized neural tube that is characteristic of Hh pathway over activation (20, 21). PKA functions at the base of the cilium to regulate the processing of Gli-FL into Gli-R (21). However, the upstream signals that mediate the activation or repression of PKA in Hh signaling remain largely elusive.
A role for GPCRs that can modulate cAMP levels to regulate the activity of PKA in Hh signaling had been speculated based on the finding that uncoupling of Gαi by pertussis toxin in zebrafish resulted in disrupted muscle development, recapitulating a partial Hh loss of function phenotype (22). Subsequently human SMO was shown to activate Gαi-mediated signaling in frog melanophores, consistent with GPCR-like activity (23). Both Gαs and Gαi have been proposed to function downstream of Smoothened in Hh signaling (23–27).
Furthermore, G protein-coupled receptor kinases (GRKs) and β-arrestin 2, two classical GPCR-interacting proteins, were shown to interact with Smo to facilitate Hh signaling (28, 29). Recently, it was shown that constitutive activation of the GPCR Gpr161 increases cellular cAMP levels via Gαs, thus increasing activation of PKA and enforcing repression of Hh signaling (30). This negative Hh pathway regulator Gpr161 is particularly interesting because it relocates from the cilium to the cell body upon Hh stimulation. However, none of these studies addressed whether there is a direct role for a GPCR besides Smo in positive modulation of Hh signaling.
The orphan GPCR Gpr175 (also known as Tpra40 or Tpra1), whose physiological functions are unknown, was first cloned as a GLP-1 receptor homolog in 3T3-L1 adipocytes and is also expressed in tissues whose development requires Hh signaling, including heart, brain, lung, pancreas, and muscle (31, 32). Gpr175 is predicted to have seven putative transmembrane domains, but does not fit into any of the four GPCR subclasses, thus its function as a GPCR remains to be confirmed. Furthermore, GPR175 was proposed to regulate early mouse embryogenesis via its interaction with Sjögren's syndrome-associated protein NA14 (SSNA1) (33), a protein that putatively regulates ciliary transport and Hh signaling (34). We therefore investigated whether Gpr175 plays a role in the Hh pathway and show that it has GPCR-like properties and can positively modulate Hh signaling by inhibiting PKA activity by lowering cAMP levels via Gαi1.
Experimental Procedures
siRNA and cDNA Transfections
ON-TARGETplus siRNAs (all pools of four) for human Gpr175 (catalog number L-005746-00), and murine Gpr175 (and individual Gpr175 siRNAs 09-12, catalog number L-047743-01), Smo (catalog number L-041026-00), Ptch1 (catalog number L-041027-00), Gαi1 (catalog number L-051423-00), Ift88 (catalog number L-050417-00), SuFu (catalog number L-047171-00), Gli3 (catalog number L-045798-00), and non-targeting control (NTC; catalog number D-001810-10) were purchased from Dharmacon Inc. (Lafayette, CO). S12 cells were seeded into 96-well white wall clear-bottom plates at 6000 cells/well; 8-well LabTekII microscope slides at 20,000 cells/well; or 60-mm plates at 4 × 105 cells/well and reverse-transfected with 50 nm final siRNA pools (or 25 nm for individual siRNAs), following 20 min preincubation of siRNAs with DharmaFECT-2 (Dharmacon) in Opti-MEM (Gibco) at room temperature. For epistasis-type experiments, two siRNA pools at 50 nm each were used.
For cDNA transfection, C3H10T1/2 cells were seeded into 6-well plates at 1 × 105 cells/well and transfected the next day with 2 μg of plasmid preincubated with 6 μl of GeneJuice transfection reagent (Novagen, Madison, WI) in Opti-MEM (Gibco) at room temperature. After 24 h at 37 °C the medium was changed to 0.5% serum HG-DMEM ± 200 ng/ml of octyl-Shh (Genentech) for another 24 h prior to quantitative PCR analysis as described below. COS7 cells plated at 2.4 × 104 cells/well in 8-well slides were reverse transfected with 0.3 μg of HA-Gpr175 cDNA and 0.9 μl of FuGENE 6 (Roche) for 66 h.
S12 Gli-luciferase Assay
S12 cells, which are C3H10T1/2 fibroblasts stably transfected with 8× Gli-binding sites fused to a luciferase reporter (35), were reverse transfected as above. They were mycoplasma-free but not sequence verified. After 48 h the medium was changed to 0.5% serum HG-DMEM ± 200 ng/ml of octyl-Shh (hereafter called Hh) and incubated for another 24 h to stimulate signaling. PKA was inhibited using 80 μm cell permeable 14–22 amide (Tocris Biosciences). Gli-luciferase activity was measured using an HTS-Steady Lite luciferase detection kit (PerkinElmer Life Sciences) and a TopCount luminometer; multiple assays were carried out, each in triplicate. Data were corrected for cell viability using cell Titer Glo (Promega).
Real-time Quantitative PCR (qRT-PCR)
Total RNA was extracted from cells using the RNeasy Protect Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. On-column genomic DNA digestion was performed with an RNase-free DNase set (Qiagen). cDNA synthesis from total RNA was conducted using the High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA) with random hexamer primers. Quantitative PCR was performed in triplicate on an ABI PRISM® 7500 Sequence detection system (Applied Biosystems) using murine or human ribosomal protein L19 (Rpl19 or RPL19) as the endogenous control. Gene expression was calculated using the relative quantification (2-ΔΔCt) method. PCR primers and TaqMan probes (5′ FAM and 3′ TAMRA-labeled) are as follows: mGli1, 5′ primer: GCA GTG GGT AAC ATG AGT GTC T; mGli1, 3′ primer: AGG CAC TAG AGT TGA GGA ATT GT; mGli1 probe: CTC TCC AGG CAG AGA CCC CAG C; mGpr175 5′ primer: TGC AGG AGG CCA ATG GAA; mGpr175 3′ primer: GGG CTC ACT GAT ATT GGA TGC T; mGpr175 probe: ACA GCG TGG CCA CCG CCC; hGli1 5′ primer: CGC TGC GAA AAC ATG TCA AG; hGli1 3′ primer: CCA CGG TGC CGT TTG GT; hGli1 probe: CAG TGC ATG GTC CTG ACG CCC A; hGpr175 5′ primer: CCT GGT CTA CTC TCT GGT GGT CAT; hGpr175 3′ primer: CCG AGA AGG CAG GGA GAT G; hGpr175 probe: CCC AAG ACC CCG CTG AAG GAG C.
Antibodies
Purchased antibodies were anti-GLI1 mouse monoclonal L42B10 (Cell Signaling Technology catalog number 2643); anti-GPR175 mouse monoclonal 6H2 (Santa Cruz sc-134350); mouse anti-rat Gαi1 R4 (Santa Cruz sc-13533); rabbit anti-mouse adenylyl cyclase-III C-20 (Santa Cruz sc-588); mouse anti-GFP JL-8 (Clontech); rabbit anti-ARL13b (Proteintech 11711-1-AP); mouse anti-acetylated tubulin 6–11B-1 (Sigma T6793), mouse anti-tyrosinated tubulin 1A2 (Sigma T9028), Alexa 488-conjugated anti-HA tag monoclonal 16B12 (Covance A488-101L-100), and goat anti-SUFU M-15 (Santa Cruz sc-10934). Rabbit 2676A and mouse 6F5 anti-Gli3 antibodies were previously described (15). Rabbit polyclonal 4241 to a peptide comprising the C-terminal 19 amino acids of murine Gpr175 (CHTGSINSTDSERWKAINA-COOH) was raised at YenZym (South San Francisco, CA). Rabbit polyclonal 10322B to MBP-mouse Smo amino acids 684–793 was generated as described (16) and validated for immunofluorescence by its recognition of gD-tagged transfected SMO and colocalization with S12 cell cilia in a Hh-dependent fashion (data not shown).
Constructs
Murine Gpr175 was PCR amplified from a cDNA template (NM_011906.2) using Pfu Turbo (Stratagene) and the following pairs of PCR primers: forward primer (5′-CAC CAT GGC CAG CCT GCA) and reverse primer (5′-GGC ATT GAT GGC TTT CCA GC); and forward primer (5′-CAC TCG AGG CCA GCC TGC AGG A) and reverse primer (5′-TCT GGC GCG CCT CAG GCA TTG AT).
The PCR amplified fragment was directionally cloned to generate the following constructs: 1) Gpr175-LAP5: used pENTR/D TOPO vector (Invitrogen) via TOPO cloning kit according to the manufacturer's instructions (Invitrogen). This Entry vector was further subcloned into vector pgLAP5 (36) for the C-terminal LAP (S-GFP)-tagged construct by Gateway recombination (Invitrogen). pgLAP5 was a gift from Peter Jackson (Addgene plasmid 19706). Gpr175-LAP5/3T3 stable cell lines were made by FuGENE 6® transfection and selection in 150 μg/ml of hygromycin. 2) HA-Gpr175: used pRK5-nHA via XhoI and AscI restrictions sites, yielding N terminally HA-tagged Gpr175. siRNA-resistant Gpr175, in which all the siRNA seed sites corresponding to individual Gpr175 siRNAs 10–12 have been replaced by silent mutations (GCCACCGTCGCCGATAAA, GCAATCGAACTCAGCGTC, CGCCAATTTTGGCTGGTG), was synthesized by Genscript (Piscataway, NJ) and subsequently cloned into the pgLAP5 vector as described above. Human SMO-M2 (W535L) in pRK5 vector was previously described (37).
Western Blotting
Cells were lysed in denaturing RIPA buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate) containing freshly added phosphatase inhibitor mixtures I and II (Sigma), Complete protease inhibitor mixture (Roche), and 1 mm phenylmethylsulfonyl fluoride (PMSF; Sigma). Equal amounts of protein (measured using the BCA kit (Pierce)) were separated on 4–12% reducing Tris glycine gels prior to transfer to nitrocellulose membranes (Invitrogen) and blocking in 5% (w/v) milk in TBST (Tris-buffered saline with 0.05% Tween 20). Anti-Gli3 6F5 (15) was used at 5 μg/ml, mouse anti-GPR175 at 0.2 μg/ml, and rabbit anti-Gpr175 at 0.32 μg/ml overnight at 4 °C. Detection employed HRP anti-rabbit (Jackson ImmunoResearch) or anti-mouse (GE Healthcare) and ChemiGlow West (ProteinSimple). Blots were reprobed for protein loading using 1:10,000 mouse anti-tubulin 1A2 and ECL detection (GE Healthcare). Blots were exposed to BioMax light film (Kodak).
Immunofluorescence Microscopy
S12 cells were plated on 8-well slides at 30,000 cells/well and reverse transfected for 72 h with siRNAs as above. Gpr175-LAP5/3T3 cells were plated at 15,000 cells/well for 72 h. The final 16–24 h were in serum-free media ± 200 ng/ml of octyl-Shh (Genentech), then cells were fixed with 3% paraformaldehyde for 20 min at room temperature and quenched for 10 min in 50 mm NH4Cl. Cells were then permeabilized with 0.1% Triton X-100 (Sigma) in PBS for 10 min, blocked in 1% BSA for 30 min, and incubated overnight with the following primary antibodies in PBS with 1% BSA: 2 μg/ml of 6H2 anti-GPR175, 1 μg/ml of mouse anti-GFP, 2 μg/ml of mouse anti-Gαi1, 2 μg/ml of rabbit anti-adenylyl cyclase, 1 μg/ml of rabbit anti-Smo 10322B, or 3 μg/ml of rabbit anti-Gli3 2676 together with 1:3000 mouse anti-acetylated tubulin or 1:500 rabbit anti-ARL13b. After 3 × 10-min washes with PBS, Cy3, or FITC-conjugated secondary anti-rabbit or anti-mouse antibodies (Jackson ImmunoResearch) in PBS, 1% BSA were applied for at least 1 h at room temperature, washed 3 times with PBS and coverslips were mounted using ProLong Gold with DAPI (Invitrogen). HA-Gpr175-transfected COS cells were stained with 2 μg/ml of 6H2, followed by Cy3 anti-mouse, which was subsequently blocked with excess (10 μg/ml) mouse IgG to permit co-staining with 1 μg/ml of Alexa 488-conjugated anti-HA. Slides were imaged using an Axio Imager M2 microscope with a ×63 N.A. 1.4 Oil DIC objective (PLAN-APOCHROMAT) and SlideBook 5.5 software. The percentage of cilia (as identified by acetylated tubulin or Arl13b staining) that co-stained for Gpr175 or Smo was counted manually.
293 CRE-luciferase Assay
HEK293 cells were plated at 10,000 cells/well in 96-well white walled plates. Cells were reverse transfected with 50 ng of Cre-luciferase, 10 ng of Renilla luciferase (Promega), and 90 ng of murine Gpr175 with an N-terminal HA tag (HA-Gpr175 in pRK5) or GFP control (pEGFP-N3) with 0.45 μl of FuGENE 6® in 25 μl of Opti-MEM. After 36 h, different amounts of forskolin (Cell Signaling Technologies) were added for 4 h and the firefly and Renilla luciferase were measured using Dual Glow Luciferase Substrate (Promega) and a TopCount or EnVision luminometer. Firefly values were normalized to Renilla values and expressed as a percentage of the 20 μm forskolin (Fsk) + GFP value.
FACS
EDTA-detached unpermeabilized HA-Gpr175-transfected 293 cells were incubated on ice for 1 h with 1 μg/ml of Alexa 488 anti-HA tag in 3% FBS in PBS, washed 3 times, and analyzed on a FACSCalibur (Becton Dickinson), gated on live cells using propidium iodide.
Daoy Cell Sequencing
Daoy cell RNA extraction, quality control, library preparation, and sequencing were done as described previously (38) on an Illumina HiSeq2500, generating 30 million single end 50 base pair reads.
Statistics
Comparison of Gpr175-depleted versus non-depleted cells was performed using the Student's unpaired t test (GraphPad).
Results
Gpr175 Is an N-Glycosylated GPCR
Gpr175 shows a high level of conservation from zebrafish to man and is predicted to be an orphan GPCR containing seven transmembrane domains (Fig. 1A) and an extracellular N terminus. FACS analysis of N terminally HA-tagged Gpr175 reveals the N terminus is indeed extracellular (Fig. 1B). Western blotting of endogenous Gpr175 in S12 cells (murine C3H10T1/2 osteoblasts stably expressing a Hh-inducible Gli-luciferase reporter (35)) with a novel rabbit polyclonal antibody revealed a single band around 50 kDa, which was reduced to 37 kDa following PNGase F treatment (Fig. 1C), consistent with its predicted molecular mass of 40.6 kDa (32) and N-glycosylation. The antibody was specific because Gpr175 siRNA (siGpr175) treatment, which knocked down the mRNA by 80% (Fig. 1D), rendered the 50-kDa band undetectable (Fig. 1E).
FIGURE 1.

Gpr175 is a conserved glycosylated GPCR-like protein. A, sequence alignment of Gpr175 protein showing 91% identity between human (Q86W33) and mouse (Q99MU1), and 70% identity between human and zebrafish Tpra1 (Q4V8X0). The seven putative transmembrane (TM) domains are underlined, identical residues are boxed, and putative N-glycosylation (NxS) sites are marked with an arrowhead. B, the N terminus of Gpr175 is extracellular, as predicted for a GPCR. Flow cytometry analysis of HEK-293T cells transfected with empty vector (gray) or N terminally tagged HA-Gpr175 (black) for 48 h and labeled with Alexa 488-conjugated anti-HA without permeabilization. C, endogenous murine Gpr175 is N-glycosylated. Western blot of S12 cells treated ±Hh, with lysates treated with PNGase F or buffer alone for 1 h at room temperature, probed with rabbit anti-Gpr175 polyclonal antibody 4241 or with tubulin antibody 1A2 as a loading control. M, molecular mass markers in kDa. D, 80% depletion of Gpr175 mRNA in S12 cells by treatment with siGpr175 with (black) or without Hh (gray) measured by qRT-PCR. Data were normalized to the housekeeping gene mRpl19 mRNA and expressed relative to siNTC −Hh control. Mean ± S.D. of three individual experiments are shown. E, Western blot with rabbit polyclonal anti-Gpr175 showing endogenous Gpr175 protein in S12 cell lysates (40 μg/lane) is apparently completely depleted by siGpr175 (±Hh stimulation) in parallel with an experiment shown in Fig. 2A. Similar results were obtained with an independent anti-GPR175 antibody (6H2, data not shown). Molecular mass markers in kDa are shown on the right. Quantification of Gpr175 relative to tubulin loading control and NTC-Hh is shown under the blot, which is representative of one done three times.
Gpr175 Is a Positive Modulator of Hh Signaling
To investigate the role of Gpr175 in Hh signaling, we monitored Gli-luciferase levels in S12 cells depleted of Gpr175. Hh stimulation of control cells resulted in the expected induction of reporter activity, but this was significantly diminished by Gpr175 depletion (Fig. 2A). Inhibition was reproducibly around 50%, less than the 75% caused by depletion of Ift88, a gene required for cilia function and hence Hh signaling (6, 34). There was no significant effect of siGpr175 in unstimulated cells (Fig. 2A).
FIGURE 2.

Gpr175 depletion impairs Hh signaling. A, percentage Gli-luciferase (Gli-Luc) activity remaining after transfection with siNTC (non-targeting control), siIft88 (positive control), and siGpr175 (all pools of 4 siRNAs) in S12 cells with (black) or without Hh (gray). Data were normalized to 100% for siNTC + Hh and the mean ± S.D. of four individual triplicate experiments are shown (*, p ≤ 0.05). B, endogenous murine Gli1 expression in S12 cells by qRT-PCR following Gpr175 depletion with (black) or without Hh (gray) and normalized to mRpl19 and siNTC -Hh control (n = 3; *, p ≤ 0.05). The mean fold-induction by Hh for each siRNA is indicated above each pair of bars. C, representative Western blot of cells treated as in B and probed with a Gli1-specific monoclonal antibody. Quantitation of three such blots normalized to tubulin revealed a 54.3 ± 20.5% decrease in Gli1 protein (p = 0.015). D, as in B but using mouse embryonic fibroblasts (n = 3; *, p ≤ 0.05). E, three of four individual Gpr175 siRNAs of the pool suppress signaling; n = 3 experiments performed as in A. F, rescue of murine Gpr175 knockdown (by pool of siRNAs #10–12) with siRNA-resistant mouse Gpr175-LAP5 (srGpr175) in C3H10T1/2 cells; mGli1 induction by Hh was monitored by qRT-PCR (mean ± S.D. of 3 independent experiments). Numbers above the black bars indicate the fold-increase over −Hh for that treatment; fold-increase induced by Gpr175 over empty vector (both +Hh) is indicated above the connecting lines. G, human medulloblastoma Daoy cells were depleted of GPR175 and endogenous hGLI1 levels were monitored in the absence of Hh stimulation (because this cell line is constitutively active) with and without 16 h treatment with 2 μm Smo inhibitor cyclopamine. hGLI1 mRNA levels were normalized to hRPL19. H, overexpression of Gpr175 enhances Hh signaling. Endogenous mGli1 expression by qRT-PCR following transfection of empty vector only, Gpr175-LAP5 or SMO-M2 with (black) or without Hh (gray) in C3H10T1/2 cells. Data were normalized to the housekeeping gene mRpl19 mRNA and expressed as percentage of SMO-M2 + Hh. Mean ± S.D. of three individual experiments are shown (*, p ≤ 0.05). Error bars in all panels represent mean ± S.D. of at least three independent triplicate experiments.
To ensure the inhibition of Gli-luciferase activity was not a luciferase translation artifact, we directly monitored endogenous Gli1 levels following Hh stimulation in Gpr175-depleted S12 cells. Gli1 induction was indeed about 50% lower at both the mRNA and protein levels after Gpr175 knockdown (Fig. 2, B and C). This was not a peculiarity of S12 cells because similar results (60% inhibition) were seen in Gpr175-depleted mouse embryonic fibroblasts (Fig. 2D). Inhibition of Hh signaling was not an artifact of a single siRNA, because three of the four individual Gpr175 siRNAs reproduced the effect of the original pool (Fig. 2E). Importantly, the three active siRNAs could be rescued by an siRNA-resistant form of mouse Gpr175 (Fig. 2F), suggesting that inhibition is not an off-target effect.
Furthermore, GPR175 can also modulate the Hh signaling context of cancer, because its depletion in human Daoy medulloblastoma cells (which exhibit constitutive pathway activation by virtue of inactivating PTCH1 mutations: P1315L, P1164L, and P1249L) reduced hGli1 levels by 65%, and was further reduced by treatment with the Smo inhibitor cyclopamine (Fig. 2G). Conversely, constitutive overexpression of Gpr175-LAP5 (C terminally S-GFP tagged) in C3H10T1/2 cells following Hh stimulation resulted in robust induction of Gli1 mRNA in the presence of Hh (Fig. 2H), albeit not quite as strong as that induced by an oncogenic form of Smoothened (SMO-M2 (39)) (Fig. 2H). Taken together our data suggest that Gpr175 is a positive modulator of Hh signaling in multiple cell lines.
Gpr175 Localizes to Cilia in a Hh-dependent Manner
The role of the primary cilium in Hh signaling has been well documented (40, 41). Upon Hh treatment, several components of the pathway shuttle in or out of the cilium, highlighting the importance of this organelle in orchestrating the proper Hh response. We therefore tested whether Gpr175 also localizes to cilia. Constitutive overexpression of HA-Gpr175 (not shown) or Gpr175-LAP in NIH-3T3 cells (Fig. 3A) indeed revealed Gpr175 localization in cilia, independent of Hh, reminiscent of that found with Smo overexpression (data not shown and Ref. 42). Anti-GPR175 monoclonal 6H2 was first validated for immunofluorescence on HA-Gpr175 expressed in COS cells (Fig. 3B), then used to stain S12 cells. Endogenous Gpr175 was only found in a few (∼20%) cilia in the absence of Hh stimulation, but Hh treatment significantly increased this to ∼60% (Fig. 3, C-E). Because Hh does not alter Gpr175 levels (Fig. 1, D and E), Gpr715 likely translocates into cilia, behavior very similar to that of Smo, despite only 9% sequence identity (Fig. 3E and data not shown). Gpr175 similarly localized to cilia in other Hh-signaling cell types, including MEFs and Daoy (data not shown), consistent with a conserved function of Gpr175 in different cell lines and suggesting Gpr175 requires Hh for localization and activity.
FIGURE 3.
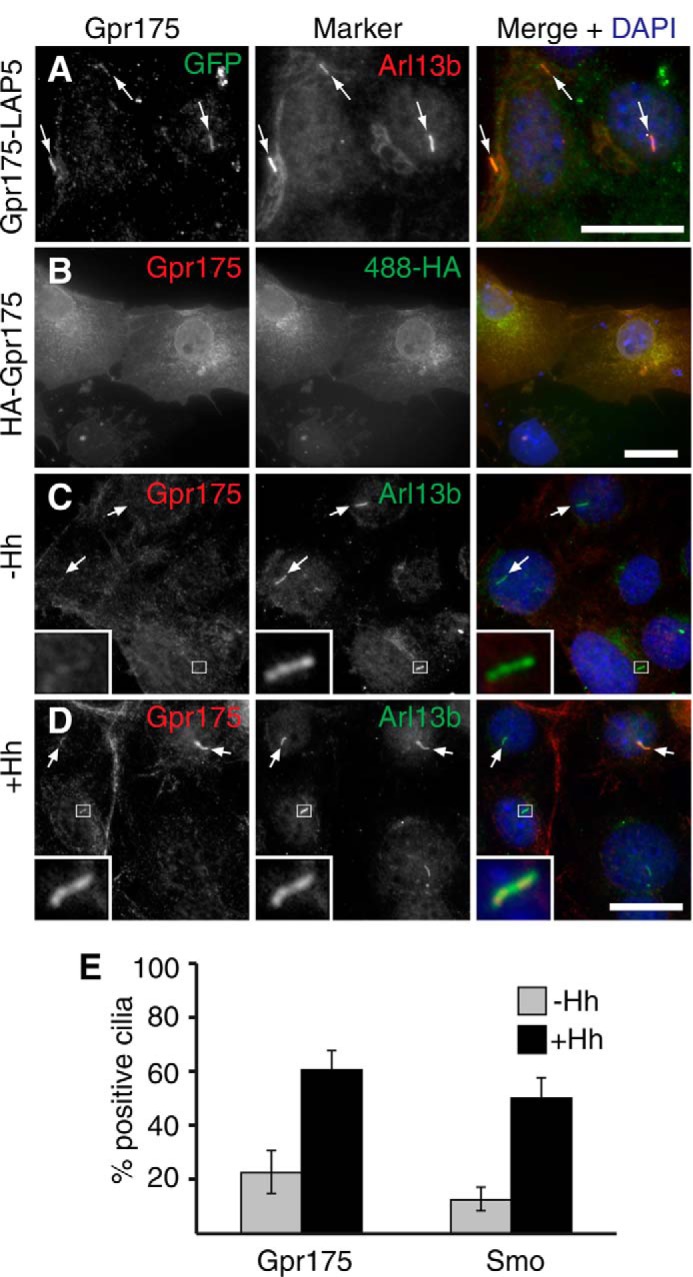
Gpr175 localizes to primary cilia of S12 cells in a Hh-dependent manner. A, NIH-3T3 cells stably expressing Gpr175-LAP5 were grown to confluence and serum starved for 16 h (without Hh) to induce ciliation. Cells were fixed and co-stained with mouse anti-GFP to detect Gpr175 (green, left) and rabbit anti-ARL13b (red, center) to label cilia. B, anti-human GPR175 monoclonal antibody 6H2 cross-reacts with HA-tagged murine Gpr175 transiently transfected in COS cells. 6H2 (red, left) colocalizes with anti-HA (green, center) at the plasma membrane and in the secretory pathway. Similar 6H2 staining was obtained in the absence of the anti-HA antibody, confirming it was not a cross-reactivity artifact (data not shown). C and D, S12 cells were fixed and stained with anti-GPR175 6H2 (red) and anti-ARL13b (green) following 24 h serum starvation alone (C) or with Hh stimulation (D). Scale bars are all 20 μm and the insets show ×5 magnification of the boxed area. The merged channels with nuclear counterstain DAPI (blue) are shown in the right column and arrows indicate cilia. Gamma levels were adjusted over the whole image to optimize the appropriate signals where necessary. E, the percentage of Arl13b-demarcated cilia positive for Gpr175 staining was scored, revealing that Gpr175 was found in only about 20% of cilia in the absence of Hh (gray), but in 60% following overnight Hh stimulation (black), very similar to the behavior of Smoothened (mean ± S.D. of 2 experiments, each counting ≥100 cilia, is shown).
Gpr175 Functions Downstream of Smo
We next wanted to test at which step Gpr175 can modulate activity of the Hh signaling pathway. Because Hh signaling requires primary cilia (5, 6) and changes in cilia length affect Hh signaling (34), a simple hypothesis would be that Gpr175 could affect Hh signaling via regulation of ciliogenesis. However, Gpr175-depleted cells showed no changes in either cilia number or length (data not shown), ruling out this possibility.
Hh activation involves Hh binding to its receptor Ptch, which relieves its inhibition of Smo, thus dissociating SuFu-bound Gli2/3 and suppressing the kinase activity of PKA, hence inhibiting Gli2/3 repressor formation (Fig. 4A). Activated Gli2/3 then translocates to the nucleus to turn on target gene transcription. To study the interaction of Gpr175 with other core components within the Hh pathway, we attempted epistasis experiments in S12 cells by knocking down Gpr175 along with depletion or activation of other pathway components and monitoring Gli-luciferase activity. Partial depletion (by 80% at the mRNA level) of the negative regulator Ptch1 (which cannot be completely knocked down because its depletion stimulates the pathway and it is itself a target gene) led to the expected ligand-independent pathway activation (5-fold induction of Gli1 compared with NTC). This was partially suppressed by co-depletion of Gpr175 (Fig. 4B), hinting that Gpr175 may function at the level of or downstream of Ptch1.
FIGURE 4.
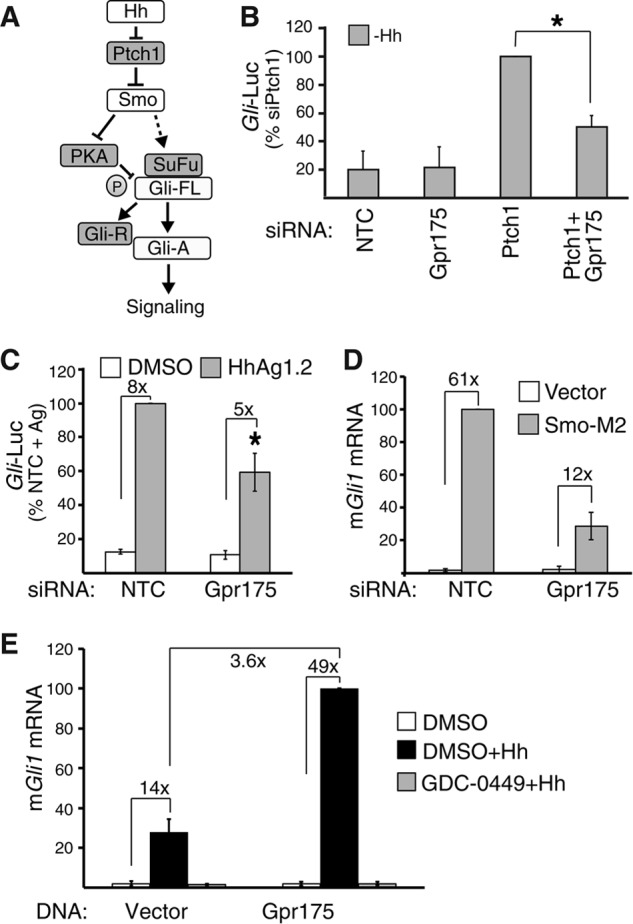
Gpr175 requires Smoothened. A, schematic of the key Hh pathway components, with positive regulators Hh, Smo, and Gli-A (active) in white and negative regulators Ptch1 (Patched 1), SuFu, Gli-R (repressor), and PKA in gray. T-bars indicate inhibition; arrows indicate activation. B, Gli-luciferase (Gli-Luc) activity in S12 cells depleted of Ptch1 or Gpr175 (both using siRNA pools) alone or in combination in the absence of Hh (expressed as a percentage of siPtch1). Mean ± S.D. of three independent experiments are plotted (*, p ≤ 0.05). C, Gli-luciferase induction in siRNA-transfected S12 cells stimulated with 100 nm of the Smoothened agonist Hh-Ag1.2 (gray bars) or vehicle (white) instead of Hh. Error bars represent S.D. of the mean of three independent experiments (*, p ≤ 0.05). D, Gli1 induction in C3H10T1/2 cells first transfected for 24 h with siNTC or siGpr175, then re-transfected with either SMO-M2 (gray) or empty vector (white) for another 24 h cells, followed by serum starvation for 16 h without Hh; mean ± S.D. of 3 independent experiments normalized to SMO-M2 + siNTC. E, mGli1 levels by qRT-PCR following 48 h transfection of C3H10T1/2 cells with empty vector or Gpr175-LAP5 construct and treated with either DMSO alone (white), DMSO +Hh (black), or 100 nm GDC-0449 +Hh (gray) in serum starvation medium for the last 16 h. Data were normalized to the housekeeping gene mRpl19 mRNA and expressed as percentage of Gpr175 +Hh. Mean ± S.D. of four independent experiments are shown.
To confirm this, we examined Gli induction by direct activation of Smo with the Smo agonist Hh-Ag1.2 (35) (Fig. 4C) or by overexpression of SMO-M2 (Fig. 4D). Gpr175 depletion suppressed Smo activation in both cases (by 41 and 72%, respectively), suggesting Gpr175 might function at the level of or downstream of Smo. Gpr175 is dependent on Smo activity because transfected Gpr175 cannot stimulate Gli1 transcription in the presence of Hh if Smo is inhibited by GDC-0449 (vismodegib (43); Fig. 4E). Cyclopamine treatment and Smo knockdown similarly prevented Gpr175-mediated pathway stimulation, ruling out the unlikely possibility that GDC-0449 inhibits Gpr175 as well as Smo (data not shown).
Gpr175 Acts at the Level of PKA
Stimulation of cells by knocking down the negative regulator SuFu (giving 4-fold induction of Gli1) was also suppressed by siGpr175 (Fig. 5A). However, Western blotting revealed a substantial amount of SuFu remaining after knockdown (Fig. 5B), consistent with its reportedly long (24 h) half-life in the absence of Hh (44). Similarly, Gpr175 knockdown partially suppressed the 7-fold pathway activation induced by removal of Gli3 repressor, although again knockdown was incomplete (albeit very efficient for the more relevant Gli3-R, which was only detectable at higher exposures; Fig. 5, C and D).
FIGURE 5.
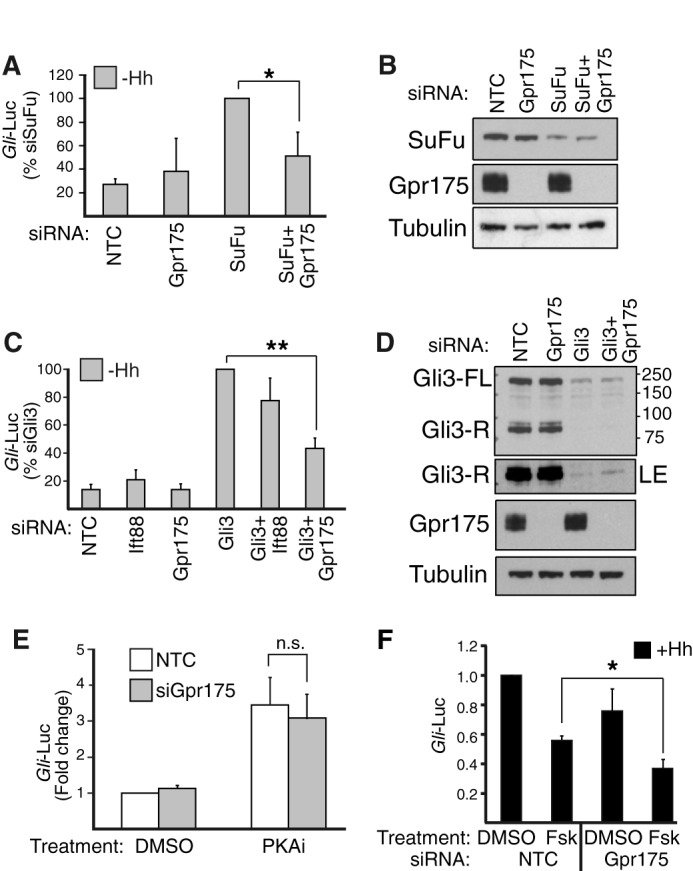
Gpr175 functions at the level of PKA. A, Gli-luciferase (Gli-Luc) activity in S12 cells depleted (with siRNA pools) of SuFu or Gpr175 alone or in combination in the absence of Hh (expressed as a percentage of siSuFu). Mean ± S.D. of three independent experiments are plotted (*, p ≤ 0.05). B, representative Western blot (of n = 3) with goat anti-SUFU, rabbit anti-Gpr175, and anti-tubulin (loading control) antibodies following SuFu and/or Gpr175 knockdown in S12 cell lysates (40 μg/lane). Note that knockdown of each protein (100% for Gpr175 and 57 ± 5.7% for SuFu; n = 3) was unaffected by the presence of the second siRNA. C, as in A but with siGli3 and siIft88 (pools of 4 siRNAs) as a positive control. Error bars represent S.D. of the mean of five independent experiments (**, p ≤ 0.01). D, a representative of 3 Western blots with mouse anti-Gli3, rabbit anti-Gpr175, and anti-tubulin (loading control) antibodies showing endogenous expression of Gli3 (full-length (73 ± 23% depleted) and repressor form (96 ± 2.8% depleted)) and Gpr175 (100% depleted) in S12 cell lysates (40 μg/lane) that were treated with siGli3 or siGpr175 alone or in combination. LE, longer exposure. E, S12 cells were stimulated with 80 μm PKA inhibitor 14–22 amide (or DMSO vehicle control) for 24 h in the absence of Hh following siGpr175 (gray) or non-targeting control (white) transfection (n.s., not significant). F, as in E except Hh-stimulated S12 cells were inhibited with 200 nm Fsk or DMSO control for the last 24 h. Mean ± S.D. of 3 independent experiments normalized to siNTC + DMSO (*, p ≤ 0.05).
Because Gli3 cleavage into Gli3-R is initiated by PKA phosphorylation, we tested the interaction of Gpr175 and PKA. The cell permeable PKA inhibitor 14–22 amide caused the expected ligand-independent activation of Hh reporter activity, but this was not rescued by Gpr175 depletion (Fig. 5E), placing Gpr175 upstream of or at the level of PKA. Gpr175 knockdown conversely enhanced the inhibition of Gli-luciferase activity by the adenylyl cyclase activator Fsk (which increases cAMP, hence stimulating PKA; Fig. 5F). Taken together these data suggest Gpr175 may be able to modify the activity of PKA to affect Gli2/3 processing.
Gpr175 Acts Upstream of the Gαi1 Subunit of Heterotrimeric G Proteins
To address the molecular mechanism(s) by which Gpr175 could affect Hh signaling, we first attempted to determine to which class of Gα proteins this orphan GPCR is coupled. Because of its potential interaction with PKA, which is regulated by cAMP via adenylyl cyclase, we tested the effect of Gpr175 overexpression in a cAMP response element reporter assay. Treatment of cells with Fsk led to the expected dose-dependent increase in reporter activity (Fig. 6A). However, co-expression of Gpr175 suppressed this increase, suggesting that Gpr175 functions via Gαi, which has been previously implicated in Hh signaling (24, 26, 27). If Gpr175 interacts with Gαi, both proteins should also localize to cilia in the presence of Hh. Indeed Gαi1 was found in most cilia, albeit in a Hh-independent manner (Fig. 6B). The ciliary staining was not increased by Hh (data not shown) and was specific, as Gαi1 depletion diminished the signal (Fig. 6C). Adenylyl cyclase (AC-III), a downstream effector of Gαi1, also localized to cilia in S12 cells (Fig. 6D), consistent with its known ciliary localization in neuronal cells (45). Thus several essential components of GPCR activation are localized to the cilia. Furthermore, S12 cells depleted of Gαi1 show compromised Hh signaling and co-depletion of Gpr175 did not further decrease the signal, suggesting that Gpr175 acts upstream of Gαi1, as would be expected for a GPCR (Fig. 6E).
FIGURE 6.
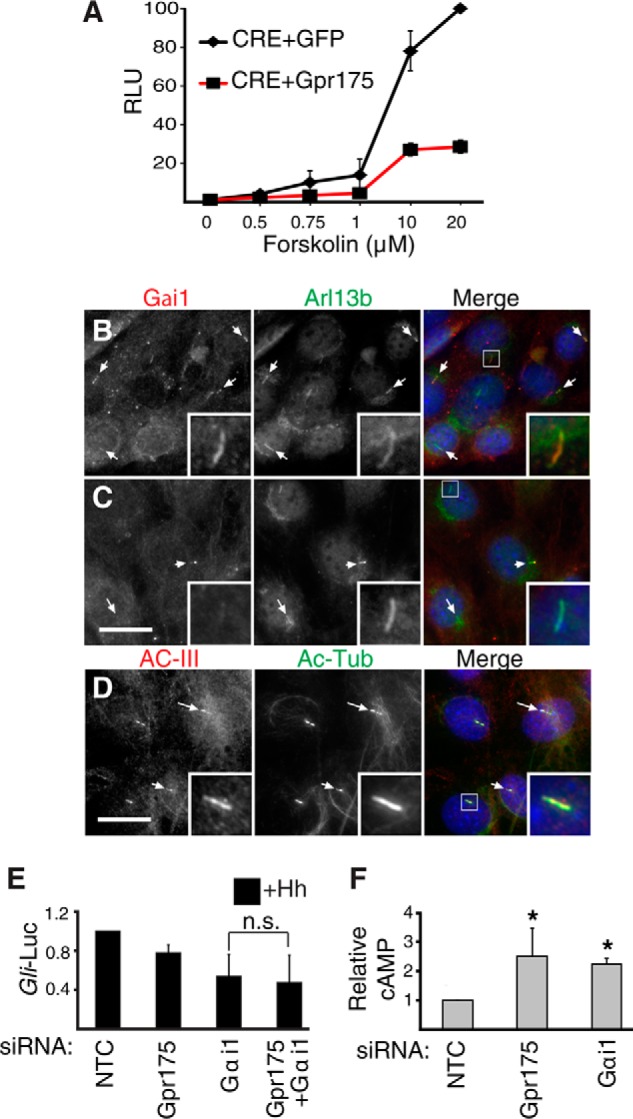
Gpr175 modulates cAMP levels via Gαi signaling. A, HEK-293T cells expressing CRE (cAMP response element)-luciferase along with a GFP (negative control, diamonds) or Gpr175 expression construct (squares, red line) were treated with varying doses of forskolin, thus increasing intracellular cAMP. The mean ± S.D. of four independent experiments normalized to the maximum tested (20 μm) Fsk in GFP-transfected cells are shown. RLU, relative luminescence units. B, S12 cells were serum starved for 24 h in the absence of Hh, fixed, and processed for immunofluorescence using mouse anti-Gαi1 antibody (red) and rabbit anti-ARL13b (green); merge with DAPI nuclear stain (blue) is shown in the right panel. C, as in B following siGαi1 transfection. D, S12 cells co-stained with rabbit anti-adenylyl cyclase-III(red), mouse anti-acetylated tubulin antibody (green), and DAPI (blue) in the merged right panel. Scale bars are 20 μm and insets show ×4 magnification of the boxed cilia. E, Gli-luciferase (Gli-Luc) levels in S12 cells following depletion of Gαi1 with or without siGpr175 in the presence of Hh (normalized to siNTC +Hh). Mean ± S.D. of 3 independent experiments are shown (n.s., not significant). F, S12 cells transfected with siNTC, siGpr175, or siGαi1 were serum starved for 24 h and then subjected to total cAMP measurement. Mean ± S.D. of three independent experiments are normalized to NTC control (*, p ≤ 0.05).
If Gpr175 indeed functions to activate Gαi1 and decrease cAMP levels, then Gpr175 or Gαi1 depletion should both increase cAMP. Indeed, total cAMP levels increased 2.5- and 2-fold upon Gpr175 and Gαi1 depletion, respectively (Fig. 6F). This increase was very similar to cells treated with Fsk to activate PKA (not shown).
Gpr175 Inhibits Gli3 Processing
In the absence of Hh, PKA initiates a phosphorylation cascade on Gli2 and Gli3 that culminates in proteasomal processing into their respective Gli repressors (19). If Gpr175 inhibits PKA activity in the presence of Hh, it should inhibit Gli3 repressor production and so Gpr175 depletion should increase Gli3-R levels. We therefore examined Gli3 in S12 cells by Western blotting. Hh stimulation drastically inhibited Gli3-R formation, but indeed less so following Gpr175 depletion (Fig. 7, A and B). Although the increase in Gli3-R appears modest, it was similar to that following Ift88 depletion, which is well established to inhibit both Gli3 activation and processing (40). The levels of Gli3-R in the absence of Hh were not significantly different between control and Gpr175-depleted cells (Fig. 7B). Although we cannot rule out that the increased processing is a feedback effect from dampened overall pathway activity, or from altered degradation of Gli3-R, our results are consistent with a role of Gpr175 in Gli3 processing via modulation of PKA activity. Consistent with Gαi1 acting in the same pathway as Gpr175, Gαi1 depletion also modestly increased Gli3 processing, albeit not as much (Fig. 7, B and D), perhaps due to the presence of other Gαi family members.
FIGURE 7.

Gpr175 knockdown modestly increases levels of Gli3 repressor. A–C, representative Western blot of endogenous Gli3 showing Gli3-FL and Gli3-R bands in S12 cells transfected for 72 h with NTC, siIft88 (positive control), or siGpr175 with and without 24 h Hh treatment. Graphs show quantitations of Gli3-R (B) and Gli3-FL (C) normalized to NTC-Hh and tubulin (1A2) loading controls from three or more independent Western blots (*, p ≤ 0.05; **, p ≤ 0.01, n.s. not significant versus respective NTC). D, as in A but with siGαi1, also quantitated in B and C. E, representative images of control and Gpr175-depleted S12 cells showing localization of Gli3 (red) at the cilia tips (distal end of acetylated tubulin signal, green) from serum-starved cells without (left panel) or with 60 min Hh treatment (right panel) and counterstained with DAPI (blue). Scale bar is 1 μm. F, quantitation of the data in E as a percentage of cilia with Gli3 accumulated at the tips (mean ± S.D. of three independent experiments of ≥100 cilia each).
Gli3-FL that escapes PKA processing upon Hh stimulation quickly accumulates at cilia tips in multiple cell types and is activated into Gli3-A (15). Because there was a similar transcription-independent increase in Gli3-FL in Gpr175 and Ift88-depleted cells (Fig. 7C and data not shown) and Gli3 accumulation at cilia tips is inhibited by Ift88 knockdown (34), we examined Gli3 transport in Gpr175-depleted cells. Gli3 significantly accumulated in ∼80% cilia tips in control cells within an hour, as previously published (15) (Fig. 7, E and F), but there was no obvious decrease in the absence of Gpr175. These data are consistent with Gpr175 regulating inhibition of Gli3 processing but not Gli3 translocation to the cilia or activation in the presence of Hh.
In summary, based on our data we propose the following working model for the role of Gpr175 in the mammalian Hh pathway (Fig. 8). In the absence of Hh ligand, Gpr175 (like Smo) is excluded from primary cilia, whereas negative regulators Ptch1 and Gpr161 (not shown) are mostly in the cilium. PKA is active and initiates Gli-R production, turning off Hh target gene transcription. In the presence of Hh, Ptch1 and Gpr161 exit, whereas Gpr175 (and Smo, SuFu/Gli2/Gli3) moves into the cilium and presumably becomes activated. There, Gpr175 along with Smo engages the activity of the constitutively ciliary heterotrimeric Gαi1 component, leading to inhibition of enzyme adenylyl cyclase (also ciliary) and hence a reduction in cAMP levels. This dampens the activity of centrosomal PKA, which represses Gli3-FL processing into Gli3-R, thereby relieving transcriptional inhibition, which (in addition to the Hh-mediated activation of Gli2, which is also augmented by PKA inactivation) enhances overall pathway activity.
FIGURE 8.
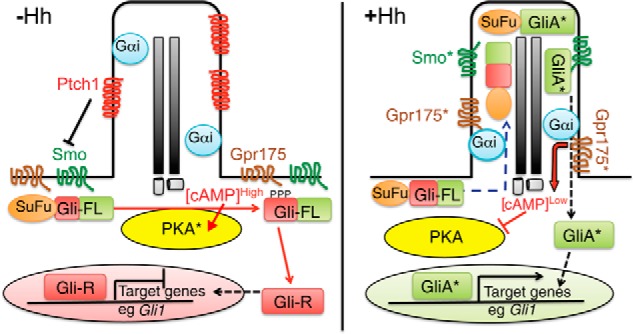
Working model for Gpr175 function as an enhancer of Hh signaling. Left panel, in the absence of Hh, Ptch1 in the primary cilium suppresses the activity of Smo and prevents its localization to cilia. Gpr175 is not present in cilia (its depiction on the plasma membrane is speculative) and so does not interact with ciliary Gαi, leading to high local concentrations of cAMP. cAMP stimulates PKA (asterisks denote active forms of all proteins) to phosphorylate full-length Gli3 (and Gli2; Gli-FL), which triggers proteasome-dependent cleavage of Gli3 into its repressor form (Gli-R). Gli3-R represses transcriptional activation of the Hh pathway target genes, thus the pathway is off. T-bars indicate inhibition; arrows indicate stimulation. Right panel, in the presence of Hh stimulation, Ptch1 is removed from cilia, allowing Smo to enter cilia and become activated. This leads to accumulation of Gpr175 in cilia, where it interacts with Gαi to inhibit local cAMP production, preventing PKA activity and Gli3 cleavage. The SuFu·Gli-FL complex accumulates at the tips of primary cilia, dissociates, and activated Gli-FL (GliA*) then exits cilia and enters the nucleus to promote transcription of Hh target genes such as Gli1. It is not clear if Gpr175 undergoes an activation step (like Smo) in addition to ciliary translocation to exert its activity.
Discussion
In this study, we show for the first time a potential role for the orphan GPCR Gpr175 in Hh signaling, demonstrating it enhances pathway activity in several mammalian cell lines. The inhibition of signaling upon Gpr175 depletion was relatively modest (only 50%), suggesting it may have a regulatory rather than an essential role. Endogenous Gpr175 localizes to cilia in a Hh-dependent manner, where it can interact with (constitutively) ciliary Gαi1 to inhibit the local production of cAMP by adenylyl cyclase. Gpr175 thus acts to inhibit PKA, a known cAMP-stimulated negative regulator of the Hh signaling pathway. In the absence of Hh, active PKA phosphorylates six conserved serine residues on Gli2 and Gli3, leading to partial processing of Gli-FL into Gli-R (7, 8, 46). In the presence of Hh, PKA activity is inhibited, thus allowing Gli-FL to accumulate and become activated following ciliary accumulation, but exactly how PKA inhibition occurs remains unclear. Although the effect of siGpr175 on Gli3 processing appeared modest, it was similar to that of siIft88, which renders cilia non-functional. Because Gpr175 and Gαi1 depletion similarly increase Gli3 processing, we propose that both proteins may act sequentially to turn off Gli3 processing to maximize Hh signaling upon pathway stimulation (47). An additional possibility is that PKA inactivation enhances Gli2 activation to maximize pathway activity, because PKA has been shown to restrain Gli2 activation (21).
Our working model is not the only possible scenario, particularly with respect to the placement of Gpr175 within the pathway, given that our epistasis attempts were performed with siRNA knockdown rather than complete knock-out cells. We propose inactivation of PKA to be its site of action, which is consistent with the well established cAMP regulation of this kinase, but our understanding of the sequence of events between PKA, SuFu, and Gli3 is still murky (16, 21). Gli3 is phosphorylated by PKA in the absence of Hh when it is complexed with SuFu, therefore it is conceivable that inhibition of PKA by Gpr175 requires prior SuFu dissociation. PKA inactivation could also destabilize SuFu to release Gli3 and promote its activation, because PKA phosphorylation has been shown to stabilize SuFu and retain it in cilia, and cilia are required for SuFu degradation (47).
Our genetic interaction experiments place Gpr175 downstream of Smo activity, which is consistent with the lack of known regulation of mammalian Smo by PKA and the fact that phosphorylation (by PKA in Drosophila (48) and casein kinase 1α/GRK2 in mammals (47, 49)) activates rather than inactivates Smo. However, it remains possible that Gpr175 is recruited to cilia by Smo, because both have similar ciliary accumulation in the absence and presence of Hh. GPCRs are well known to heterodimerize as part of their normal trafficking to attain proper surface expression and functional activity (50, 51). Because Gpr175 does not contain any known ciliary targeting motifs, it might need to dimerize with a ciliary-targeted protein to get there, although we did not investigate this. Gpr175 could alternatively translocate to cilia independently of Smo via a novel ciliary targeting motif and dimerize with it once there. However, our attempts to co-immunoprecipitate endogenous Smo with Gpr175 have been unsuccessful (data not shown), thus how Gpr175 is targeted to cilia is an area for future investigation.
The Hh dependence of Gpr175 translocation to cilia (Fig. 3), its requirement for Smo activity (Fig. 4E), and the lack of effect of Gpr175 depletion on basal signaling (−Hh) argue against a parallel role for this GPCR independent of Hh signaling. However, this does not preclude Gpr175 from also responding independently to an unknown ligand to regulate the pathway in a parallel fashion. Indeed, in embryonic neural tube explants there is evidence that PKA can modulate Gli activity independently of Hh via PI3 kinase and Akt (52).
Why Is Gpr175 Needed in Addition to Smo as a Positive Regulator of Hh Signaling?
The role of GPCRs in the Hedgehog field has long been controversial (53). Vertebrate Smo has the ability to couple to G-proteins, so it was proposed that Smo, being similar to the Frizzled family of seven pass transmembrane proteins, functions via heterotrimeric G-proteins (54). Indeed, in insect and mammalian cells, expression of Smo results in activation of several Gαi family members (55) and is as effective as a classical Gαi-coupled GPCR (the serotonin receptor) (27). Smo-mediated Hh pathway activation is dependent on Gαi and Gαi-independent mechanisms involving its C-terminal tail (52). Furthermore, studies in vertebrate cell models have highlighted the role of GPCR-associated proteins such as GRK2 and β-arrestin in Hh signaling via biochemical interaction with Smo (29, 56). Intriguingly, β-arrestin (1 and 2) and GRK2 knock-out mice are embryonic lethal, but whether these lethalities are associated with Hh signaling remains debated (53, 57, 58). Furthermore, in chick neural tube, Smo-mediated Gαi activity has been shown to play an insignificant role in Gli3 processing (59), so it is possible that other GPCRs may also be involved. Because knockdown of the orphan GPCR Gpr175 decreases Smo-mediated Hh pathway activation but does not enhance siGαi1 inhibition, we think Gpr175 likely acts between Smo and Gαi to modulate PKA activity. We thus propose that Gpr175 enhances Hh signaling by Smo via Gαi in certain contexts (such as cells needing high levels of signaling) in mammalian cells, albeit probably as a non-essential regulator (50, 51).
Gαi1 Can Modulate Hh Signaling Downstream of Gpr175
Our finding that Gαi1 can regulate Hh signaling is not unprecedented, because Smo has been shown to signal through Gαi in Drosophila (26). In vertebrates, proliferation of rat cerebellar granular neuronal precursors was strongly enhanced by overexpression of the active forms of Gαi2 and Gαi3 proteins (24), which localized to the primary cilium, and their knockdown significantly reduced Hh-induced proliferation (24). Furthermore, the Gi family was implicated downstream of Smo in C3H10T1/2 cells by the 50% inhibition in purmorphamine-stimulated Gli-luciferase induction following Gi inactivation by pertussis toxin (27, 52), a similar magnitude of effect to what we found in this cell type upon Gαi1 knockdown (Fig. 6E).
Gpr175 Function Is Distinct to That of Gpr161
Gpr161, a Gαs-coupled GPCR, was recently shown to stimulate PKA activity by increasing cellular cAMP levels via adenylate cyclase (30) in the absence of Hh (30). Gpr161 thus functions as a negative regulator to help keep the Hh pathway off via increased Gli processing, and knock-out mice are embryonic lethal with a ventralized neural tube phenotype (30). Gpr161 is found in cilia in the absence of Hh and its function is relieved when it exits cilia upon Hh stimulation. Because Gpr175 does the converse, namely entering cilia upon Hh stimulation and functioning as a positive regulator via Gαi, it may counteract Gpr161 activity to repress adenylyl cyclase and hence PKA to maximize pathway stimulation. We hypothesize that with both GPCRs, the cAMP changes are most pronounced in the cilium, ideally situated for regulating centrosomal PKA at its base.
Unlike Gpr161, however, the role of Gpr175 is likely regulatory rather than essential, consistent with its absence in Drosophila (which does not require cilia to mediate Hh signaling), because Gpr175 knock-out mice are viable with no developmental defects (data not shown). This could be explained by possible compensation in vivo, because cAMP is a crucial effector of several GPCR signaling pathways, and can thus be modulated by various Gα-coupled GPCRs, thus negating any changes in cAMP levels during development. It is also worth noting that Smo+/− heterozygotes exhibit no phenotype, suggesting that 50% loss of signaling (as seen with Gpr175 knockdown) is well tolerated (60). This supports the idea that Gpr175 has a permissive rather than instructive role in Hh signaling, in other words acts as a booster of Smo activity in cells requiring high levels of signaling rather than being an essential component. In this respect, it is interesting to note that GPR175 is up-regulated in certain Hh ligand-dependent cancers (data not shown), such as colorectal cancers that are clinically unresponsive to Smo inhibitors (61, 62). Because GPR175 depletion inhibited Hh signaling in medulloblastoma cells (Fig. 2G), it could be of interest to further study the effect of Gpr175 in cancer.
Author Contributions
J. S. performed all experiments except one, analyzed data and wrote the paper; X. W. conducted the Gli3 translocation experiment (Fig. 7, E and F). S. J. S supervised the study, analyzed data, and wrote the paper.
Acknowledgments
We are indebted to Mark Solloway, Lisa Lima, Willie Ortz, Zhiqiang Dong, and Su Guo for their help and suggestions; Mamie Yu, Zora Modrusan, Richard Bourgon, Pete Haverty, and Christiaan Klijn for deep sequencing the Daoy cells; Zahira Begum, James Ernst, and Henry Maun for octyl-Shh, and Peter Jackson for the pgLAP5 vector. We also thank the Scales, de Sauvage, and Peterson labs for all their help, reagents, and discussions.
All authors are employed by Genentech, a member of the Roche group, and hold shares in Hoffman La Roche.
- Hh
- Hedgehog
- GPCR
- G-protein coupled receptor
- qRT PCR
- quantitative RT-PCR
- GRK
- G protein-coupled receptor kinase
- Fsk
- forskolin
- SuFu
- Suppressor of Fused.
References
- 1.Cohen M. M., Jr. (2003) The hedgehog signaling network. Am. J. Med. Genet. Part A 123A, 5–28 [DOI] [PubMed] [Google Scholar]
- 2.Varjosalo M., and Taipale J. (2008) Hedgehog: functions and mechanisms. Genes Dev. 22, 2454–2472 [DOI] [PubMed] [Google Scholar]
- 3.Bale A. E., and Yu K. P. (2001) The hedgehog pathway and basal cell carcinomas. Hum. Mol. Genet. 10, 757–762 [DOI] [PubMed] [Google Scholar]
- 4.Jiang J., and Hui C. C. (2008) Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davenport J. R., and Yoder B. K. (2005) An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am. J. Physiol. Renal Physiol. 289, F1159–1169 [DOI] [PubMed] [Google Scholar]
- 6.Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., and Anderson K. V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 [DOI] [PubMed] [Google Scholar]
- 7.Wang B., and Li Y. (2006) Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. U.S.A. 103, 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan Y., Bai C. B., Joyner A. L., and Wang B. (2006) Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki H., Nishizaki Y., Hui C., Nakafuku M., and Kondoh H. (1999) Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924 [DOI] [PubMed] [Google Scholar]
- 10.Tempé D., Casas M., Karaz S., Blanchet-Tournier M. F., and Concordet J. P. (2006) Multisite protein kinase A and glycogen synthase kinase 3β phosphorylation leads to Gli3 ubiquitination by SCFβTrCP. Mol. Cell. Biol. 26, 4316–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohatgi R., Milenkovic L., and Scott M. P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- 12.Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., and Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 13.Endoh-Yamagami S., Evangelista M., Wilson D., Wen X., Theunissen J. W., Phamluong K., Davis M., Scales S. J., Solloway M. J., de Sauvage F. J., and Peterson A. S. (2009) The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 19, 1320–1326 [DOI] [PubMed] [Google Scholar]
- 14.Hsu S. H., Zhang X., Yu C., Li Z. J., Wunder J. S., Hui C. C., and Alman B. A. (2011) Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 138, 3791–3801 [DOI] [PubMed] [Google Scholar]
- 15.Wen X., Lai C. K., Evangelista M., Hongo J. A., de Sauvage F. J., and Scales S. J. (2010) Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 30, 1910–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tukachinsky H., Lopez L. V., and Salic A. (2010) A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191, 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai P., Akimaru H., Tanaka Y., Maekawa T., Nakafuku M., and Ishii S. (1999) Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 274, 8143–8152 [DOI] [PubMed] [Google Scholar]
- 18.Mo R., Freer A. M., Zinyk D. L., Crackower M. A., Michaud J., Heng H. H., Chik K. W., Shi X. M., Tsui L. C., Cheng S. H., Joyner A. L., and Hui C. (1997) Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123 [DOI] [PubMed] [Google Scholar]
- 19.Wang B., Fallon J. F., and Beachy P. A. (2000) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 [DOI] [PubMed] [Google Scholar]
- 20.Hammerschmidt M., Bitgood M. J., and McMahon A. P. (1996) Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes Dev. 10, 647–658 [DOI] [PubMed] [Google Scholar]
- 21.Tuson M., He M., and Anderson K. V. (2011) Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 138, 4921–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammerschmidt M., and McMahon A. P. (1998) The effect of pertussis toxin on zebrafish development: a possible role for inhibitory G-proteins in hedgehog signaling. Dev. Biol. 194, 166–171 [DOI] [PubMed] [Google Scholar]
- 23.DeCamp D. L., Thompson T. M., de Sauvage F. J., and Lerner M. R. (2000) Smoothened activates Gαi-mediated signaling in frog melanophores. J. Biol. Chem. 275, 26322–26327 [DOI] [PubMed] [Google Scholar]
- 24.Barzi M., Kostrz D., Menendez A., and Pons S. (2011) Sonic Hedgehog-induced proliferation requires specific Gα inhibitory proteins. J. Biol. Chem. 286, 8067–8074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He X., Zhang L., Chen Y., Remke M., Shih D., Lu F., Wang H., Deng Y., Yu Y., Xia Y., Wu X., Ramaswamy V., Hu T., Wang F., Zhou W., Burns D. K., Kim S. H., Kool M., Pfister S. M., Weinstein L. S., Pomeroy S. L., Gilbertson R. J., Rubin J. B., Hou Y., Wechsler-Reya R., Taylor M. D., and Lu Q. R. (2014) The G protein α subunit Gαs is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat. Med. 20, 1035–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogden S. K., Fei D. L., Schilling N. S., Ahmed Y. F., Hwa J., and Robbins D. J. (2008) G protein Gαi functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456, 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen F., Cheng L., Douglas A. E., Riobo N. A., and Manning D. R. (2013) Smoothened is a fully competent activator of the heterotrimeric G protein Gi. Mol. Pharmacol. 83, 691–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W., Ren X. R., Nelson C. D., Barak L. S., Chen J. K., Beachy P. A., de Sauvage F., and Lefkowitz R. J. (2004) Activity-dependent internalization of smoothened mediated by β-arrestin 2 and GRK2. Science 306, 2257–2260 [DOI] [PubMed] [Google Scholar]
- 29.Wilbanks A. M., Fralish G. B., Kirby M. L., Barak L. S., Li Y. X., and Caron M. G. (2004) β-Arrestin 2 regulates zebrafish development through the hedgehog signaling pathway. Science 306, 2264–2267 [DOI] [PubMed] [Google Scholar]
- 30.Mukhopadhyay S., Wen X., Ratti N., Loktev A., Rangell L., Scales S. J., and Jackson P. K. (2013) The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223 [DOI] [PubMed] [Google Scholar]
- 31.Ingham P. W., and McMahon A. P. (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087 [DOI] [PubMed] [Google Scholar]
- 32.Yang H., Egan J. M., Rodgers B. D., Bernier M., and Montrose-Rafizadeh C. (1999) Differential expression of a novel seven transmembrane domain protein in epididymal fat from aged and diabetic mice. Endocrinology 140, 2859–2867 [DOI] [PubMed] [Google Scholar]
- 33.Aki T., Funakoshi T., Nishida-Kitayama J., and Mizukami Y. (2008) TPRA40/GPR175 regulates early mouse embryogenesis through functional membrane transport by Sjogren's syndrome-associated protein NA14. J. Cell. Physiol. 217, 194–206 [DOI] [PubMed] [Google Scholar]
- 34.Lai C. K., Gupta N., Wen X., Rangell L., Chih B., Peterson A. S., Bazan J. F., Li L., and Scales S. J. (2011) Functional characterization of putative cilia genes by high-content analysis. Mol. Biol. Cell 22, 1104–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank-Kamenetsky M., Zhang X. M., Bottega S., Guicherit O., Wichterle H., Dudek H., Bumcrot D., Wang F. Y., Jones S., Shulok J., Rubin L. L., and Porter J. A. (2002) Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J. Biol. 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres J. Z., Miller J. J., and Jackson P. K. (2009) High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics 9, 2888–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murone M., Rosenthal A., and de Sauvage F. J. (1999) Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr. Biol. 9, 76–84 [DOI] [PubMed] [Google Scholar]
- 38.Klijn C., Durinck S., Stawiski E. W., Haverty P. M., Jiang Z., Liu H., Degenhardt J., Mayba O., Gnad F., Liu J., Pau G., Reeder J., Cao Y., Mukhyala K., Selvaraj S. K., Yu M., Zynda G. J., Brauer M. J., Wu T. D., Gentleman R. C., Manning G., Yauch R. L., Bourgon R., Stokoe D., Modrusan Z., Neve R. M., de Sauvage F. J., Settleman J., Seshagiri S., and Zhang Z. (2015) A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 33, 306–312 [DOI] [PubMed] [Google Scholar]
- 39.Xie J., Murone M., Luoh S. M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C. W., Hynes M., Goddard A., Rosenthal A., Epstein E. H. Jr., and de Sauvage F. J. (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92 [DOI] [PubMed] [Google Scholar]
- 40.Eggenschwiler J. T., and Anderson K. V. (2007) Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 23, 345–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong S. Y., and Reiter J. F. (2008) The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 85, 225–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohatgi R., Milenkovic L., Corcoran R. B., and Scott M. P. (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. U.S.A. 106, 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robarge K. D., Brunton S. A., Castanedo G. M., Cui Y., Dina M. S., Goldsmith R., Gould S. E., Guichert O., Gunzner J. L., Halladay J., Jia W., Khojasteh C., Koehler M. F., Kotkow K., La H., Lalonde R. L., Lau K., Lee L., Marshall D., Marsters J. C. Jr., Murray L. J., Qian C., Rubin L. L., Salphati L., Stanley M. S., Stibbard J. H., Sutherlin D. P., Ubhayaker S., Wang S., Wong S., and Xie M. (2009) GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 19, 5576–5581 [DOI] [PubMed] [Google Scholar]
- 44.Yue S., Chen Y., and Cheng S. Y. (2009) Hedgehog signaling promotes the degradation of tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 28, 492–499 [DOI] [PubMed] [Google Scholar]
- 45.Bishop G. A., Berbari N. F., Lewis J., and Mykytyn K. (2007) Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 505, 562–571 [DOI] [PubMed] [Google Scholar]
- 46.Niewiadomski P., Kong J. H., Ahrends R., Ma Y., Humke E. W., Khan S., Teruel M. N., Novitch B. G., and Rohatgi R. (2014) Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 6, 168–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y., Yue S., Xie L., Pu X. H., Jin T., and Cheng S. Y. (2011) Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3β regulates its stability and localization in the primary cilium. J. Biol. Chem. 286, 13502–13511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia J., Tong C., Wang B., Luo L., and Jiang J. (2004) Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050 [DOI] [PubMed] [Google Scholar]
- 49.Chen Y., Sasai N., Ma G., Yue T., Jia J., Briscoe J., and Jiang J. (2011) Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 9, e1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordan B. A., and Devi L. A. (1999) G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399, 697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prinster S. C., Hague C., and Hall R. A. (2005) Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 57, 289–298 [DOI] [PubMed] [Google Scholar]
- 52.Riobó N. A., Lu K., Ai X., Haines G. M., and Emerson C. P. Jr. (2006) Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 4505–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayers K. L., and Thérond P. P. (2010) Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 20, 287–298 [DOI] [PubMed] [Google Scholar]
- 54.Alcedo J., Ayzenzon M., Von Ohlen T., Noll M., and Hooper J. E. (1996) The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86, 221–232 [DOI] [PubMed] [Google Scholar]
- 55.Riobo N. A., Saucy B., Dilizio C., and Manning D. R. (2006) Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meloni A. R., Fralish G. B., Kelly P., Salahpour A., Chen J. K., Wechsler-Reya R. J., Lefkowitz R. J., and Caron M. G. (2006) Smoothened signal transduction is promoted by G protein-coupled receptor kinase 2. Mol. Cell. Biol. 26, 7550–7560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohout T. A., and Lefkowitz R. J. (2003) Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 63, 9–18 [DOI] [PubMed] [Google Scholar]
- 58.Kohout T. A., Lin F. S., Perry S. J., Conner D. A., and Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Low W. C., Wang C., Pan Y., Huang X. Y., Chen J. K., and Wang B. (2008) The decoupling of Smoothened from Gαi proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev. Biol. 321, 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X. M., Ramalho-Santos M., and McMahon A. P. (2001) Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 105, 781–792 [PubMed] [Google Scholar]
- 61.Berlin J., Bendell J. C., Hart L. L., Firdaus I., Gore I., Hermann R. C., Mulcahy M. F., Zalupski M. M., Mackey H. M., Yauch R. L., Graham R. A., Bray G. L., and Low J. A. (2013) A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 19, 258–267 [DOI] [PubMed] [Google Scholar]
- 62.Goldman J., Eckhardt S. G., Borad M. J., Curtis K. K., Hidalgo M., Calvo E., Ryan D. P., Wirth L. J., Parikh A., Partyka J., Faessel H., Gangolli E., Stewart S., Rosen L. S., and Bowles D. W. (2015) Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin. Cancer Res. 21, 1002–1009 [DOI] [PubMed] [Google Scholar]