

Background: Because of the mobile nature of the opd island, identical opd and orf306 sequences are found among soil bacteria.
Results: In E. coli, Orf306 suppresses glycolysis and the TCA cycle and promotes up-regulation of alternate carbon catabolic operons.
Conclusion: The up-regulated hca and mhp operons contribute to PNP-dependent growth of E. coli.
Significance: Together with opd, orf306 contributes to the complete mineralization of OP residues.
Keywords: biodegradation, Escherichia coli (E. coli), gene expression, gene knock-out, metabolic regulation, microarray
Abstract
In previous studies of the organophosphate degradation gene cluster, we showed that expression of an open reading frame (orf306) present within the cluster in Escherichia coli allowed growth on p-nitrophenol (PNP) as sole carbon source. We have now shown that expression of orf306 in E. coli causes a dramatic up-regulation in genes coding for alternative carbon catabolism. The propionate, glyoxylate, and methylcitrate cycle pathway-specific enzymes are up-regulated along with hca (phenylpropionate) and mhp (hydroxyphenylpropionate) degradation operons. These hca and mhp operons play a key role in degradation of PNP, enabling E. coli to grow using it as sole carbon source. Supporting growth experiments, PNP degradation products entered central metabolic pathways and were incorporated into the carbon backbone. The protein and RNA samples isolated from E. coli (pSDP10) cells grown in 14C-labeled PNP indicated incorporation of 14C carbon, suggesting Orf306-dependent assimilation of PNP in E. coli cells.
Introduction
Bacterial phosphotriesterases (PTEs)3 are a group of structurally unrelated enzymes that cleave the triester linkage found in both organophosphate (OP) insecticides and OP nerve agents (1). Because of their broad substrate range and high catalytic efficiency, they have been exploited for detection and decontamination of OP compounds (2). The PTEs have been classified into three main groups: (i) the organophosphate hydrolases (OPHs), (ii) methyl parathion hydrolases (MPHs), and (iii) organophosphate acid anhydrases. Among the PTEs, only the organophosphate acid anhydrases have known physiological substrates: they have been shown to be dipeptidases that cleave dipeptides with a prolyl residue at the carboxyl terminus and hence are described as prolidases (3). The OP hydrolyzing activity of prolidases is considered to be an ancillary activity due to the structural similarity of OP compounds to their usual substrates (3).
The physiological substrates for OPH and MPH enzymes are unknown. These enzymes are believed to have evolved in soil bacteria to counter the toxic effects of OP insecticide residues released into agricultural soils (4, 5). Bacterial OPH enzymes, besides showing high structural similarities with the quorum-quenching lactonases, possess weak lactonase activity (6, 7). Consequently the quorum-quenching lactonases are considered to be the possible progenitors of the bacterial OPH enzymes (7). Unlike the OPH enzymes, the MPHs have no structural similarity with quorum-quenching lactonases but instead are highly similar to β-lactamases (8). The structurally diverse PTEs are therefore assumed to have evolved independently in response to OP residues accumulated in agricultural soils (9, 10).
The genetics of organophosphate degradation has attracted considerable attention among soil microbiologists. Both the OPH-encoding organophosphate degradation (opd) genes and the MPH-encoding methyl parathion degradation (mpd) genes have been shown to be part of mobile genetic elements (11–13). The lateral transfer of opd and mpd genes is evidenced by the existence of identical opd and mpd genes among taxonomically unrelated soil bacteria (14, 15). Even dissimilar indigenous plasmids found in bacteria collected from diverse geographical regions contained identical opd gene clusters (14). There are four indigenous plasmids in OP-degrading Sphingobium fuliginis ATCC 27551. Of these four plasmids, the opd containing pPDL2 has been shown to be a mobilizable plasmid within which the opd region has unique organizational features (11). Along with an operon that contributes to protocatechuate degradation, the opd gene forms part of an active transposon (11). In addition to the degradation module, pPDL2 contains genes for plasmid mobility and site-specific integration, and the plasmid has been shown to integrate site-specifically at an artificially created attachment (attB) site (11). Based on such experimental observations, the opd island carried on the mobilizable plasmid pPDL2 has been designated as an integrative mobilizable element.
A novel open reading frame (ORF), orf306, has been identified within the opd island. It is found in between the opd gene and the truncated tnpA gene of a defective transposon, Tn3 (13). A canonical catalytic triad typically seen in esterases and lipases was identified in Orf306 (16), and the esterase activity of the protein has been demonstrated using phenyl acetate as a substrate. As Orf306 shows very weak homology to the aromatic hydrolases such as TodF and CumD, we tried to evaluate its role in degradation of aromatic compounds and their meta-fission products (16, 17). More specifically, because p-nitrophenol is the lone aromatic compound generated during the OPH/MPH-mediated hydrolytic cleavage of OP insecticides such as methyl parathion, parathion, and sumithion, we attempted to find a role for Orf306 in p-nitrophenol degradation.
These studies showed unexpectedly that Escherichia coli MG1655 cells expressing Orf306 were capable of growth on p-nitrophenol as the sole carbon source. While investigating the molecular basis for this unusual phenomenon, we observed an Orf306-dependent metabolic shift in E. coli (pSDP10) cells expressing Orf306. The catabolic pathways involved in hydroxyphenylpropionate (mhp) and the phenylpropionate (hca) operons are up-regulated, and there is dramatic down-regulation of the conventional glycolysis and TCA cycles. These novel observations offer a rationale for the presence of orf306 within the opd island.
Experimental Procedures
Strains and Plasmids
The bacterial strains and plasmids used in the study are listed in Table 1, and primers used in the study are shown in Table 2. E. coli strains and S. fuliginis ATCC 27551 were grown either in LB medium or in minimal salt medium at 37 and 30 °C, respectively. When necessary, PNP was added to the culture medium as the sole source of carbon. The PNP concentration in the culture medium was determined spectrophotometrically (18). As more than 50 μm PNP is toxic to the cells, after the supplemented PNP was consumed, a fresh aliquot of PNP was added from the stock solution to keep the final concentration of PNP in the culture medium below 50 μm. Nitrite estimation in the spent medium of E. coli (pSDP10) was done following protocols described elsewhere (18).
TABLE 1.
Strains and plasmids used in this study
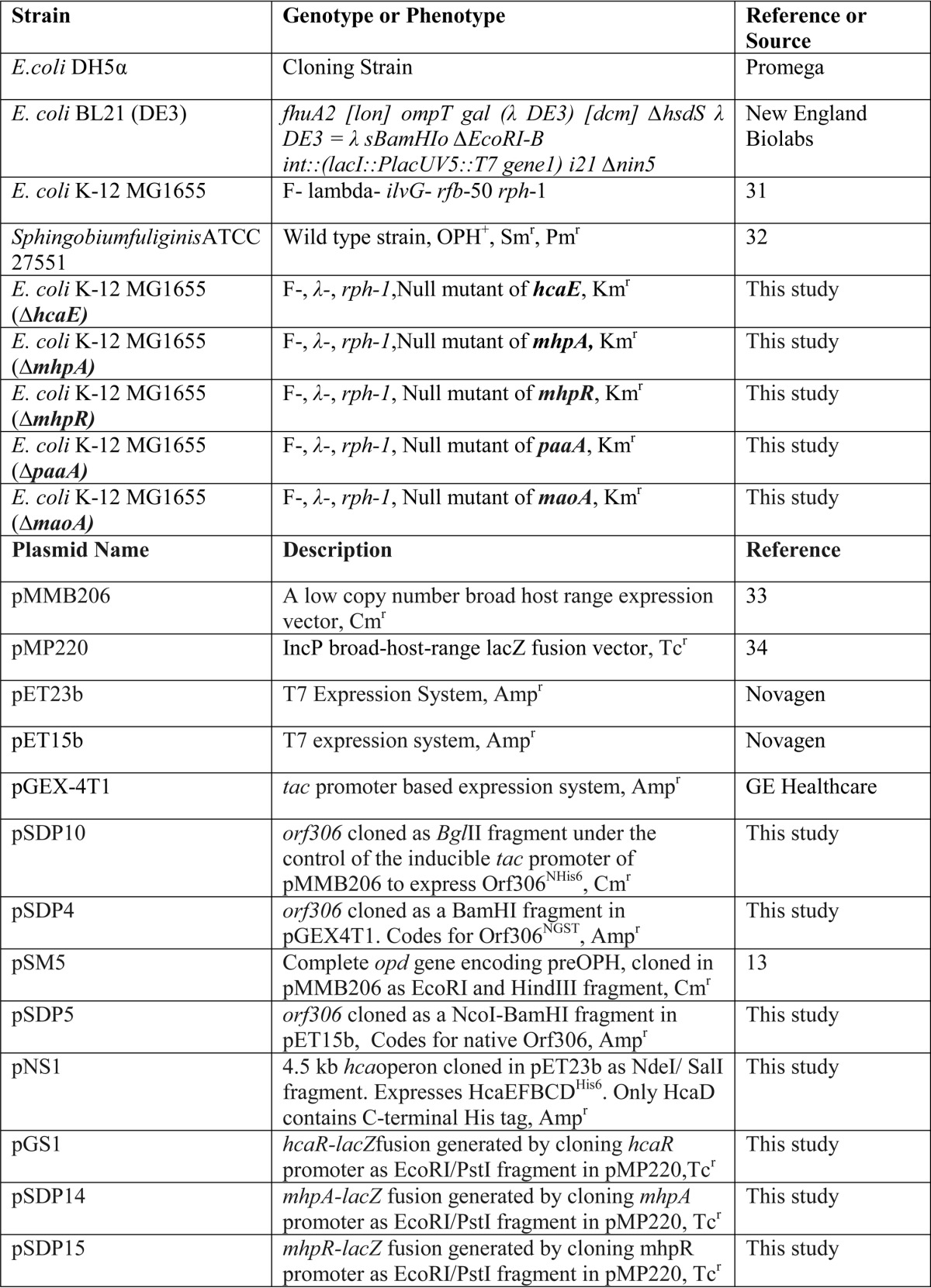
TABLE 2.
Primers used in this study
F, forward; R, reverse. The underlined nucleotides designate respective restriction sites.
| Primer name | Sequence (5′–3′) | Purpose |
|---|---|---|
| SDP13 F | ATCTTTCAGGATTAAAAAAT | hcaE knock-out |
| SDP14 R | CTCTAGTGAAACTTGCGCAC | |
| SDP15 F | GAACCGAGTCGTGAGGTACT | mhpA knock-out |
| SDP16 R | AGTGAAGATAAGCGTGCATA | |
| SDP17 F | ATTTTGTTGTTAAAAACATGTAA | mhpR knock-out |
| SDP18 R | CCACCAGAATAGCCTGCGAT | |
| SDP19 F | GCTATCGAGCCACAGGACTG | paaA knock-out |
| SDP20 R | ATTACTCATTTTGAATCTCC | |
| SDP21 F | AACATCTGACGAGGTTAATA | maoA knock-out |
| SDP22 R | CGTTTTTTTGTCTGAAACAA | |
| SDP31 F | AGTCGAACGGTAACAGGAAGA | 16S rRNA qPCR |
| SDP32 R | GCAATATTCCCCACTGCTG | |
| SDP33 F | GAAAGAGCACGATCGCGTGG | aceB qPCR |
| SDP34 R | TTGCTCGGAATAAACGCCGC | |
| SDP35 F | GAAAGAGCACGATCGCGTGG | aceK qPCR |
| SDP36 R | GCAAGCTGGCGAATAGCGTT | |
| SDP37 F | TAAGGACACGTTGCAGGCCA | acnA qPCR |
| SDP38 R | GGCGGTAGGCAATTTCACGG | |
| SDP39 F | TGGTTCCCGTGAAGCTCTGG | eno qPCR |
| SDP40 R | TGCAGCAGCTTTGGCGTTAG | |
| SDP41 F | CAATTCGCGGGGTTGTTCGT | pfkA qPCR |
| SDP42 R | GGTTTTCGATAGCCACGGCG | |
| SDP43 F | GCAGAAATGCTGGCGCGTAT | fadA qPCR |
| SDP44 R | ATGTGCCCGCCGTTACCATA | |
| SDP45 F | ACCGCTAATGAAGTGGCCGA | fadR qPCR |
| SDP46 R | CTGCACAACGCCGACAGTTT | |
| SDP47 F | TGCTCGGTATGGCACCGAAT | glcB qPCR |
| SDP48 R | GGCTTTGATCCAAGGCGTCG | |
| SDP49 F | TACTGAAGGTCGGTCAGCCG | glcC qPCR |
| SDP50 R | CCTCCAGTAATGCGCGAACG | |
| SDP51 F | CGAAGGCGGCACCGAAATTG | hcaE qPCR |
| SDP52 R | CGGGCGGTTTGTCCATCACC | |
| SDP53 F | CAGACAGCCAGACACCTTGA | hcaR qPCR |
| SDP54 R | CGGATCGGTACTGACGAAA | |
| SDP55 F | CCCGGTTGGGCTGATGATGG | mhpA qPCR |
| SDP56 R | CGGCGTAGTGTGCGGCAGAA | |
| SDP57 F | GCGGTACGGCGATAGAGGCG | mhpR qPCR |
| SDP58 R | ACCTGGCTGGCCTTTTGCCC | |
| SDP59 F | CCACCGACGAATTACGCAGC | prpB qPCR |
| SDP60 R | TTCACCTGGCTACGGGCAAA | |
| SDP61 F | TATTTTGCCCGCCACGATGC | prpR qPCR |
| SDP62 R | GCGGCATTTCGCCAATCTCA | |
| Orf306F2 | GGCTGCCATCATCCCATGGGAACACCCGCTA | Amplification of orf306 as NcoI-BamHI fragment to be cloned in pET15b |
| Orf306R3 | CGCGGATCCCTATTCCCCGTCAAGATAC | |
| Orf306F5 | GGCTGCCATCATCGGATCCGAACACCCGCTA | Amplification of orf306 as BamHI fragment using |
| Orf306R3 as reverse primer to be cloned in pGEX4T1 | ||
| HcaRFP1 | AGGAATTCAAGCTCCAGTTGGTAA | Amplification of hcaR promoter to be cloned in pMP220 |
| HcaRRP1 | ACCTGCAGACGGGTAAAGTTCAGT | |
| HcapET23bFP1 | ATACGCATATGACCACACCCTCAGATTTGAA | Amplification of hca operon as NdeI-SalI fragment to be cloned in pET23b |
| HcapET23bRP1 | ACTACGTCGACCAGTGATTTAAGCGCGATGTT | |
| MhpRF | CCGAGCTGCAGATTAATTGACATTTCTATA | Amplification of mhpR promoter to be cloned in pMP220 |
| MhpRR | CCGGAGAATTCTTCAGTACCTCACGAC |
The oxygen consumption in the resting E. coli (pSDP10) cells was estimated using a Gilson oxygraph. Extraction and separation of the PNP metabolites in the culture medium of MG1655 (pSDP10) were done using standard procedures (19). The metabolites were identified using Bruker Daltonics mass spectrometer systems. Data pertaining to the growth, nitrite estimation, and oxygen consumption are the average values of three independent experiments. The restriction and other modifying enzymes were purchased from Fermentas, India. Biochemicals used for enzyme assays were procured from Sigma-Aldrich. The 14C-labeled PNP was obtained from American Radiolabeled Chemicals, Inc., St. Louis, MO. All DNA manipulations were done following standard procedures (20).
Growth of E. coli MG1655 (pSDP10) on 14C-Labeled PNP
MG1655 (pSDP10) cells were grown to midlog phase in minimal salt medium containing glucose as the carbon source. The culture was then induced for 3 h by adding 0.5 mm IPTG. After induction, the cells were harvested and thoroughly washed with minimal salt solution. The washed cells were dissolved in fresh minimal salt solution to a final OD of 0.05. Sterile PNP was added as a carbon source to a final concentration of 50 μm. Taking total counts into consideration, an aliquot of 14C-labeled PNP was added to the culture medium to get a final count of 1000 Bq. The culture was allowed to grow until the culture reached 0.1 OD units. Once added PNP was consumed, a fresh aliquot of unlabeled and labeled PNP (1000 Bq) was added to keep the concentration of added PNP at 50 μm. Protein samples were extracted from 1 ml of culture and analyzed by SDS-PAGE along with protein samples prepared from the cultures grown using unlabeled PNP. The gel was dried, and the autoradiogram was developed following standard procedures. The total RNA isolated from the labeled and unlabeled PNP grown cultures was analyzed on an agarose gel, and the autoradiogram was developed after transferring the RNA onto a nylon membrane.
Transcriptome Analysis
Total RNA was isolated from E. coli cells using TRIzol reagent (Sigma-Aldrich) following standard procedures and stored at −80 °C in 70% (v/v) ethanol until further use. When required, the RNA was precipitated and used for RT-PCR and quantitative PCR (qPCR) experiments following standard procedures (21).
Probe Design
The Agilent Custom Gene Expression E. coli MG1655 8 × 15K array (GT_CAT_11; AMADID number 019439) designed by Genotypic Technology Pvt. Ltd., Bengaluru, India with probes having 45–60-mer oligonucleotides from coding DNA sequences was downloaded from NCBI. The 8 × 15K array comprised 15,744 features including 15,208 probes and 536 Agilent controls. All the oligonucleotides were designed and synthesized in situ according to the standard algorithms and methodologies used by Agilent Technologies for 45–60-mer in situ oligonucleotide microarray. On average, three probes were designed for 4294 coding regions, and one probe was designed for each of the 172 structural RNA sequences. The array also contains two probes for each of the 2240 non-coding regions. For non-coding regions, two probes were designed in both sense and antisense directions. Blast was performed against the coding DNA sequence databases to check the specificity of the probes. Finally, 15,208 probes were designed.
Labeling and Hybridization
E. coli MG1655 (pSDP10) cells were grown to midlog phase in minimal salt medium containing glucose as the sole carbon source. The culture was then induced by adding 0.5 mm IPTG, and the total RNA was isolated from the induced culture at 0, 1.5, and 3.0 h after induction. MG1655 (pMMB206) cells grown under similar conditions served as controls. The samples were labeled using an Agilent Quick Amp kit (catalog number 5190-0442). About 500 ng of total RNA was reverse transcribed using random hexamer primer tagged to T7 promoter sequence. cDNA thus obtained was converted to double-stranded cDNA in the same reaction. The cDNA was then converted to cRNA in an in vitro transcription step using T7 RNA polymerase enzyme and Cy3 dye added into the reaction mixture. During cRNA synthesis, Cy3 dye was incorporated into the newly synthesized cRNA strands. cRNA obtained was cleaned up using Qiagen RNeasy columns (catalog number 74106). The concentration and amount of dye incorporated were determined using a Nanodrop. Samples that passed the quality control for specific activity were taken for hybridization. 2 μg of Cy3-labeled cRNA samples were mixed and hybridized on the array using the Gene Expression Hybridization kit (catalog number 5190-0404, Agilent Technologies) in Sure hybridization chambers (Agilent Technologies) at 65 °C for 16 h. Hybridized slides were washed using Agilent Gene Expression wash buffers (catalog number 5188-5327). The hybridized, washed microarray slides were then scanned at 5-μm resolution on a G2505C scanner (Agilent Technologies).
Microarray Data Analysis
Images were quantified using Agilent Feature Extraction software. Feature extracted raw data were analyzed using Agilent GeneSpring GX software. Normalization of the data was done in GeneSpring GX using the 75th percentile shift method. Significantly up- and down-regulated genes (1-fold and above within the samples with respect to the control sample) were identified.
Quantitative Real Time PCR
Isolation of total RNA and cDNA synthesis was done as described in the aforementioned sections. When multiple gene expression analysis was performed, 16S rRNA gene was taken as an internal control for the expression calibration of other genes. DNA sequences of target genes of E. coli were retrieved from GenBankTM. The Primer3 program available online was used for primer design. For each target gene, a primer pair capable of amplifying a DNA fragment of about 150–250 bp was chosen and commercially synthesized at Sigma-Aldrich. qPCRs were conducted in an Eppendorf qPCR machine operating with Realplex 2.2 software using 30 ng of cDNA template, 0.25 μm primers, and Brilliant SYBR Green reagents (Bio-Rad). Data were normalized to 16S rRNA and analyzed by absolute quantification by comparing the Ct value of the test sample with a standard curve (21).
Absolute Quantification of Target Genes
To quantify the expression levels of a target gene, the amplicons of each target gene were cloned into vector pTZ57R/T (Thermo Scientific). Using these constructs as templates, a 10–60-fold dilution series was made, resulting in a set of standards containing 102–108 copies of the target gene. The standards and test samples were assayed in the same run. A standard curve was constructed with the logarithm of the initial copy number of the standards plotted along the x axis and their respective Ct values plotted along the y axis. Finally, the copy number of the target gene in the test sample was obtained by interpolating its Ct value against the standard curve.
E. coli Knock-outs
Phage P1 particles were prepared by infecting appropriate mutant strains of E. coli K12 BW1153. These P1 particles were then incubated with an overnight culture (2 ml) of MG1655 cells. After adding 5 mm CaCl2, the contents were incubated at 37 °C to facilitate absorption of P1 particles. The unabsorbed phage particles were removed by centrifugation, and the cell pellet was resuspended in 5 ml of LB broth containing 5 mm sodium citrate. The culture was then incubated for a further period of 45 min at 37 °C with mild shaking. After incubation, the cells were harvested and plated on an LB plate containing 5 mm sodium citrate and 30 μg/ml kanamycin. The kanamycin-resistant colonies were screened to confirm mutation by performing PCR amplification with gene-specific primers. MG1655 cells having deletions in hcaR, mhpA, mhpR, paaA, and maoA were transformed with pSDP10 and tested for their ability to grow using PNP as the sole source of carbon.
Proteomics
MG1655 cultures carrying the Orf306 expression plasmid pSDP10 or the control vector pMMB206 were grown in minimal medium containing glucose as the sole source of carbon. When the cultures reached midlog phase, they were induced for 1.5 h with IPTG (0.5 mm). The total soluble proteins extracted from these cultures were subjected to isoelectric focusing and second dimension electrophoresis (22). The proteome maps generated for both strains were compared, and the protein spots that were either up-regulated or down-regulated were identified by image analysis using ImageMaster 2D Platinum software (GE Healthcare). The differentially and differently expressed proteins were identified by MALDI-TOF/TOF Autoflex (Bruker Daltonics) and the Mascot search engine (Matrix Science) against Swiss-Prot and NCBI databases following a procedure optimized in our laboratory (22).
Reciprocal Pulldown Assays
The orf306 and opd genes were cloned in compatible plasmids to code for Orf306 and OPHHis6. E. coli BL21 carrying either pSM5 or pSM5 and pSDP5 were induced following standard procedures. Clear lysates were prepared and incubated with 100 μl of MagneHisTM (Promega) beads for 4 h. After incubation, the beads were collected and washed thoroughly with buffer containing 50 mm imidazole. Finally, protein bound to the beads was eluted by adding buffer containing 500 mm imidazole. The eluted sample was mixed with 2× Laemmli buffer and analyzed by SDS-PAGE. Similarly, OPHHis6 (pSM5) and Orf306GST (pSDP4) were expressed in E. coli BL21. The protein lysate was passed through a glutathione-Sepharose column (GE Healthcare) and after thorough washing eluted using buffer containing glutathione. The samples were analyzed by SDS-PAGE, and Western blotting was performed by probing with either anti-OPH antibodies or anti-GST antibodies. The cell lysates prepared from BL21 (pSM5 + pGEX-4T1) cells served as controls.
Promoter Assays
Both quantitative and qualitative assays were performed to assess promoter activity of hcaR, mhpR, and mhpA genes. Initially, the promoters of these genes were amplified using primer sets shown in Table 2 and cloned in the promoter test vector pMP220 to obtain plasmids pGS1, pSDP15, and pSDP14. The respective promoter-lacZ fusions were transformed into lacZ-negative strains of E. coli MG1655 and used for performing both quantitative and qualitative promoter assays. For qualitative assays, minimal medium plates were prepared by supplementing M9 medium with X-gal (40 μg/ml) and propionate (20 mm) as the sole source of carbon. E. coli MG1655 as well as cells containing vector pMP220 and promoter-lacZ fusions were plated on propionate plates before incubating them for 72 h at 37 °C. Similar cultures grown on M9 medium containing glucose as the source of carbon served as controls. The E. coli MG1655 cells with promoter-lacZ fusions and the vector were grown to midlog phase in propionate medium containing tetracycline (10 μg/ml). Cells from 1 ml of culture were harvested, and promoter activity was quantified by performing a β-galactosidase activity (23).
Cloning and Expression of hca Operon
The HcaEFBCD ORFs were amplified from the hca operon as an NdeI/SalI fragment and cloned in pET23b to generate expression plasmid pNS1. Plasmid pNS1 codes for HcaEFBCDHis6. Plasmid pNS1 was transformed into BL21 and induced following standard procedures (20). Metal affinity chromatography using a nickel column was performed to purify the HcaEFBCDHis6 complex.
Results
PNP Metabolism in E. coli (pSDP10)
E. coli has not previously been observed to use PNP as a sole source of carbon (24). We therefore conducted a series of in vitro and in vivo studies to gain better insights into this unusual process. In these studies, we used plasmid pSDP10, which expresses orf306 under the control of an inducible tac promoter.
To assess whether PNP is a direct substrate of Orf306, we performed in vitro studies by incubating purified Orf306 with PNP. The concentration of PNP in the reaction mixture remained unaltered even after prolonged incubation (12 h), suggesting that PNP is not a direct substrate of Orf306. However, when assayed for esterase activity, Orf306 was active and showed a specific activity of 0.236 nmol/mg/min. Although purified Orf306 failed to degrade PNP, when the growth properties of E. coli MG1655 (pSDP10) cells were examined, the influence of Orf306 on PNP catabolism was apparent (Fig. 1A). If PNP supports Orf306-dependent growth, its degradation products should gain entry into the carbon backbone of MG1655 (pSDP10) cells. Providing solid evidence of Orf306-dependent PNP assimilation in MG1655 (pSDP10) cells, the 14C carbon associated with 14C-labeled PNP became incorporated into proteins and RNA molecules (Fig. 1, B and C). This is direct evidence showing that the catabolic intermediates generated from PNP gain entry into central metabolic pathways and generate precursor molecules necessary for synthesis of macromolecules like proteins and RNA.
FIGURE 1.

PNP supported growth of E. coli MG1655 (pSDP10). A indicates growth of MG1655 (pSDP10), MG1655 (pMMB206), and MG1655 cells in PNP-containing minimal medium. Closed (growth) and open (PNP) circles represent growth and PNP concentration in culture medium containing MG1655 (pSDP10). Similar parameters are indicated with closed (growth) and open (PNP) squares for MG1655 (pMMB206) and closed (growth) and open (PNP) rhombuses for MG1655 cultures. Proteins extracted from MG1655 (pSDP10) cells grown in the presence of normal and 14C-labeled PNP were analyzed by SDS-PAGE, and incorporation of 14C into proteins is shown in the corresponding autoradiogram (B). RNA extracted from similarly grown cultures and the corresponding autoradiogram are shown in C. The intense sharp signal shown by the arrow indicates incorporation of 14C into 5S rRNA. D indicates a decrease in concentration of PNP and concomitant release of stoichiometric amounts of nitrite in resting cells of E. coli (pSDP10). HPLC profiles showing the time-dependent decrease of PNP in the culture medium and the appearance of nitrocatechol (4.5 min) and benzenetriol (8.3 min) are shown in E and F. AU, absorbance units; ND, not detected.
Resting cells of MG1655 (pSDP10) consumed 3 mol of oxygen for every 1 mol of p-nitrophenol degraded (data not shown), indicating involvement of Orf306-induced oxygenases in the degradation of PNP. The cells also released nitrite in a stoichiometric relationship to PNP depletion, suggesting hydroxylation of PNP at the para position (Fig. 1D). Furthermore, PNP metabolites such as nitrocatechol (4.5 min) and benzenetriol (8.3 min) were also detected in the culture medium (Fig. 1, E and F). Detection of nitrocatechol and benzenetriol indicated involvement of typical hydroxylation steps during PNP degradation in MG1655 (pSDP10) cells (19, 24). Because Orf306 showed no direct activity on PNP, it seemed possible that its influence on PNP catabolism might be through induction of novel proteins or pathways.
Phenylpropionate (PP) and Hydroxyphenylpropionate (HPP) Pathways Contribute to PNP Catabolism
To investigate the catabolome involved in PNP degradation, we performed two-dimensional electrophoresis of the proteome extracted from MG1655 (pSDP10) cells. When compared with a similar map generated from a control strain MG1655 (pMMB206) that did not express Orf306, the proteome map of these cells revealed up-regulation of several protein spots. Most notable among them were HcaR (Fig. 2B), the transcriptional activator of the phenylpropionate degradation operon (hca), and 3-hydroxyphenylpropionate oxygenase (MhpA) (Fig. 2B). These two proteins play a key role in PP and HPP catabolism (24). Up-regulation of the PP and HPP pathway enzymes suggests a possible role for the PP and HPP pathway-specific oxygenases in the oxidation of PNP in MG1655 (pSDP10) cells.
FIGURE 2.

The Orf306-dependent induction of the hca and mhp operons. A, heat map showing expression of the hca and mhp operons at 0, 1.5, and 3.0 h after induction of orf306. The portion of the two-dimensional gel showing Orf306-specific induction of HcaR and MhpA proteins is shown in B. The quantification (qPCR) of mhpA-, mhpR-, hcaE-, and hcaR-specific transcripts under similar growth conditions is shown in C with the error bars representing the S.D. Growth of the MG1655 (pDS10) (●) and hcaE (■), mhpA (▴), and mhpR (♦) mutants in PNP-containing minimal medium is shown in D. E represents agarose gel pictures indicating deletion of hcaE, mhpA, and mhpR genes. The PNP-dependent growth of MG1655 (pDS10) (●) and paaA (■) and maoA (▴) mutants (F) and an agarose gel showing the deletion of the paaA and maoA genes (G) are shown.
The pp and hpp operons are normally tightly regulated in MG1655 and are only induced in the presence of their cognate substrates (24–26). However, the presence of Orf306 induced expression of these two tightly regulated operons in MG1655 (pSDP10) cells grown in minimal medium containing glucose as the only source of carbon. Anticipating a major shift in carbon catabolism in MG1655 (pSDP10) cells, we carried out genome-wide expression profiling for MG1655 cells with and without orf306. Both strains were grown in glucose-containing minimal medium with neither PNP nor any other aromatic compound to serve as an alternative carbon source. Total RNA was extracted from these induced cultures 0, 1.5, and 3.0 h after induction and used to synthesize labeled cDNA. Analysis of the global transcription expression profiles revealed up-regulation of the hca and mhp operons in MG1655 (pSDP10) cells (Fig. 2C). Furthermore, expression of other two operons, paa and mao, involved in aromatic compound utilization remained unaltered, suggesting possible involvement of the hca and mhp operons in PNP degradation (Fig. 2A). To test this hypothesis, MG1655 null mutants were generated by deleting key genes in each of the four operons involved in aromatic compound degradation (Fig. 2, E and G). These mutants were then independently tested for growth on minimal medium containing PNP. PNP-dependent growth was not observed in hca- and mhp-null mutants of MG1655 (pSDP10) (Fig. 2D). However, PNP-dependent growth was seen in MG1655 (pSDP10) paaA- and maoA-negative mutants, providing conclusive evidence for the involvement of the hca and mhp operons in Orf306-dependent degradation of PNP (Fig. 2F).
Up-regulation of the Alternate Carbon Utilization Pathways in MG1655 (pSDP10)
The aforementioned results implicated Orf306-dependent induction of the hca and mhp operons and their involvement in PNP degradation. However, when the heat map generated from microarray data was examined, it indicated major alterations in the expression profile of other genes involved in carbon catabolism. In particular, a significant decrease was observed in the transcription of genes coding for glycolysis and TCA cycle enzymes. The decrease in the expression pattern of these pathway-specific genes started soon after induction of Orf306, and the repressive trend increased up to 3 h after induction (Fig. 3). To gain supportive evidence for the microarray data, qPCR experiments were performed to quantify expression of certain key glycolysis- and TCA cycle-specific genes. As expected, there was greater than a 3-fold reduction in the expression of phosphofructokinase (pfkA), enolase (eno), and aconitase (acnA) genes, consistent with repression of glycolysis and the TCA cycle in MG1655 (pSDP10) cells (Fig. 3).
FIGURE 3.

Orf306-induced metabolic diversion in MG1655 (pSDP10). Heat maps represent differential expression of genes encoding glycolysis, glyoxylate, and TCA cycle enzymes at 0, 1.5, and 3.0 h. The green arrows represent down-regulated pathways. The up-regulated pathways are shown with red arrows. Up-regulation of the phenylpropionate pathway and down-regulation of glycolysis are shown with dotted green and red arrows, respectively. Quantitative PCR results showing either an Orf306-dependent decrease (pfkA, eno, and acnA) or increase (glcB, glcC, and prpB) in the concentration of specific mRNAs are inserted at places showing the corresponding enzyme reactions with the error bars representing the S.D. The two-dimensional gel portions indicate Orf306-dependent induction of MhpA, HcaR, GlcC, PrpB, and SdhA. Eno, enolase; PEP, phosphoenolpyruvate.
Interestingly, the heat map also revealed a significant increase in the expression of genes coding for alternative carbon catabolic pathways. The genes coding for the propionate catabolic pathway, the methylcitrate cycle, and glyoxylate pathway showed significant induction in the presence of Orf306. The qPCR performed for glcC and glcB supported the microarray findings and is consistent with up-regulation of the glyoxylate pathway (Fig. 3). A similar trend was seen in the proteomics data. The two-dimensional gels clearly indicated Orf306-dependent up-regulation of methylisocitrate lyase (PrpB) and the transcription factor GlcC in MG1655 (pSDP10) cells (Fig. 3). The transcription activator GlcC activates the glc operon and contributes to induction of glycolate oxidase (GlcD, -E, and -F) and malate synthase G (GlcB), providing conclusive evidence for the up-regulation of the glyoxylate pathway. Taken together with up-regulation of PP- and HPP-specific enzymes, this shift in carbon catabolic pathways is very significant because the end products generated from PNP degradation such as succinyl-CoA and acetyl-CoA gain direct entry into the TCA and glyoxylate pathways.
Orf306 contains a lipase/esterase domain, and its esterase activity is apparent when assayed following standard procedures (27). E. coli cells have been shown to use endogenous fatty acids as a source of carbon in an FadD-dependent manner (28). In such cells, as seen in the present study, glyoxylate pathway enzymes are up-regulated (29). One possible hypothesis was that the lipase activity of Orf306 in E. coli could generate odd chain fatty acids that when oxidized to propanoyl-CoA could lead to induction of the propionate catabolic operon. If this hypothesis is correct the propionate should also serve as a signaling molecule for induction of the hca and mhp operons that have been shown to play a key role in PNP degradation. The hpp and mhp operons are positively regulated by transcription factors HcaR and MhpR (26). To examine the link between propionate generation and the induction of the pp and hpp operons, we assayed promoter activity of hcaR, mhpR, and mhpA genes in the presence of propionate. Interestingly, propionate induced transcription of both the hcaR and mhpR genes. However, the propionate-dependent activity of the hcaR promoter was 5-fold higher than that of the mhpR promoter (Fig. 4, A and B). Nonetheless, when grown in glucose, such induction of the hcaR and mhpR genes was seen only in IPTG-induced cells of MG1655 (pDS10) expressing orf306 (Fig. 4E) and not in an orf306-negative background (Fig. 4, C and D). The HcaR concentration in E. coli is negligible in early log phase and midlog phase cultures. Its concentration dramatically increases during stationary phase in an RpoS-independent manner (25). To rule out the possibility of RpoS involvement in hcaR induction, we assayed promoter activities of hcaR and mhpR in early log phase cultures (25). The data obtained through quantitative and qualitative assays clearly indicated propionate-dependent induction of hcaR and mhpR, suggesting that the propionate generated due to the esterase/lipase activity of Orf306 is responsible for the induction of hcaR and mhpR.
FIGURE 4.

Propionate-dependent induction of hcaR and mhpR genes. The lacZ-negative strains of MG1655 containing promoter test vector pMP220 and hcaR-, mhpR-, and mhpA-lacZ fusions were grown either on a propionate + X-gal (A) or a glucose + X-gal plate (B). The quantification of promoter activity for propionate- and glucose-grown cultures is shown in C and D, respectively, with the error bars representing the S.D. The Orf306-dependent induction of the hcaR, mhpR, and mhpA genes in MG1655 (pSDP10) cells is shown in E and F, respectively.
Genetic evidence gathered in this study clearly suggested the requirement of both hca and mhpA operons for degradation of PNP (Fig. 2). Because propionate induces expression of HcaR, the transcriptional activator of the hca operon, we designed further experiments to gain biochemical evidence linking hca operon and PNP degradation. The products of the hca operon convert PP to dihydroxyphenylpropionate. The HcaEFCD complex, otherwise known as PP dioxygenase, converts PP to PP dihydrodiol (24, 25). PP-dihydrodiol dehydrogenase, the product of hcaB, converts PP dihydrodiol to dihydroxyphenylpropionate. Hydroxylation is the first step in biodegradation of PNP. Assuming that the hca operon has a role in hydroxylation of PNP, we attempted to conduct in vitro studies using purified HcaEFBCD complex. Our attempts to obtain pure HcaEFBCDHis6complex from the induced BL21 (pNS1) cells gave no positive results. The cell lysate when analyzed by SDS-PAGE showed the existence of all other subunits except HcaC and HcaDHis6 (data not shown). To gain further insights into these unexpected results, the gel was used to perform immunoblotting using anti-His antibodies to detect HcaDHis6. Surprisingly, we obtained signal at a position far below the predicted mass (43 kDa) of HcaDHis6, suggesting degradation of HcaDHis6 in BL21 (pNS1) cells. Changes in induction conditions such as IPTG concentration gave no positive results. In the absence of HcaDHis6, it was not possible to obtain pure HcaEFBCDHis6 complex. Therefore, we failed to gain biochemical support for our genetic evidence that suggests involvement of hca operon in degradation of PNP.
In the sequence of plasmid pPDL2, we noticed a lig operon coding for a 4,5-dioxygenase along with an ORF that codes for a LysR-type transcription factor. These two genes are located in the upstream region along with the opd gene and orf306, and all of them are part of the opd island (11). It is not known whether there is a link between Orf306 and expression of this dioxygenase and transcription factor. The existence of all these genes as part of the opd island and the considerable sequence homology between HcaR and the opd island-borne LysR homologue may be significant with respect to the mechanism underlying esterase-dependent activation of hcaR.
Having established the influence of Orf306 on p-nitrophenol catabolism in E. coli and observed a related Orf306-dependent shift in carbon metabolism, we conducted further experiments to examine the expression and subcellular localization of Orf306 in S. fuliginis ATCC. Membrane-associated OPH, the product of opd gene, initiates degradation by cleaving the triester linkage found in organophosphates. The genetically linked opd and orf306 are flanked by mobile elements IS21 and Tn3 to facilitate their lateral transfer (13); hence, we were interested in their potential co-localization within the cell.
The Orf306-specific signal was observed in protein extracts prepared form S. fuliginis ATCC 27551, and on closer examination of the Western blot, the protein appeared to be a doublet. The two forms of Orf306 show an ∼2-kDa size difference and suggest the possibility of posttranslational modification of Orf306 when expressed in S. fuliginis ATCC 27551. Studies of the subcellular localization of these two forms of Orf306 showed the modified form to be exclusively found in the membrane, whereas the unmodified version was only seen in the cytoplasm (Fig. 5, A and B). Hence, both OPH and the presumptively modified form of Orf306 are membrane-associated, and assuming that this may have functional relevance, we conducted experiments to ascertain whether there are any physical interactions between these two proteins. The reciprocal pulldown experiments were performed by co-expressing these two proteins in E. coli with two different affinity tags. The pulldown experiments gave a clear indication of possible interactions between these two proteins (Fig. 5, C, D, E, and F). If these interactions are seen together with Orf306-induced metabolic reprogramming in E. coli it points toward the existence of an OPH-Orf306-mediated signaling mechanism in S. fuliginis ATCC 27551. The existence of hcaR and hcaA functional homologues lysR and lig as part of the opd island and Orf306-dependent induction of these genes in E. coli add strength to this proposition (11).
FIGURE 5.

Expression and subcellular localization of Orf306 in S. fuliginis ATCC 27551. The proteins extracted from whole cells (W), membrane (M), and cytoplasm (C) were analyzed by SDS-PAGE (A). PM, protein molecular weight marker. The corresponding Western blot developed using Orf306-specific antibodies is shown in B. Arrows indicate the existence of Orf306 in two different forms. The asterisk (*) indicates the exclusive presence of posttranslationally modified Orf306 in the membrane fraction. Reciprocal pulldown assays performed to show OPH and Orf306 interactions are shown in C–F. C indicates proteins extracted from BL21 (pSM5) and BL21 (pSM5 + pSDP5) loaded in lanes 1 and 2. The corresponding pulldown samples are loaded in lanes 3 and 4. Co-elution of Orf306 along with OPHHis6 is shown with an arrow (lane 4). D, lanes 1 and 3 represent protein extracts prepared either from BL21 (pSM5 + pSDP4) or BL21 (pGEX4T1 + pSM5) as input. Lane 3 and 4 represent proteins in pulldown samples. The Western blots developed with either OPH-specific antibodies or GST-specific antibodies are shown in E and F, respectively.
Discussion
Organophosphates were introduced as pest control agents to replace the most persistent organochloride insecticides, and they are now the most predominant insecticides used in world agriculture. Although they are less persistent in the environment, their degradation products such as PNP are highly persistent and toxic to soil microbes. Therefore, metabolic diversion towards use of compounds like PNP as a source of carbon, as we observed when Orf306 was expressed in E. coli, is advantageous and can contribute to organismal fitness. We set out to establish a link between Orf306 and the induction of alternate carbon catabolic pathways, and this study has revealed novel regulatory mechanisms hitherto unknown in E. coli. Interestingly, our investigations have revealed propionate-dependent induction of the transcription factors HcaR and MhpR. Considerable attempts have been made to understand regulation of hcaR and mhpR expression (25), and none of them have shown propionate as an inducer of the hcaR and mhpR genes. Although the mechanistic details are still unclear, our studies clearly demonstrate the existence of a positive influence of propionate on the expression of these two genes. Apparently, these transcription factors contribute to the up-regulation of the hca (phenylpropionate) and mhp (hydroxyphenylpropionate) degradation operons (24). The results presented in this study clearly demonstrate involvement of these two operons in degradation of PNP. If propionate-dependent induction of the hca and mhp operons is considered together with the esterase/lipase activity of Orf306, the endogenous propionate generated in E. coli (pSDP10) cells appears to be the precise link between Orf306 and the induction of these two operons. The existence of a PNP monooxygenase activity in HcaEFCD-expressing E. coli cells provides conclusive evidence to show a link between ORf306 expression and utilization of PNP as a carbon source by E. coli (pSDP10) cells.
In the opd island on plasmid pPDL2, orf306, which codes for the esterase, is located between the opd gene and the tnpA gene, which codes for a truncated transposase of a defective transposon, Tn3. There is no detectable terminator motif between orf306 and tnpA, which originally led us to deduce that these two genes are co-transcriptional. However, RT-PCR and promoter assays have clearly shown that orf306 is an independent transcriptional unit (data not shown). The dissimilar opd plasmids pCMS1 and pPDL2 are mobilizable in nature and possess absolute sequence identity in the DNA region containing the opd gene, the IS element IS21, and orf306. However, in pCMS1, the sequence coding orf306 is extended to give an ORF coding for a 345-amino acid carboxyesterase that appears to form an operon along with an ORF coding for amidase (30). In pPDL2, insertion of Tn3 in orf345 appears to have created orf306 and generated the genetic organization seen in this plasmid. That organization, which resembles a complex transposon, suggests possible reasons for the existence of identical opd regions in taxonomically diverse microbes (30).
Orf306-dependent PNP catabolism has a significant advantage for soil microbes. PNP is the lone aromatic compound generated during PTE-mediated hydrolysis of OP insecticides. Bacterial strains having exclusive PNP degradation pathways are known both in Gram-positive and Gram-negative soil bacteria (18, 19). However, most of them fail to show PTE activity. Unless PNP degradation capability is laterally transferred along with PTE coding sequences its contribution to the total elimination of OP residues is compromised. The existence of orf306 as part of a PTE-coding integrative mobilizable element is therefore advantageous to soil microbes as it enables the recipient cells to use OP residues as a source of carbon.
Author Contributions
D. C., R. G., A. K. M., and A. N. conducted the experiments. E. V. P. P. and S. P. analyzed the microarray data. D. S. designed the experiments and prepared the manuscript.
Acknowledgments
The Department of Animal Biology is a Department of Science and Technology-Fund for Improvement of Science and Technology Infrastructure in Higher Educational Institutions supported Department. The School of Life Sciences, University of Hyderabad is supported by the Department of Biotechnology-Centre for Education and Research in Biology and Biotechnology and University Grants Commission-Center of Advanced Study. Funding provided by CSIR, DST, and DBT to the D. S. laboratory is respectfully acknowledged. We thank Dr. M. J. Merrick for critical reading of the manuscript and Dr. Manjula Reddy for useful suggestions and providing E. coli mutants. Genotypic Technology Pvt. Ltd. is acknowledged for help in transcriptome data analysis.
This work was supported by Indian Council of Medical Research and Council for Scientific and Industrial Research (CSIR) research fellowships (to D. C., R. G., and A. K. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- PTE
- phosphotriesterase
- PNP
- p-nitrophenol
- OP
- organophosphate
- orf306
- open reading frame 306
- OPH
- organophosphate hydrolase
- MPH
- methyl parathion hydrolase
- opd
- organophosphate degradation gene
- mpd
- methyl parathion degradation gene
- TCA
- tricarboxylic acid
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- qPCR
- quantitative PCR
- PP
- phenylpropionate
- HPP
- hydroxyphenylpropionate
- Tn
- transposon
- IS
- insertion element.
References
- 1.Singh B. K., and Walker A. (2006) Microbial degradation of organophosphorus compounds. FEMS Microbiol. Rev. 30, 428–471 [DOI] [PubMed] [Google Scholar]
- 2.Singh B. K. (2009) Organophosphorus-degrading bacteria: ecology and industrial applications. Nat. Rev. Microbiol. 7, 156–164 [DOI] [PubMed] [Google Scholar]
- 3.Štěpánková A., Dušková J., Skálová T., Hašek J., Koval' T., Østergaard L. H., and Dohnálek J. (2013) Organophosphorus acid anhydrolase from Alteromonas macleodii: structural study and functional relationship to prolidases. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 346–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar-Rogovsky H., Hugenmatter A., and Tawfik D. S. (2013) The evolutionary origins of detoxifying enzymes: the mammalian serum paraoxonases (PONs) relate to bacterial homoserine lactonases. J. Biol. Chem. 288, 23914–23927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roodveldt C., and Tawfik D. S. (2005) Shared promiscuous activities and evolutionary features in various members of the amidohydrolase superfamily. Biochemistry 44, 12728–12736 [DOI] [PubMed] [Google Scholar]
- 6.Afriat L., Roodveldt C., Manco G., and Tawfik D. S. (2006) The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry 45, 13677–13686 [DOI] [PubMed] [Google Scholar]
- 7.Merone L., Mandrich L., Rossi M., and Manco G. (2005) A thermostable phosphotriesterase from the archaeon Sulfolobus solfataricus: cloning, overexpression and properties. Extremophiles 9, 297–305 [DOI] [PubMed] [Google Scholar]
- 8.Garau G., Di Guilmi A. M., and Hall B. G. (2005) Structure-based phylogeny of the metallo-β-lactamases. Antimicrob. Agents Chemother. 49, 2778–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Afriat-Jurnou L., Jackson C. J., and Tawfik D. S. (2012) Reconstructing a missing link in the evolution of a recently diverged phosphotriesterase by active-site loop remodeling. Biochemistry 51, 6047–6055 [DOI] [PubMed] [Google Scholar]
- 10.Copley S. D. (2009) Evolution of efficient pathways for degradation of anthropogenic chemicals. Nat. Chem. Biol. 5, 559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandeeti E. V., Longkumer T., Chakka D., Muthyala V. R., Parthasarathy S., Madugundu A. K., Ghanta S., Medipally S. R., Pantula S. C., Yekkala H., and Siddavattam D. (2012) Multiple mechanisms contribute to lateral transfer of an organophosphate degradation (opd) island in Sphingobium fuliginis ATCC 27551. G3 2, 1541–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei M., Zhang J. J., Liu H., Wang S. J., Fu H., and Zhou N. Y. (2009) A transposable class I composite transposon carrying mph (methyl parathion hydrolase) from Pseudomonas sp. strain WBC-3. FEMS Microbiol. Lett. 292, 85–91 [DOI] [PubMed] [Google Scholar]
- 13.Siddavattam D., Khajamohiddin S., Manavathi B., Pakala S. B., and Merrick M. (2003) Transposon-like organization of the plasmid-borne organophosphate degradation (opd) gene cluster found in Flavobacterium sp. Appl. Environ. Microbiol. 69, 2533–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulbry W. W., Kearney P. C., Nelson J. O., and Karns J. S. (1987) Physical comparison of parathion hydrolase plasmids from Pseudomonas diminuta and Flavobacterium sp. Plasmid 18, 173–177 [DOI] [PubMed] [Google Scholar]
- 15.Zhang R., Cui Z., Zhang X., Jiang J., Gu J. D., and Li S. (2006) Cloning of the organophosphorus pesticide hydrolase gene clusters of seven degradative bacteria isolated from a methyl parathion contaminated site and evidence of their horizontal gene transfer. Biodegradation 17, 465–472 [DOI] [PubMed] [Google Scholar]
- 16.Khajamohiddin S., Babu P. S., Chakka D., Merrick M., Bhaduri A., Sowdhamini R., and Siddavattam D. (2006) A novel meta-cleavage product hydrolase from Flavobacterium sp. ATCC27551. Biochem. Biophys. Res. Commun. 351, 675–681 [DOI] [PubMed] [Google Scholar]
- 17.Khajamohiddin S., Repalle E. R., Pinjari A. B., Merrick M., and Siddavattam D. (2008) Biodegradation of aromatic compounds: an overview of meta-fission product hydrolases. Crit. Rev. Microbiol. 34, 13–31 [DOI] [PubMed] [Google Scholar]
- 18.Kadiyala V., and Spain J. C. (1998) A two-component monooxygenase catalyzes both the hydroxylation of p-nitrophenol and the oxidative release of nitrite from 4-nitrocatechol in Bacillus sphaericus JS905. Appl. Environ. Microbiol. 64, 2479–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pakala S. B., Gorla P., Pinjari A. B., Krovidi R. K., Baru R., Yanamandra M., Merrick M., and Siddavattam D. (2007) Biodegradation of methyl parathion and p-nitrophenol: evidence for the presence of a p-nitrophenol 2-hydroxylase in a Gram-negative Serratia sp. strain DS001. Appl. Microbiol. Biotechnol. 73, 1452–1462 [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J., and Russell David W. (1989) Molecular Cloning: A Laboratory Manual, Vol. 3, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21.Longkumer T., Parthasarathy S., Vemuri S. G., and Siddavattam D. (2014) OxyR-dependent expression of a novel glutathione S-transferase (Abgst01) gene in Acinetobacter baumannii DS002 and its role in biotransformation of organophosphate insecticides. Microbiology 160, 102–112 [DOI] [PubMed] [Google Scholar]
- 22.Pandeeti E. V., Chinnaboina M. R., and Siddavattam D. (2009) Benzoate-mediated changes on expression profile of soluble proteins in Serratia sp. DS001. Lett. Appl. Microbiol. 48, 566–571 [DOI] [PubMed] [Google Scholar]
- 23.Miller J. H. (1972) Experiments in Molecular Genetics, p. 352, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24.Díaz E., Ferrández A., Prieto M. A., and García J. L. (2001) Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 65, 523–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turlin E., Perrotte-piquemal M., Danchin A., and Biville F. (2001) Regulation of the early steps of 3-phenylpropionate catabolism in Escherichia coli. J. Mol. Microbiol. Biotechnol. 3, 127–133 [PubMed] [Google Scholar]
- 26.Manso I., Torres B., Andreu J. M., Menéndez M., Rivas G., Alfonso C., Díaz E., García J. L., and Galán B. (2009) 3-Hydroxyphenylpropionate and phenylpropionate are synergistic activators of the MhpR transcriptional regulator from Escherichia coli. J. Biol. Chem. 284, 21218–21228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huggins C., and Lapides C. (1947) Chromogenic substrates: IV. acyl esterases of p-nitrophenol as substrates for the colorimetric determination of esterase. J. Biol. Chem. 170, 467–482 [Google Scholar]
- 28.Pech-Canul Á., Nogales J., Miranda-Molina A., Álvarez L., Geiger O., Soto M. J., and López-Lara I. M. (2011) FadD is required for utilization of endogenous fatty acids released from membrane lipids. J. Bacteriol. 193, 6295–6304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walsh K., and Koshland D. E. Jr. (1984) Determination of flux through a branch point of two metabolic cycles. The tricarboxylic acid cycle and the glyoxylate shunt. J. Biol. Chem. 259, 9646–9654 [PubMed] [Google Scholar]
- 30.Pandeeti E. V., Chakka D., Pandey J. P., and Siddavattam D. (2011) Indigenous organophosphate-degrading (opd) plasmid pCMS1 of Brevundimonas diminuta is self-transmissible and plays a key role in horizontal mobility of the opd gene. Plasmid 65, 226–231 [DOI] [PubMed] [Google Scholar]
- 31.Blattner F. R., Plunkett G. 3rd, Bloch C. A., Perna N. T., Burland V., Riley M., Collado-Vides J., Glasner J. D., Rode C. K., Mayhew G. F., Gregor J., Davis N. W., Kirkpatrick H. A., Goeden M. A., Rose D. J., Mau B., and Shao Y. (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 [DOI] [PubMed] [Google Scholar]
- 32.Kawahara K, Tanaka A, Yoon J, and Yokota A (2010) Reclassification of a parathione-degrading Flavobacterium sp. ATCC 27551 as Sphingobium fuliginis. J. Gen. Appl. Microbiol. 56, 249–255 [DOI] [PubMed] [Google Scholar]
- 33.Morales V. M., Bäckman A., and Bagdasarian M. (1991) A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97, 39–47 [DOI] [PubMed] [Google Scholar]
- 34.Spaink H. P., Okker R. J., Wijffelman C. A., Pees E., and Lugtenberg B. J. (1987) Promoters in the nodulation region of the Rhizobium leguminosarum Sym plasmid pRL1JI. Plant Mol. Biol. 9, 27–39 [DOI] [PubMed] [Google Scholar]