

Abstract
Ribonucleotides are incorporated into the genome during DNA replication. The enzyme RNase H2 plays a critical role in targeting the removal of these ribonucleotides from DNA, and defects in RNase H2 activity are associated with both genomic instability and the human autoimmune/inflammatory disorder Aicardi-Goutières syndrome. Whether additional general DNA repair mechanisms contribute to ribonucleotide removal from DNA in human cells is not known. Because of its ability to act on a wide variety of substrates, we examined a potential role for canonical nucleotide excision repair in the removal of ribonucleotides from DNA. However, using highly sensitive dual incision/excision assays, we find that ribonucleotides are not efficiently targeted by the human nucleotide excision repair system in vitro or in cultured human cells. These results suggest that nucleotide excision repair is unlikely to play a major role in the cellular response to ribonucleotide incorporation in genomic DNA in human cells.
Keywords: autoimmune disease, autoimmunity, DNA damage response, DNA repair, DNA replication, genomic instability, inflammation, mutagenesis, nucleotide excision repair, ribonucleotides, DNA damage, innate immunity
Introduction
The high cellular level of ribonucleotides relative to deoxyribonucleotides and the lack of absolute selectivity of replicative DNA polymerases for dNTPs result in the incorporation of ribonucleotide monophosphates (rNMPs)2 into genomic DNA (1–3). In mammals, this translates into the introduction of more than 1 million rNMPs into the genome during each cell cycle (4), suggesting that rNMPs can be considered the most prevalent form of endogenous DNA “damage.” In the absence of 3′-exonucleolytic proofreading by DNA polymerases, these rNMPs can be removed from DNA through a process termed ribonucleotide excision repair, which is initiated by RNase H2-mediated incision of the phosphodiester linkage immediately 5′ to the rNMP (5). Strand displacement synthesis by DNA polymerase δ or ϵ and flap cutting by FEN1 or Exo1 then removes the ribonucleotide damage from DNA to complete the repair reaction (5).
Mutations in RNase H2 are found in patients with Aicardi-Goutières syndrome (6), a rare inflammatory condition with symptoms that overlap with the autoimmune disorder lupus erythematosus. Studies from a variety of model eukaryotic systems, including yeast (7–9), mice (4, 10), and Aicardi-Goutières syndrome patient-derived cells (11, 12), have shown that the disruption of RNase H2 results in an accumulation ribonucleotides in genomic DNA. The presence of ribonucleotides in DNA is problematic for cellular and organismal function, as evidenced by the replication stress, mutagenesis, genomic instability, and developmental defects that are observed in these experimental systems upon disruption of RNase H2 function.
In yeast strains lacking the RNase H2-mediated ribonucleotide excision repair pathway, rNMPs in DNA are targeted for removal by a Topoisomerase I-dependent process that generates a nick containing 5′-OH and cyclic 2′-3′ phosphate ends (13, 14). Because these ends cannot be ligated, additional as yet unknown factors are required to complete repair. Nonetheless, although this Topoisomerase I-mediated process helps RNase H2-deficient yeast cells to cope with rNMPs in DNA, the repair is thought to be largely mutagenic (14–18).
Canonical nucleotide excision repair (excision repair) is another potential mechanism for removing rNMPs from the genome (19). This general repair system is capable of excising a variety of bulky and non-bulky DNA base adducts and also DNA with backbone modifications through dual incisions that bracket the lesion, which releases the damaged base in the form of a 12- to 13-mer oligonucleotide in prokaryotes (20) and a 24- to 32-mer oligonucleotide in eukaryotes (21–24).
Consistent with the hypothesis that nucleotide excision repair may target rNMPs embedded in DNA, genetic disruption of excision repair in RNase H2-deficient budding yeast was shown to result in an increase in transcription-associated mutagenesis of quasi-palindromic repeats (25). Similarly, an increase in base substitution mutagenesis was observed upon disruption of excision repair function in RNase H-defective Escherichia coli strains expressing a mutant version of the translesion synthesis polymerase UmuC (pol V) that readily incorporates rNMPs into DNA (26). Employing prokaryotic excision repair factors from thermophilic eubacteria and an elevated reaction temperature, this later study also showed that the thermostable prokaryotic UvrABC system is able to make incisions on DNA duplexes containing ribonucleotides nearly as well as DNA containing a bulky fluorescein adduct (26).
However, other studies have not found a role for nucleotide excision repair in the response to rNMPs in DNA. For example, a yeast strain lacking the excision repair factor XPA/Rad14 and RNase H1/2 was found to be no more sensitive than an excision repair-proficient strain to the ribonucleotide reductase inhibitor hydroxyurea (8), which results in the increased incorporation of rNMPs into genomic DNA. Similarly, an in vitro study using cell-free extracts from yeast strains lacking various nucleases reported that loss of the excision repair factor XPF/Rad1 did not affect incision on a rNMP-containing DNA substrate (27).
To clarify the role of nucleotide excision repair in the processing of ribonucleotide-containing DNA and to extend such analyses from simple prokaryotes and yeast to more complex human cells, we examined a potential role for the human nucleotide excision repair system in the removal of rNMPs from DNA. Reconstituted biochemical reactions containing defined DNA substrates and the six core human nucleotide excision repair factors XPA, XPC, TFIIH, RPA, XPG, and XPF-ERCC1 revealed that rNMP-containing DNA is a very poor substrate for excision repair in vitro relative to standard UV photoproduct-containing substrates. Furthermore, through the use of a highly sensitive assay for measuring the dual incision reaction of excision repair in cultured human cells, we find no evidence that nucleotide excision repair targets rNMPs for removal from genomic DNA in living human cells.
In an attempt to reconcile our findings with a recent study employing recombinant proteins from thermophilic eubacteria (26), we also analyzed the activity of the canonical E. coli UvrABC system but were unable to detect significant levels of excision on rNMP-containing DNA. We conclude that rNMPs are very poorly recognized by both the human and E. coli nucleotide excision repair systems.
Experimental Procedures
Cell Culture and RNA Interference
Human A375 melanoma cells were cultured at 37 °C in a 5% CO2 humidified incubator in DMEM supplemented with 10% FBS and penicillin/streptomycin. Cells were placed under a GE germicidal lamp that emits primarily 254-nm UV light (UV-C) and that was connected to a digital timer to irradiate with the indicated fluences of UV light. Non-Targeting siRNA Pool #1 and ON-TARGETplus Human RNase 2HB siRNA SMARTpool (Dharmacon) were transfected into cells using Lipofectamine RNAiMAX (Invitrogen). Cells were transfected twice with siRNAs (20 nm) over a period of 2 days and harvested 48 h following the first transfection.
Alkaline Agarose Gel Electrophoresis
Genomic DNA was purified from siRNA-transfected cells using the QIAamp DNA mini kit (Qiagen). Genomic DNA (1 μg) was then either incubated on ice in TE (10 mm Tris-HCl, 1 mm EDTA, pH 8) or heated for 2 h at 55 °C in the presence of 0.3 m NaOH. After the addition of 6× alkaline loading buffer (300 mm NaOH, 6 mm EDTA, 18% (w/v) Ficoll (Type 4000), 0.25% (w/v) bromphenol blue, 0.25% (w/v) xylene cyanol), DNA samples were electrophoresed at 1 V/cm for 18 h on alkaline agarose gels (50 mm NaOH, 1 mm EDTA) as described previously (4, 7). Gels were neutralized in 1 m Tris-HCl, pH 8, 1.5 m NaCl for 1 h, stained with SYBR Gold (Invitrogen), and visualized using a Molecular Imager ChemiDoc XRS+ system (Bio-Rad) with a UV-C light source.
Immunoblotting
Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose, and then probed by immunoblotting using standard procedures. Primary antibodies included anti-RNase H2C (Proteintech catalogue number 16518-1-AP; 1:2000 dilution) and anti-actin (Santa Cruz Biotechnology sc-1616; 1:5000 dilution) antibodies. Secondary antibodies included horseradish peroxidase-linked anti-rabbit IgG (GE Healthcare catalogue number NA934V) and anti-goat IgG (Pierce catalogue number 31402). Chemiluminescence was visualized with Clarity Western ECL substrate (Bio-Rad) using a Molecular Imager ChemiDoc XRS+ system (Bio-Rad).
Measurement of Nucleotide Excision Repair in Cultured Cells
Cells in 6-cm plates were harvested 30 min following UV irradiation or 48 h after transfection with siRNA. Following centrifugation and washing with cold PBS, the cell pellets were resuspended in 250 μl of lysis buffer (25 mm HEPES, pH 7.9, 100 mm KCl, 12 mm MgCl2, 0.5 mm EDTA, 12.5% glycerol, 0.5% Nonidet P-40) and incubated for 10 min on ice with occasional vortexing. Following centrifugation at 16,873 × g for 30 min at 4 °C, the soluble cell lysates were transferred to a new tube. Anti-TFIIH (XPB subunit) antibody (Santa Cruz Biotechnology sc-293; 2 μg) was added to the lysates, which were then rotated for 2 h at 4 °C. After adding protein A/G PLUS-agarose (Santa Cruz Biotechnology sc-2003; 5 μl pelleted volume) to the lysates, samples were rotated again for 2 h at 4 °C. The TFIIH immunoprecipitates were pelleted by centrifugation, washed three times with lysis buffer, and then resuspended in elution buffer (50 mm Tris, pH 7.4, 200 mm NaCl, 10 mm EDTA, 0.5% SDS). A fraction (20%) of the TFIIH immunoprecipitate was taken for immunoblot analysis with anti-XPB antibody. Following the addition of proteinase K (4000 Units; New England Biolabs P8107), the immunoprecipitates were incubated for 20 min at 55 °C. Following a brief centrifugation to pellet the agarose resin, DNAs associated with TFIIH were isolated by phenol-chloroform extraction and precipitated in ethanol with glycogen (20 μg) as a carrier. TFIIH-bound DNAs were resuspended in 10 μl of water. Half of this DNA (5 μl) was 3′-end labeled in a 10-μl reaction containing 6 units of terminal deoxynucleotidyl transferase (New England Biolabs), 0.25 mm CoCl2, and 3′-[α-32P]deoxyadenosine 5′-triphosphate (cordycepin 5′-triphosphate; PerkinElmer Life Sciences) in 1× terminal deoxynucleotidyl transferase buffer (New England Biolabs) for 1 h at 37 °C. Following ethanol precipitation, the radiolabeled, TFIIH-associated excised oligonucleotides were separated by urea-PAGE and detected with a PhosphorImager. Radiolabeled oligonucleotides of known length were resolved on all gels as size markers. Excision repair activity was quantified using the ImageQuant 5.2 software (GE Healthcare) as described previously (28).
Protein Purification
Human nucleotide excision repair factors XPA (containing maltose-binding protein tag), XPC-HR23B, XPG, RPA, XPF-ERCC1, and TFIIH were purified as described previously (29, 30). A cDNA encoding the E. coli repair factor UvrA was cloned into the expression vector pET28a to generate plasmid pET28a-UvrA, which encodes a His-tagged UvrA construct. Plasmids pUNC211 and pDR3274 were used to express UvrB and UvrC, respectively (29, 30). All three E. coli repair factors were expressed in the E. coli strain DR153. UvrA was purified using nickel-nitrilotriacetic acid-agarose (Qiagen) and MonoQ column chromatography. UvrB was purified using DEAE-Sepharose and phenyl-Superose. UvrC was purified using heparin-agarose.
Excision Repair DNA Substrates
Internally 32P-labeled DNA substrates (140 bp) containing a single (6-4) UV photoproduct, cyclobutane pyrimidine dimer (CPD), or ribonucleotide were constructed by annealing and ligating six oligonucleotides as described previously (30). The 12-mer oligonucleotides (sequence 5′-AGGAATTAAGGA-3′) containing the UV photoproduct or ribonucleotide were radiolabeled with [γ-32P]ATP and polynucleotide kinase (New England Biolabs) prior to annealing and ligation with the five other oligomers. For the substrates used in Figs. 1 and 4A, the 12-mer oligomer contained an adenine ribonucleotide at position 8. Adenine or cytosine ribonucleotides were present at position 5 within the 12-mer for the substrates used in Fig. 4B. Sequences of the remaining oligonucleotides are available upon request. The Synthetic Organic Chemistry Core at the University of Texas Medical Branch (Galveston, TX) prepared the UV photoproduct-containing oligonucleotides. The ribonucleotide-containing oligonucleotides were purchased from Sigma.
FIGURE 1.

Ribonucleotide-containing DNA is a poor substrate for the human nucleotide excision repair system in vitro. A, silver- or Coomassie-stained SDS-PAGE of the human nucleotide excision repair proteins used in this study. MW, molecular weight markers. B, the indicated DNA substrates (UM, unmodified; rA, adenine ribonucleotide; CPD, cyclobutane pyrimidine dimer; (6-4)PP, (6-4) photoproduct) were incubated with or without the human nucleotide excision repair proteins (Exc. Nuc) for 90 min and then analyzed by urea-PAGE and phosphorimaging. C, quantification of repair assays shown in B. Repair of each substrate was normalized to the level of repair of the (6-4)PP-containing substrate, which was set to a value of 100. Data show the average and standard deviation from up to eight independent experiments. The level of excision of the rA-containing substrate was approximately twice that observed with the unmodified DNA substrate (p = 0.01).
FIGURE 4.

Ribonucleotide-containing DNA is a poor substrate for the E. coli nucleotide excision repair system in vitro. A, the E. coli excision repair factors were incubated with the indicated DNA substrates (UM, unmodified; rA, adenine ribonucleotide; CPD, cyclobutane pyrimidine dimer; (6-4)PP, (6-4) photoproduct) for various periods of time and then analyzed by urea-PAGE and phosphorimaging. The graph shows the quantitation of the representative experiment in A. B, the indicated substrates were incubated with or without the E. coli repair factors (Exc. Nuc.) for 1 h and then analyzed as in A. Quantitation shows the repair of each substrate normalized to the level of repair with the (6-4)PP-containing substrate, which was set to an arbitrary value of 100. Data show the average and standard deviation from five independent experiments.
Measurement of Nucleotide Excision Repair in Vitro
The in vitro excision repair assays with defined DNA substrates were performed as described previously (30) using the human and E. coli nucleotide excision repair factors with reactions taking place at either 30 °C or 37 °C, respectively.
Results
Ribonucleotide-containing DNA Is a Poor Substrate for the Human Nucleotide Excision Repair System
We previously showed that the human excision nuclease is capable of acting on a wide range of DNA substrates, including DNA mismatches, DNA with minor backbone modifications, and even undamaged DNA (19, 31, 32). However, there is significant variation in the efficiency by which this general repair system is able to target different DNA lesions (32). To determine whether the human nucleotide excision repair system is capable of detecting and excising a ribonucleotide that is embedded in DNA, we first used a reconstituted in vitro system composed of defined DNA substrates and purified excision repair proteins. The 140-bp-long DNA substrates were internally radiolabeled and included unmodified DNA (UM), DNA containing a single adenosine mononucleotide (rA), and DNA containing either a CPD or (6-4) pyrmidine-pyrimidone ((6-4))PP) UV photoproduct. All substrates were internally radiolabeled with [γ-32P]ATP to allow for detection of the 24–32-nt-long oligonucleotide products of nucleotide excision repair, which are generated by dual incision events that occur 20 ± 5 phosphodiester bonds 5′ and 6 ± 3 phosphodiester bonds 3′ to the DNA lesion (21–24). The six core excision repair proteins, XPA, XPC-HR23B, XPF-ERCC1, XPG, TFIIH, and RPA, which are shown in Fig. 1A, were purified from bacteria, baculovirus-infected insect cells, and/or mammalian cells.
To measure excision of the various lesions, we incubated the indicated DNA substrates with or without the nucleotide excision repair factors, separated the reaction products by urea-PAGE, and then used phosphorimaging to detect the canonical 24–32-nt-long oligonucleotide products of nucleotide excision repair. As shown in Fig. 1B, excision on DNA substrates containing the two UV photoproducts was readily observed. In contrast, the level of excision with the rAMP-containing DNA substrate was very weak and only slightly higher than that observed with unmodified DNA.
Quantification of excision from several individual experiments with the different substrates is shown in Fig. 1C and is normalized to the level of excision with the (6-4)PP substrate, which is a classical excision repair substrate that distorts the helical nature of DNA. As expected, CPDs, which distort the helix to a lesser extent and are recognized less efficiently by the human excision nuclease, yielded a lower level of excision. Unmodified DNA containing no known DNA damage was excised at ∼1/100th the level of the (6-4)PP, which was consistent with previous work (32). We found that the level of repair of the rAMP-containing DNA substrate was ∼2-fold higher than the unmodified DNA. These results suggest that a ribonucleotide embedded in DNA is a poor substrate for the human nucleotide excision repair system in vitro.
Nucleotide Excision Repair Does Not Efficiently Remove Ribonucleotides from Genomic DNA in Human Cells
To determine whether nucleotide excision repair contributes to the removal of rAMPs from the genome in cultured human cells, we used RNA interference to silence RNase H2, which normally initiates the process of ribonucleotide excision repair. In the absence of RNase H2 activity, more than a million ribonucleotides have been reported to be incorporated into the genome during DNA replication (4).
As shown in Fig. 2A, RNase H2 protein levels were reduced by greater than 98% in A375 human melanoma cells upon transfection with a pool of siRNAs targeting the B subunit of RNase H2. Analysis of the genomic DNA from these cells by alkaline hydrolysis and alkaline gel electrophoresis demonstrated a high level of incorporation of ribonucleotides (Fig. 2B) that was comparable with a previous study employing a genetic knock-out approach of RNase H2 in mice (4).
FIGURE 2.
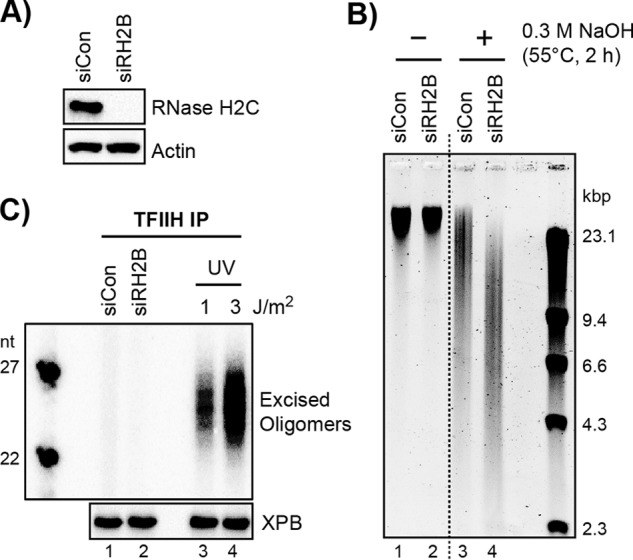
Ribonucleotide-containing DNA is a poor substrate for the human nucleotide excision repair system in cultured human cells. A, A375 cells were transfected with control siRNA (siCon) or siRNAs targeting the B subunit of RNase H2 (siRH2B). Cell lysates were prepared and analyzed by immunoblotting with antibodies against the indicated proteins. B, genomic DNA was prepared from cells transfected with control or RNase H2 siRNA, incubated or not for 2 h at 55 °C with 0.3 m NaOH, and then analyzed by alkaline agarose gel electrophoresis. The gel was neutralized and stained with SYBR Gold. The dashed line indicates that the two portions of the image were taken from non-adjacent parts of the same gel. C, cells were transfected with control or RNase H2 siRNA or were irradiated with the indicated dose of UV. TFIIH was then immunoprecipitated from the cell lysates. A portion of the immunoprecipitate (IP) was analyzed by immunoblotting against the XPB subunit of TFIIH, and the remaining material was processed for detection of the bound oligonucleotide products of nucleotide excision repair. Data show the average and standard deviation,
We next determined whether this incorporation of ribonucleotides into genomic DNA led to detectable levels of repair by the human nucleotide excision repair system in living cells. We took advantage of the fact that during nucleotide excision repair, the 24–32-nt-long excised oligonucleotide repair products are released from DNA in a stable complex with the repair factor TFIIH (33), which can be readily isolated from cultured human cells by immunoprecipitation with antibodies against TFIIH subunits and then detected by radiolabeling with terminal transferase (24, 28, 34). Thus, the presence of DNA oligonucleotides of this length in complex with TFIIH is a defining feature of the human nucleotide excision repair system and can be used to monitor excision repair in cultured cells. Moreover, we have shown that this general methodology is useful for monitoring excision repair kinetics in response to a wide variety of DNA-damaging agents that generate DNA adducts that are targeted by the nucleotide excision repair system (35).
We therefore examined whether the increased incorporation of ribonucleotides in genomic DNA in RNase H2-depleted cells led to their removal by the canonical nucleotide excision repair pathway. As a positive control, we irradiated cells with UV light. As shown in Fig. 2C (lanes 3 and 4), excised oligomers were readily observed in TFIIH immunoprecipitates from cells irradiated with low doses of UV radiation. In contrast, we did not detect any excised oligomers in TFIIH immunoprecipitates from cells transfected with control or RNase H2 siRNAs (Fig. 2C, lanes 1 and 2). These results suggest that the human nucleotide excision repair system may not efficiently excise rNMPs in living cells.
After their release from TFIIH, excised oligonucleotides associate with RPA (33), which can similarly be observed following immunoprecipitation of RPA from cell lysates and 3′-end radiolabeling of bound DNAs (24, 28). However, we did not detect RPA-bound excised oligomers in cells depleted of RNase H2 (data not shown).
We note that RNase H2 knockdown is expected to yield roughly 1.6 million ribonucleotides throughout the diploid human genome (4) and that 1 J/m2 of UV yields ∼60,000 UV photoproducts (36). Thus, although there are ∼26 times more rAMPs than UV photoproducts present in the genomes of the cells in these experiments, we did not detect any evidence for excision of ribonucleotides from human cells by canonical human nucleotide excision repair. We therefore conclude that ribonucleotides are not efficiently targeted for removal by the nucleotide excision repair system in human cells.
Ribonucleotide-containing DNA Is a Poor Substrate for the E. coli Nucleotide Excision Repair System
Using thermophilic bacterial nucleotide excision repair proteins and an elevated reaction temperature, a previous study showed robust incision activity of the bacterial excision repair system on rNMP-containing DNA (26). Because our data thus far showed that rNMP-containing DNA is at best a very weak substrate for the human nucleotide excision repair system in vitro, we decided to examine the action of the canonical E. coli excision nuclease on DNA substrates containing a ribonucleotide.
We therefore purified the E. coli nucleotide excision repair factors UvrA, UvrB, and UvrC from E. coli using conventional chromatographic approaches (Fig. 3A). To ensure that the activity of our bacterial enzyme preparation was comparable with the activity of our human repair system, we measured the activities of both repair systems on the (6-4)PP-containing DNA substrate (Fig. 1A). As shown in Fig. 3B, the E. coli UVR system was ∼1.5 to 2 times more efficient at excising the damage than the human system under our in vitro conditions. In contrast to the human system, which excises 24–32-nt-long oligomers (21–24), we note that the bacterial system generates a 12–13-nt long oligonucleotide (19, 20). We conclude that our E. coli repair factors are active at carrying out the dual incision reaction of nucleotide excision repair.
FIGURE 3.

Comparison of the activities of the human and E. coli nucleotide excision repair systems. A, Coomassie-stained SDS-PAGE of the E. coli nucleotide excision repair proteins used in this study. The repair proteins UvrA, UvrB, and UvrC, respectively, are indicated. MW, molecular weight markers. B, the E. coli and human excision repair factors were incubated with the (6-4)PP-containing substrate for the indicated periods of time and then analyzed by urea-PAGE and phosphorimaging. C, quantitation of experiments performed as described in B. Data show the average and standard deviation from up to eight independent experiments.
We next compared the activity of the E. coli repair proteins on the UV photoproduct and ribonucleotide-containing DNA substrates. As shown in Fig. 4A (lanes 9–15), the DNA substrates containing a CPD or (6-4)PP were efficiently targeted by the E. coli UvrABC proteins such that 50–70% of the damage was excised after a 90-min incubation. In contrast, we did not detect significant levels of excision of the rAMP-containing substrate under identical reaction conditions (Fig. 4A, lanes 5–8).
We next considered that a ribonucleotide embedded in DNA may be a better substrate for nucleotide excision repair if mispaired with an incorrect deoxyribonucleotide. We therefore prepared an additional DNA substrate containing a cytosine ribonucleotide opposite a thymine deoxyribonucleotide within the same sequence as with the other DNA substrates. Although we observed approximately three times more excision of the rCMP-mispaired substrate than with the rAMP-containing or unmodified DNA substrates (Fig. 4B, compare lanes 2 and 4 with lane 6), the level was barely detectable relative to the UV photoproduct-containing substrates (Fig. 4B).
In an additional attempt to detect excision of the rNMP-containing substrates with our E. coli excision repair proteins and to mimic the elevated reaction temperature that was employed with excision repair factors from thermophilic bacteria (26), we also varied the reaction temperature from 37 °C up to 55 °C. However, the higher temperature inhibited excision with our E. coli excision repair proteins (data not shown), suggesting that the proteins are sensitive to reaction temperature.
Discussion
Using a variety of model systems, including bacteria, yeast, mice, and cultured human cells, the incorporation of ribonucleotides into genomic DNA has been shown to be mutagenic and/or detrimental to cellular and organismal growth and viability (2, 4, 7, 10, 12, 14, 25). The RNase H2-mediated ribonucleotide excision repair system is thought to be the primary mechanism for removing rNMPs from DNA in both prokaryotes and eukaryotes (5). The importance of this repair system in mammals is demonstrated by the fact that RNase H2 is essential for embryonic development in mice (4, 10) and by the observation that mutations in the RNase H2 gene are seen in human patients with the autoimmune inflammatory disorder Aicardi-Goutières syndrome (6, 11, 12). Characterizing the cellular pathways that recognize and remove rNMPs from DNA therefore has the potential to provide a better understanding of a number of human pathologies and disease processes, including autoimmune disorders such as Aicardi-Goutières syndrome and lupus and also carcinogenesis and aging.
Nucleotide excision repair is a general DNA repair system that can act on virtually any DNA substrate (19, 31, 32). Genetic analyses in E. coli and yeast have led to conflicting reports regarding the role of the canonical nucleotide excision repair system in the repair of ribonucleotides that remain in DNA when RNase H2 is deleted. Although there is some evidence that loss of nucleotide excision repair capacity in simple single-celled organisms promotes mutagenesis in specific reporter genes (25, 26), another study showed that the loss of excision repair in budding yeast lacking all RNase H activity did not increase the sensitivity of the strains to hydroxyurea, a ribonucleotide reductase inhibitor that increases rNMP incorporation into genomic DNA (8). Similarly, an in vitro biochemical assay exploring a role for various yeast nucleases in targeting rNMP-containing DNA found no evidence that the excision repair nuclease XPF/Rad1 acted on ribonucleotides in DNA (27).
In contrast, a recent biochemical study showed that the bacterial UvrABC enzyme system from thermophilic bacteria can make incisions on DNA containing ribonucleotides nearly as efficiently as on DNA containing a classical bulky fluorescein adduct (26). Nonetheless, the relevance of these various genetic and biochemical studies in bacteria and yeast to higher eukaryotes including humans has not previously been examined.
We show here that the human excision repair system is unable to efficiently target ribonucleotide-containing DNA in vitro (Fig. 1). Moreover, using a highly sensitive assay for detecting nucleotide excision repair events in living cells, we similarly find no evidence for excision of ribonucleotides from genomic DNA in vivo (Fig. 2). To reconcile these findings with evidence that the thermophilic eubacterial excision nuclease is able to efficiently target rNMP-containing DNA (26), we made use of the canonical E. coli UvrABC protein system. However, we observed that a ribonucleotide in DNA is also a very poor substrate for excision by the E. coli repair factors relative to common UV photoproducts (Figs. 3 and 4). Together, our data show that the canonical human and E. coli nucleotide excision repair systems do not readily excise ribonucleotides from DNA, which suggests that the nucleotide excision repair system may not be a major mechanism for removing ribonucleotides from genomic DNA in human cells.
The difference in relative repair efficiency of rNMP-containing DNA in our study and in the previous work with the thermostable enzymes (26) may be due to a number of potential factors, including differences in substrate DNA sequence and length and the source of excision repair factors. We speculate that the ability of the UvrABC proteins from thermostable bacteria to make incisions on rNMP-containing DNA at a comparable level as on fluorescein-adducted DNA may be due to the high reaction temperature (55 °C) that was used (26) with the thermostable enzyme system. The high reaction temperature would be expected to partially melt the DNA substrate and thus allow for greater helical distortion on which the thermostable nucleotide excision repair system can act. In contrast, our reactions with the classical E. coli (37 °C) and human (30 °C) excision repair systems were performed at lower but optimal temperatures and with relatively thermolabile enzymes.
Nonetheless, it seems plausible that the nucleotide excision repair system may have the capacity to excise ribonucleotide-containing DNA under certain experimental or cellular conditions. However, under physiological conditions that are most relevant to common experimental model organisms, including cultured human cells, our work here suggests that nucleotide excision repair is unlikely to be a major mechanism for removing ribonucleotides from the genome.
Author Contributions
A. S. conceived of the idea for this project. J. H. prepared some of the DNA substrates used in the experiments. L. A. L.-B. and M. G. K carried out the experiments. M. G. K, L. A. L.-B., and A. S. analyzed the results, and M. G. K. wrote the paper.
This work was supported by National Institutes of Health Grants R01GM32833 and R21ES024425 (to A. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
- rNMP
- ribonucleotide monophosphate
- CPD
- cyclobutane pyrimidine dimer
- (6-4)PP
- (6-4) pyrimidine-pyrimidone
- TFIIH
- transcription factor II-H
- RPA
- replication protein A
- XP
- Xeroderma pigmentosum
- nt
- nucleotide
- rAMP
- ribonucleotide AMP
- rCMP
- ribonucleotide CMP.
References
- 1.Nick McElhinny S. A., Watts B. E., Kumar D., Watt D. L., Lundström E. B., Burgers P. M., Johansson E., Chabes A., and Kunkel T. A. (2010) Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U.S.A. 107, 4949–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams J. S., and Kunkel T. A. (2014) Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst.) 19, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaisman A., and Woodgate R. (2015) Redundancy in ribonucleotide excision repair: Competition, compensation, and cooperation. DNA Repair (Amst.) 29, 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reijns M. A., Rabe B., Rigby R. E., Mill P., Astell K. R., Lettice L. A., Boyle S., Leitch A., Keighren M., Kilanowski F., Devenney P. S., Sexton D., Grimes G., Holt I. J., Hill R. E., Taylor M. S., Lawson K. A., Dorin J. R., and Jackson A. P. (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sparks J. L., Chon H., Cerritelli S. M., Kunkel T. A., Johansson E., Crouch R. J., and Burgers P. M. (2012) RNase H2-initiated ribonucleotide excision repair. Mol. Cell 47, 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reijns M. A., and Jackson A. P. (2014) Ribonuclease H2 in health and disease. Biochem. Soc. Trans. 42, 717–725 [DOI] [PubMed] [Google Scholar]
- 7.Nick McElhinny S. A., Kumar D., Clark A. B., Watt D. L., Watts B. E., Lundström E. B., Johansson E., Chabes A., and Kunkel T. A. (2010) Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 6, 774–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazzaro F., Novarina D., Amara F., Watt D. L., Stone J. E., Costanzo V., Burgers P. M., Kunkel T. A., Plevani P., and Muzi-Falconi M. (2012) RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell 45, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arana M. E., Kerns R. T., Wharey L., Gerrish K. E., Bushel P. R., and Kunkel T. A. (2012) Transcriptional responses to loss of RNase H2 in Saccharomyces cerevisiae. DNA Repair (Amst.) 11, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiller B., Achleitner M., Glage S., Naumann R., Behrendt R., and Roers A. (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 209, 1419–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pizzi S., Sertic S., Orcesi S., Cereda C., Bianchi M., Jackson A. P., Lazzaro F., Plevani P., and Muzi-Falconi M. (2015) Reduction of hRNase H2 activity in Aicardi-Goutieres syndrome cells leads to replication stress and genome instability. Hum. Mol. Genet. 24, 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Günther C., Kind B., Reijns M. A., Berndt N., Martinez-Bueno M., Wolf C., Tüngler V., Chara O., Lee Y. A., Hübner N., Bicknell L., Blum S., Krug C., Schmidt F., Kretschmer S., Koss S., Astell K. R., Ramantani G., Bauerfeind A., Morris D. L., Cunninghame Graham D. S., Bubeck D., Leitch A., Ralston S. H., Blackburn E. A., Gahr M., Witte T., Vyse T. J., Melchers I., Mangold E., Nöthen M. M., Aringer M., Kuhn A., Lüthke K., Unger L., Bley A., Lorenzi A., Isaacs J. D., Alexopoulou D., Conrad K., Dahl A., Roers A., Alarcon-Riquelme M. E., Jackson A. P., and Lee-Kirsch M. A. (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Invest. 125, 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sekiguchi J., and Shuman S. (1997) Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol. Cell 1, 89–97 [DOI] [PubMed] [Google Scholar]
- 14.Kim N., Huang S. N., Williams J. S., Li Y. C., Clark A. B., Cho J. E., Kunkel T. A., Pommier Y., and Jinks-Robertson S. (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho J. E., Kim N., and Jinks-Robertson S. (2015) Topoisomerase 1-dependent deletions initiated by incision at ribonucleotides are biased to the non-transcribed strand of a highly activated reporter. Nucleic Acids Res. 43, 9306–9313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams J. S., Smith D. J., Marjavaara L., Lujan S. A., Chabes A., and Kunkel T. A. (2013) Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol. Cell 49, 1010–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams J. S., Clausen A. R., Lujan S. A., Marjavaara L., Clark A. B., Burgers P. M., Chabes A., and Kunkel T. A. (2015) Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading-strand specific. Nat. Struct. Mol. Biol. 22, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sparks J. L., and Burgers P. M. (2015) Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J. 34, 1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reardon J. T., and Sancar A. (2005) Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79, 183–235 [DOI] [PubMed] [Google Scholar]
- 20.Sancar A., and Rupp W. D. (1983) A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell 33, 249–260 [DOI] [PubMed] [Google Scholar]
- 21.Huang J. C., Svoboda D. L., Reardon J. T., and Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J. C., and Sancar A. (1994) Determination of minimum substrate size for human excinuclease. J. Biol. Chem. 269, 19034–19040 [PubMed] [Google Scholar]
- 23.Svoboda D. L., Taylor J. S., Hearst J. E., and Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 24.Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., and Sancar A. (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim N., Cho J. E., Li Y. C., and Jinks-Robertson S. (2013) RNA:DNA hybrids initiate quasi-palindrome-associated mutations in highly transcribed yeast DNA. PLoS Genet. 9, e1003924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaisman A., McDonald J. P., Huston D., Kuban W., Liu L., Van Houten B., and Woodgate R. (2013) Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet. 9, e1003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rydberg B., and Game J. (2002) Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc. Natl. Acad. Sci. U.S.A. 99, 16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kemp M. G., Gaddameedhi S., Choi J. H., Hu J., and Sancar A. (2014) DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J. Biol. Chem. 289, 26574–26583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsey-Boltz L. A., Kemp M. G., Reardon J. T., DeRocco V., Iyer R. R., Modrich P., and Sancar A. (2014) Coupling of human DNA excision repair and the DNA damage checkpoint in a defined in vitro system. J. Biol. Chem. 289, 5074–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reardon J. T., and Sancar A. (2006) Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems. Methods Enzymol. 408, 189–213 [DOI] [PubMed] [Google Scholar]
- 31.Huang J. C., Hsu D. S., Kazantsev A., and Sancar A. (1994) Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl. Acad. Sci. U.S.A. 91, 12213–12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branum M. E., Reardon J. T., and Sancar A. (2001) DNA repair excision nuclease attacks undamaged DNA: a potential source of spontaneous mutations. J. Biol. Chem. 276, 25421–25426 [DOI] [PubMed] [Google Scholar]
- 33.Kemp M. G., Reardon J. T., Lindsey-Boltz L. A., and Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi J. H., Kim S. Y., Kim S. K., Kemp M. G., and Sancar A. (2015) An integrated approach for analysis of the DNA damage response in mammalian cells: nucleotide excision repair, DNA damage checkpoint, and apoptosis. J. Biol. Chem. 290, 28812–28821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vreeswijk M. P., van Hoffen A., Westland B. E., Vrieling H., van Zeeland A. A., and Mullenders L. H. (1994) Analysis of repair of cyclobutane pyrimidine dimers and pyrimidine 6-4 pyrimidone photoproducts in transcriptionally active and inactive genes in Chinese hamster cells. J. Biol. Chem. 269, 31858–31863 [PubMed] [Google Scholar]